
コンテンツ
Neural Mechanisms of Inflammation-Induced Fever
www.ncbi.nlm.nih.gov/pmc/articles/PMC6047205/
オンラインで公開 2018 Mar 20
アンダース・ブロムクヴィスト1,デビッド・エングブロム1
要旨
発熱は感染症や炎症性疾患によく見られる症状である。発熱の最終的なメディエーターはプロスタグランジンE2であり、視床下部前部のEP3受容体サブタイプに結合することで熱発生を引き起こすことがよく知られている。ここでは、末梢から放出された発熱物質がどのようにして脳に伝達され、発熱を引き起こすのかについて、さまざまな仮説を検討した。
その結果、炎症性サイトカインが脳内皮細胞の受容体に結合することで、これらの細胞でプロスタグランジンE2の合成を誘発し、発熱を引き起こすという体液性のシグナル伝達経路が明確に証明された。
炎症性サイトカインが脳内皮細胞の受容体に結合することで、プロスタグランジンE2が脳内皮細胞で合成されることで発熱が引き起こされるという説もあるが、脳内皮細胞以外にも、脳循環器や末梢神経を介したシグナル伝達や、末梢で合成されたプロスタグランジンE2の脳内への移行があることを示す証拠はまだない。また、発熱経路の出口についても検討した。結論として、視床前部でのプロスタグランジンE2の結合が、脳幹の交感神経前部を抑制することで発熱を引き起こすことはよく知られているが、栄養状態や周囲の温度などの要因が、末梢の免疫チャレンジに対する反応を形成するメカニズムについては、まだほとんど理解されていない。
キーワード:発熱、サイトカイン、プロスタグランジンE2,脳内皮細胞、視索前中央核、EP3受容体
はじめに
今から 20年余り前、Clifford SaperとChristopher Brederは、The New England Journal of Medicine誌に、当時知られていた「発熱の神経学的基礎」についての権威ある総説をまとめた(Saper and Breder 1994)。末梢から放出されるサイトカインが発熱反応に重要な役割を果たしていることは認識されていたが、これらの物質は血液脳関門を通過できないため、どのようにして脳に信号を送るのかは明らかではなかった。また、プロスタグランジンが発熱に関与していることは知られてたが、発熱を引き起こすプロスタグランジンがどこで、どの細胞によって作られるのかは明らかではなかった。さらに、体温の上昇は、エネルギー生産の増加と(末梢血管収縮による)エネルギー損失の減少によって生じることが知られていたが、中枢神経回路が関与していることはほとんど知られていなかった。この総説では、これらの問題に関する現在の知識を紹介するとともに、さらなる調査を必要とする未解決の問題を指摘する。
発熱は、感染症や炎症性疾患の特徴である。発熱は、末梢血管収縮や発汗の減少などの様々な自律神経反応が協調して作用し、熱損失を減少させ、戦慄を伴う熱産生(場合によっては非戦慄を伴う熱産生も)を行うことで生じる。発熱は、体温の上昇が免疫細胞の活動を高めると同時に、多くの微生物の複製を損なうため、有益であると考えられているが(Evansなど2015;Kluger 1991)発熱の有益性に関する対照的な臨床研究は不足している(Hardenなど2015)。免疫チャレンジ時の体温上昇は、poikilothermsを含むすべての脊椎動物に見られるステレオタイプの反応であり、感染症にかかったときにはより暖かい環境を好むことが示されている(Boltana et al 2013)。
発熱には脳の関与が必要であることは、すでに19世紀末に実証されていた(Atkins 1982参照)。アメリカの病理学者/細菌学者であるウィリアム・H・ウェルチは、頸髄を切断した動物に発熱物質を静脈内注射しても発熱しないことを示した。当時は、炎症の過程で発熱を引き起こす物質が放出されることも理解されていた。しかし、後に内因性パイロジェンと名付けられ、その後サイトカインと同定されたこれらの物質が、どのようにして脳に影響を与えるのかは長い間不明であった。なぜなら、脳は20世紀初頭に記述された血液脳関門によって保護されていたからである(Goldman 1913)。それにもかかわらず、内因性パイロゲンを頸動脈に注射すると、迅速かつ強い発熱反応が得られることが実証され、脳内の体温調節中枢に直接作用していることが示唆された(King and Wood 1958)。さらに、内因性パイロゲンを脳内に直接注射すると、視床下部前部/視索前野に投与した場合には発熱が誘発されるが、他の脳部位に注射した場合には発熱が誘発されないことが判明し、この考えが裏付けられた(Cooperら 1967)。その後、Eシリーズのプロスタグランジンを脳室に注入すると発熱するという観察結果(Milton and Wendlandt 1970)から、内因性パイロゲンはプロスタグランジンを放出することで作用すると考えられ(Feldberg and Saxena 1971)アスピリンなどの解熱剤がプロスタグランジン阻害作用を示すことで、この考えはさらに強化された(Vane 1971)。
発熱における免疫-脳間のシグナル伝達経路
長年にわたり、末梢の免疫シグナルが血液脳関門を通過または回避して脳に影響を与え、発熱を誘発する方法について、いくつかの異なる仮説が浮上した。これらの仮説には、パイロジェンが終末膜の血管系器官に直接作用する、血液脳関門の細胞が活性化される、血液中のプロスタグランジンE2(PGE2)が脳に移行する、免疫シグナルが末梢神経を活性化する、などがある(図1)。それぞれを批判的に検討していく。
図1 末梢から放出された炎症性シグナルが血液脳関門(BBB)をバイパスして中枢神経系を活性化する、示唆されるさまざまな経路
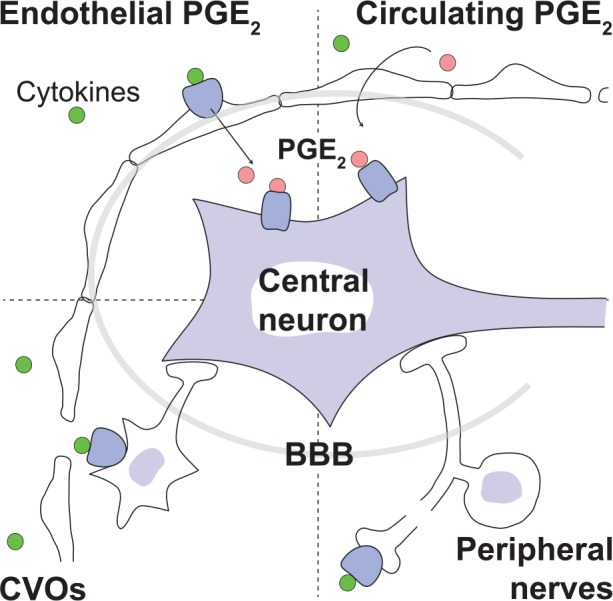
末梢から放出された炎症性サイトカイン(緑丸)は、(i)脳血管の細胞上の受容体に結合してプロスタグランジンE2(PGE2;ピンク丸)の合成を誘導し、脳実質に輸送される(左上)(ii)柵状の毛細血管を含む脳室周囲器官(CVO)の神経細胞を活性化する(左下)(iii)末梢神経を活性化する(右下)。(iv)末梢の炎症により、循環しているPGE2が放出され、それが脳に到達することもある(右上)。
発熱に対する末端葉状体の血管内器官の役割
末端膜血管組織(OVLT)は、通常の血液脳関門を持たない脳の一部である、脳室周囲器官(CVO)に属する(Siso and others 2010)。CVOの毛細血管は柵状になっており、血液中の高分子が構造体内の細胞に到達できるため、体液バランス(アンジオテンシンII、ナトリウム利尿ペプチド、浸透圧)代謝制御(アミリン、グレリン、レプチン)生殖(リラキシン)などの重要な情報をモニターすることが可能である。CVOのニューロンは、その求心性投射によって、これらの情報を視床下部や脳幹にある恒常性維持のための制御センターに伝えることができる(Ferguson 2014)。
CVOは早くから、炎症シグナルを脳深部構造のニューロンに伝達する候補として示唆されていた。この考えを裏付けるように、OVLTに加えて角膜下器官や後頭葉領域を含む感覚的なCVOが、病原体関連分子パターン(PAMPs)やインターロイキン(IL)-1β、IL-6,腫瘍壊死因子α(TNF-α)などのサイトカインの受容体を発現していることが示された(Ericsson et al 1995。Konsmanら 2004; Laflamme and Rivest 2001; Nadeau and Rivest 1999a, 1999b; Vallieres and Rivest 1997)。しかし、受容体を発現する細胞は神経細胞ではなく内皮細胞であり、優先的にCVOの近傍に位置していると思われる(Konsmanら 2004; Rummelら 2006も参照)。また、感覚器CVOはPGE2合成の末端酵素であるミクロソーム型プロスタグランジンE合成酵素-1(mPGES-1)を構成的に発現しており(Eskilsson et al 2014b)これらの構造で局所的にPGE2が合成されていることを示唆している。また、感覚器CVOの細胞は、脳内の他の構造に比べて、末梢の免疫刺激に対して低い閾値で反応すること(Lacroix and Rivest 1997)、末梢の免疫チャレンジが感覚器CVOでサイトカインの発現を誘導すること(Brochu and others 1999; Nadeau and Rivest 1999b; Quan and others 1999)が実証されている。さらに、病変研究では、領域後野がIL-1βによる視床下部-下垂体-副腎軸の活性化(Lee et al 1998)や、末梢のサイトカイン放出を含む様々な疾患パラダイムにおける食欲不振反応に寄与している可能性が示されている(Borner et al 2017;Tsai et al 2014)。
発熱に対する感覚CVOの役割については、視床前部の体温調節ニューロンに隣接する位置にあることから、OVLTが注目されている。Blatteisらによるモルモットを用いた研究では、OVLTを含む前腹側第三脳室の病変は、末梢の炎症モデルとして広く用いられている細菌壁のリポポリサッカライド(LPS)を末梢に注射することで誘発される発熱を抑制した(Blatteisら 1983)。しかし、その後のOVLTに病変のある動物を用いた研究では、発熱が減衰した、つまりBlatteisら(1983)の所見を支持する研究もあれば、発熱が増強した、あるいは効果がなかったという矛盾した結果が出ている(文献のレビューは、Romanovsky et al 2003年を参照)。OVLTはサイズが小さく、位置的にも問題があるため、隣接する構造物を損傷することなくOVLTをアブレーションすることは困難であり、OVLTの病変はいくつかの急性および慢性的な影響を誘発することが報告されているが、これらはそのような追加的な損傷によるものである可能性が高く、発熱反応に影響を及ぼす可能性も高い。したがって、慎重に実施されたある研究では、OVLT病変を持つラットは、やせ細り、浸透圧刺激の反応性低下、慢性的な高ナトリウム血症、高浸透圧、そして最も重要なことに高体温を示すことがわかった(Romanovsky and others 2003)。
免疫誘発性発熱に対する角膜下臓器の切除の効果を検討した研究が1件ある(Takahashi他1997)。この研究では、角膜下臓器の切除により、LPSの末梢注射による発熱が抑制されたが、OVLTや後頭葉の切除では効果がなかった。この研究は、独立した実験で繰り返し行う必要があることは明らかである。
OVLTのアブレーションに見られる「副作用」が認識された後(Romanovsky et al 2003年)発熱を誘発するCVOの潜在的な役割に対する関心は薄れていた。しかし、LPSとIL-1βのシグナル伝達のアダプタータンパク質であるMyD88を脳内皮細胞で欠失させたパターンが異なるマウス系統で発熱反応を比較した最近の報告(Xu et al 2000)では、脳室に注入したIL-1βに対する発熱にはCVOの柵状毛細血管が重要であることが示唆された(Knoll et al 2017)。しかし、柵状毛細血管でのMyD88欠失に関するマウス系統の違いとは別に、脳内皮や末梢の免疫細胞での組み換え効果など、これらのマウス系統の間にはいくつかの潜在的な違いがあり、それが結果に影響を与えている可能性があった。さらに、IL-1βは脳室内(i.c.v.)に投与されたため、今回の知見が末梢から脳へのIL-1シグナル伝達に関連するかどうかは不明である。
脳への免疫シグナルの伝達装置としての血液脳関門
1980年代後半に行われた試験管内試験の研究では、脳の微小血管系でPGE2が産生されていることが示された(Bishai et al 1987。しかし、血液脳関門細胞が脳への免疫シグナルの伝達物質であることを示す決定的な証拠は、生体内でLPSによるPGE2の免疫反応の発現(Van Dam et al 1993)と、これらの細胞におけるプロスタグランジン合成酵素シクロオキシゲナーゼ-2(Cox-2)の誘導が証明されたことであった(BrederとSaper 1996年、Cao et al 1995)。しかし、後者の発見の直後から、プロスタグランジン産生細胞の正体は論争の的となった。いくつかの研究では内皮細胞であると示唆されたが、他の研究では血管周囲細胞(内皮細胞の実質側の血管壁に位置し、基底膜の2枚のシートに包まれた免疫細胞)であると同定されたからである(文献のレビューについては、Rivest 1999を参照)。現在、ほとんどの研究者が、中用量および高用量のLPSに反応してCox-2を発現する脳血管細胞の大部分は、投与経路とは無関係に内皮細胞であるという見解で一致しているが、IL-1βおよび低用量のLPSに対する反応に関してはまだ意見が分かれており、ある研究室では、これらの条件下では血管周囲の細胞がPGE2の主な供給源であると報告している(Schiltz and Sawchenko 2002; Serrats and others 2010)。
脳血管系でCox-2の発現が誘導されることを示す知見は、血液脳関門細胞がPGE2産生の場であることを強く示唆しているが、Cox-2はPGH2の生成を触媒し、PGH2は他のいくつかのプロスタノイドに変換されるため、これらの細胞が実際にPGE2を産生しているという証拠がないため、明確な結論を出すことができなかった。最後の重要なステップは、誘導可能な末端PGE2合成酵素であるミクロソーム型プロスタグランジンE合成酵素1(mPGES-1)(Jakobsson et al 1999)が脳血管細胞に発現していることが明らかになったことである(Ek et al 2001,井上 et al 2002,Yamagata et al 2001)。これらの研究から、mPGES-1は、調べた種(ラット)のナイーブな脳では最小限の発現しかしていないが、低用量のIL-1βをi.v.で注射した場合(Ek et al 2001)中~高用量のLPSを腹腔内(i.p.)で注射した場合(井上 et al 2002,山形 et al 2001)それぞれ脳血管系で強く誘導されることが明らかになった。mPGES-1の血管内での発現は、IL-1タイプ1受容体(IL-1R1)Cox-2および内皮細胞マーカーを発現する細胞に局在していた(井上 et al 2002,山形 et al 2001,Ek et al 2001,Engblom et al 2002bも参照)。その後、関節炎、熱傷、肉球の炎症など、より自然な疾患モデルを用いた研究(Engblom et al 2002a;伊吹 et al 2003;岡 et al 2007;尾崎・岡山 et al 2004;Rummel et al 2011;竹宮 et al 2010)や、マウスを用いた研究(Eskilsson et al 2014b;Vasilache et al 2015)でも、脳内皮細胞ではmPGES-1の誘導・発現が認められたが、血管周囲の細胞では認められなかった(図2)。我々の知る限り、ラットで行われた研究では、血管周囲の細胞におけるmPGES-1の誘導発現を報告したものが1件あるのみである(Serrats et al 2010)。
図2 プロスタグランジンE2合成酵素であるシクロオキシゲナーゼ-2(Cox-2)とミクロソーム型プロスタグランジンE合成酵素-1(mPGES-1)および血管周囲の細胞に発現するマクロファージマーカーであるCD206に対する抗体で染色した、マウス脳内の血管の共焦点顕微鏡写真
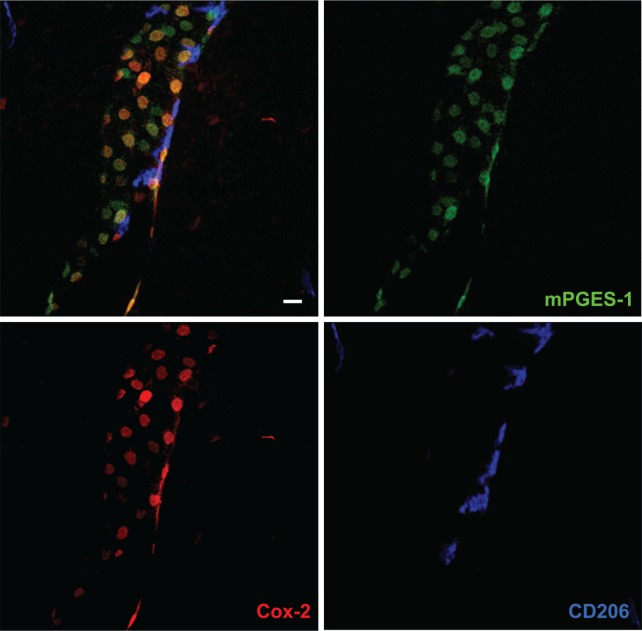
左上のパネルは、これらのタンパク質を3重に標識したもので、他のパネルは各タンパク質を1重に標識したものである。Cox-2を発現している細胞のほとんどがmPGES-1も発現しており、その逆もまた然りであることに注目。また、Cox-2/mPGES-1を発現している細胞にはCD206の染色が見られないことから、この集団には血管周囲の細胞は含まれていないと考えられる。スケールバー=20μm。
Cox-2およびmPGES-1を全欠失させた動物を用いた研究により、これらの酵素が発熱に重要であるという考えが裏付けられた(Engblom et al 2003,Li et al 1999年、Nilsberth et al 2009b、Saha et al 2005)。その後、脳血管系でのプロスタグランジン合成が発熱反応に果たす役割についての機能的研究が行われた。これまでに得られたデータは、脳内皮細胞が重要な役割を果たしていることを示しているが、それだけではないかもしれない。最初の機能的証拠は、Chingらの研究で得られた。脳内皮細胞のIL-1R1をノックダウンすると、i.v.投与されたIL-1βによる病気の症状(発熱および運動能力の低下)が消失し、血液脳関門のCox-2発現や視床下部傍室のFos発現が誘発されなくなることから、これらの現象に内皮細胞のIL-1R1が重要な役割を果たしていることが示された。その後、Ridder et al 2011)は、Cox-2遺伝子の転写を制御する転写因子である核内因子-κB(NF-κB)およびc-Junの上流でIL-1βのシグナル伝達に重要な役割を果たすMAP(mitogen-activated protein)キナーゼキナーゼTAK1を脳内皮に特異的に欠失させると、i.v.注射されたIL-1βに対する発熱反応が鈍化することを明らかにした。PGE2合成酵素mPGES-1を造血細胞(血管周囲のマクロファージを含む)または非造血細胞(脳内皮細胞を含む)のいずれかに発現させたキメラマウスを用いた。Engströmらは、非造血系細胞でのmPGES-1の発現は、末梢の免疫チャレンジに対する発熱反応を引き起こすのに十分であるのに対し、血管周囲の細胞を含む造血系細胞に限定してmPGES-1を発現させると、発熱が抑制されることを示した(Engströmら 2012)(図3)。3). 最後に、Wilhelms et al 2014)は、内皮細胞でのPGE2合成が発熱反応に決定的に関与していることを直接証明した。Wilhelmsらは、Ridder et al 2011)の研究と同じ組織特異的なCre媒介組換えを用いて、脳内皮特異的にCox-2およびmPGES-1を欠失させると、IL-1βおよびLPSのi.p.注射後の発熱が鈍化することを実証した(Wilhelms et al 2014)(図4)が、神経細胞や骨髄系細胞などの他の細胞タイプでの欠失は影響を及ぼさないことが示されている(Nilsson et al 2017)。Wilhelms et al 2014)の研究における発熱の減衰は、LPSチャレンジ後の血中のIL-1βレベルが影響を受けなかっただけでなく、脳内皮の炎症遺伝子(Cxcl10,Ccl2,Lcn2)の誘導も影響を受けなかったことから、炎症プロセスに対する非特異的な影響によるものではないと考えられる。このような特異性は、発熱を抑制する内皮特異的な操作を行っても、他の多くの全身性炎症の症状には影響がないことからも明らかである。このように、炎症によって誘発される食欲不振、運動不足、視床下部-下垂体-副腎軸の活性化は、発熱を減衰させる内皮操作を行ったマウスでは無傷であることが示されており、一方、炎症によって誘発される場所嫌悪も脳内皮シグナルを必要とする(Fritzなど2016,Fritzなど2018,Nilssonなど2017,Ridderなど2011,Wilhelmsなど2014)。Wilhelmsらの知見に加えて、脳内皮細胞上のIL-6受容体α(IL-6Rα)およびIL-1R1を欠失させると、LPSに対する発熱反応が減弱することも実証された(Eskilsson et al 2014a;Matsuwaki et al 2017;後述)。これらを総合すると、細胞型特異的な遺伝子操作を用いた機能研究から得られた知見は、発熱の発生における脳内皮の役割を強く支持するものである。細胞型特異的な操作は必ずしも100%の特異性や選択性があるわけではないので、脳内皮の役割は2つの異なる内皮特異的プロモーター、すなわちSlco1cプロモーターとTie2プロモーターを用いて示されていることに注意する必要がある。したがって、これらの研究で見られた効果をオフターゲット効果が説明する可能性は極めて低い。むしろ、脳内皮の重要性が過小評価されている可能性の方が高い。なぜなら、使用したCre-lineがすべての脳内皮細胞を標的としていない可能性があるからである(例えば、Eskilsson et al 2017を参照)。
図3 全身照射を行った後、WT(+/+)またはKO(-/-)の骨髄を移植した野生型(WT)およびmPGES-1ノックアウト(KO)マウスにおける細菌壁のリポ多糖(LPS)の腹腔内注射に対する体温反応
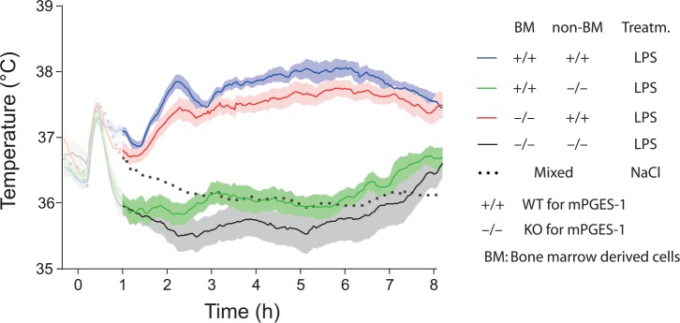
WTまたはKOの骨髄を移植したWTマウス(non-BM +/+)は顕著な発熱反応を示し(上の2つの発熱曲線)WTまたはKOの骨髄を移植したKOマウス(non-BM -/-)は無熱であることがわかる(下の発熱曲線)。すべてのグループで見られる初期の温度ピーク(影付き)は、ストレス誘発性の高熱を処理している。この実験では、白血球と脳のマクロファージ(血管周囲細胞)の約90%、肝臓(クッパー細胞)と肺のマクロファージの約70%が生来の造血由来細胞に置き換わっていた。NaCl処理群は平均値を、その他は平均値とSEM(Standard Error of the Mean)を示した。Engström and others (2012)より引用。
図4 プロスタグランジンE2合成酵素であるシクロオキシゲナーゼ-2(Cox2ΔbEnd)とミクロソーム
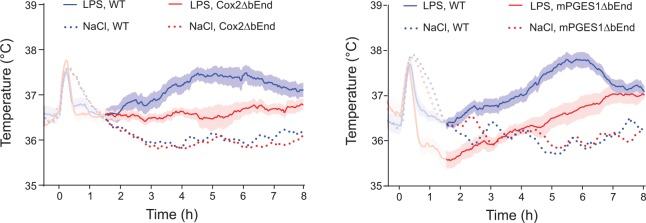
プロスタグランジンE合成酵素-1(mPGES1ΔbEnd)を脳内皮細胞で選択的に欠失させたマウスでは、腹腔内に注入した細菌壁のリポポリサッカライド(LPS)に対する発熱反応が鈍化する。WT, 野生型マウス。NaCl処理群では平均値を、LPS処理群では平均値とSEM(平均値の標準誤差)を示した。Wilhelms et al 2014)より引用。
熱反応に対する血管周囲の細胞の役割
内皮細胞の炎症性シグナル分子の遺伝子欠失を用いた研究は、したがって、これらの細胞が発熱反応に重要な役割を果たしていることを示す説得力のある証拠を提供しているが、脳の血管周囲のマクロファージの発熱への貢献は限定的であると思われる。IL-1βによる中枢神経系の活性化には、血管周囲の細胞が重要な役割を果たしているが、一方で、これらの細胞は、内皮細胞におけるPGE2の合成や、このPGE2の生成によって引き起こされる急性期反応を抑制する役割を果たしていることが示唆されている。Serratsらは、クロドロネートリポソーム(Van Rooijen 1989)を脳内に注入して血管周囲の細胞を切除したモデルを用いて、IL-1βを静脈内に注入した際の脳内Cox-2誘導、ACTHおよびコルチコステロンの放出を減少させたが、LPSを静脈内に注入した際には同様の反応を増強させたと報告した(Serratsら 2010)。一方、発熱反応は、LPSとIL-1βのいずれに対しても、そのまま、あるいは適度に増強された。このように、本研究では、血管周囲の細胞におけるCox-2の誘導と視床下部-下垂体-副腎軸の活性化との関係について多くの疑問が残されているが、発熱反応の誘発に血管周囲の細胞が重要でないことは明らかであると思われる。
血液脳関門を通過するIL-1およびIL-6シグナルの発熱反応への役割
前述のように、脳内皮細胞におけるIL-1シグナルは、外因性のIL-1β投与による発熱反応に重要である。しかし、LPSのようなより自然な刺激に対する発熱反応に、この経路がどのように寄与しているかは、あまり明らかになっていない。IL-1βを欠損させたマウスでは、急性期反応が損なわれないばかりか(Fantozzi and Dinarello 1996)、LPSを免疫的に投与すると発熱が増強される(Alheim and others 1997)。さらに、IL-1R1を欠損させたマウスや、IL-1受容体アンタゴニストを投与したマウスを用いた初期の研究では、これらのマウスはLPSに反応しても無傷であるか、わずかに発熱が抑えられただけであったことが報告されており(Bluthé et al 2000,Labow et al 1997年、Leon et al 1996年、Luheshi et al 1996年、Smith and Kluger 1992年)IL-1シグナル伝達経路がこの刺激に対する発熱反応に重要ではないことが示唆されている。我々は最近、この問題を再検討し、IL-1R1のグローバルな欠失、およびIL-1受容体アンタゴニストによるマウスの処理が、i.p.注射されたLPSに対する発熱反応を減少させるが、廃止しないことを発見した(Matsuwaki and others 2017)。さらに、IL-1R1を細胞特異的に欠失させたマウスを用いて、脳内皮細胞ではIL-1R1を欠失させると熱反応が減衰するが、神経細胞や末梢神経ではこの受容体を欠失させても熱反応が起こらないことを確認した。グローバルノックアウトマウスでは、発熱の残存・減衰は遅延反応であり、野生型マウスの約3時間後に比べ、LPS注入後約5〜6時間後に見られた(図5)。同様の結果は、Chingらが内皮細胞のIL-1R1ノックダウンを用いて報告している。この方法では、i.v.およびi.c.v.のIL-1β注射に対する反応は完全に消失したが、i.p.のIL-1β注射に対する発熱反応は弱まり、遅延するだけであった(Chingら 2007)。これらの結果から、LPSやIL-1βをi.p.で注射すると、脳内皮細胞のIL-1R1シグナルとは一部独立した形で発熱が誘発されると考えられる。
図5 サイトカイン受容体を欠失させたマウスにおける細菌壁のリポ多糖(LPS)に対する発熱反応
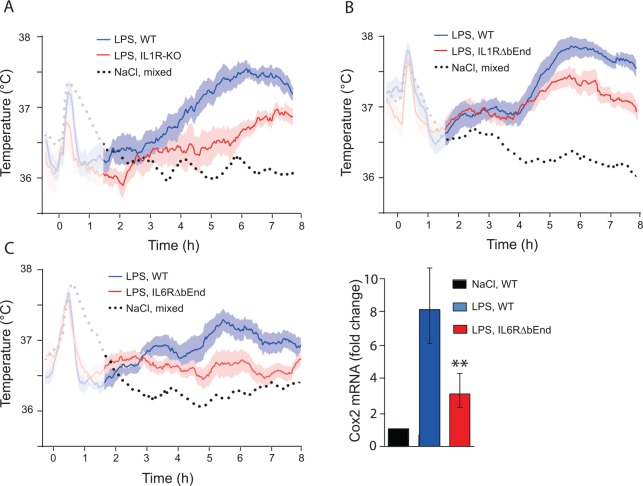
(A)インターロイキン1型受容体を欠損させたマウス(IL1R-KO)では発熱が軽快したが、発熱の発現が遅いことに注意が必要である。(B)脳内皮細胞で選択的にIL-1R1を欠失させたマウス(IL1RΔbEnd)では、約5時間後に見られる発熱が軽快した。(C)脳内皮細胞に選択的にインターロイキン6受容体αを欠失させたマウス(IL-6RΔbEnd)では、発熱が減弱した(左)。この反応は、視床下部におけるシクロオキシゲナーゼ-2(Cox-2)の誘導の減弱と関連していた(右)。**はP < 0.01を示す。Matsuwaki et al 2017)およびEskilsson et al 2014)から引用した。
IL-1βとは対照的に、IL-6は、例えばLPSチャレンジ時にも放出されるが、LPSによる発熱には重要であると思われる。IL-6ノックアウト動物や、IL-6に対する中和抗体を投与した動物は、IL-6自体には発熱性がないか、あるいは弱い発熱性しかないにもかかわらず、発熱反応を起こすことができない(Chai et al 1996年、Kozak et al 1998年、Rummel et al 2011)(LeMay et al 1990年、Nilsberth et al 2009a、Rummel et al 2008,Wang et al 1997)。IL-6Rαを内皮特異的に欠失させたマウスでは、上述のように末梢投与されたLPSに対する発熱が強く抑制され、下流のシグナル分子であるSTAT3を欠失させたマウスでも同様の結果が得られていることから、IL-6の作用は脳内皮細胞のシグナルを介して発揮されていると考えられる。また、IL-6Rαを欠失させると、脳内皮細胞におけるLPS誘導性のCox-2発現が強く抑制された(図5)(Eskilsson et al 2014a)。まだ答えの出ていない興味深い問題は、LPSに対する発熱反応に両方のサイトカインが必要と思われることを考慮すると、脳内皮細胞においてIL-6シグナル経路がIL-1シグナル経路(図6)とどのように相互作用しているのか、そしておそらくToll-like receptor 4シグナルとも相互作用しているのではないかということである。なお、内因性のTNF-αは、IL-1βやIL-6と同様にLPSによって放出されるが、おそらく他のサイトカインを放出することで、外因性に投与すると発熱を誘発するものの、クライオジェンである(Cao et al 1998年、Matsuwaki et al 2017)。
図6 末梢に放出された炎症性メディエーターによって惹起される血液脳関門における伝達機構
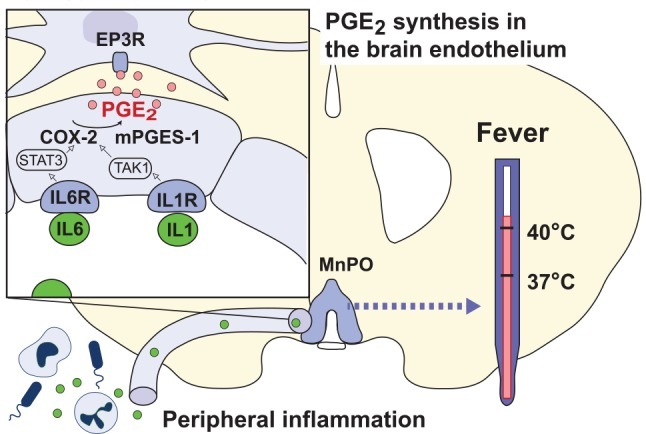
サイトカインIL-1βとIL-6(緑丸)は、視床下部前葉の脳内皮細胞に存在する受容体(IL1R、IL-6R)に結合し、TAK1とSTAT3を介してそれぞれシクロオキシゲナーゼ-2(COX-2)とミクロソームプロスタグランジンE合成酵素-1(mPGES-1)を転写させる。その後、新合成されたPGE2(ピンクの丸)が、視床下部の正中部視索前核(MnPO)にあるPGE2 EP3受容体(EP3R)を発現する細胞に結合すると、発熱が誘発される。
血液中のPGE2に対する発熱反応?
マウスやラットにLPSをi.p.で注射すると、ハンドリングストレスによる最初の体温ピークの後に、二相性の発熱反応が起こる(Romanovskyら 1998)。しかし、動物を扱わない条件で静脈留置カテーテルから注射すると、3相性の発熱反応が見られ、最初の相は注射後60分に見られるため、i.p.で注射した場合に起こるハンドリングストレスによって不明瞭になる(Romanovsky et al 1998年、Rudaya et al 2005)。この最初の発熱期のメカニズムについては多くの議論があり、後述するように、末梢神経、特に迷走神経の活性化が関与していることが示唆されている(「発熱に対する末梢神経の役割」の項を参照)。しかし、ロマノフスキーらによって提案された有力な考えは、発熱の第一段階は、肺のマクロファージから血液中にPGE2が放出されることによって誘発され、血液中のPGE2が視索前野で脳に入り、そこで発熱反応を誘発するというものである(Steinerら 2006)。この仮説は主に2つの観察結果に基づいている。第一に、発熱の第一段階は、ウエスタンブロット法による視床下部でのプロスタグランジン合成酵素の誘導に先行するが、末梢組織、特に肺のマクロファージで見られるシクロオキシゲナーゼの誘導と同時であることがわかった(Romanovsky他2006,Steiner他2006)。第二に、PGE2に対する中和抗体を静脈内に注射すると、末梢に投与したLPSに対する初期の発熱反応が弱まった(消失はしなかった)(Steiner et al 2006)。この2つの観察結果は、若干の注意を払って解釈する必要がある。少数だが重要な位置にある血管(すなわち視床下部前部)で、例えばCox-2の合成が誘導されても、視床下部全体のタンパク質分析では検出されなかったかもしれない。自律神経系の重要な部位の血管では、他の部位の血管に比べて、IL-1受容体や下流の細胞内シグナル分子の発現が顕著であることが示されている(Konsmanら 2004)。さらに、当研究室の最近の研究では、マウスにLPSをi.v.注射した後、すでに30分後に視床下部で強いCox-2 mRNAの誘導があり、脳液中のPGE2も高濃度であることが示され、視床下部の血管ではCox-2タンパク質が発現していることが明らかになった(Eskilssonら、未発表)。中和抗体を用いた実験では、その効果は部分的なものにとどまったことに留意すべきである。この観察結果は、抗体がすべての循環PGE2を完全に中和できなかったことによるものかもしれないが、循環PGE2とは別の発熱開始メカニズムが存在している可能性もある。さらに、注入された抗体のごく一部が血液脳関門を部分的に通過し、内皮細胞が産生するPGE2を中和した可能性も否定できない。Steiner et al 2006)は、i.c.v.に注入された少量の抗体が発熱の初期段階に影響を与えないことを示すことで、全身に注入された微量の抗体が脳に浸透し、脳実質で作用を発揮した可能性をコントロールしようとした。しかし、この実験では、視索前野のEP3受容体発現ニューロンを取り巻く細胞外液中の抗体濃度が、局所的に産生されるプロスタグランジンを遮断するのに十分な高さであったかどうかは不明であり、結論は出ていない。
しかし、最も重要なことは、末梢のマクロファージが産生するPGE2が発熱の初期段階をもたらすという考えが直接検証され、反証されたようだということである。Engblom et al 2003年)Eskilsson et al 2017年)Nilsberth et al 2009b)Engström et al 2012)は、mPGES-1を造血細胞で欠失させたマウスは正常な発熱初期を示すのに対し、mPGES-1を造血細胞のみで発現させたマウスは発熱しない(代わりに低体温)ことを示した(図7)。また、後者のマウスでは、LPS投与40分後に、血漿中のPGE2代謝物が有意に上昇したが、脳脊髄液(CSF)中のPGE2は上昇しなかったことから、移植された造血細胞がプロスタグランジン産生能に関して機能していたことが示唆された。この実験により、LPS投与後早期に見られる循環PGE2の少なくとも一部が造血系由来であることが確認されたが、このPGE2は発熱反応を誘発せず、さらに、少なくともCSF中のPGE2濃度の上昇に反映されるほどには脳に侵入しないようである。同様の結果は、腫瘍を持つマウスを用いた研究でも報告されている(Ruud and others 2013)。この研究では、血漿中に高濃度のPGE2およびPGE2代謝物が記録されたが、発熱はなく、CSF中のPGE2濃度も上昇しなかった。ただし、血液脳関門の伝染性を変化させる可能性のある炎症状態でPGE2濃度が上昇した場合は、条件が異なる可能性があることに留意する必要がある。
図7 WT(+/+)およびKO(-/-)の骨髄を移植した全身照射野生型(WT)およびmPGES-1ノックアウト(KO)マウスにおける、静脈内注射したリポ多糖(LPS)に対する発熱反応
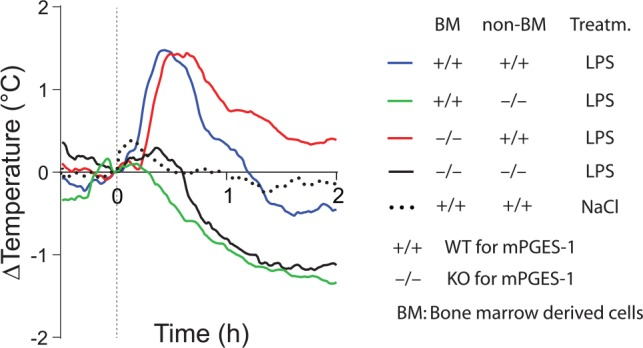
WTマウス(non-BM +/+)は、WTまたはKOの骨髄を移植したかどうかに関わらず、第一段階の発熱を示した(上の2つのトレース;生理食塩水を注入したマウスの温度曲線を参照)。一方、WT骨髄を移植したKOマウス(non-BM -/-)は、KO骨髄を移植したKOマウスと同様に低体温反応を示す(下段2つのトレース)。すべてのトレースの平均値を示す。縦の破線は注入した時間を示す。これらの実験の詳細については、図3を参照のこと。Engström et al 2012)から引用した。
発熱に対する末梢神経の役割
末梢性炎症のセンサーとしての末梢神経の役割は、広範な研究の対象となっており、迷走神経が最も研究されている。迷走神経の切断を用いた初期の研究では、痛みの感受性の変化、摂食行動、社会的探索、睡眠、ストレスホルモンの放出など、さまざまな病気の症状に迷走神経が関与していることが報告されている(Blutheなど1994年、Bret-Dibatなど1995年、Gaykemaなど1995年、Hansen and Krueger 1997年、Kentなど1996年、Luheshiなど2000,Watkinsなど1994)。これらの観察結果を裏付けるように、結節神経節にはIL-1R1とEP3受容体のmRNAが発現しており(Ek et al 1998年、中村 et al 2000年)熱反応に重要なPGE2受容体サブタイプ(Ushikubi et al 1998年)i.v.または大動脈内にIL-1βを注入すると迷走神経求心性の放電活性が増加することが示された(Ek et al 1998年、新島 et al 1996)。下流のシグナル伝達機構についてはほとんど検討されていないが、下脳幹のスライス標本を用いた解析では、PGE2がEP3を介して迷走神経のシナプス伝達を抑制し、アデニルシクラーゼと結合したGi/oタンパク質が関与することが示されている(Marty et al 2008)。
発熱における迷走神経の役割については、入手可能なデータは相反するものである。迷走神経を外科的に切断すると、LPSやIL-1βをi.p.に注射したときの発熱反応が弱まったり、消失したりするという初期の観察結果がある。迷走神経を外科的に切断すると、LPSやIL-1βのi.p.注射による発熱反応が弱まったり、消失したりするという初期の観察結果(Hansen and Krueger 1997; Sehic and Blatteis 1996; Watkins and others 1995)は、迷走神経切断によって生じる栄養失調が動物に発熱反応を起こさせなくする可能性があるため、その後疑問視された(Caldwell and others 1999; Hansen and others 2000; Luheshi and others 2000)(Hoffman-Goetz and Kluger 1979; Inoue and others 2008; Krall and others 2010; Shido and others 1989)。また、カプサイシンを用いて内臓求心性線維を化学的に変性させると発熱が抑制されることが報告されているが(Szekely and others 1997)、その後、この効果はカプサイシンによる肝臓でのエンドトキシン作用の変化など、神経以外のメカニズムによるものであることが示唆されている(Dogan and others 2004; Petervari and others 2005)。
しかし、IL-1βやLPSの閾値用量に反応して見られる単相性発熱が迷走神経を介している可能性も残されている。この反応は、ハンドリングストレスを伴わない条件下で免疫原を静脈注射したときに見られる1℃以下の短時間の発熱ピークであるが、栄養失調を防いだラットでも迷走神経切断後に消失することが報告されている(Romanovsky et al 1997a; Romanovsky et al 1997b)。これは迷走神経の肝枝に依存しており、他の迷走神経の枝や脾臓神経には依存していないようである(Dogan and others 2003; Simons and others 1998)。迷走神経の肝枝がこのような役割を果たすということは、多相性発熱の第一相が迷走神経の活性化を介しているという指摘に合致する(Blatteis 2007; Li and others 2006; Romanovsky and others 2000)。この活性化は、構成的なシクロオキシゲナーゼによって合成され、デノボのタンパク質合成を必要としないPGE2が、補体によって肝臓のクッパー細胞から放出されることに起因すると考えられてきた。しかし、この考えは否定されている。横隔膜下および頸部迷走神経切断後(後者は麻酔ラット)内因性の末梢放出を模倣していると考えられるPGE2のi.v.注射を行ったところ、やはり発熱が生じた(Ootsuka and others 2008)。発熱は、延髄鞘核の交感神経前駆細胞を阻害することで消失したことから、末梢投与されたPGE2の発熱作用は、迷走神経を介さないPGE2の脳への直接作用(前述)によるものと考えられた(大塚 et al 2008)。
迷走神経に加えて、体性求心性線維も発熱反応に関与していることが示唆されている。結節神経節と同様に、後根神経節にもIL-1R1およびEP3受容体が発現していることが報告されており(Binshtok et al 2008,中村 et al 2000年)末梢神経はIL-1βおよびPGE2に反応することが示されているが、これらの炎症性メディエーターはそれ自体が放電を引き起こすというよりも、他の刺激に対して神経を感作することがわかっている(Binshtok et al 2008,Derow et al 2007)。全身の微細な求心性線維が体内環境を感知し、その求心性放電を介して様々な自律神経中継構造に影響を与え、ホメオスタシスを維持しているという考えに沿って(Craig 2002)、体性求心性線維が局所的な末梢の炎症に対する発熱反応を媒介することは考えられる。しかし、LPSを空気袋や人工皮下腔に注入するモデルでは、ほとんどの場合、サイトカイン、特にIL-6が局所炎症部位から循環系に漏出し(Cartmell et al 2000,Miller et al 1997年、Ross et al 2003,Rummel et al 2006,Zhang et al 2008年)IL-6抗血清による全身治療で発熱反応が消失する(Cartmell et al 2000)ことから、神経経路ではなく体液経路が発熱の原因であることが示唆される。局所麻酔薬をLPSと一緒に皮下チャンバーに注入すると、発熱は減少したが、循環IL-6レベルは減少しなかったことから、局所的な神経活性化が発熱の原因であると解釈された(Ross et al 2000)。しかし、著者らが認めているように、局所麻酔薬は免疫反応を阻害する可能性があり(Schmidt and others 1997; Sinclair and others 1993)例えば、IL-6の発熱作用の重要な補因子であるIL-1β(Sinclair and others 1993)を阻害することで(Cartmell and others 2000)発熱反応を減弱させた可能性がある。
局所的な炎症を利用したいくつかの研究では、いくつかの条件下で、発熱があるにもかかわらず、脳内でCox-2のアップレギュレーションが見られないことが報告されている。Rummelらは、モルモットの皮下に埋め込んだ人工的なテフロンチャンバーに低用量のLPSを注射したが、脳内のCox-2のハイブリダイゼーションシグナルは検出されなかった(Rummelら 2005)。また、Zhangらは、ラットの皮下の空気袋にカゼインを注射したが、同様に視床下部にCox-2が誘導されないことをイムノアッセイとリアルタイムPCRで報告した(Zhangら 2008)。これらの観察結果は、局所的な炎症部位から循環系にIL-6が漏出しても、中枢のプロスタグランジン合成は活性化されないことを示唆している。これらのデータは興味深いものであり、発熱の神経求心性経路の存在を指摘していると解釈されるかもしれないが、そのような経路は、視床前部のEP3受容体の活性化を伴わずに、つまり、末梢の温度受容器と同じように中枢の体温調節系に供給される可能性も示唆している(Blatteis 2007; Morrison 2016)。Rossと共同研究者によって議論されたように、強力な候補は明らかに冷感繊維である。ラットやマウスの有害な寒さによって活性化され(Chen et al 2013)侵害受容性求心性線維に発現している化学センサーであるTRPA1イオンチャネル(Julius2013)が、LPSによって活性化され、急性の神経原性炎症と痛みを媒介することに注目すべきである(Meseguer et al 2014)。これらの観察結果を念頭に、我々は最近、LPSによる発熱へのTRPA1の関与を検討した。しかし、TRPA1 KOマウスは、WTマウスと同様にi.p.LPSに対する発熱反応を示した(未発表)。
発熱に対する体性求心性線維の役割をさらに裏付けるものとして、末梢神経を切断すると発熱が抑制されることが報告された2つの研究がある。そのうちの1つは、上顎の歯肉袋にLPSを注入することで誘発された発熱が、眼窩下孔から出ているすべての三叉神経の枝を切断すると(局所麻酔薬やシクロオキシゲナーゼ阻害剤を局所的に注入した場合と同様に)軽減されることを示した(Navarro and others 2006)。しかし、この報告では、動物が適切に咀嚼する能力に影響を与えると思われる外科的処置が、動物の体重、ひいては熱影響能力に影響を与えていないことの確認が不足している。もう一つの研究では、舌咽神経の切断は、軟口蓋に注入されたLPSまたはIL-1βに対する発熱反応を減少させたが、i.p.に注入された場合は減少しなかった(Romeo and others 2001)。しかし、その効果は小さく、神経を切断した群と偽手術を行った群の温度差は10分の1度に過ぎず、温度反応は明らかに注射を行ったときの全身麻酔の影響を受けていた。
これまでに、末梢神経上のサイトカイン受容体が熱反応に果たす役割を調べるために、遺伝子を欠損させた研究がいくつか行われている。末梢神経を含む神経堤誘導体やvanilloid受容体を発現する微細な求心性神経(すなわち侵害受容性C線維)でIL-6Rαを欠失させたマウスは、i.p.に注射したLPSに対して無傷の発熱反応を示し(Eskilsson et al 2014a)IL-1R1を同様に欠失させたマウスでも同様の結果が得られた(Matsuwaki et al 2017)。いずれの研究でも、脳内皮細胞のIL-6RαおよびIL-1R1を欠失させると、熱反応が減弱した(上述)。これらのデータは、末梢に放出されたIL-6およびIL-1βによる末梢神経の直接的な活性化を否定するものであるが、高レベルの循環サイトカインが存在しない場合にのみ、免疫-脳間コミュニケーションの神経細胞ルートが役割を果たす可能性があることに留意すべきである(Quan 2014; Ross and others 2000; Rummel and others 2005)。上述の2つの研究で使用した量のLPSを注射すると、かなりの量のサイトカインが循環に放出されるため(松脇 et al 2017)感覚神経の寄与が検出されなかった可能性が考えられるのである。
以上のように、発熱における末梢神経の役割に関するデータは矛盾している。すべての研究の明らかな弱点は、末梢神経が発熱に必要かどうか、つまりシグナル伝達を遮断することで発熱反応が減衰するかどうかを検討していることである。この方法では、上述のように、神経のシグナル伝達が、例えば体液性のシグナル伝達と並行して存在しているかどうかを検出することが困難であることを意味する。免疫性発熱に関与するシグナル分子を末梢神経に特異的に発現させた動物を用いた研究は、この問題の解決に役立つだろう。そのような動物モデルは現在利用可能になりつつある(例えば、Liu et al 2015)。
発熱反応に重要な中枢神経細胞
PGE2が中枢神経系のEP3受容体の活性化を介して、LPSやサイトカインによる全身免疫チャレンジによって誘発される発熱の最終的なメディエーターであることはよく知られている(Engblom et al 2003,Lazarus et al 2007,Nilsberth et al 2009b、Saha et al 2005,Ushikubi et al 1998)。末梢の炎症では、末梢だけでなく脳内でも発熱性サイトカインが強く転写されるが、脳内で産生されたサイトカインはPGE2の合成が誘導されていなければ発熱を起こさない(Nilsberth et al 2009b)。視床下部前野が発熱の誘発に重要な役割を果たしていることは早くから認識されており(Cooperなど1967年)その後、この領域はPGE2の発熱作用に最も敏感な部位であり(Scammellなど1996年、Scammellなど1998年)EP3受容体発現ニューロンが高濃度に発現していることが判明した(Ekなど2000,Okaなど2000,Vasilacheなど2007)。また、PGE2産生を局所的に回復させることで、他の方法では発熱しない動物でもLPSに対する体温反応が得られる部位でもある(Eskilsson et al 2017)。
では、視床下部前部はどのようにして発熱するのだろうか。健康な動物の視床前部のニューロンは、脳幹の吻側髄質瀬状核(RMR)の発熱性交感神経前部ニューロンに緊張性の抑制性GABA作動性入力を与えており、PGE2がEP3受容体に結合すると視床前部ニューロンがサイレンシングされ、交感神経前部ニューロンの抑制が解除されるという考えが有力である(図8)。この考えは、中村らがラットで発見した、(1)視索前野のEP3受容体発現ニューロンはRMRに投射される、(2)視索前野のEP3受容体発現ニューロンの大部分は、GABA合成酵素GAD67の転写産物を共発現している、(3)GAD67を注射すると、GABA合成酵素GAD67はRMRに投射される、という知見に基づいている。(3)GABAアゴニストであるムシモールをRMRに注射すると、視床下部前野へのPGE2注射による熱発生が阻害される(中村ほか2002)。また、視床前野のアブレーションや視床前野の尾側をナイフで切断すると高体温になることから(Almeida他2006,Chen他1998年、Romanovsky他2003年)視床前野が尾側に位置する熱産生ニューロン群を緊張的に抑制していることが示唆されている。しかし、以下に詳述するように、この記述は非常に単純化された見方である。ここで注意すべきは、RMRに投射する視床下部細胞のうち、EP3受容体を発現しているのはごく少数であるということである。ほとんどはそうではなく(中村 et al 2002)代わりに興奮性のグルタミン酸系細胞である可能性がある(Abbott et al 2016)。さらに、視索前野のすべてのEP3受容体が抑制性Gタンパク質に接続されているわけではない。視索前野におけるEP3受容体サブタイプの解析により、かなりの割合で刺激性Gタンパク質に結合したスプライス変異体を表すことが示されている(Vasilache et al 2007)。PGE2によって興奮する視床下部ニューロンが同定されているが(Ranels and Griffin 2003)一部の細胞がこのような受容体亜種を独占的に発現しているのか、あるいは異なる受容体亜種が同じ細胞に存在しているのかは不明である。最後に、視索前野ニューロンにおけるEP3活性化の下流の分子イベントについては、ほとんど知られていない(Vasilache and others 2013)。これらのニューロンの緊張性抑制活動がPGE2によってサイレンシングされるという考えは、PGE2投与時にその下流の標的で観察された放出現象(Nakamura et al 2002;Nakamura et al 2005)や、他の細胞タイプにおけるこれらの受容体の抑制的役割の観察(例えば、Breyer and Breyer 2001)から推論される。これまでの直接観察では、培養した視床下部前部ニューロンの記録から、PGE2がEP3受容体陽性のGABA作動性ニューロンの発火率を低下させることが示されている(Tabareanら 2004)。また、PGE2で抑制される視床前部ニューロンは、温熱感受性を持つことも示されており(Ranels and Griffin 2003)温熱感受性ニューロンはGABA作動性であり、下行性投射を介して熱発生を促すことがわかっている(Tan and others 2016)。しかし、温熱感受性ニューロンとPGE2応答性ニューロンの集団がどの程度重なっているのかは明らかになっていない。
図8 視床下部前部からのパイロジェン経路の回路図
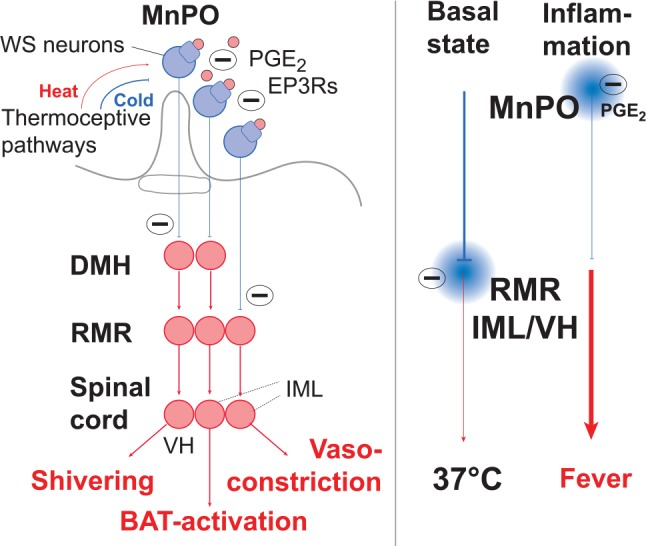
中央視床前核(MnPO)のEP3受容体を発現するニューロンは、視床背内側(DMH)のニューロンだけでなく、吻側髄質瀬状核(RMR)の交感神経前駆細胞にも緊張性の抑制を及ぼし、その結果、サイレンシングが行われる。安静時には、中間外側細胞列(IML)からの交感神経性の熱産生出力、および脊髄腹角(VH)の震動熱産生を担う運動ニューロンへの興奮性出力を抑制する。免疫誘導によりPGE2が放出され(ピンクの丸)EP3受容体(EP3R)に結合すると、MnPOニューロンはサイレンシングされ、その結果、DMHとRMRのニューロンの抑制が解除され、発熱回路が活性化される。EP3発現ニューロンはPGE2に反応するだけでなく、温熱感受性(WS)を有しているため、熱によって活性化されて熱発生を抑制し、寒さによって抑制されて熱発生を促進する。なお、MnPOからRMRへの直接的な投射は血管収縮を制御し、DMHを介した間接的な経路は戦慄および非戦慄(褐色脂肪組織[BAT]の活性化)の熱生成を制御する。
この構造は、視床前部からRMRへの直接投射に加えて、背内側視床下部(DMH)のリレーを介して視床前部からの入力を受ける(図8)。視床前部からRMRへの投射は抑制性であるのに対し、DMHからの入力は、視床前部からの抑制的な制御を受けているにもかかわらず、興奮性である(中村 et al 2005)。逆行性トレーシング実験により、DMHとRMRへのそれぞれの視床前部の投射神経は、大部分がインターカレーションされた異なる集団を構成していることが明らかになっている(中村 et al 2009,吉田 et al 2009)。さらに、この投射ニューロンは、視床下部の2つの異なる核グループ、すなわち、正中部視床前核(MnPO)と背外側視床前野(DLPO)に存在する。両核群は、DMHおよびRMRの標的ニューロンに緊張性の抑制性入力を与えるが、PGE2の作用に反応するのはMnPOのみであり、DLPOは皮膚温度の変化に対応した温度感覚の調節を媒介することが示唆されている(Yoshida and others 2009)。皮膚の冷却や視床前部でのPGE2投与による皮膚血管収縮は、主に視床前部からRMRへの直接投射の抑制に依存しているが、褐色脂肪組織の交感神経活性化による熱発生には、DMHからRMRへの投射の抑制解除が必要であるというデータがある(Rathner他2008)。後者の構造は、RMRへの投射を介して、戦慄熱発生を媒介することも報告されている(Nakamura and Morrison 2011)(図8)。
NakamuraとMorrisonは、寒冷時に誘発される熱発生反応や局所PGE2注射による熱発生反応の研究から、DMHに緊張性の抑制を与えるPGE2感受性ニューロンはMnPOではなく内側視索前核(MPO)に存在し、後者の構造が周囲の寒冷時にMPOにGABA作動性の抑制を与えることを示唆している(Nakamura and Morrison, 2008, 2011)。しかし、Yoshida et al 2009)が指摘しているように、MPOからDMHやRMRの交感神経前部への投射は知られておらず、視床前部にEP3受容体を発現するニューロンが密集していてもMPOは関与していない。
発熱時には体温調節の閾値が変化し、寒冷防御反応と熱溶解反応の両方の閾値が最初に上方にシフトし、その後、閾値間温度帯が拡大することに注意する必要がある(Vybiral et al 1987)。閾値間の拡大は体温の安定性を低下させ、LPSのi.v.注入後に見られるような急激な体温の変化を可能にする。また、麻酔をかけると同様の低体温状態になることもよく知られている(Díaz and Becker 2010)。なぜなら、これらの論文の所見は、麻酔をかけて人工的に常温に保った状態で得られたものだからである(例えば、Nakamura and Morrison 2008, 2011; Nakamura and others 2002; Nakamura and others 2005)。
末梢の炎症に対する発熱反応と体温変化に対する体温調節反応の間には、解剖学的に密接な関係があり、これらの現象の間には密接な機能的関係があると考えられる。発熱反応が周囲の温度によって変化することはよく知られている。周囲温度が低い場合、ラットはLPSを末梢に注射すると、特に高用量の免疫原を投与した場合には、発熱ではなく低体温を示する(Almeida and others 2006; Romanovsky and others 2005)。
熱反応は動物の栄養状態によっても調節される。体温低下を引き起こす飢餓状態は、中性以下の環境温度で飼育されている動物にLPSを投与すると、おそらくレプチン依存性のメカニズムにより、熱反応を強く抑制したり、低体温を引き起こしたりする(井上 et al 2008,Krall et al 2010,Shido et al 1989)。熱反応に対する環境温度と栄養状態の影響は、いずれもDMHによって媒介されていると考えられる。DMHは、LPSによる体温調節にも、敗血症に対する行動的な寒冷探索にも重要であり(Almeida et al 2006,Wanner et al 2017年)DMHのレプチン受容体発現ニューロンは寒冷曝露により活性化され、RMRとのシナプス結合を介して褐色脂肪組織とつながっていることが示されている(Zhang et al 2011)。
空腹時の発熱の減衰には、脳内PGE2産生の減衰は関与していないようである。なぜなら、空腹時には、中枢でのPGE2合成や脳内のPGE2レベルに重要な、末梢から脳への発熱性シグナルが変化しないからである。しかし、空腹時には、脳内に注入されたPGE2に対する反応が弱まる(井上 et al 2008)。また、免疫誘導性低体温のメカニズムはPGE2とは独立しているようで、おそらく別のプロスタノイド合成経路が介在していると考えられているが(Krall et al 2010;Steiner et al 2009)この低体温もまた、ポキロサーマル状態や行動的な体温調節の結果であると示唆されている(Wanner et al 2017)ことにも注意が必要である。mPGES-1を欠損したマウスに見られるように、低体温を促進するメカニズムはパイロジェン反応と共存しているが、一般的には後者によってマスクされたり抑制されたりすることが考えられる(図7)。しかし、LPSの静脈注射後に起こる3相性発熱の最初の体温ピークの後など、通常の発熱時にも急速で活発な体温低下のエピソードが見られることがある。仮に、DMHまたはDMHに接続された構造体が、代謝状態や周囲の温度に関連して免疫刺激の強さ(すなわち、免疫チャレンジの重さ)を評価し、利用可能なエネルギー資源が発熱を引き起こすのに十分かどうか、または低体温がより適応的な反応になるかどうかを決定する統合装置として機能する可能性がある(Liu and others 2012)。
おわりに
SaperとBrederの総説(1994)から 20年にわたる研究の結果、発熱のメカニズムについて深い洞察が得られた。しかし、まだいくつかの重要な問題が残っている。これらは特に、発熱反応の求心性肢に関連している。末梢で進行中の炎症に関する情報を脳に伝達する経路、OVLTに対するパイロジェンの作用、免疫シグナルによる末梢神経の活性化、末梢で合成されたPGE2の脳への輸送、血液脳関門の細胞の活性化などの可能性のうち、血液脳関門を介した体液性シグナルのみが明確な観察によって裏付けられている。これらの観察結果は、サイトカイン受容体またはPGE2合成酵素のいずれかを脳内皮細胞で選択的に欠失させた研究で得られ、そのような欠失が末梢から注入された免疫原に対する発熱反応を弱めることを実証している。しかし、これらの研究では発熱が軽減されたものの、少なくとも免疫原(LPS)をi.p.で投与した場合には、発熱が完全に消失することはなかった。末梢性発熱経路の研究に関しては、栄養状態や環境温度などの要因が末梢性免疫チャレンジに対する反応を形成するメカニズムや、反応が発熱になるか低体温になるかを決定する神経回路がどのように構成されているかについては、まだほとんど理解されていない。また、解熱経路の機能研究のほとんどが、正常な体温調節ができない麻酔下で行われていることも、これらの経路を理解する上での注意点となっている。