
コンテンツ
ouncebiotech.substack.com/p/are-we-getting-closer-to-curing-alzheimers
Are we getting closer to curing Alzheimer’s Disease?
2023/02/21
2023年1月6日、FDAはアルツハイマー病(AD)の新薬レケンビ(旧名レカネマブ)を承認した。これは、症状を一時的に和らげるのではなく、病態の生物学的根源をターゲットとする、FDAが承認した2番目のAD治療薬である。レケンビは現在、エーザイによって商品化されており、価格は26,500ドル (370万円)1年間の治療でである。 アメリカ人の約6百万人がADに苦しんでいる。この薬はブロックバスターになる可能性を秘めているが、メディケアによる償還決定が今年後半になるため、エーザイは現在のところ売上予測を控えめにしている。
レカネマブは、AD患者の脳内で凝集するタンパク質であるβアミロイド(Aβ)を標的とするモノクローナル抗体(mAb)である。ADの治療薬ではないが、レカエンビは認知機能の低下や記憶喪失を遅らせることにより、早期AD患者の時間を稼ぐことができる。レッケンビの成功はAβ説を支持する証拠となったが、Aβ標的薬の臨床試験での度重なる失敗の後、この分野はAβ説から離れつつある。
ADの治療に近づいているということだろうか?
この疑問に光を当てるため、AD治療開発について掘り下げてみたい。この投稿では、βアミロイド仮説と、レカネマブがAβ標的治療薬として今のところ最高である理由に焦点を当てる。次回は、代替経路と創薬ターゲット、有望な治療薬、そして神経変性創薬を促進するために現在のテックバイオとデジタルバイオプラットフォームをどのように活用できるかについて述べる。
ベータアミロイド説
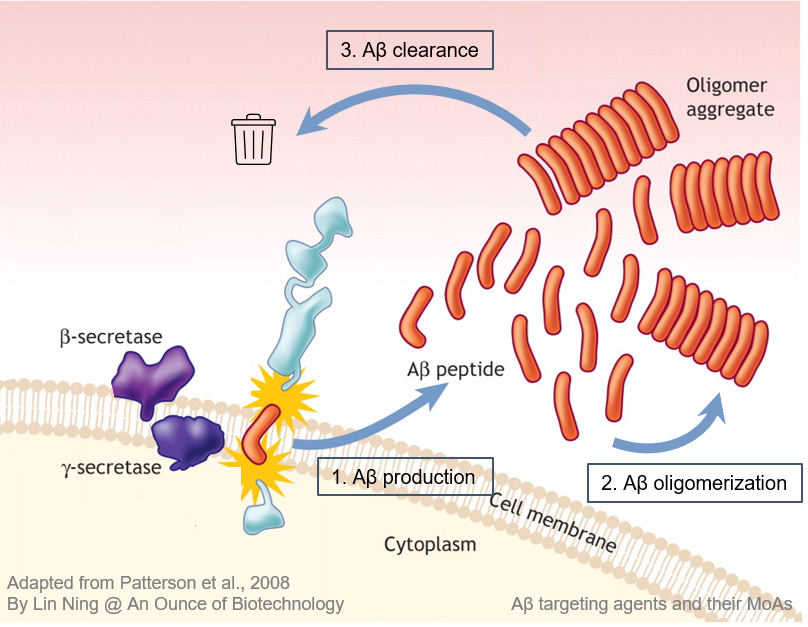
ADが同定されて1世紀以上経つが、この病気を引き起こす生物学的メカニズムはいまだに不明である。Aβ仮説は、神経病理学的および遺伝学的データによって十分に支持されており、最も有力な説となっている。
Aβはアミロイド前駆体タンパク質(APP)由来のペプチドであり、神経細胞表面の受容体としてシナプス形成や神経細胞の可塑性を制御している。βセクレターゼやγセクレターゼなどの細胞酵素は、APPの複数の切断部位を認識し、APPを様々な長さのAβペプチドに消化する。Aβモノマーは、その産生とクリアランスが平衡状態にある健康な脳では毒性はない。しかし、加齢やその他のAD危険因子のために一旦クリアランスが阻害されると、過剰に産生されたAβモノマーが蓄積してオリゴマーに凝集し、最終的には高密度で不溶性のプラークを形成する。
Aβ仮説は、 脳内でのAβタンパク質の凝集が、神経変性とAD病態を引き起こす中心的な出来事であると仮定している。 しかし、神経細胞死や認知機能低下に直接関係するのはAβではなくタウの沈着であることを示唆するデータが蓄積されている。その代わりに、Aβ凝集体は、下流の神経毒性シグナル伝達カスケードをゲートする「閾値アーカイバー」であると 提唱された。Aβ負荷が閾値以下の場合、タウ沈着は局所的に発現し、良性と考えられる。脳内Aβ濃度が上昇し、アミロイド負荷が閾値を超えると、タウ病態が増強され、複数の脳領域に広がる。
この理論に基づけば、脳内Aβを閾値以下まで減少させることで、AD患者の疾患進行を止めるか遅らせることができると期待される。ADの生物学は本質的に複雑であり、脳内Aβの組成も不均一であるため、Aβは治療法開発にとって困難な標的である。効果的な抗Aβ療法を開発するためには、いくつかの疑問に対処する必要がある。2) Aβ標的薬の治療効果の上限はどこか?3) AD患者が最大限の臨床効果を得るための最適な投与時間枠は何か?
アミロイド標的薬
過去20年の間に、 Aβの恒常性を回復させるために開発された40以上の治療薬が 臨床段階に達したが、これらの臨床試験の95%以上は失敗に終わっている。 すべてのAβ標的治療薬の中で、3つのクラスの戦略が採用されてきた。
-
Aβ産生を防ぐ
APPをAβに変換する酵素を阻害すれば、Aβの産生を遅らせ、凝集を防ぐことができる。BACE-1とγセクレターゼを標的とする阻害剤は、後期臨床試験に達している。しかし、これらの薬剤はすべて、有効性の欠如や安全性の問題により、第III相試験前あるいは第III相試験で失敗している。その主な原因は、健康な脳機能にとって基本的な他のシグナル伝達経路に対するオフターゲット効果である。
-
Aβのオリゴマー化を阻害する
抗体や低分子薬はAβモノマーに結合し、他のAβモノマーとの結合部位をブロックすることで、Aβの凝集を防ぐ。その中でもALZ801は、現在第III相試験が行われている最先端のプログラムである。ALZ801は第II相試験で有望な臨床効果を示しており、Aβのオリゴマー化を阻害することが有効な戦略であることを示唆している。他のプログラムは、まだ前臨床段階あるいは初期臨床段階にある。
-
既存のAβ凝集体のクリアランスを促進する
抗Aβ免疫療法は、ミクログリアや他の免疫細胞を活性化し、Aβタンパク質の貪食を促進することによって、既存のAβ凝集体を除去するために採用された。これらの薬剤の中で、抗Aβモノクローナル抗体(mAbs)が広く研究されてきた。現在、いくつかの抗Aβモノクローナル抗体は、良好な特徴と有望な臨床転帰を有する後期臨床開発に到達している。
抗Aßモノクローナル抗体
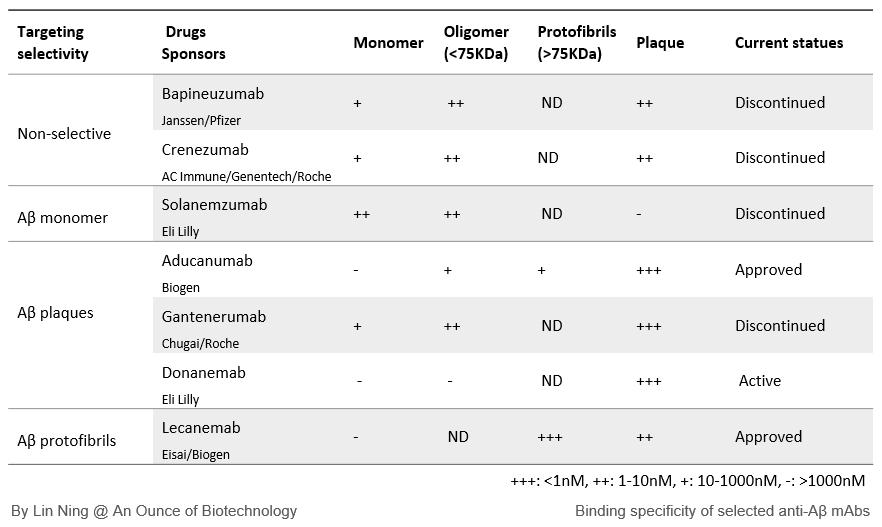
ここでは、バピネズマブ、ソラネズマブ、クレネズマブ、ドナネマブ、ガンテルマブ、そしてFDAに承認されたアデュカヌマブとレカネマブを含む7つの後期抗AβmAbについて、そのAβ結合選択性、安全性プロファイル、臨床成績について概説する。
非選択的抗Aβ mAbs
BapineuzumabとCrenezumabは、あらゆる凝集状態のAβを非選択的に除去するようにデザインされた。In vitroの結合アッセイでは、Aβプラークと可溶性オリゴマーに対する高い親和性と、モノマーに対する中程度の親和性が確認された。
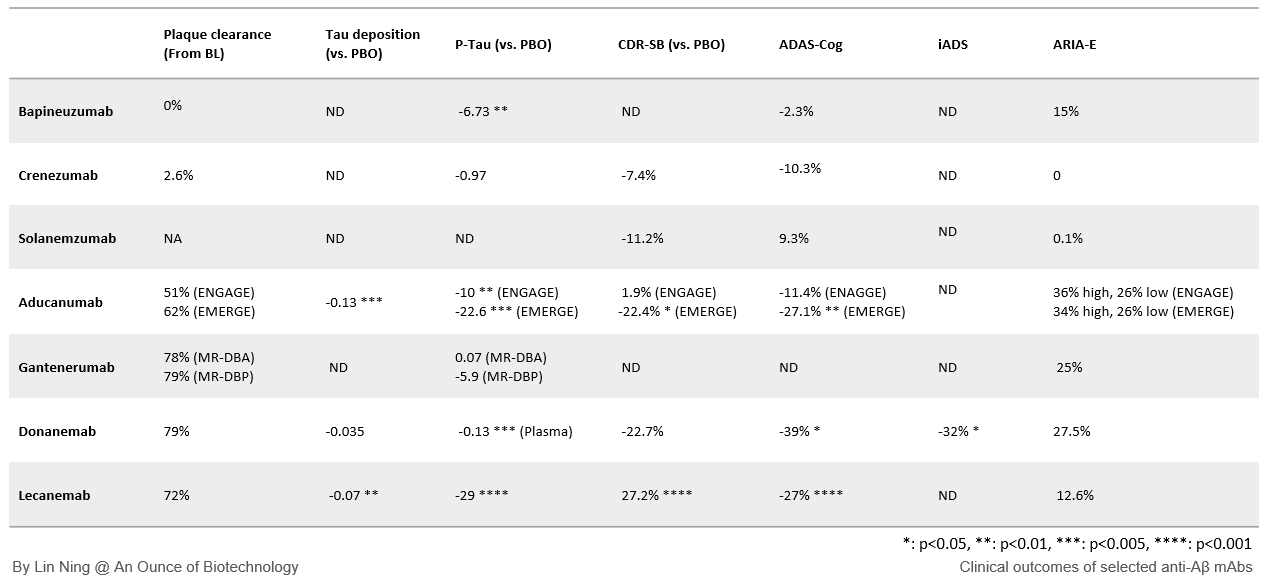
ベースライン時に高レベルのアミロイド負荷があった2つの第III相試験において、バピヌズマブは治療期間中Aβプラークレベルを変化させなかったが、プラセボ対照群ではAβ沈着が急速に増加した。興味深いことに、バピヌズマブは髄液中のp-タウ濃度を有意に低下させ、タウの過リン酸化の制御がAβプラーク負荷と相関していないことを示した。クレネズマブもまた、第II相試験ではAβプラークの除去を誘導できなかった。しかし、CSFのAβオリゴマーは約50%減少し、一方Aβモノマーレベルは増加した。これは、ターゲットがAβオリゴマーにうまく関与し、モノマーから新たなオリゴマー形成を阻害したことを示唆している。クレネズマブは髄液中のp-tauおよびt-tauレベルには影響を与えなかった。
軽度・中等度AD患者において、バピネズマブおよびクレネズマブは、ADAS-CogおよびCDR-SBで測定される最高耐容量において、認知機能低下を予防する臨床的有効性を示さなかった。安全性プロファイルに関しては、Aβプラーク除去による一般的な副作用であるARIA(アミロイド関連画像異常)をバピヌズマブ投与患者の最大15%が発症したが、クレネズマブはARIAを誘発しなかった。Aβプラークに対する抗体の高い親和性を考えると、プラーククリアランスが非効率的なのは、抗体が可溶性Aβで飽和し、プラークにおける標的の関与が低い結果かもしれない。バピネズマブがAβプラークのクリアランスに失敗したのは、アミロイド負荷が高く、プラセボ群では急速に悪化したことから明らかなように、ベースライン時のAβ病態がより重症であったことが部分的に原因かもしれない。また、クレネズマブの失敗は、オリゴマーAβの除去が必ずしもAD患者の臨床的利益につながらないことを示唆している。
Aβモノマー標的mAb
ソラネムズマブは 可溶性Aβモノマーおよびオリゴマーを標的として開発された。この仮説は、Aβプラークがリザーバーとして機能し、末梢のAβレベルを下げるとリザーバーからのAβの放出が促進され、プラークが分解されるというものである。 In vitroの結合アッセイでは、Aβ単量体およびオリゴマーへの強い親和性が確認されたが、プラークには親和性がなかった。
3つの第III相臨床試験において、ソラネムズマブは標的との結合に成功し、脳内Aβ平衡の不溶性プールから可溶性プールへのシフトを誘導したが、脳内Aβ斑を有意に減少させることはできなかった。また、ソラネムズマブは髄液中のp-tauおよびt-tauレベルには影響を与えなかった。
これらの試験において、ソラネムズマブは、ADAS-ADLに 症状の軽い患者のサブグループにおいてわずかな効果がみられた以外は、臨床的有効性を示さなかった。 これらの結果は、可溶性Aβ単量体の除去はAβ斑の十分な崩壊を誘導せず、臨床的利益はないことを示唆している。
Aβプラーク標的mAbs
ガンテネルマブ、アデュカヌマブ、ドナネマブは いずれもプラークを標的とする抗AβmAbで、不溶性のAβプラークに対してサブナノモルアフィニティーを有する。 ドナネマブはプラーク排他的であると考えられている。AducanumabとgantenerumabはともにAβオリゴマーに異なる親和性で結合する(Aducanumab KD=4-138 nM、gantenerumab KD=4nM)。注目すべきことに、ガンテネルマブはAβ単量体にも弱い結合を示した。
前駆期AD患者に105mgと225mgを4週間ごとに投与した場合、ガンテネルマブは 脳内のAβ斑を有意に減少させなかったが、髄液中のp-tauとt-tauを有意に減少させた。3つの非盲検試験で 1200mgまで投与した場合、Aβ斑の66-78%の減少が検出され、24カ月の治療終了時までに50%以上の患者がアミロイド陰性(脳のAβ PET信号が24センチロイド以下)に転じた!プラークの十分な除去にもかかわらず、ガンテネルマブは認知機能の改善にはあまり効果がないようで、アデュカヌマブ、ドナネマブ、レカネマブの試験に参加した患者に比べて、治療した患者は認知機能の低下が早かった。2022年11月に報告されたように、最新の第III相GRADUATE試験では、1200mgのガンテネルマブは期待された半分のプラークしか除去せず、2年間の治療後も期待された臨床的利益は得られなかった。さらに、この用量ではARIAの発生率が25%近くと高かった。これらの結果からガンテネルマブの投与は中止された。
アデュカヌマブは、 認知症を発症しにくい高齢者の血清成分から開発された。 得られたmAbはAβプラークに高い親和性を示し、オリゴマーには中程度の親和性を示し、モノマーには結合しない。 2つの並行第III相臨床試験において EMERGEとENGAGEの、アデュカヌマブは18カ月後にAβプラークをそれぞれ62%と51%減少させた。 さらに、CSFのp-tauとt-tauだけでなく、タウ沈着物の有意な減少も検出された。残念ながら、アミロイド負荷とタウ病態の減少は、一貫した臨床的改善には結びつかなかった。EMERGE試験では、アデュカヌマブは高用量でプラセボ群と比較してCDR-SBを22%緩徐に減少させたが、ENGAGE試験では有意な改善を示さなかった。矛盾した結果と結論の出ない臨床的有効性にもかかわらず、アデュカヌマブは2021年6月にFDAによって承認され(商品名アデュヘルム)、最初の疾患修飾性AD治療薬となった。
ドナネマブは プラーク特異的mAbとして開発され、Aβ p3-42に結合する。Aβ p3-42は、N末端から最初の2残基が切り取られ、3残基にピロール環が形成された修飾Aβペプチドである。 Aβ p3-42は積極的に凝集する性質があるため、プラークと原線維にのみ 存在する。In vitro実験では、Aβプラークへの強い親和性が示された。予想されたように、ドナネマブはプラークのクリアランスにおいてはるかに優れている。第II相Trailblazer-ALZ試験では、毎月1400mgのドナネマブ投与により、アミロイド斑は6カ月で64%、18カ月で79%減少し、60%以上の患者がAβ陰性となった。重要なことは、有効群の患者はAβ陰性になった時点でプラセボに切り替えられ、治療中止後もプラークレベルは低いままであったことである。この試験でEli Lilly社は、統合ADRS(iADRS認知機能を測定する主要評価項目として)を用いた。有効群では、18カ月の試験終了時までに認知機能の低下がプラセボ群より31%遅かった。抗Aβ mAbが第II相臨床試験で統計的に有意な効果を示したのはこれが初めてである。現在、より大規模な集団を対象としたドナネマブの第III相臨床試験が3件進行中であり、2024年までに結果が出る見込みである。2022年10月、第III相臨床試験Trailblazer-ALZ 4の中間解析で、 ドナネマブがアデュカヌマブと比較した場合、プラーククリアランスにおいて優れていることが確認された。
Aβプロトフィブリル標的mAb
レカネマブは、 Aβプラークよりも毒性が強く、AD病理の真犯人かもしれないと考えられている可溶性の高分子Aβオリゴマーの一種であるプロトフィブリルを標的として開発された。 スウェーデンのバイオベンチャー企業Bioarctic社は、arctic変異(E693G)を持つAβはプロトフィブリルを形成しやすいことを発見した。そこで、この変異型Aβ42をマウスに免疫し、mAb m158を同定した。この抗体はプロトフィブリルのエピトープに対して強い構造特異性を持っている。アデュカヌマブと並べて比較すると、レカネマブはプロトフィブリルに対して80-105倍 高い親和性を示す。 レカネマブはまた、ナノモル領域の親和性でAβプラークに結合する。第III相CALRITY-AD試験では、軽度認知障害患者に10mg/kgを隔週投与したところ、レカネマブは3カ月後にすでに有意なアミロイドクリアランスを示し、プラークの減少は18カ月までに72%に達した。さらに、タウ沈着、CSFのp-tauとt-tauは12-18カ月の間にすべて有意に減少した。さらに重要なことは、CDR-SB(26.7%)とADAS-Cog(27.2%)の有意な改善が6カ月後に観察されたことである。有害事象は、ARIA-Eが12.6%、ARIA-Hが17.3%と、他のプラークを標的とするmAbに比べてはるかに低い。優れた臨床効果と安全性プロファイルにより、レカネマブは最高のAD治療薬となった。
プラーク除去mAbsの上限
上記で検討したすべての抗Aβ mAbsの中で、4つのmAbsがAβプラークを効果的に除去した:ガンテネルマブが最も高いプラーク除去効果を示し(ΔAβ=-90.3CL、-74.9CL)、次いでドナネマブ(ΔAβ=-84.5CL)、レカネマブ(ΔAβ=-58CL)、アデュカヌマブ(ΔAβ=-31.4CL、-27.4CL)であった。プラーククリアランスレベルはかなり印象的であったので、認知機能の改善も期待された。しかし、4つのmAbのうち認知機能の低下を遅らせる効果を示したのは、ガンテネルマブを除いて3つだけであった。特筆すべきは、ガンテネルマブがAβクリアランスレベルを達成するのに36カ月かかったのに対し、他のものは18カ月しかかからなかったことである。プラークのクリアランスレベルを時間で正規化すると、ドナネマブは1カ月あたり11.3CL、レカネマブは5.67CL、ガンテネルマブは3.5CLと3.9CLであった。プラークのクリアランス率が低いことが、ガンテネルマブが臨床的有効性を達成できなかった一因かもしれない。
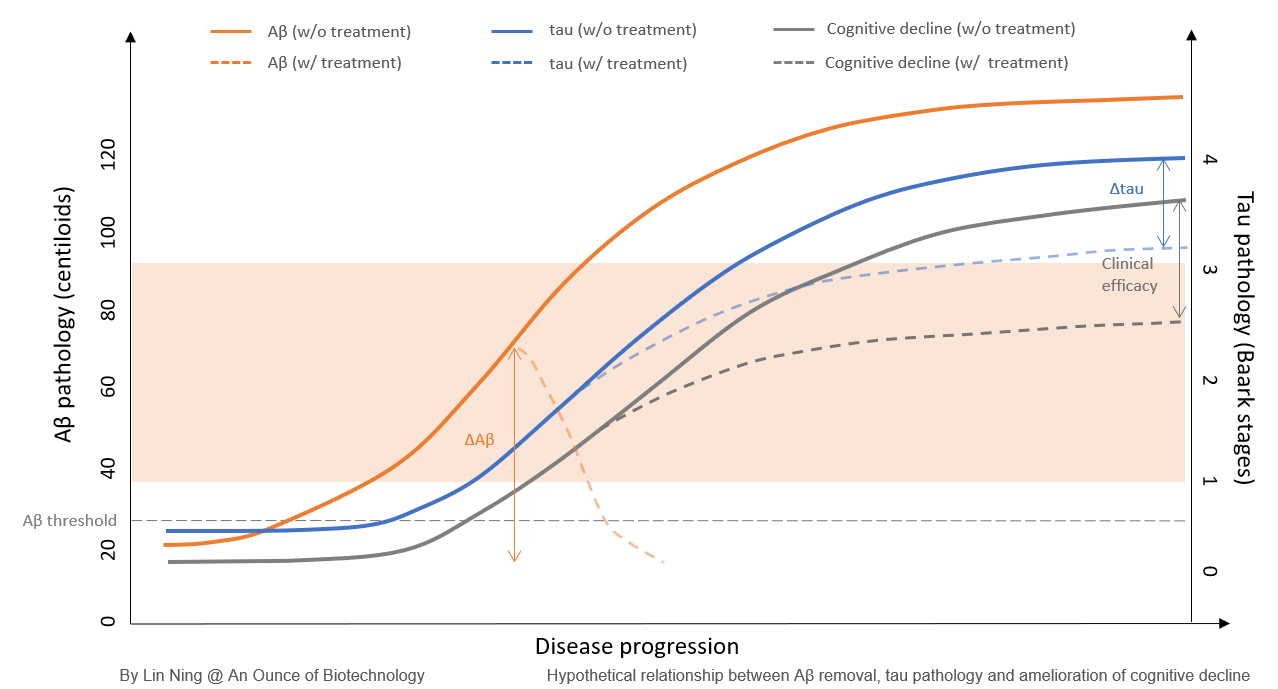
これらのデータは、「Aβ閾値」モデルを支持するものである。アミロイドを標的とする薬剤の場合、アミロイド負荷を速やかに閾値以下のレベルにする能力が、期待される臨床効果を達成するために極めて重要である。閾値以下のAβレベルに到達するのに時間がかかりすぎると、制御不能で自己増殖するタウ病理を伴う、より進行した病期へとすでに進行している可能性がある。
Aβクリアランス率を高めることによって、抗AβプラークmAbの有効性をさらに向上させることは可能だろうか?親和性と標的への関与を最適化することによってAβクリアランス率を高めることが可能であると仮定すると、プラークを迅速に除去することは、患者をARIA発症のリスクにさらすことになるかもしれない。Aβプラークを標的とするmAbを開発する際には、Aβクリアランス率と安全性プロファイルのバランスをとる必要がある。ドナネマブはAβプラークを標的とするmAbの上限であり、〜30%の臨床的有効性がこのクラスの治療薬で達成できる最高値であろう。
オリゴマーはどうだろう?
前臨床研究で蓄積された証拠は、神経細胞死とシナプス神経伝達を媒介する可溶性オリゴマーAβの役割を支持している。この仮説は、Aβプラークレベル、タウ病態、認知機能低下の間に相関がないという問題をほぼ解決するものであり、魅力的である。オリゴマーAβは神経細胞に対して直接的な毒性を引き起こし、シナプスの消失や神経細胞死を引き起こすかもしれない。しかし、これは依然として非常に複雑な科学分野であり、臨床試験による裏付けデータが必要である。
すべてのバイオマーカーエンドポイントをマークし、臨床的有効性を達成した最初のAβオリゴマー標的mAbとして、レカネマブは、Aβオリゴマー、特にプロトフィブリルがAD発症にどのように関与しているかについての洞察を提供することが期待される。レカネマブはAβプラークにも中程度の親和性で結合することから、臨床効果はAβプロトフィブリルとプラークの両方を除去することによる混合効果かもしれない。ドナネマブとアデュカヌマブが18カ月まで臨床効果を示さなかったのに対し、レカネマブは治療開始後6カ月という早い時期に臨床効果を示した。さらに、ドナネマブでは27.5%、アデュカヌマブでは35%であったのに対し、レカネマブは12.6%とはるかに低いARIA率を示した。さらに、レカネマブは6カ月という早い時期に、タウ病態、グリア細胞の活性化、シナプス機能障害のレベルを有意に低下させた。これは、グリアシグナル伝達のエラーとシナプス毒性を修正する直接的な効果を示している。レカネマブのこれらの異なる挙動は、その臨床的利益が少なくとも部分的には原線維除去に起因することを示唆している。Aβオリゴマーを標的とする治療効果は、Aβプラークの閾値とは無関係に、発症初期のタウ病理と認知機能低下の曲線を平坦化する可能性が高い。レカネマブの標的への関与と治療メカニズムを評価するためには、脳と髄液中の原線維の変化を定量的に測定する、原線維を検出する新しい方法が必要である。
ALZ-801(valiltramiprosate)は、脳内浸透性の低分子薬剤であり、Aßモノマーのオリゴマー化部位を阻害し、凝集を防ぐが、プラーククリアランスを誘発しない薬剤としてAlzheon社により開発された。二つの並行した第II相試験において、ALZ-801は軽度のAD症状を有するAPOE4ホモ接合体のADAS-CogおよびCDR-SBにおいて、プラセボに対してそれぞれ40-66%および25-45%の臨床的改善を示した。これらの結果は、まだ大規模な患者集団で確認されるには至っていないが、第II相試験の予備的データから、高分子量AßオリゴマーがADの好ましい標的であることが確認された。
アミロイドβ治療薬が効果を発揮する最適なタイミング
治療に適した患者集団を選択することは、臨床試験の成功に重要な役割を果たす。臨床スコアは、目標とする病期内の患者を定義するために最も使用されるマトリックスである。ADの臨床試験では、症状が軽い患者に介入した方が臨床成績が良い。バイオマーカーもまた、より良い効果を得るために患者集団をさらに豊かにするために採用される。バイオマーカーの変化は臨床症状よりずっと前に起こるし、アミロイド沈着はタウ病態や認知機能低下との相関が低いので、これはADの臨床試験において特に重要である。
Aβプラークを標的とする治療薬の場合、治療開始の理想的なタイミングは、Aβとタウの病理学的レベルによってマッピングできる:
-
アミロイド陽性の状態
いくつかのヒトを対象とした研究で、Aβ負荷量が30CL未満の高齢者では、一般にAD病態や認知機能障害がないことが示された。そのため、業界では20-25CLを Aβ閾値としており、これを超えるとAβ斑が下流の病理学的事象を引き起こす可能性がある。 抗Aβ治療薬が有効であるためには、脳のAβ負荷量が閾値より高くなければならない。
-
タウ病理は陽性だが、まだ自己増殖していない
タウ病態をモデル化するためにBaarkスケールが提案され、認知機能低下との関係が確立されている。タウPETのSUVR値に基づいて5つのBaark病期がある。ステージ0(0-1.129)は、タウ病理や認知機能異常の徴候がないとされる。ステージ1-2(1.129-1.523)は、タウ病態が始まり、中側頭葉から他の脳領域へと広がり、記憶や認知機能の低下が始まる時期である。これらの病期では、タウ病態だけでは認知機能低下を起こすには不十分であり、アミロイド病態が必要となる。ステージ3-4(>1.523)は、タウ病態が広範囲に広がり、自己増殖する時期である。認知機能の低下はアミロイドがなくても起こる。したがって、ステージ1-2は抗Aβ抗体の理想的なターゲットとなる。ドナネマブTrailblazer-ALZ試験では、タウPETのSUVRが1.1-1.46の間であることが患者選択基準の一つとなっており、これはBaark病期1-2に該当する。
Aβオリゴマーを標的とする治療薬については、Aβオリゴマー、タウ病態、認知機能の相互作用を説明するモデルを構築するのに十分な臨床データがない。もしAβオリゴマーが直接タウ病態を促進するのであれば、Aβオリゴマーのレベルを下げることで、早期にタウ病態を発症する確率を下げ、発症を遅らせることができるかもしれない。
ベータアミロイドはまだADの有望な標的なのか?
薬物標的が疾患に対する決定論的モデルに適合する場合、症状発現前の段階での治療は、疾患プロセスを中断し、臨床症状を予防するだろう。一方、症状発現段階での治療は、特に標的がうまく関与している場合には、少なくとも臨床症状の悪化を食い止めることが期待される。
BCR-ABLチロシンキナーゼの低分子阻害剤であるイマチニブは、慢性骨髄性白血病の臨床的悪化を確かに食い止めた。ほとんどすべてのCML患者がイマチニブに反応し、どの病期においても慢性骨髄性白血病の治療にはイマチニブによる治療が必要かつ十分である。
最近の抗Aβ薬の臨床データは、別のモデルを示唆している:Aβは依然としてAD病態生理の重要な因子だが、ADカスケードを駆動する唯一の原因ではなく、臨床症状とも比例しない。抗Aβ薬は、認知機能低下を30%程度遅らせることができ、患者にとって有意義である。しかし、抗Aβ薬は病気の進行を止める特効薬にはならない。別の経路や薬物標的が探索され、そのうちのいくつかは有望な結果を示している。