
コンテンツ
Dysregulated brain-gut axis in the setting of traumatic brain injury: review of mechanisms and anti-inflammatory pharmacotherapies
https://pubmed.ncbi.nlm.nih.gov/38730498
オンライン公開 2024年5月10日.
AI 解説
一般読者向け解説
この論文は、外傷性脳損傷(TBI)が脳だけでなく腸にも影響を与え、脳と腸の間の双方向のコミュニケーション(脳腸相関)が損なわれることで、脳の炎症や神経変性が悪化することを示唆している。
わかりやすく説明すると、脳と腸は密接につながっており、お互いに影響を及ぼし合っている。脳の損傷は腸の機能を乱し、腸内細菌のバランスを崩したり、腸の炎症を引き起こしたりする。一方で、腸の変化は脳の炎症を悪化させ、脳の損傷からの回復を妨げる。つまり、脳と腸の間で悪循環が生じている。
外傷性脳損傷の患者にとっては、この研究から以下のような実践的な意味合いが考えられる。
- 腸の健康管理の重要性:脳損傷後は、腸内細菌叢を整えたり、腸の炎症を抑えたりすることが大切である。プロバイオティクス(善玉菌)の摂取や、食物繊維の豊富な食事が役立つかもしれない。
- 薬物療法の可能性:この論文では、セロトニンやグレリンなどのホルモン、β遮断薬などの自律神経を調節する薬、スタチンなどのコレステロール低下薬が、脳と腸の炎症を抑える可能性があると述べられている。将来的に、これらの薬が脳損傷の治療に役立つかもしれない。
- 長期的な健康管理:脳損傷の影響は長期間続くことがあるので、腸の健康を含めた全身的な健康管理を継続することが大切である。定期的な検診や、症状の変化に注意を払おう。
- 脳腸相関を意識した治療アプローチ:脳損傷の治療では、脳だけでなく腸の状態も考慮することが重要だと考えられる。医療者と良くコミュニケーションを取り、全人的な治療を心がけよう。
この研究は、外傷性脳損傷の患者のケアと治療における新たな視点を提供している。脳と腸の健康は密接に関係しているため、両方に目を向けることが大切だというメッセージが込められていると言えるだろう。
AI 医療専門家向け
本総説では、外傷性脳損傷(TBI)後の脳腸相関の障害と、それに対する様々な抗炎症薬の役割について包括的に論じた。TBIは腸に重大な変化を引き起こし、全身性免疫系、自律神経系、腸神経系、神経内分泌系、腸内細菌叢の複雑なネットワークを介して脳損傷を悪化させる悪循環を開始する。
主な知見は以下の通りである:
- TBIは腸管バリアの崩壊、腸内細菌叢の変化、腸の運動障害など、腸に多面的な影響を及ぼす。これらの変化は、腸管由来の炎症性メディエーターや病原性細菌の全身循環への移行を促進し、脳の炎症と神経変性を悪化させる。
- セロトニン、グレリン、プロゲステロンなどの腸管由来ホルモンは、抗炎症作用を介して脳損傷を軽減する可能性がある。これらのホルモンを補充することで、TBI後の転帰を改善できる可能性がある。
- β遮断薬やα-アドレナリン作動薬などの自律神経調節薬は、TBI後の交感神経の過活動を抑制し、全身性および局所的な炎症反応を緩和することができる。
- スタチンは、多様な抗炎症・免疫調節作用を介して、TBI後の脳損傷と腸管障害の両方を緩和する可能性がある。
- プロバイオティクスや抗生物質などの腸内細菌叢調節薬は、腸管バリア機能を改善し、病原性細菌の増殖を抑制することで、TBI後の全身性炎症と脳損傷を軽減できる可能性がある。
本総説の知見は、TBI後の脳腸相関の障害が、全身性の炎症反応や腸管バリア機能の破綻を介して、脳損傷の進行と神経学的転帰の悪化に寄与している可能性を示唆している。したがって、TBI患者の管理において、脳だけでなく腸の健康にも目を向け、腸管由来の炎症性メディエーターや病原性因子の全身循環への移行を抑制することが重要と考えられる。
今後は、TBI後の脳腸相関の障害メカニズムをさらに解明するとともに、本総説で取り上げた各種薬物療法の有効性と安全性を検証するための臨床研究が必要である。また、脳腸相関を標的とした新たな治療戦略の開発も期待される。
TBI治療における脳腸相関の重要性を認識し、腸管の健康に配慮した全人的なアプローチを取り入れることで、TBI患者の予後改善につながることが期待される。
要旨
外傷性脳損傷(TBI)は慢性的で衰弱性の疾患であり、精神疾患や神経変性疾患の高いリスクを伴う。予後を改善するための大きな進歩にもかかわらず、効果的な治療法の欠如は、革新的な治療戦略の緊急の必要性を強調している。脳-腸軸は、神経経路、ホルモン経路、免疫経路の複雑なネットワークを通じて、脳と消化器系をつなぐ重要な双方向経路として浮上してきた。このクロストークには、全身性免疫系、自律神経系、腸神経系、神経内分泌系、マイクロバイオームなど、主に4つの経路が関与している。
TBIは腸に重大な変化を引き起こし、脳腸軸を通じて脳損傷を悪化させる無制限な悪循環を開始する。腸における変化には、栄養素/電解質の吸収不良に伴う粘膜損傷、腸管バリアの崩壊、全身性免疫細胞の浸潤増加、運動障害、腸内細菌症、腸内分泌細胞(EEC)の機能障害、腸神経系(ENS)と自律神経系(ANS)の混乱などがある。これらの変化は総体的に、腸-脳軸を介して脳の神経炎症と神経変性にさらに寄与している。
この総説では、TBIにおける脳-腸軸に沿った炎症反応の調節異常を減弱させることができる様々な抗炎症薬物療法の役割を明らかにする。これらの薬物には、セロトニン、グレリン、プロゲステロンなどのホルモン、β遮断薬などのANS調節薬、スタチンなどの脂質低下薬、プロバイオティクスや抗生物質などの腸内細菌叢調節薬などが含まれる。これらの薬剤は、TBI後の脳と腸の両方における明確な炎症経路を標的とすることで、神経炎症を抑制する。これらの治療薬は、脳-腸軸に沿った炎症を緩和し、TBI患者の神経認知の転帰を改善する有望な可能性を示している。
キーワード
TBI、脳腸軸、腸炎症、マイクロバイオーム、腸内分泌細胞
背景
TBIは米国における公衆衛生上の問題であり、毎日約190人の米国人がTBIに関連した傷害で死亡している。[1]。その臨床経過は十分に確立されているにもかかわらず、根本的なメカニズムはまだ完全には解明されていない。[2]。TBIは罹患率と死亡率のリスクを著しく高め、アルツハイマー病、パーキンソン病、慢性外傷性脳症、うつ病、てんかんを含む様々な神経変性疾患や精神疾患の一因となっている。[3]。近年、腸がTBIの病因における重要な役割を果たすことが明らかになっており、TBIが消化管に有害な変化を引き起こすことが示されている。これらの変化は、最終的には腸-脳軸を介して脳の炎症と認知機能を悪化させる。本総説では、TBI後の脳-腸軸機能障害の機序を、脳と腸の間のこの調節不全のサイクルを断ち切ることができる様々な抗炎症薬物療法の役割に特に焦点を当てながら探っていく。
脳神経炎症に対するTBIの影響
TBIは、脳の構造的完全性を破壊するような外部からの機械的障害によって生じる後天的な脳損傷の一形態である。TBIは主に一次損傷と二次損傷に分類される。不可逆的な一次損傷は、転倒、自動車事故、銃創などの鈍的外傷や貫通性外傷によるもので、その後、二次的に調節不全の炎症反応が起こり、組織損傷や神経細胞損傷を悪化させる。[5]。TBIは、即座の臨床的悪化からより重篤な認知的転帰に至るまで、幅広い結果をもたらす。臨床症状には、局所性およびびまん性の脳腫脹、血管攣縮 [6]、血行動態の乱れ(低血圧 [7]、低酸素 [8,9])、代謝異常(低血糖/高血糖) [10]、頭蓋内圧(ICP)の上昇、凝固障害 [11]などがある。さらに、TBIは急性および慢性の認知障害を引き起こし、注意欠陥、記憶障害、実行機能障害を特徴とする。細胞レベルでは、TBI直後の損傷関連分子パターン(DAMPs)の急速な放出により、調節不全の神経炎症反応が始まる。これには、常在脳細胞(ミクログリア、アストロサイト、オリゴデンドロサイト、ニューロン)の活性化と、損傷部位への様々な免疫細胞(単球、好中球、B細胞、T細胞)の動員を伴う。DAMPsは、ヌクレオチド結合オリゴマー化ドメイン様受容体やtoll様受容体などの「パターン認識受容体」に結合し、炎症性ケモカイン、サイトカイン、活性酸素種(ROS)を増加させる。これが正のフィードバックループを引き起こし、さらに免疫細胞を傷害部位に動員して炎症反応を激化させる。[13, 14]。TBIの特徴として、神経細胞傷害、軸索損傷、ミトコンドリア機能障害、興奮毒性、酸化ストレス、血液脳関門(BBB)破壊などが挙げられる。[15, 16]。BBBの破壊は、内皮細胞の死、タイトジャンクションタンパク質の分解、基底膜の損傷、アクアポリン4(AQP4)チャネルの再分布、アストロサイト終末の膨潤によって起こり、脳浮腫とICPの上昇を引き起こす。[17]。興奮毒性は、グルタミン酸の細胞外腔への放出の亢進と、それに続くカルシウム(Ca2+)の細胞内への侵入を伴い、プログラムされた細胞死を引き起こす。[18]。さらに、代謝要求の増大とミトコンドリアのアデノシン三リン酸(ATP)産生量の減少との間の不均衡が、炎症反応をさらに悪化させる。ミトコンドリアの機能不全は、カスパーゼ、B細胞リンパ腫2(Bcl-2)ファミリータンパク質、アポトーシス誘導因子の発現の変化に加え、活性酸素の増加、Ca2+過剰負荷、興奮毒性の組み合わせから生じる。[19]。TBIはまた、前臨床モデルと臨床モデルの両方において、脳、脳脊髄液、血清中の多価不飽和脂肪酸レベルを増加させることが示されている。[20, 21]。これらの脂肪酸はアラキドン酸代謝経路を活性化し、プロスタグランジンとロイコトリエンの放出につながり、脳損傷をさらに助長する。[22]。
アストロサイトとミクログリアは、TBI後の炎症反応を引き起こすCNSの重要な細胞である。[13]。これらの細胞は、炎症性または抗炎症性の表現型を獲得する能力を持ち、様々なケモカイン、サイトカイン、成長因子を分泌することができる。[23, 24]。これらの変化は局所的な組織微小環境に影響を与え、二次的な細胞損傷や組織修復を調節する。[25]。アストロサイトは、分子的、構造的、機能的変化を伴う反応性アストログリオーシスと呼ばれる過程を経る。[26]。アストロサイトは、神経炎症反応、瘢痕形成、血液脳関門伝染性、シナプスリモデリングを制御している。同時に、活性化したミクログリアは病変部位に移動して残骸を貪食し、様々なサイトカインやケモカインを分泌して炎症プロファイルを調節する。ミクログリアは、その活性化状態(炎症促進性/抗炎症性)によって、神経変性と組織修復において異なる役割を果たすことができる。炎症性ミクログリアは、神経傷害を悪化させるサイトカインの産生を促進する一方、抗炎症性ミクログリアは貪食の役割を獲得し、向神経性因子を放出することで修復を促進する。[30, 31]。最近では、TBI後の腸内細菌叢の変化が脳内のミクログリアの表現型を変化させ、最終的にTBIの転帰に影響を及ぼすことが示され、その役割が拡大している[32]。アストロサイトとミクログリアの両方が慢性的に活性化すると、伝染性BBBを介した末梢免疫細胞の浸潤が増加し、認知の転帰をさらに悪化させ、罹患率と死亡率のリスクを高める。
TBI後の脳腸軸障害
TBIは全身性の疾患であり、肺、消化管、肝臓、腎臓などさまざまな末梢臓器に影響を及ぼす。[34-37]。TBI生存者は健常者に比べて、敗血症 [38]、肺炎 [39]、消化器疾患 [40]による死亡リスクが高い。さらに、TBIは胃不全麻痺と腸運動障害を誘発し、摂食不耐性をもたらし、重度のTBI患者の50%が受傷後1週間以内に摂食不耐性を経験する。[41]。McConnochieらによって実施された単一施設の観察研究では、TBI患者の約52%が排便遅延をしばしば経験し、このことが集中治療室(ICU)滞在期間の延長や胃残量の増加と関連していることが示された。[42]。また、あるレトロスペクティブ研究では、TBI患者は一般集団と比較して、消化器疾患関連疾患で死亡する可能性が2.5倍、敗血症で死亡する可能性が12倍高いことが明らかにされている。[43]。別のレトロスペクティブ研究では、リハビリテーションセンターに入院した後天性脳損傷患者の便失禁率が最大70%であることが示されている。[44]。便失禁はまた、前頭葉病変や血行動態の不安定性の有無とも有意に相関していた。さらに、腹痛、腹部膨満感、便秘は、最初の脳損傷から2年経過した患者にもよくみられる症状であった。[46]。したがって、現在のガイドラインでは、TBIによる腸機能障害を軽減し、患者の予後を改善するために、24~48時間以内に経腸栄養を開始することが推奨されている。[47]。TBIによる腸機能障害には、全身性免疫調節障害、ANS機能障害、腸内細菌叢異常、神経内分泌系障害など、さまざまな異なる重複経路も関与している(図1)。
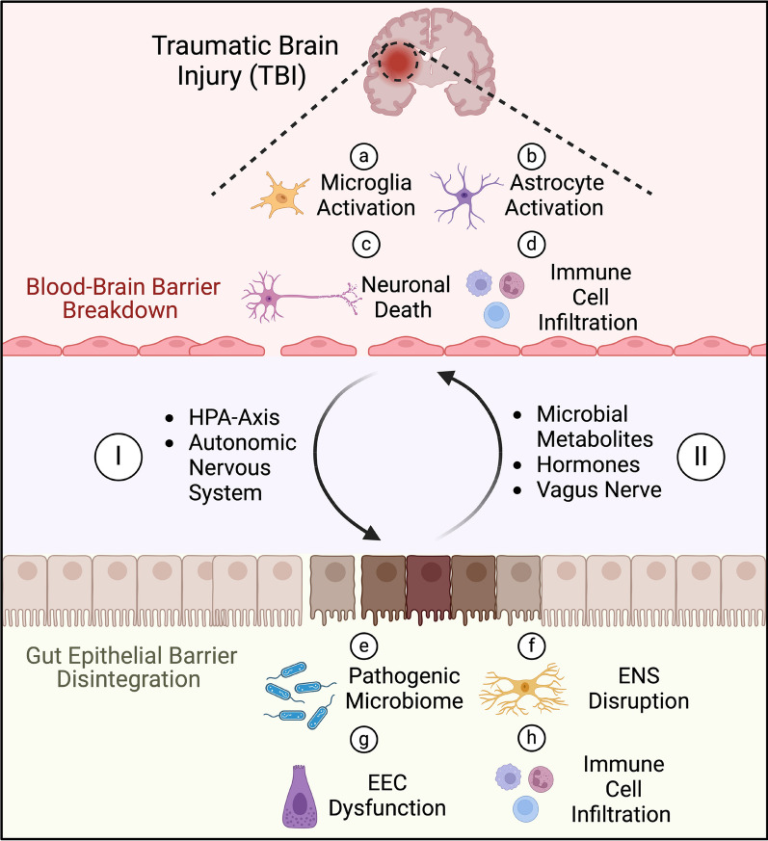
図1
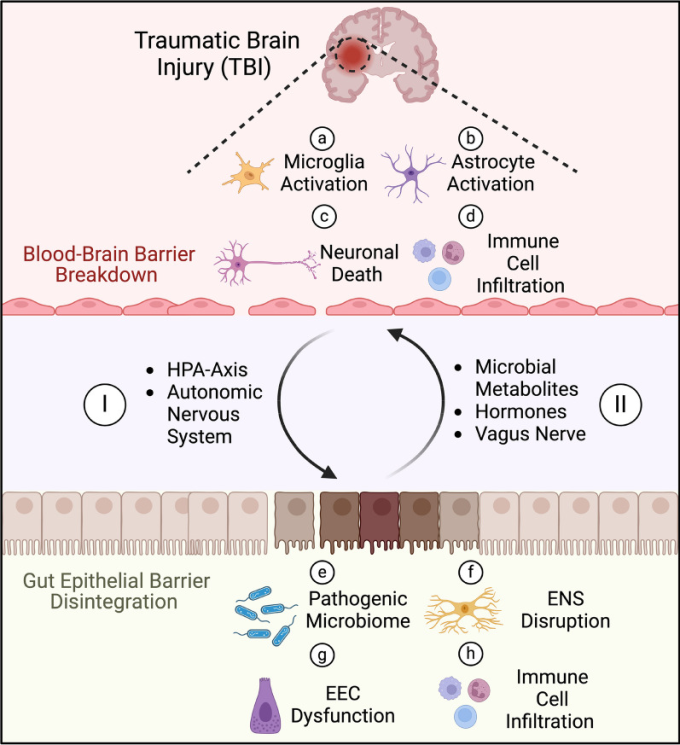
TBIにおける脳と腸の双方向クロストーク。TBIは組織と細胞の破壊を引き起こし、さまざまな炎症性サイトカイン、ケモカイン、補体因子、損傷関連パターン(DAMPs)を放出させ、多様な細胞応答を促進する:a 脳の常在免疫細胞であるミクログリアが活性化し、損傷部位に向かって移動して破片を貪食し、炎症性サイトカインを放出する。c 活性酸素種(ROS)の産生増加、興奮性グルタミン酸の過剰放出、脳への血流低下により、神経細胞のアポトーシスと神経変性が促進される。 d 血液脳関門の破壊により、好中球、単球、T細胞などの様々な免疫細胞が脳実質に浸潤し、破片の除去を助ける。しかし、過剰な浸潤は組織損傷を悪化させ、神経学的転帰を悪化させる。I TBIは、コルチゾールを放出する視床下部-下垂体-副腎(HPA)軸の活性化や、カテコールアミンを放出する自律神経系(ANS)の交感神経の活性化など、いくつかの経路を通じて腸に影響を及ぼし、腸管バリアーの崩壊をもたらす。 e これにより、病原性細菌が腸管内腔から腸実質に移行し、微生物異常症を悪化させる。h T細胞や単球などの炎症性免疫細胞が腸管上皮に浸潤し、腸の炎症をさらに悪化させる。(Ⅱ)短鎖脂肪酸(SCFA)や胆汁酸などの微生物代謝産物の放出の減少、EECからの抗炎症ホルモン分泌の減少は、この有害なサイクルを悪化させる。さらに、求心性迷走神経経路と遠心性迷走神経経路の障害は、脳-腸のホメオスタシスを破壊し、傷害を受けた脳の神経炎症反応を悪化させる。これらの重なり合い、相互に関連する経路は、TBIによる神経炎症を緩和し、神経学的転帰を改善するための潜在的な治療標的となる。www.Biorenderで作成
全身性免疫調節異常
TBIは、末梢免疫系を調節することにより、全身経路を通じて消化管に有害な影響を及ぼす。[48]。TBIは、視床下部-下垂体-副腎(HPA)軸および交感神経系の活性化を誘発し、その結果、グルココルチコイド/コルチゾールおよびカテコールアミンのレベルがそれぞれ上昇する。[49]。このようなコルチゾールの急増は、腸管バリアの伝染性を高めることにより、最終的にリーキーガットにつながる可能性がある。[50, 51]。その後、崩壊したバリアを越えて内腔の病原性細菌が移動することで、全身性炎症反応症候群(SIRS)が誘発され、多数のサイトカインやケモカインが全身循環に放出されることで、全身性炎症がさらに悪化する。[52]。TBIは、TNF-α、IL-1β、IL-6などの炎症性メディエーターの急性増加を引き起こし、これらのメディエーターは認知予後の悪化と関連している。IL-1βレベルの上昇は、急性期におけるワーキングメモリーの障害と関連しており、ケモカインリガンド2(CCL2)レベルの上昇は、脳震盪後の症状の重症度と相関している。さらに、受傷直後のサイトカインスコア負荷の増加は、TBI後6カ月および12カ月のGlasgow Coma Scale(GCS)スコアの悪化と相関することが、さらなる研究によって示されている。さらに、血中コルチゾールレベルの上昇は、末梢の免疫抑制を引き起こすことにより、感染症のリスクを上昇させる可能性がある。[56]。TBIの結果、循環好中球が増加し、循環単球、T細胞リンパ球、ナチュラルキラー(NK)細胞が減少し、貪食能の欠損を伴う。[57, 58]。これは、好中球や単球の呼吸バーストや貪食能の障害、パーフォリン陽性NK細胞の割合の減少によって特徴づけられる。[59, 60]。さらに、TBIは胸腺の退縮を引き起こし、慢性的なT細胞リンパ球減少を引き起こす。これらの変化は、全身免疫の抗炎症状態への移行をもたらし、TBI後の入院患者における院内感染リスクの上昇を説明しうる。[61]。このような変化は、TBIの影響と急性的に闘うためには必要かもしれないが、慢性的なストレス反応は、多臓器機能不全を引き起こし、最終的には死に至る可能性のある炎症亢進状態を長期化させる。[62, 63]。興味深いことに、TBIはまた、加齢に関連したミクログリアの表現型を増加させることが示されており、ミクログリアにおける脂質の蓄積によって、受傷後1年まで描かれている。[64]。神経免疫反応におけるこれらすべての急性、亜急性、慢性の変化は、炎症を悪化させ、免疫老化と神経変性を加速させる。
TBI後の腸の炎症に対する全身免疫系の影響を評価するために、我々のグループは、末梢の炎症性C-Cケモカイン受容体2型(Ccr2)依存性単球をノックアウトすると、腸の腫瘍壊死因子α(TNF-α)、インターロイキン-1β(IL-1β)、リポカリン-2のレベルが低下することを証明した。さらに、TBIは損傷直後に腸管toll-like receptor 4(TLR4)の発現を有意に増加させるが、これは腸管炎症の悪化を説明する可能性がある。注目すべきことに、Ccr2koマウスでは、受傷後1日目と3日目に腸管TLR4のレベルが低下していた[66]。これらの知見は、TBI後の脳と腸の双方向コミュニケーションを調節する上で、全身の自然免疫系が重要な役割を担っていることを示唆している。自然免疫系に加えて、適応免疫、特にT細胞が脳-腸軸に沿って神経炎症反応を調節することが示されている。[67]。T細胞はTBI後5日目から脳に浸潤し、[68]、グランザイム+ CD8+ T細胞はTBI後8カ月目にも検出されている。[69]。Daglasらは、CD8+ T細胞の薬理学的および遺伝学的枯渇が神経学的転帰を改善し、神経保護的なTh2/Th17免疫学的変化をもたらすことを示した。最近の研究では、腸内細菌叢がエフェクターT細胞の腸からレプトメン膜への輸送を調節し、脳卒中とTBIの両モデルにおける傷害の転帰に影響を与えることも示されている。[70]。T細胞輸送は、中枢神経系損傷後のマイクロバイオームの変化によって調節され、最終的に脳内のミクログリア機能を制御すると考えられている。[71]。腸の自然免疫と適応免疫、特にマクロファージ/単球とT細胞が、TBI後の神経認知の転帰を調節する役割を理解するために、さらなる研究が必要である。
自律神経系および腸管神経系の機能障害
HPA軸に加えて、中枢神経系はANSの分枝を通じて腸に影響を及ぼす。副交感神経系、交感神経系、固有ENSなど、ANSのすべての部門が消化管を支配している。[72]。脳と腸の双方向軸は、ANSがENSに影響を及ぼす導管として機能する。TBIは交感神経の亢進と関連しており、その結果、循環カテコールアミン濃度が急上昇し、様々な末梢臓器、特に消化管に重大な影響を及ぼす。[73]。カテコールアミンの増加は、受傷後数週間持続し、全身的な免疫抑制を引き起こし、臨床転帰を悪化させ、罹患率と死亡率を増加させる重要な役割を果たす。TBIは全身のエピネフリンによる交感神経ストームを誘発し、血液を消化管から遠ざけ、その結果、腸の運動障害と胃不全麻痺を引き起こす。腸内のカテコールアミン濃度の上昇も、主にコリン作動性であるENSの恒常性を乱す。Maらによる研究では、TBI後4週間で、大腸の腸グリア細胞(EGC)の活性化が亢進し、ENSの調節異常が証明された。反応性グリオーシスとして知られるこの過程は、腸の運動障害をさらに助長する可能性がある。副交感神経系に関しては、迷走神経が脳と腸の双方向の神経免疫相互作用の主要な調節因子である。[78]。迷走神経は、脳から腸へ、神経節後性求心性ニューロンを介してシグナルを送り、アセチルコリン(Ach)を分泌することによって腸の分泌運動機能を調節し、それによって腸の運動を促進する。[79]。さらに、Achは腸管マクロファージに存在するニコチン性アセチルコリン受容体のα7サブタイプ(α7nAChR)にも結合し、炎症性サイトカインの産生を低下させる。[80]。最近の知見では、Ach生合成の律速酵素である細胞外コリンアセチルトランスフェラーゼ(ChAT)が、全身の炎症を抑え、炎症性サイトカインの放出を抑制することも明らかにされている。この効果は、迷走神経刺激またはChATの生理活性組換え型投与により観察され、受傷後2週間のDSS大腸炎モデルマウスにおいて、血清中の炎症性マーカーTNF-αとIL-6のレベルが低下したことで証明された。[81]。一方、迷走神経は、その求心性ニューロンを介して腸から脳へ情報を伝達し、脳の神経炎症性転帰を制御している。内臓求心性神経は、マイクロバイオームの多様性の変化 [83]、腸内分泌細胞(EEC)から分泌される神経ペプチドやホルモン [84]、炎症性メディエーターに対する感作など、様々な機械的・化学的刺激 [82]に反応する。迷走神経機能の重要性は、迷走神経刺激(VNS)の前臨床試験で強調されており、プラセボと比較して、TBI後の認知アウトカムの改善が実証されている。[85]。したがって、自律神経失調症は受傷後のTBI転帰を媒介する上で重要な役割を担っており、β遮断薬を使用する可能性がある交感神経ストームを減弱させることを治療目標とすべきである。さらに、副交感神経の迷走神経腕を刺激し、腸管伝染性、腸管運動、神経行動学的転帰などの機能的転帰に加え、脳と腸の免疫細胞反応への影響を研究することに、より焦点を当てるべきである。
腸内細菌叢異常症
腸内細菌叢が行動転帰や認知に及ぼす影響は、急速に拡大している研究分野であり、腸内の変化がTBIの病態生理学において重要な役割を果たしていることが示唆されている。[86]。TBIがマイクロバイオームの多様性と豊かさに及ぼす影響については、前臨床 [87, 88] および臨床 [89, 90] で広く研究されている。TBIは病原性細菌の増加と防御集団の減少をもたらす。このような変化は、TBI後早ければ2時間で現れ [92]、最初の受傷から数年間 [89] 持続することが示されている。腸の運動性の変化やパネス細胞の発現の変化など、様々な因子が微生物異常に関与している可能性がある。腸の蠕動運動が低下すると、微生物組成が病原性の状態に移行し、その結果、腸の運動性が悪化し、運動障害とディスバイオシスの間の有害なサイクルが引き起こされる。[93]。さらに、パネス細胞は腸の陰窩に存在する別のタイプの上皮細胞で、リゾチームやα-ディフェンシンなど、様々な抗菌ペプチドを分泌する。[94]。また、炎症性腸疾患(IBD)や過敏性腸症候群(IBS)などの様々な腸疾患において、細菌マイクロバイオームの組成を制御することも示されている。TBIの設定において、ヤン(Yang)らは、リゾチーム抗菌ペプチドの発現低下と、崩壊した上皮バリアを通過する病原性マイクロバイオームの移動増加との間に有意な相関関係があることを示した。[96]。これらの変化は、TBI後の微生物叢-腸-脳軸の調節におけるパネス細胞の重要な役割を示唆している。
腸内細菌叢が脳に及ぼす影響は、主に血管、神経、免疫経路を介して媒介される。TBIは粘膜損傷と上皮バリア伝染性の亢進を引き起こし、これにより管腔内の病原性細菌の腸実質への移行が促進される。[96]。腸内細菌叢はまた、食事成分を短鎖脂肪酸(SCFA)、トリプトファン代謝産物、胆汁酸代謝産物などの様々な代謝産物に変化させる。[97]。これらの微生物代謝産物は、迷走神経求心性神経の受容体に結合することによって脳に直接影響を及ぼすか、あるいは全身循環に入り、腸内微生物と中枢神経系(CNS)の細胞(主にアストロサイト、ミクログリア、ニューロン)との間の細胞間相互作用を調節することによって間接的に影響を及ぼす可能性がある。[98, 99]。Xiongらの研究では、TBI後6カ月間、酪酸や酢酸などのSCFAを投与すると、ミクログリアの活性化が抑制され、抗炎症性ミクログリアの表現型が促進されるとともに、T細胞の活性化や細胞障害関連経路も抑制されることが実証された[100]。同様に、抗生物質、特にアモキシシリン-クラブラン酸の投与は、TBIの2日後に脳へのT細胞の浸潤を減少させた。[70]。様々なマイクロバイオームの代謝産物とミクログリアやT細胞などの脳免疫細胞との相互作用は、最終的に認知や神経行動学的転帰に影響を及ぼす可能性がある。したがって、健全なマイクロバイオームを促進することを目標とした治療法は、脳腸軸に沿った炎症反応を改善し、TBI後の認知転帰を改善する上で不可欠である。
神経内分泌系の障害
これまでの研究では、TBI後の脳腸双方向コミュニケーションの研究に焦点が当てられ、HPA軸、ANS、細胞性免疫反応、マイクロバイオーム軸の役割が強調されてきた。しかし、最終的に認知の転帰に影響を与えうる多様なホルモンを放出する腸の能力を考えると、神経内分泌軸の徹底的な検討が必要である。[101]。EECは腸管上皮を覆う特殊な細胞で、体内で最大の内分泌系を形成している。EECは、体内のセロトニンの90%、グレリン、グルカゴン様ペプチド1(GLP-1)、ニューロペプチドY、コレシストキニンを含む様々なホルモンやペプチドを放出することにより、脳と腸のクロストークを調整する極めて重要な役割を果たしている。[101, 102]。EECはまた、迷走神経との興奮性シナプス結合 [103]を介して脳に直接影響を与えたり、様々なホルモンを全身循環に放出する。[104] ことによって間接的に影響を与えたりすることもあり、脳と腸の間のインターフェースを形成している。EECの主要マーカーであるクロモグラニンA(ChgA)の発現が有意に低下していることからわかるように、TBI後3日目にEECの発現が大幅に低下していることを示したのは、われわれのグループが初めてである(図2A-C)。また、ノッチ受容体1(Notch1)、atonal bHLH転写因子1(Atoh1)、ニューロジェニン3(Neurog3)など、ロイシンリッチリピート含有Gタンパク質共役型受容体5(Lgr5+)腸管幹細胞の成熟ChgA+細胞への分化に関与する転写因子の減少も示した(図2D、E)。脳と腸のコミュニケーションにおけるEECの関与は、パーキンソン病や統合失調症などの様々な神経疾患や精神疾患でも研究されている[105, 106]。パーキンソン病では、EECがα-シヌクレインのミスフォールドタンパク質を発現し、それがα-シヌクレインを含む神経に直接つながり、腸と神経系の間に神経回路を形成していることが示されている[106]。EECが産生する様々なホルモン、特にセロトニン、グレリン、GLP-1は主に抗炎症作用がある。
図2
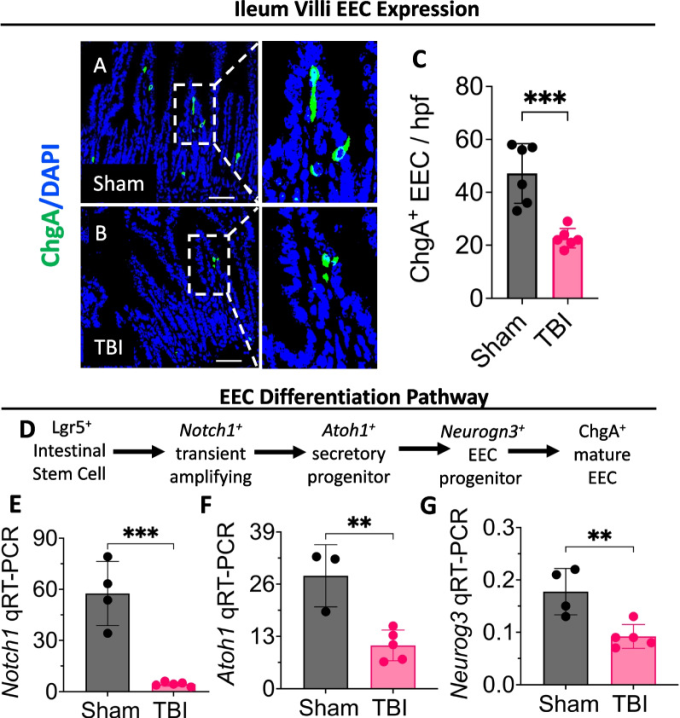
TBIは腸内分泌細胞の消失を誘導し、EECの分化を低下させる。雄性C57BL/6マウスを4-6週齢で用い、既述のように中等度-重度のTBIを誘導した[66]。3日後にマウスを犠牲にし、回腸を採取してEECの発現を調べた。腸管組織は4%パラホルムアルデヒドで一晩固定し、パラフィン包埋用に処理した。その後、CUT 6062ミクロトーム(SLEE Medical GmbH, D-55129 Mainz, Germany)を用いてパラフィンブロックから5μmの組織切片を切り出し、DAPI(青)とクロモグラニンA(ChgA)(緑)で染色した。ChgA+細胞はImageJ2ソフトウェアを用いてカウントした。スケールバーは50μmである。A-C 受傷後3日目(PID)において、シャムと比較してTBIマウスの腸管ChgA発現が減少していることを示す代表的な共焦点画像。 D 腸におけるEEC分化経路の説明図。TBIは、Lgr5+腸管幹細胞からChgA+成熟EECへの分化に関与する主要転写因子(E Notch1、F Atoh1、G Nuerog3など)の発現を低下させることが、定量的逆転写ポリメラーゼ連鎖反応(qRT-PCR)によって測定された。mRNAレベルはハウスキーピング遺伝子Rplp0発現に対する相対値として表した。統計的有意性は、GraphPad Prism 10ソフトウェアを用いたstudent’s t-testによって決定した。グラフ上の各点は異なるマウスを表す。エラーバーは平均値±SEMを示す。*p< 0.05, **p< 0.01, ***p< 0.001
まとめると、脳と腸の間の双方向コミュニケーションは、全身の免疫経路、神経ネットワーク、内分泌ホルモン、微生物叢軸を通して起こり、それによってTBI後の消化管に有害な変化を引き起こす。これらの変化は、図3に示すように、脳損傷をさらに悪化させる。このような有害なサイクルを断ち切り、脳と腸における明確なメカニズムを通じて炎症を軽減する上で、多様な治療薬が果たす役割を支持する強力な証拠がある。この包括的な総説では、(1)セロトニン、(2)グレリン、(3)プロゲステロンなどのホルモン、(4)β遮断薬などのANS調節薬、(5)スタチンなどの脂質低下薬、(6)プロバイオティクス/抗生物質などの腸内細菌叢調節薬など、様々な治療手段の抗炎症的役割に焦点を当てる。これらの治療的介入が、TBI後の脳腸軸に沿った炎症亢進反応をどのように減弱させるかについて述べる。
図3
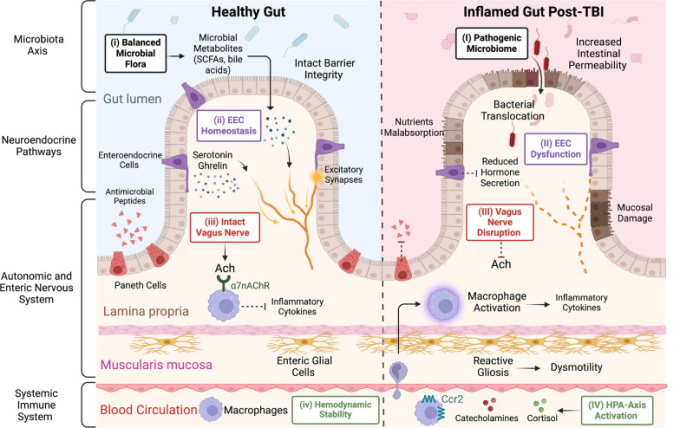
TBIは脳腸軸を介して腸機能障害を引き起こす。この図は、TBIが脳腸軸を介して腸の機能障害を引き起こす複雑な経路を示している。脳と腸の間のクロストークには、全身性免疫系、自律神経系、腸神経系、神経内分泌系、微生物叢軸を含む4つの主要経路が関与している。健康な腸(左)では、(i) ランスのとれた微生物叢が、食事成分を短鎖脂肪酸(SCFA)、トリプトファン代謝産物、胆汁酸などの様々な代謝産物に変換する。SCFA、特に酢酸、プロピオン酸、酪酸は、ニューロンやミクログリアのエネルギー基質として働くことで、脳機能に様々な有益な作用を及ぼす。(ii)さらに、腸内分泌細胞(EEC)は、体内最大の内分泌系であり、管腔刺激に応答して、セロトニン、グレリン、グルカゴン様ペプチド1(GLP-1)などの様々なホルモンを分泌することにより、消化プロセスを調節している。これらの細胞は、血流を介してホルモンのシグナルを脳に送ったり、迷走神経求心性経路を介して神経のシグナルを送ったりすることで、中枢神経系と双方向の情報伝達を行っている。(iii) 無傷の迷走神経はアセチルコリン(Ach)も放出し、このアセチルコリンは腸マクロファージにあるニコチン性アセチルコリン受容体のα7サブタイプ(α7nAChR)に結合し、炎症性サイトカインの産生を低下させる。Achはまた、消化管の平滑筋細胞に存在するムスカリン受容体にも結合し、蠕動運動を促進する。Achはまた、ENSの興奮性運動ニューロンを刺激し、腸の運動性をさらに高める。(iv) 正常な恒常性では、免疫細胞の活性化はバランスよく行われ、コルチゾールの分泌は日内リズムに従い、カテコールアミンのレベルは生理的範囲内に保たれ、全体として安定した循環と組織灌流が維持される。しかし、TBIによる腸管機能障害は、一連の変化を引き起こす(右図)。(I)マイクロバイオームが病原性状態へと移行し、細菌が腸関門を通過して腸実質へと移動する。(II)EECが機能不全に陥り、その発現、分化、抗炎症ホルモンの分泌が低下する; (III)同時に迷走神経の機能障害が起こり、マクロファージを炎症亢進状態に偏向させ、炎症を亢進させるAchが減少する。(IV)TBI中の視床下部-下垂体-副腎(HPA)軸の活性化は、循環カテコールアミンとコルチゾールを増加させ、リーキーガットと粘膜損傷を引き起こす。さらに、全身性の単球やT細胞が腸に浸潤し、炎症性サイトカインをアップレギュレートすることで炎症を悪化させる。さらに、腸グリア細胞(EGC)の反応性グリオーシスが腸の運動障害を引き起こす。最後に、パネス細胞は抗菌ペプチドの分泌を減少させ、マイクロバイオーム異常症をさらに悪化させる。これらの複雑な変化は、腸-脳軸を通じて、脳の炎症と神経変性を悪化させる。www.Biorenderで作成
TBI後の脳腸軸の混乱を抑制する治療法
セロトニン
脳内では、セロトニンは脳幹にあるラペ核に由来するニューロンによって産生される。セロトニンは、気分を調節する役割で知られる神経伝達物質として働くが、神経新生や可塑性 [107-109]、認知 [110,111]、記憶 [112]、炎症 [113]、腸の運動 [114,115]などの他の過程にも関与していることが示されている。炎症状態で発現が増加するセロトニンとキヌレニンは、どちらもトリプトファンから合成され、その利用可能性をめぐって競合する。体内のトリプトファンのほとんどはキヌレニンの合成に使われ、セロトニンに使われるのは少数である。トリプトファン水酸化酵素(TPH)は5-HT(セロトニン)合成の律速酵素であり、中枢神経系と末梢器官の両方に存在する。セロトニンはBBBを通過できないため、体内のセロトニンの90%の合成を担う腸のEECではTPH1を介して、CNSではTPH2を介して合成される。[107, 112, 113, 115]。5-HT1~5-HT7の7つのクラスに分けられる合計14のセロトニン受容体が知られている。5-HTが活性化する受容体、体内の位置、活性化する濃度によって、その作用が決定される。[107, 112, 113, 115]。
TBI患者ではセロトニンレベルの変化が観察されているが、これはトリプトファン代謝の変化によって説明できる。[109]。Zhangらは、制御された皮質衝撃を受けたウサギが炎症を増加させ、キヌレニン合成を担う酵素の発現を上昇させ、トリプトファンをセロトニン合成からシフトさせたことを示した。セロトニンレベルは有意に減少しなかったが、セロトニン/トリプトファン比が有意に減少し、偽薬と比較してセロトニン線維の平均と全長が減少した。[109]。トリプトファン枯渇の臨床試験では、認知障害が海馬の5-HTによって媒介されることが証明されていることから、5-HTレベルのこれらの変化は脳機能障害に寄与する可能性がある。[112]。このような証拠を考慮すると、TBIの治療または転帰の変更におけるセロトニンの役割は研究中である。[66, 116, 117]。TBI患者の治療における選択的セロトニン再取り込み阻害薬(SSRI)の有効性に関するデータは、前臨床モデルから得られている。Weaverらは、制御された重度の皮質衝撃を受けたマウスに、直接腹腔内(IP)フルオキセチン(5mg/kg)を単回注射したところ、プラセボを投与したマウスと比較して、TBI後4日目から大腸伝染性が低下し、運動協調性が改善したことを示した[117]。Craineらは、セロトニン・ノルエピネフリン再取り込み阻害薬(SNRI)であるミルナシプランの慢性IP投与(30mg/kg/日)が、前頭葉損傷を受けた雄ラットの認知改善において果たす役割を研究した。TBI後、ミルナシプランを投与したラットは、プラセボを投与したラットと比較して、注意セットシフト課題において有意に良好な成績を示し、その成績は偽ラットで見られた成績と同等であった。[111]。ヒトでは、Yueらによる系統的レビューで、SSRIの投与がTBI後の抑うつ症状の改善に関連することが示された。[118, 119]。TBIに加えて、多くの中枢神経系疾患が5-HTレベルの変化と関連している。例えば、アルツハイマー病(AD)患者では、5-HTおよび5-HT受容体の総レベルが低下していることが判明しており、[120-122]、キヌレニンの増加とともに5-HTが低下することは、認知機能障害および炎症の増加と関連していた。[123]。同様に、パーキンソン病患者においても、5-HTの枯渇は認知機能の悪化と関連していた。[124-126]。健常者では、トリプトファン枯渇による急性セロトニン枯渇が、言語記憶や感情処理の障害と関連している。[127, 128]。この証拠は、SSRIがこれらの病態のいくつかを緩和していることからも裏付けられる。前臨床研究では、SSRIによる治療がADの雌マウスのアミロイドβ斑を減少させ、認知機能、学習、記憶の改善と関連することが示された。[129]。また、SSRIの投与期間が長いほど認知症予防効果が高いという証拠もある。[130, 131]。前臨床モデルでは、異なる5-HT受容体のアゴニズムが記憶や認知にプラスにもマイナスにも影響することが示されているため、セロトニンがこれらの状態を改善するメカニズムは完全には解明されていない。[132, 133]。
腸は体内の5-HTの主な供給源であるため、TBI患者においてセロトニンレベルが変化することは驚くべきことではない。Mercadoらは、中等度TBIを受けた雄性マウスモデルでは、十二指腸と結腸のTPH1がそれぞれ0.9倍と0.5倍と有意に減少していることを示している。さらに、免疫蛍光染色で結腸のセロトニン発現が減少し、偽薬と比較して結腸セロトニン再取り込みトランスポーター(Sert)が有意に増加した。これは、末梢循環における5-HTレベルの全体的な有意な減少として反映された。[116]。さらに我々のグループは、雄マウスの重度の片側TBI後、腸上皮、特に回腸に沿ってEECの発現が有意に低下することを初めて証明した。その結果、セロトニン合成遺伝子、特にセロトニン合成を担うもう一つの必須酵素であるドーパ脱炭酸酵素のレベルが低下した。[66]。TBIにおける炎症と運動障害のために起こる長期的な腸内細菌異常症は、5-HTレベルの変化を説明するもう一つの要因である。[4, 134]。腸内細菌叢の変化に関する研究は、誘発されたものであれ、IBSのような疾患状態であれ、5-HTレベルおよび行動の変化と相関している。[135-137]。同様に、統合失調症 [138]やうつ病 [139]を患う人々は、対照群よりも病原性微生物叢を保有し、5-HTレベルが低下している。[140, 141] ことから、双方向の相互作用が強調されている。
消化器系では、セロトニンは5-HT3および5-HT4を介して運動性を調節し、カハール間質細胞(ICC)の5-HT2Bに作用して蠕動運動を調節する役割を果たすことが示されている。TBIやその他の神経疾患に加え、IBD [142, 143]、セリアック病 [144, 145]、大腸炎 [146]、憩室炎 [147]などの腸疾患においても、5-HTレベルの変化が広く報告されている。セロトニンは、機能不全の症例において食道運動を改善し、[148]、便秘を伴う炎症性腸症候群の患者において便の回数を増やし、その硬さを改善することが示されている。[149]。興味深いことに、SSRIの慢性投与は、マウスおよびヒトを対象とした研究 [150, 151]において、腸管運動の低下および通過遅延と関連しており、セロトニンシグナル伝達の複雑さを浮き彫りにしている。5-HTはまた、免疫細胞上に存在する様々な受容体を介して、炎症促進作用と抗炎症作用の両方を発揮することにより、腸における炎症を制御することが示されている。[152]。腸上皮細胞はその頂膜にセロトニン再取り込みトランスポーター(Sert)を発現しており、[153]、TBIが結腸絨毛の腸上皮におけるこれらの受容体の発現を増加させることを考慮すると [116]、フルオキセチンのようなSSRIの投与は、腸脳軸相互作用およびTBI後の神経行動学的転帰に対する腸セロトニンの影響を探索するための有望な手段となる。
グレリン
1999年に初めて同定されたグレリンは、主に胃のP/D1細胞から産生されるが、少量は小腸、膵臓、脳からも放出されるオレキシジェニックペプチドホルモンである。グレリンは、下垂体の調節、空腹感、満腹感に作用することでよく知られている。[154-156]。さらに最近では、その抗炎症特性がよりよく理解されるようになってきた。[157, 158]。脳損傷モデルにおける外因性グレリンの役割に関する研究により、グレリンが同時に消化管機能障害を改善し、さまざまな神経保護効果を発揮するメカニズムがいくつか明らかになった。グレリンの神経保護作用は、BBBの保全、酸化的損傷の減少、および他の神経保護機構と関連している。TBIの体重減少(WD)モデルにおいて、雄マウスにグレリン(合計20μg)をIP投与すると、神経細胞の変性が減少し、血管伝染性とAQP-4の血管周囲発現が減少することがわかった[159]。AQP-4の増加は、TBI後の細胞浮腫と関連していることが示されており、[160]、TBI誘発性の神経細胞損傷に寄与している。この研究では、脳損傷の神経生化学的マーカーであるS100Bも、TBIと同時にグレリンを投与されたマウスの血清中で減少していることが判明しており[159, 161]、グレリンがBBBの保全を通じて外傷後の神経細胞損傷とアポトーシスを予防するという証拠が追加された。TBIにおけるグレリンの保護的役割は、線維芽細胞増殖因子(FGF)と関連しており、Shaoらの研究では、TBIラットモデルでグレリンを静脈内投与(20μg/kg)すると、同側半球でFGF結合蛋白(FGF-BP)と塩基性FGF(bFGF)の両方がダウンレギュレートされることを発見した。研究グループは、グレリンがbFGF/FGF-BP誘導性の新生血管形成を競合的に阻害することにより、脳損傷を減弱させることを提唱した。しかしながら、グレリンとbFGFおよびFGF-BPの両方との間に発見されたこの関係が、因果関係なのか競合関係なのかは、まだ調査されていない。敗血症時の脳損傷に対するグレリンの保護的役割に関連して、別のメカニズムが同定されている。Sunらは、PI3K/Aktシグナルの活性化が、グレリンの脳浮腫、神経細胞アポトーシス、およびBBB完全性の亢進を抑制する能力を媒介することを発見した。[163]。ホスホイノシチド3-キナーゼ(PI3K)の活性化は、ホスファチジルイノシトール3,4,5三リン酸(PIP3)とホスファチジルイノシトール3,4二リン酸(PIP2)の産生をもたらし、プロテインキナーゼB(Pkb/Akt)の活性化につながる。Aktはグリコーゲン合成酵素キナーゼ3β(GSK-3β)とBcl-2ファミリーの両方をリン酸化することにより、細胞の生存を促進する。[164, 165]。これら2つの分子は神経細胞の生存と密接に結びついており、グレリンが敗血症の際に神経保護作用を発揮する潜在的な機序を強調している。最後に、グレリンは虚血再灌流障害(IRI)の大脳モデルにおいて神経保護作用を示すことが示されている。[166]。IRI対照と比較すると、ラットおよびマウスの大脳IRIモデルにおいて、非アシル化グレリンを投与すると、酸化的損傷の減少を介して、傷害、アポトーシス、炎症およびBBBの破壊が減少した。[167, 168]。グレリンはまた、脊髄損傷(SCI)後の好中球浸潤を減少させ、その結果、損傷後の脂質過酸化とDNA損傷が減少することが判明している。[169, 170]。しかしながら、このような潜在的な保護作用があるにもかかわらず、グレリンに関する研究は、普遍的に肯定的な所見をもたらしたわけではない。例えば、Ersahin らは、IPグレリン(10μg/kg/day)はラットのSCI後の神経学的悪化を軽減しないことを見いだした[169]。利用可能なデータにおけるこのような乖離は、脳-腸軸に作用し、脳-腸軸を通して作用する保護剤としてのグレリンの潜在的役割について、さらなる調査が必要であることを強調している。
中枢神経系損傷後の消化管機能不全を軽減するグレリンの能力は、迷走神経との関連、腸関門の保全、虚血損傷後の哺乳類ラパマイシン標的(mTOR)経路の活性化など、いくつかのメカニズムに起因している。TBI後の腸脳軸の保護反応を媒介するグレリンの役割を支持する証拠がある。Bansalらは、重度のWD TBIの直前と直後に2用量のグレリン(合計20μg/kg)をIP投与すると、腸管バリアの完全性が維持され、絨毛構造が回復し、回腸のTNF-αレベルが低下することを示した[171]。また、TBI後の迷走神経刺激(VNS)の有益な効果は、グレリンを介して媒介される可能性があることを示した[172,173]。VNSは、TBI後に血清グレリン濃度を上昇させ、血清炎症性サイトカインを減少させることが示された。外因性グレリンは、循環TNF-αを減少させることにより、VNSにみられる反応を模倣することが判明した。重要なことは、グレリン受容体アンタゴニストをVNSと同時に投与すると、VNSの保護効果が消失したことで、TBIの際にVNSが作用するグレリン依存的メカニズムの可能性が強調された。グレリン受容体が脳の迷走神経背側運動核に局在していることは、この関連を支持している[174]。さらに、雄ラットの重度のTBI後30分にグレリン(20μg/kg)を1回静脈内投与すると、腸の運動が改善し、回腸粘膜の構造が維持された[175]。グレリンは、その重要かつ確立された抗炎症作用のほかに、マウスにおける脳内出血(ICH)後の腸管バリア機能障害を抑制することが示されている。[176]。雄性脳出血モデルマウスにおいて、グレリン2回投与(合計20μg)は、組織学的および超微細構造学的レベルで回腸粘膜傷害を顕著に減少させた。グレリンはまた、腸のタイトジャンクション関連タンパク質であるZonula occludens-1(ZO-1)とclaudin-5をアップレギュレートすることで、ICH後によく見られる腸伝染性の上昇を抑制した[176]。mTORの活性化もまた、グレリンの胃保護メカニズムに関与している。Zhangらは、グレリンが上腸間膜動脈(SMA)閉塞後のmTORのリン酸化を増加させることを発見した[177]。この所見は、特異的グレリン拮抗薬である成長ホルモン放出ペプチド6を併用した場合には起こらなかった。グレリンのmTORを活性化する能力は、最終的に、SMA閉塞と同時にグレリン治療を受けたマウスにおける臓器傷害の軽減と生存率の増加という所見と相関していた[177]。グレリンが脳と腸の傷害を改善するメカニズムはいくつかある。強調された論文を通して一貫したメカニズムがないことから、グレリンの多面性がその有益な影響に寄与しているという仮説が支持される。
プロゲステロン
プロゲステロンは、主に生殖腺と副腎皮質で産生されるステロイドホルモンで、子宮機能と女性の生殖を調節する。[178]。しかし、プロゲステロンが、神経細胞やグリア細胞から分泌される中枢神経系など、女性の生殖器官以外でも役割を果たしていることが、研究から示唆されている。その作用には、神経新生と髄鞘形成の促進、学習と記憶の改善などがある。[179]。プロゲステロンの役割は、TBI、パーキンソン病、ADを含む複数の中枢神経系疾患において広く報告されており、抗炎症特性により神経保護作用があるとされている。[180]。プロゲステロンの治療効果は、TBI [181]、脳卒中 [182]、神経変性疾患の動物モデル [183]で検討され、プロゲステロン受容体は、末梢神経系のシュワン細胞に加え、ニューロン、アストロサイト、オリゴデンドロサイトなど様々な中枢神経系細胞に発現している。[184]。研究によると、TBI後のプロゲステロンの早期投与は、炎症性サイトカインの発現を減少させ [185]、BBBを修復し、[186]、ミエリンと軸索の再生を促進し、[187]、ミトコンドリアの酸化的損傷を抑制し、[188]、認知および運動の転帰を改善する。[189]。プロゲステロンはまた、神経成長因子と脳由来向神経性因子(BDNF)の発現を増加させることにより、シナプス形成と神経新生を刺激する。[190]。
Guoらは、プロゲステロン(16mg/kg)の皮下(SQ)投与が、病変部位近傍のAQP4水チャネルの発現を増加させることにより、雄ラットのTBI後3日目の脳浮腫を減少させることを示した[186]。他の研究でも、プロゲステロンは、転写因子NF-κBの活性化と炎症性サイトカインTNF-αおよびIL-1βの発現を低下させることにより、TBI後の神経炎症反応を減弱させることが明らかにされている。[185, 191]。最終的には、アポトーシスと炎症を促進することが知られている転写因子であるcFosの活性化を減少させることになる[192, 193]。これらの結果と一致して、Yaoらは、雄ラットの中等度側方打撲損傷(FPI)後にプロゲステロンを投与すると、プロアポトーシス遺伝子BadとBaxの発現が減少し、抗アポトーシス遺伝子Bcl-2が増加することを示した[194]。さらに、プロゲステロンは循環内皮前駆細胞(CD31とCD34)を増加させ、神経の再生と血管のリモデリングを促進し、認知の転帰を改善する[187]。プロゲステロンが治療効果を発揮する別のメカニズムとしては、神経炎症カスケードの強力な阻害因子である補体崩壊促進因子(CD55)の活性化がある。[195]。
ある系統的レビューでは、実験的脳損傷直後にプロゲステロンを投与すると病変容積が減少することがわかったが、[196]、より最近の系統的レビューでは、TBI後の死亡率や有害転帰は減少しなかった。[197]。多くの前臨床研究で有望な結果が得られたため、研究者たちは、急性TBI後のヒト被験者におけるプロゲステロンの有効性を研究するために、第II相臨床試験を実施した。ジョージア州アトランタで行われたProTECTと名付けられた最初の第Ⅱ相臨床試験では、プロゲステロンが受傷後30日目の死亡率を低下させることが示された。[198]。その後、中国の杭州でより長期間の臨床試験が実施され、受傷後3カ月での機能的転帰の改善と回復が示された。[199]。結局、2つの第III相臨床試験(ProTECT IIIとSyNAPSe)は、以前の有望な再生促進効果を確認することはできなかった。それにもかかわらず、研究の批評家たちは、その失敗の原因を主観的な測定に頼っていたことと、結果を正確に評価する客観的な測定法がなかったことにあるとした。薬の投与量に関する問題や、前臨床試験における偽陽性の結果を過大評価する傾向も指摘された。[200]。しかしながら、臨床試験を開始する際の注目すべき見落としは、プロゲステロンが腸の運動性にどのように影響するのか [201]、そしてそれに続く腸の免疫プロファイルの時間的変化 [66, 202]を理解できなかったことであり、これは神経学的転帰に影響を及ぼす可能性がある。第III相臨床試験は期待外れであったとはいえ、プロゲステロンは、特にTBIの治療薬としてFDAに承認された薬剤がない現状では、重要な標的薬理療法であると考えられる。
プロゲステロンには神経保護作用があることから、研究者たちはTBIに関連した腸機能障害におけるプロゲステロンの役割を研究している。Chenらは、SQプロゲステロン(16mg/kg)の5日間投与が、炎症性サイトカインIL-1βとTNF-αを低下させ、細胞アポトーシスを減少させることにより、回腸粘膜の完全性を維持し、TBI後の炎症を減少させることを示した[203]。そして、プロゲステロンが転写因子NF-κBの活性化を低下させることも発見され、これが治療効果の原因であると考えられた[204]。その後の調査で、抗酸化転写因子である核赤血球2関連因子2(Nrf2)もまた、NF-κBの活性化を低下させることにより、TBI後の腸の炎症を緩和することが明らかになり[205]、プロゲステロンがNrf2経路を調節することにより、NF-κBの活性化を低下させる可能性が導かれた。さらに、Jinらは、Nrf2欠損マウスにおいて、TBI後の腸管伝染性の上昇を観察し、その結果、血漿中のエンドトキシン濃度が上昇した。その結果、抗酸化酵素と解毒酵素のレベルが低下した。[206]。さらに、Nrf2欠損マウスは細胞間接着分子-1(ICAM-1)発現レベルの上昇を示したが、プロゲステロン療法はTBI後のICAM-1発現を減少させることに成功したことも特筆に値する。[204, 205]。これらの研究は、粘膜の完全性を維持する上で、プロゲステロンとNrf2経路の回復相乗効果を実証している。プロゲステロン受容体を介したNrf2経路に対するプロゲステロンの作用の根底にある正確なメカニズムを完全に解明するには、さらなる研究が必要である。その後、Zhouらは試験管内試験実験を行い、プロゲステロンがタイトジャンクションのオクルジンの発現をアップレギュレートすることにより、腸の伝染性を効果的に低下させることを示した。[207]。さらに、腸の機能障害は、くも膜下出血(SAH)やパーキンソン病などの脳疾患で観察され、プロゲステロンの使用は腸の炎症を緩和することが示されており、脳腸軸機能障害における保護的役割を示唆している。雄性SAHモデルラットにおいて、プロゲステロン(16mg/kg)を5日間IP投与すると、炎症性サイトカインであるIL-1β、TNF-α、IL-6の発現が減少し、回腸の粘膜の完全性が回復した。[208]。パーキンソン病モデルマウスにおいて、プロゲステロンの使用も回腸組織における抗炎症作用を示した。[209]。結論として、これらの研究は、TBI後の脳と腸の炎症を緩和するプロゲステロンの相乗的役割を実証している。
ANS調節薬
β遮断薬
カテコールアミン、すなわちノルエピネフリン(NE)は、β受容体:β1-3およびα受容体:α1-2の活性化を通じてその作用を発揮する。[210, 211]。β1-3受容体は、体内の様々な臓器に存在し、心収縮力の増大、心拍数の増加、血管拡張、気管支拡張をもたらす。β1-3受容体はまた、白色脂肪組織における脂肪分解、褐色脂肪組織における熱発生、造血前駆細胞の動員を調節する役割も担っている。[212]。TBI患者は交感神経の亢進を経験し、カテコールアミンの急増を引き起こすことが示されている。[213-218]。このサージは、外傷後高熱 [216, 218]、炎症と白血球遊走の亢進 [219, 220]、大脳の恒常性と灌流の障害 [217]、心血管機能障害 [216]などの二次障害を引き起こし、死亡率の上昇 [221]と関連している。このようなエビデンスを踏まえて、TBIによる二次的障害を軽減するためのβ遮断薬の使用が注目されるようになった。β遮断薬には、メトプロロール(β1受容体遮断薬)のように1種類の受容体に選択的に作用するものと、プロプラノロールのようにすべてのβ受容体に非選択的に作用するものがある。ラベタロール(β-1およびβ-2受容体遮断薬)のように、α-1受容体にも作用し、それによって期待される結果が変化するものもある。[212]。
Lopezらによる研究では、雄マウスに制御された皮質衝撃を与えて重度のTBIを行い、非選択的β遮断薬であるプロプラノロールを2日間 [219] および14日間投与した場合の効果を検証した。[220]。プロプラノロールの投与は、用量依存的に陰茎白血球の遊走を減少させ、両時点で体重の回復を改善し、2日後には脳浮腫とアルブミン漏出を減少させた。他の前臨床試験では、β遮断薬投与によるp-tau蓄積の減少[222]、脳酸素化と灌流の改善[212]、脳自動調節の回復[217]、海馬壊死の減少[217]、神経学的スコアの上昇[212, 219]、行動[222]、認知・記憶[223]の改善が明らかにされた。β遮断薬は、(a) ショック蛋白70(HSP-70)などの保護蛋白を増加させ、酸化的損傷に関与するユビキチンカルボキシル末端ヒドロラーゼL1(UCHL-1)をダウンレギュレートすることにより、神経保護環境を提供することによって作用すると仮定されている。[223]; (b) 胞内へのカルシウム侵入を減少させ、アポトーシスを減少させる。[223]、(c) クログリア機能とシナプス可塑性を調節する。[224]、(d) 症性IL-6シグナル伝達を阻害する。[217]。
前臨床試験におけるβ遮断薬治療の成功は、臨床試験にも十分に反映された。観察研究では、β遮断薬、特にプロプラノロールを投与したTBI患者では、プラセボを投与した患者と比較して死亡率が有意に減少し、[225]、長期的な神経学的機能が改善した。[215]。結果は入院期間の延長と関連しており、ICU滞在期間については一貫した結果が得られていない。[214, 216, 225]。β遮断薬を使用した場合の有害作用としては、人工呼吸器装着時間の延長や感染率の上昇などがあった。[214, 216] ;しかしながら、β遮断薬群もプラセボ群も、低血圧、徐脈、ハートブロック、停止、気管支痙攣などの心肺有害事象の発生率は同程度であった。[215]。Asmarらは、β遮断薬、特にプロプラノロールが、高体温エピソードの数を有意に減少させ、体温の中央値を低下させ、エピソードの間隔をあけることによって、TBI後の体温調節障害を緩和する役割を果たすことを示した。この結果は、重度の頭部外傷患者においてより顕著であり、これらの患者はさらに、ICU滞在期間の短縮と退院時のグラスゴー・コーマ・スケール得点の上昇を示した。[218]。Khaliliらは、前向きランダム化比較試験において、重度の孤立性TBI患者をプロプラノロールで治療すると、プラセボと比較して6カ月後の死亡率が低下し、機能的転帰が改善することを示した(証拠レベルII) [226]。Eastern Association for the Surgery of Trauma(EAST)は、禁忌のない成人TBI患者にβ遮断薬を使用することを推奨しており、主に低血圧や徐脈などの心血管系の副作用を監視し、速やかに治療できるICU環境でのみ使用することを推奨している。[213]。
TBIは腸管伝染性の亢進と関連しており、これは死亡率の上昇と相関しているが、β遮断薬治療により有意に減少することを示す証拠がある。[221, 227]。Langらはまた、TBIを受けた雄ラットでは、偽ラットと比較して、エピネフリンレベルが上昇し、腸管TNF-aレベルが高く、腸のタイトジャンクションを形成するZO-1タンパク質の発現が低下し、回腸組織病理学上の粘膜損傷が悪化することを示した。これらの変化はすべて、TBI直後にラベタロール(β1・2受容体遮断薬)をIP投与すると有意に回復した[227]。NEは脳-腸軸において双方向的な役割を果たしているため、TBIでみられる腸の機能障害は、NEシグナルを介して脳にも変化を引き起こし、腸の機能障害をさらに悪化させる可能性がある。例えば、ラットモデルでは、胃拡張により、ストレスへの反応に関与することが知られている終末線条体腹側床核(vBNST)のNEが有意に増加することが示されている。vBNSTのβ受容体のアゴニズムは、胃排出と小腸通過の有意な減少につながり、これはβ遮断薬の投与で逆転する。[228]。
α-アゴニスト/アンタゴニスト
脳-腸軸の設定において、α-アドレナリン受容体の調節は比較的研究されていない。α受容体調節薬の治療作用に関する双方向のデータが不足しているにもかかわらず、TBI後の脳に対するα受容体調節薬の作用は、トランスレーショナルリサーチと臨床研究の両面で大きな関心を集めている。[229, 230]。雄ラットにおいて、α2アドレナリン作動薬であるマフェジンは、TBI後7日目に、離れた脳領域における半球間結合と、影響を受けていない半球内の半球間結合の回復を促進した。[231]。著者らはまた、マフェジンが光刺激と体性感覚刺激に対する皮質反応を改善することも発見した。[231]。α2アゴニストと並んで、α1アンタゴニストのTBIにおける役割も活発に研究されている。Koboriらは、α-1アンタゴニスト(2-[b-(4-Hydroxyphenyl)ethyl]aminomethyl-1-tetralone hydrochloride (HEAT))のIP投与(0. (0.1mg/kg)は、オスSprague DawleyラットのTBI後14日目のワーキングメモリーを改善した。[232]。このような研究から、αアドレナリン受容体の調節、ひいてはノルエピネフリンシグナル伝達の調節が、TBI患者に有益である可能性が示唆される。これらの知見は、カテコールアミンの上昇が中等度から重度の孤立性TBI後の好ましくない転帰と関連することを示すCOMA-TBI研究から得られた既存の臨床データに加えている。[73, 233]。COMA-TBI研究では、エピネフリン(Epi)とノルエピネフリン(NE)の予測的使用における違いが強調されており、入院時のEpi値は好ましくない転帰と死亡率の高さと関連し、NEの変化は死亡リスクの高さと関連していることが重要である。これらのカテコールアミンの予測値の違いは、アドレナリン受容体のサブタイプに対する親和性によると考えられる。
臨床の場では、脳損傷後の交感神経亢進を抑制することが期待されるα2作動薬の役割が、重度のTBIにおいて広く研究されている。[234]。クロニジン(α2作動薬)とプロプラノロールの併用は、循環カテコールアミンのレベルを低下させ、脳血流を変化させることなく脳血管収縮のレベルを低下させるため、重症TBI患者を対象に研究されている。[235]。現在までに、”DASH After TBI Study” [236]やNordnessら [237]のパイロット研究を含むいくつかの研究で、プロプラノロールとクロニジンによるアドレナリン作動性遮断は安全で実行可能であるが、重度のTBI患者においてこれらの薬剤は人工呼吸器無操作日数を変化させないことが明らかにされている。
スタチン
1970年代初頭にスタチンが発見されて以来、スタチンはコレステロール値の高い患者に対する主な治療薬となっている。[238]。研究が進むにつれ、スタチンの抗炎症作用が冠動脈疾患 [239]、関節炎 [240]、脳損傷 [241]、腸炎 [242]の管理に有効であることが示され、スタチンの使用が拡大した。脳損傷後のCNSにおけるスタチンの保護的役割については、広く述べられている。[243-245]。スタチンは、神経細胞死、アポトーシス、ミクログリアの活性化、アストログリオーシスを減少させることにより、神経変性を抑制することが示されている。[246]。スタチンはまた、脳、特に病変の境界領域と海馬における神経新生、シナプス形成、血管新生を促進する。[247]。研究によると、TBI後のスタチンの使用は、死亡リスクの低下、受傷後12カ月における機能と能力の改善と関連している。[248]。無作為化二重盲検臨床試験において、スタチンは脳挫傷容積には影響を及ぼさなかったが、受傷後3カ月の機能回復を改善した。[249]。ラットの動物モデルでも、TBI後のアトルバスタチン治療により空間記憶機能の亢進が認められた。[250, 251]。Sultanらによるシステマティックレビューでは、スタチンは神経保護作用を示し、特に認知機能の改善と認知症リスクの軽減に効果があることが示唆された。[243]。スタチンはまた、TBI後のβアミロイドペプチド濃度を低下させることが示されており、TBI患者の認知機能の改善やADリスクの低下に関与している可能性がある。[252]。
スタチンの抗炎症作用は数多くあり、例えば、炎症性マーカーであるTNF-αおよびIL-1βの発現の減少や、ミクログリアおよびアストロサイトの活性化の減少が、それぞれ分化クラスタ68(CD68)およびグリア線維酸性蛋白(GFAP)の発現の減少によって証明されている。[253, 254]。スタチンは、アストロサイトの脂質ラフトにおけるカベオリン-1の発現と上皮成長因子受容体を調節することにより、アストログリオーシスを抑制する。[255]。スタチンはまた、Akt依存性のシグナル伝達経路を介して、海馬歯状回における血管内皮増殖因子(VEGF)とBDNFの発現をアップレギュレートし、GSK-3βとcAMP応答エレメント結合タンパク質の発現をアップレギュレートし、最終的に細胞増殖を増加させ、神経新生を促進する。[256]。さらに、シンバスタチンを毎日経口投与(1mg/kg/日)すると、雄性ラットのTBI後の神経細胞アポトーシスが、Aktリン酸化の増加およびその下流標的であるForkhead転写因子1、inhibitory-κB、内皮一酸化窒素合成酵素の活性化を通じて減少し、同時にカスパーゼ-3の活性化が抑制される。これらの作用はすべて、神経細胞の生存と機能の改善につながる。[257]。さらに、スタチンは、ICAM-1と好中球浸潤の減少によりBBBの伝染性を低下させることで、傷害後の脳浮腫を減少させる。[258]。これらの所見を総合すると、スタチンはTBI後の神経炎症反応を抑制し、転帰を改善する上で有益な役割を果たすことが示唆される。
脳-腸軸に沿ったスタチンの保護的役割が明らかになりつつある。まず、腫瘍壊死因子受容体ファミリーの膜貫通型受容体であるCluster of Differentiation 40(CD40)が、TBI、IRI、IBDなど様々な疾患における腸の炎症に強く関与していることに注目することは重要である。[259]。ラットのTBIモデルにおいて、Huらは、CD40の発現が空腸で増加し、それがNF-κBの活性やTNF-α、血管細胞接着分子-1(VCAM-1)、ICAM-1のレベルの増加と正の相関があることを示した[260]。最終的に、TBI直後にロスバスタチンを1回IP投与(30mg/kg)すると、TNF-αとIL-1βの発現が減少し、CD40/NF-κB経路が遮断されることで、空腸障害が軽減されることが明らかになった。スタチンはまた、正常な粘膜構造を維持し、絨毛の完全性を維持することにより、TBIによる腸の形態学的変化を緩和した[261]。さらに、生体内試験(実験的マウス大腸炎モデル)および試験管内試験(腸上皮細胞)の研究では、NF-κBの活性を阻害し、IκBのリン酸化を抑制し、最終的にTNF-αの発現を低下させることにより、スタチンの抗炎症特性が再確認された[262]。スタチンはまた、ヒト血管細胞におけるCD40の発現を低下させることが示されている。[263]。さらに、Ozacmacらは、IRIの3日前にアトルバスタチン(10mg/kg)を連日経口投与すると、酸化ストレス、好中球の蓄積、組織のマロンジアルデヒド発現が減少し、還元型グルタチオンレベルが上昇するためか、腸の運動性と回腸の収縮性が促進されることを明らかにした[264]。スタチンが腸で保護作用を発揮するもう一つのメカニズムは、腸内細菌叢を調節することである。TBIが腸内細菌叢の組成を変化させ、主に腸管運動障害と細胞間伝染性の亢進によって常在菌よりも病原性細菌を促進し、その結果、疾患の進行を悪化させることを示唆する証拠が増えている。[4]。さらに迷走神経は、その求心性線維を通じて微生物産物を感知することで、中枢神経系に相互に影響を及ぼす。このことは、傷害を受けた脳と腸内細菌異常の間の有害なサイクルを引き起こす。この炎症サイクルを止める一つの方法として、スタチン療法が考えられる。スタチンは健康な腸内細菌叢を促進し、腸内細菌異常症を改善することが示されている。[265]。また、スタチンはIBDにおいて抗炎症作用を示し、ステロイドの必要量の減少につながることが研究で示されている。[242, 266]。これらの薬剤の使用の可能性は有望であり、興味深い。スタチン療法がTBIにおいて脳腸軸をどのように制御しているかを解明するために、さらなる研究が望まれる。
マイクロバイオーム調節薬
抗生物質
最近の研究から、腸内微生物の異常は、IBSやIBDのような疾患だけでなく、TBIや脳卒中などの神経疾患においても腸の機能に影響を及ぼすことが示されている。[267, 268]。TBIでは、常在菌よりも病原性細菌の存在が促進され、腸内炎症の亢進、バリア伝染性の亢進、運動障害につながる。[93, 268]。TBI後のマイクロバイオームの変化と脳炎症への影響をより深く理解するために、健康な腸内細菌叢を促進する有益なプロバイオティクス株の投与に加えて、マイクロバイオームを減少させるための様々な抗生物質の併用が研究されてきた。多くの動物実験では当初、微生物異常と宿主の生理学的反応との関係を解明するために、無菌(GF)マウスが用いられていた。[71, 269]。Simonらによる研究では、雄マウスにTBIの2週間前に抗生物質(アンピシリン、メトロニダゾール、ネオマイシン、バンコマイシンの1g/L)を組み合わせて投与すると、盲腸上皮に沿ったZO-1染色の増加から明らかなように、腸管バリア伝染性が改善することが示された。その結果、ミクログリアの活性化が減少し、海馬の神経細胞密度が増加し、病変容積が減少し、学習成績が改善した[269]。逆に、Celorrioらによる別の研究では、傷害の2週間前から同じ抗生物質を使用したところ、海馬の神経細胞損失が悪化し、成体雄マウスにおいてより顕著な恐怖反応を引き起こした。[202]。このような相反する結果は、異なる動物施設におけるマウスの腸内細菌叢の違い、宿主マイクロバイオームに影響を与える食餌の違い、傷害の重症度や行動分析の時期の違いなど、様々な要因に起因すると考えられる[270, 271]。Celorrioらの研究は、腸内細菌叢と免疫系の複雑な相互作用も明らかにした。TBI後1週間の抗生物質投与は、末梢単球とTリンパ球の脳への浸潤を減少させたが、一方で受傷3日後にはミクログリアの炎症マーカーを増加させた。ミクログリアによる炎症性表面マーカー(TLR4とMHCII)の高発現は、アメーバ状への形態の変化とともに、歯状回における神経新生の減少と海馬のCA3領域における神経細胞変性の増加と相関していた[71, 202]。Benakisらはまた、アモキシシリン-クラブラン酸(1g/L)を2週間投与することによって雄マウスの腸内細菌叢が変化し、虚血性脳損傷が減少することを示した。この減少は、腸管制御性T細胞の増加と、IL-17+γδT細胞の脳への浸潤の減少に起因しており、傷害後16時間で観察された。[70]。腸-微生物-免疫軸を標的とすることは、TBI後の回復を改善する有望なアプローチとなる。
プロバイオティクス
マイクロバイオームの変化をよりよく理解するために、プロバイオティクスの投与がTBIモデルで広く実験されている。プロバイオティクスは、有益な細菌のコロニー形成を促進し、病原性の細菌集団を減少させることにより、腸管内腔の微生物集団を制御する。[272]。プロバイオティクスは、上皮細胞の分化と増殖を促進し、上皮バリアの完全性を維持することで、有害細菌の腸実質への移行を抑制する。[273]。Maらによる研究では、プロバイオティクス、特にLAを体重減少TBI後7日間補充することで、TBI後の回腸に保護効果があることが示された。これは、腸管バリア機能の改善と、栄養素および電解質に対する腸の吸収能の改善によって明らかになった[274, 275]。LAの効果には、ミオシン軽鎖キナーゼタンパク質のレベルを増加させ、平滑筋の収縮を促進することにより、カハール間質細胞(ICC)として知られる腸のペースメーカー細胞を回復させることによる回腸絨毛構造の改善と回腸運動性の向上も含まれていた。[276]。ラクトバチルス・ロイテリは、軽度のTBIを受けた退役軍人を対象とした臨床試験にも利用された。その結果、ラクトバチルス・ロイテリを8週間毎日摂取した患者では、プラセボ群と比較して血清中の炎症性C反応性タンパク質値が有意に低下したことが示された。[277]。さらに、Duらによって行われたシステマティックレビューとメタアナリシスでは、TBI後のプロバイオティクスによる早期経腸栄養が死亡率を低下させ、便秘、腹部膨満、逆流、ストレス潰瘍などの消化器合併症を減少させることが示された。[278]。
プロバイオティクスはまた、酢酸、酪酸、プロピオン酸などのSCFAsの末梢循環への放出を刺激し、最終的にBBBを通過して、炎症性サイトカインの発現を減少させ、神経新生を促進することにより、脳の炎症を改善することができる。[279-281]。Liらの研究では、酪酸産生プロバイオティクスであるClostridium butyricum(Cb)が、大腸のGLP-1およびグルカゴン様ペプチド1受容体レベルを増加させることにより、脳腸軸を介してTBI後に神経保護効果を示すことが示された。Cb(109CFU/ml)を1日1回、TBIの前後2週間にわたって胃内に補給すると、神経変性を抑制し、アポトーシスを減少させ、BBBの完全性を高めることによって、雄マウスの神経学的転帰が改善した[282]。これと並行して、プロバイオティクスであるラクトバチルス・アシドフィルス(LA)(1×1010 CFU)を体重減少TBI後7日間投与すると、雄マウスのマイクロバイオームがより健康的なプロファイルにシフトする。LAは、アストロサイトとミクログリアの活性化を減少させることによって神経学的転帰を改善し、BBBの完全性を維持することによって脳浮腫を減少させた。LAの補充はまた、脳内のグラム陰性菌受容体、主にTLR4の発現を減少させた[275]。最後に、無作為化臨床試験により、重度のTBI後21日間、プロバイオティクス(ビフィドバクテリウム・ロンガム、ラクトバチルス・ブルガリクス、ストレプトコッカス・サーモフィルス)を組み合わせて毎日投与することで、IL-4とIL-10のレベルを低下させ、院内感染のリスクを低下させることにより、全身性の炎症が抑制されることが示された。抗生物質/プロバイオティクスの利用は、TBI管理における実質的な治療的可能性を有しており、脳の炎症および行動の転帰に対する効果をより明確にするために、さらなる研究が必要である。
微生物代謝産物
研究により、TBIがSCFAや胆汁酸を含む腸内微生物の代謝産物に変化を引き起こすことも明らかになっている。これらの代謝産物の中でも、酪酸、プロピオン酸、酢酸などのSCFAは重要な役割を果たしている。[284]。これらは傷害を受けた脳の代替エネルギー源となり、ミトコンドリアのホメオスタシスを調節するのに役立っている。[285]。Opeyemiらの研究では、成体の雄マウスにおいて、TBI後24時間と28日の糞便中のSCFAs濃度が有意に減少することが示された。この減少は学習成績の悪化と相関していたが、SCFAsの投与はこれらのマウスの空間学習を改善した[286]。さらに、胆汁酸の役割は、頭蓋内出血や虚血性脳卒中などの様々な神経疾患においても研究されている[287, 288]。タウロソデオキシコール酸の投与は、神経機能を改善し、再灌流から2日後と7日後の梗塞サイズを減少させることが示されている。[289]。TBIに関連して、Youらは、成体雄性マウスにおいてTBI後1日目の血清および糞便サンプル中の胆汁酸レベルが低下していることを示した。[268]。さらに、Zhuらは、TBI患者における血漿胆汁酸レベルの低下を示した前向き研究を行った。[290]。したがって、胆汁酸やSCFAなどの微生物代謝産物副産物の補給は、TBI後の神経炎症を緩和する有望な治療法となる。
常在細菌叢
支配的な細菌マイクロバイオームに加えて、腸内常在細菌叢(真菌マイクロバイオーム)も、脳と腸のホメオスタシスを制御する重要な治療標的として浮上している。常在細菌叢の不均衡は、IBS [291]、IBD [292]、大腸がん。[293]など、様々な腸疾患で観察されている。IBSにおける内臓過敏症は、真菌のディスバイオシスの増加と関連している。[294]。外傷性脳損傷における真菌の役割についてはまだ十分な研究がなされていないが、Parkらによる最近のプロスペクティブ観察コホート研究では、集中治療室入院後2~3週間まで、重症外傷および敗血症における常在細菌叢バランスの調節異常が明らかになった。カンジダ属の優勢と常在真菌種の減少は、感染に対する脆弱性の高まりと相関していた。[295]。興味深いことに、Hunagらによる別の研究では、TBI後3日目のマウスの腸内真菌叢の変化が、真核生物のmRNAの転写後修飾に重要なN6-メチルアデンソイン(m6A)の機能調節障害と関連していることが示された[296]。細菌と真菌のマイクロバイオームには複雑な関係があることから、TBI患者における真菌血症、特にカンジダ血症の役割や、常在細菌叢を調節することで神経学的転帰をどのように変化させることができるかをよりよく理解するために、さらなる研究が必要である。
現在の限界
TBI後のマイクロバイオームの変化と神経学的転帰への影響を調べるには、ヒトおよび動物実験が極めて重要である。[297, 298]。これらの変化を検出するために、抗生物質の投与 [299]、プロバイオティクス [283]、SCFA [300]、糞便マイクロバイオーム移植 [301]など、様々な方法論が用いられている。開放性頭部外傷や多発外傷の患者の中には抗生物質の投与が必要な場合があることから、抗生物質の影響に関する研究は特に重要である。[302, 303]が、このような介入による神経学的影響についてはまだ十分に理解されていない。抗生物質の投与がTBIの転帰に及ぼす影響を検討した臨床研究はほとんどなく、抗生物質の投与に関する一般的なガイドラインも現在のところ存在しない。[299]。TBIによって誘発された腸内微生物異常症の文脈では、長期にわたる感染率の変化と神経学的転帰に対する抗生物質投与の効果を調べるために、十分に管理された臨床試験を開発する必要がある。
TBIにおける微生物叢研究における主要な課題は、標的シーケンスやメタゲノム・シーケンスを含むサンプル収集や処理方法における広範なばらつきに起因する。[304]。さらに、個人の食事パターンや既存のマイクロバイオームの多様性が不均一であるため、マイクロバイオームの変異を検出する上でさらなる課題がある。[305]。前臨床動物モデルは、標準化された環境などの管理された実験条件を提供するが、その知見はトランスレーショナルな取り組みに課題をもたらす。[306]。もう一つの重要な側面は、プロバイオティクスが神経認知の結果に影響を及ぼすまでに必要な時間であり、潜在的には何年にもわたる。[307]。したがって、TBIにおけるマイクロバイオームの影響を十分に理解するためには、十分に設計された長期臨床試験が必要である。これらの限界に対処することは、効果的な治療介入を開発するために不可欠である。
表11と表22に、TBI後の脳と腸の炎症を抑制する様々な抗炎症剤の概要を示す。
表1 TBI誘発神経炎症に対する各種薬物療法の影響に関する知見の要約
| セラピー | 介入 | セックス | 種 | モデル | タイムポイント | 脳炎症への影響 |
|---|---|---|---|---|---|---|
| セロトニン | IP SSRI(フルオキセチン) 5 mg/kg、傷害直後に単回投与 | 該当なし | マウス | 片側だけの重度のTBI(CCI) | TBI後7日 | ロタロッド上での運動回復と協調性が改善された[117]。 |
| SNRI(Milnacipran) 30 mg/kg/日をIP浸透圧ミニポンプで犠牲まで投与 | 男性 | ネズミ | 片側だけの中等度TBI(CCI) | TBI後5週間 | 内側前頭前皮質におけるセロトニン作動性緊張の亢進と注意セットシフト課題遂行能の改善[111] | |
| SSRI(セルトラリン)1日50mgを24週間、TBI既往歴のある患者にPO投与する。 | 男性 | 人間 | 中等度-重度のTBI | 治療後24週 | 気分と抑うつ症状の有意な改善[119]。 | |
| グレリン | TBIの1時間前と直後にIPグレリンを2回投与(合計20μg)。 | 男性 | マウス | 片側のTBI(WD) | TBI後6時間 | 大脳皮質の損傷部位の血管透過性とAQP-4発現を低下させることにより、BBBの破壊を防ぐ[159] 。 |
| 受傷30分後にグレリン(20μg/kg)を1回静脈内投与 | 該当なし | ネズミ | 片側TBI(CCI) | TBI後24、48、72時間 | 損傷した大脳皮質における塩基性線維芽細胞増殖因子(bFGF)を減少させることにより、脳の炎症を抑え、神経学的重症度スコアを改善する[162] | |
| 脳卒中後同日に脱アシル化グレリン(1mg/kg)をIP投与(3回 | 男性 | マウス | 一過性中大脳動脈閉塞症(tMCAo) | 脳卒中後24時間 | 虚血大脳半球において、タイトジャンクションタンパク質オクルジンとクローディン-5をアップレギュレートすることにより、梗塞サイズを縮小し、アポトーシスを減少させ、BBBの完全性を維持する[167] 。 | |
| プロゲステロン | TBI後6、24、48時間にSQプロゲステロン(16mg/kg)を3回投与。 | 男性 | ネズミ | 両側TBI(CCI) | TBI後72時間 | 傷害を受けた大脳皮質における灌流周囲のAQP-4チャネルの発現を減少させることにより、脳浮腫を軽減する[186] 。 |
| IPプロゲステロン(8mg/kg)およびアロプレグナノン(4mg/kg)をTBI後5日間投与 | 男性 | ネズミ | 両側性の重度のTBI(CCI) | TBI後6日 | 前頭葉における炎症性サイトカインIL-1BとTNF-aの発現を減少させる[185] 。 | |
| SQプロゲステロン(16mg/kg)をTBI後14日間毎日投与 | 男性 | ネズミ | 片側だけの中等度TBI(FPI) | TBI後2週間 | MWMテストにおける神経学的転帰を改善し、循環内皮前駆細胞を増加させることにより、損傷した頭頂葉の血管リモデリングを誘導する[187] 。 | |
| ベータ遮断薬 | TBI後1時間にプロプラノロール(1mg/kg)静注1回投与 | 男女 | 豚 | 単一の中等度TBI(FPI) | TBI後4時間 | IL-6の発現をダウンレギュレートすることにより、海馬の神経細胞死を減少させる[217] 。 |
| IPプロプラノロール(1~4mg/kg)をTBI後2日間および14日間毎日投与 | 男性 | マウス | 重度のTBI(CCI) | TBI後2日と14日 | 浮腫を軽減し、用量依存的に脳BBBの透過性を維持する[219,220] | |
| IPプロプラノロール(4mg/kg)をTBI後7日間毎日投与 | 男性 | マウス | 片側だけの中等度TBI(CCI) | TBI後7日 | 海馬-CA1領域の細胞死を減少させることにより、記憶、学習、認知を改善する[223] 。 | |
| スタチン | シンバスタチン(1mg/kg/日)を犠牲まで毎日経口投与 | 男性 | ネズミ | シングルTBI(CCI) | TBI後1、3、7、14、35日目 | 炎症性サイトカインの発現を低下させ、病変境界領域周辺のアストロサイトとミクログリアの活性化を減少させる[253] 。 |
| シンバスタチン(1mg/kg/日)を犠牲まで毎日経口投与 | 男性 | ネズミ | 片側TBI(CCI) | TBI後1、3、7、14、35日目 | Akt依存性のシグナル伝達を通じて同側の海馬におけるVEGFとBDNFの発現を増加させることにより、空間学習を改善し、神経新生を促進する[256,257] 。 | |
| TBI後1時間と6時間にシンバスタチン(37.5mg/kg)を2回経口投与 | 男性 | ネズミ | 片側だけの中等度TBI(FPI) | TBI後24時間 | クローディン-5をアップレギュレートすることによりBBBの完全性を維持し、脳浮腫を軽減した[258] 。 | |
| 抗生物質/プロバイオティクス | アンピシリン、メトロニダゾール、バンコマイシン、ネオマイシン(1g/L)をTBI前に2週間連日経口投与 | 男性 | マウス | 片側TBI(CCI) | TBI後3日 | 海馬の神経細胞密度を増加させ、病変体積を減少させる[269] 。 |
| クロストリジウム・ブチリカム(109CFU/ml)を1日1回、TBI前後2週間にわたり胃内投与した。 | 男性 | マウス | シングルTBI(WD) | TBI後2週間 | 神経機能障害を改善し、脳浮腫を減少させ、病変部位周辺の神経細胞変性を抑制する[282]。 | |
| 乳酸菌(1×1010CFU)を犠牲となるまで1日1回経口投与 | 男性 | マウス | 片側のTBI(WD) | TBI後1、3、7日 | 損傷した大脳皮質におけるアストロサイトとミクログリアの活性化を減少させ、BBBの完全性を維持することにより脳浮腫を軽減する[275] 。 |
CCI:制御された皮質衝撃;WD:体重減少;FPI:流体打撃損傷;TBI:外傷性脳損傷
表2 TBI誘発腸炎症に対する各種薬物療法の影響に関する知見の要約
| セラピー | 介入 | セックス | 種 | モデル | タイムポイント | 腸内炎症への影響 |
|---|---|---|---|---|---|---|
| セロトニン | SSRI(IPフルオキセチン) 5mg/kg、傷害直後に単回投与 | 該当なし | マウス | 片側だけの重度のTBI(CCI) | TBI後4日 | FITC-デキストランの透過性を低下させることにより、大腸バリアの完全性を維持する[117] 。 |
| グレリン | TBIの1時間前と直後にIPグレリンを2回投与(合計20μg)。 | 男性 | マウス | 片側の重度のTBI(WD) | TBI後6時間 | 回腸バリアの完全性と構造を維持し、TNF-αレベルを低下させる[171] 。 |
| TBIの30分後にグレリン(20μg/kg)を1回静脈内投与 | 男性 | ネズミ | 片側の重度のTBI(CCI) | TBI後1~3日 | 腸の運動を改善し、回腸粘膜上皮を保護する[175]。 | |
| ICH直後にIPグレリンを2回投与(合計20μg)。 | 男性 | マウス | 脳内出血(ICH) | ICH後1日 | タイトジャンクションZO-1とクローディン-5をアップレギュレートすることにより、回腸透過性を低下させる[176] 。 | |
| プロゲステロン | SQプロゲステロン(16mg/kg)をTBI後5日間毎日投与 | 男性 | ネズミ | 片側のTBI(WD) | TBI後5日 | 回腸TNF-α、IL-1β、ICAM1の発現を低下させ、粘膜のアポトーシスを減少させる[203]。 |
| SQプロゲステロン(16mg/kg)をTBI後5日間毎日投与 | 男性 | ネズミ | 片側のTBI(WD) | TBI後5日 | 回腸のNF-κB活性化と炎症性サイトカイン発現を減少させる[204] 。 | |
| プロゲステロン(16mg/kg)を1日1回SAH後5日間IP投与 | 男性 | ネズミ | くも膜下出血(SAH) | SAH後5日 | 回腸粘膜の完全性を回復し、炎症性サイトカインIL-1β、TNF-α、IL-6を減少させる[208] 。 | |
| ベータ遮断薬 | TBI直後にラベタロールをIP投与(30mg/kg)1回 | 男性 | ネズミ | 片側のTBI(WD) | TBI後3、6、12時間 | アドレナリン作動性緊張亢進を抑制し、腸管TNF-αレベルを低下させ、回腸透過性の増加を防ぐ[227] 。 |
| TBI後にラベタロール(1Mスクロース中62.5μM)をPO投与 | 男女 | ショウジョウバエ | 高衝撃外傷装置を用いた閉鎖頭部TBI | TBI後24時間 | 腸管透過性と早期死亡率を低下させる[221] | |
| スタチン | TBI直後にロスバスタチンをIP投与(30mg/kg)。 | 男性 | ネズミ | 片側のTBI(WD) | TBI後24時間 | CD40/NF-κB経路を阻害することにより、空腸のTNF-αおよびIL-1βレベルを低下させ、絨毛の組織型を改善する[261]。 |
| 虚血を誘発する3日前にアトルバスタチン(10mg/kg)を1日1回経口投与する。 | 男性 | ネズミ | 腸管虚血再灌流障害(IRI) | 再灌流後3時間 | 酸化ストレスを軽減し、グルタチオンレベルを上昇させることにより、回腸の運動を促進する[264] 。 | |
| 抗生物質/プロバイオティクス | アンピシリン、メトロニダゾール、バンコマイシン、ネオマイシン(1g/L)をTBI前に2週間連日経口投与 | 男性 | マウス | 片側TBI(CCI) | TBI後3日 | 盲腸における上皮のZO1発現を増加させることにより、腸の透過性を改善する[269] 。 |
| 乳酸菌(1×1010CFU)を犠牲となるまで1日1回経口投与 | 男性 | マウス | 片側のTBI(WD) | TBI後1、3、7日 | PKC/MLCK/MLCシグナル伝達経路を通じて、回腸の炎症を抑え、バリア機能を維持し、腸の運動を促進する[276] 。 | |
| 乳酸菌(1×1010CFU)を犠牲となるまで1日1回経口投与 | 男性 | マウス | 片側のTBI(WD) | TBI後1、3、7日 | 回腸のバリア機能と、栄養素と電解質に対する腸の吸収能力を改善する[275] 。 |
CCI:制御された皮質衝撃;WD:体重減少;FPI:流体衝撃損傷;TBI:外傷性脳損傷;N/A:入手不可能
退役軍人における脳腸軸機能障害
退役軍人は、現実的な環境で外傷性脳損傷が腸に及ぼす影響を研究するためのユニークなヒトモデルとなる。[308]。爆風曝露を伴う軍事職業訓練後、1~16時間後に腸管伝染性の有意な上昇と腸内細菌の循環中への移動の証拠が観察され、それに伴ってα微生物の多様性が段階的に増加し、ゾヌリンやオクルディン-3などの腸管伝染性タンパク質バイオマーカーのレベルが上昇した。[309]。さらに、不朽の自由作戦/イラクの自由作戦/新しい夜明け作戦(OEF/OEI/OND)の退役軍人(男性26人)を対象とした別の研究では、腸内細菌組成、特に疣贅微生物叢の違いが、疲労、抑うつ症状、PTSDの重症度、注意力、実行機能、学習、記憶の困難などの精神症状や認知症状と相関していることが明らかになった。[310]。これらの知見は、退役軍人の腸内細菌叢と精神的健康との間に複雑な関係があることを強調している。さらに、湾岸戦争退役軍人を対象とした研究では、バクテロイデーテス門、放線菌門、ユーラシア門、プロテオバクテリア門、およびバクテロイデス科、エリシペロトリス科、ビフィドバクテリウム科の存在量が多いことが、慢性疼痛、疲労、睡眠障害の報告とともに、消化器症状の重症度の増加、炎症性サイトカインTNF-αの高値と相関することが示された。[311]。US Veteran Microbiome Project(US-VMP)に参加した退役軍人は、マイクロバイオームの構成、胃腸炎のエピソード、重度のうつ症状との間に有意な関連を示した。[312]。さらに、OEF/OEI/OND退役軍人を対象に実施されたRCTでは、ロイテリ乳酸菌を8週間毎日投与したところ、未投与群と比較して血漿CRP値が低下したことが明らかにされており、TBI後の炎症を緩和する上で、マイクロバイオームを標的とした介入の可能性が示されている。[277]。逆に、米軍退役軍人や米海兵隊員を対象としたいくつかの研究では、中等度/重度のTBI後の腸内微生物の多様性と伝染性に有意差は認められなかった。[313, 314]。このような相反する知見は、実環境におけるマイクロバイオーム研究の複雑さを浮き彫りにしている。このような研究においては、真の対照となる被験者を特定することの難しさ、便の採取時期のばらつき、マイクロバイオームに大きな影響を与える食事療法や薬剤の違いなどにより、数多くの限界が存在する。[313]。これらの限界に対処し、研究方法論を洗練させる努力は、軍人の集団におけるTBIの文脈における脳腸軸の理解を進める上で極めて重要である。
なぜTBI治療は失敗し続けるのか?
広範なTBI研究と様々な治療法への多大な投資にもかかわらず、疾患の進行を抑制する効果的な治療法は依然として不足している。[315]。多くの臨床試験は、前臨床動物モデルで観察された抗炎症効果を再現できていないが、これにはいくつかの課題がある。[316]。第一に、TBIは本質的に不均一であり、複雑な発症経路を引き起こすため、管理するのが非常に複雑な疾患である。[317]。基礎科学では、ほとんどの研究が脳内の特定の細胞過程に焦点が当てられており、しばしば疾患の全体像を見逃している。[316]。加えて、それぞれのモデルは、傷害を誘発するために、 異なる傷害パラダイムと解剖学的部位を利用している。局所挫傷、脳震盪、爆風傷害は、傷害に応答して異なる炎症経路を惹起することを認識することが重要である。[318]。炎症モデルを局所挫傷から脳震盪や爆風に外挿する傾向は、それぞれの傷害タイプに特有の炎症プロファイルのため、重大な問題を提示している[319, 320]。
TBI研究におけるもう一つの重要な課題は、臨床試験と前臨床モデルの両方において、男性に偏りすぎていることである[321]。TBI反応における性差は明らかで、臨床試験では男性がより良好な回復を示し、前臨床研究では女性が優れた結果を示している。[322]。腸内細菌叢と免疫系の活性化を調節することで、脳-腸のホメオスタシスに性差が重要な役割を果たすことを示唆する証拠が登場している。[323, 324]。動物実験から、腸内細菌叢の構成には雌雄間で顕著な違いがあり、雌の方が多様性が高いことが明らかになっている。[325, 326]。TBIにおけるマイクロバイオームへの性差の影響についてはまだ十分に検討されていないが、他の疾患における研究から、女性は微生物の変化により自己免疫疾患にかかりやすいことが示唆されている。[327, 328]。同様に、女性におけるIBSの有病率の高さは、エストロゲンを介した反応や腸管バリア機能の低下によるマイクロバイオームの変化と関連している可能性がある。[329]。脳腸軸に対する性差の影響を包括的に理解するためには、女性をより多く含む臨床試験や動物モデルが必要である。
TBI後の免疫反応の複雑さは、別の課題を提示している。[330]。経時的な免疫細胞の活性化の進化には、有益な要素と有害な要素の両方が含まれるため、特定の免疫経路を標的とする試みはしばしば失敗に終わる。[331]。さらに、試験管内試験での細胞培養に基づく既存のM1/M2ミクログリアの分類は、特にTBIのような複雑な疾患において、これらの細胞が生体内試験で遭遇する多様なサイトカインや細胞シグナルを除外している。M1、M2の分類から、トランスクリプトームやプロテオミクスのプロファイリングを組み込んだより良い解析へのシフトが必要である。さらに、TBIにおける炎症は中枢神経系に限定されるものではない。最大の免疫貯蔵庫である腸は、マクロファージとT細胞の免疫プロフィールに大きな変化を示しており、腸管免疫と脳免疫細胞との相互作用について理解を深める必要がある。
ほとんどの薬剤は、傷害発生から治療開始までの間隔が長くなるにつれて効力を失う。[334]。さらに、プロバイオティクスのような特定の治療法は、認知機能の改善が観察されるまでに長期間の投与が必要であり、その期間は数年に及ぶ可能性がある。[307]。TBI治療法の開発を進め、前臨床から臨床への移行を成功させるために、TBI治療における治療時間枠は、依然として重要な研究課題である。[335]。動物モデルでは受傷直後から治療が行われることが多いが、専門の外傷センターで行われる臨床試験では、通常、中等度から重度のTBIの4~7時間後に患者が登録される。[336]。さらに、軽傷の患者は、受傷後数年経って症状が現れるまで治療が遅れることがある。[337, 338]。第III相臨床試験PROTECTおよびSYNAPSE [339]におけるプロゲステロン治療の失敗により、治療時間枠の重要性が強調された。両試験とも、それぞれTBI後4時間以内と7時間以内に患者をリクルートし、受傷後1時間以内にプロゲステロンを投与した前臨床動物モデルで観察された有益性を再現できなかった。[340]。これらの臨床試験の失敗は、治療開始における治療域の重要性を浮き彫りにしている。前臨床モデルでは、現実の状況をシミュレートするために、受傷後の投与を徐々に遅らせて薬効を調べることが奨励されている。
治療のタイミングに加えて、最適な治療期間を理解することは、長期的な転帰を解明するために不可欠である。[341]。β遮断薬のように、2日や2週間といった短期間の投与で著明な改善を示す薬剤もあるが、プロバイオティクスのように、特にTBIのような慢性疾患では、神経保護効果を発揮するために長期の治療期間が必要な場合もある。[219, 220, 277, 342]。TBI後の慢性的治療レジメンの効果を評価するためには、脳腸軸内の免疫、ホルモン、神経経路の時間的・空間的動態を包括的に理解することが不可欠である。[343]。現在の動物実験では、主に急性期の変化に焦点が当てられており、薬剤の有効性やその後の神経学的転帰に影響を及ぼす可能性のある慢性的な変化を見逃している。[15]。TBI後何年にもわたって観察される持続的なマイクロバイオームの変化は、脳損傷の影響を軽減するために、治療期間を延長する必要性があることを強調している。[89]。治療のための治療域、治療期間、脳腸内ホメオスタシスへの影響を研究することは、TBI治療法を成功させるために極めて重要である。[334]。
臨床試験では、併存疾患を有する患者の母集団が一様でないことや、無作為化に関する倫理的配慮など、多くの課題に遭遇する。[344]。さらに、軽症-中等症-重症のTBIの分類は過度に広範であり、放射線学的所見や臨床所見を考慮していないため、主観的なものとなっている。[345]。この問題に対処するためには、分類システムの改善が必要である。GFAPやUCH-L1などの血中バイオマーカーを患者のベッドサイドでわずか15分以内に同定するように設計されたアボット社のi-STAT TBIカートリッジが最近FDAに承認されたことにより、傷害の重症度を層別化し、従来の軽症-中等症-重症の分類から管理を調整するためのより客観的な結果が約束された。[346]。最後に、このような複雑な疾患を治療するために単剤療法に依存していることが、課題を複雑にしている。この複雑な疾患を効果的に管理するためには、脳と腸の様々な経路を同時に標的とすることにもっと重点を置くべきである。
結論
結論として、我々は、セロトニン、グレリン、プロゲステロンなどのホルモン、β遮断薬やα-アドレナリン作動薬などのANS調節薬、スタチンなどの抗脂血症薬、プロバイオティクスや抗生物質などの腸内細菌叢調節薬など、TBIにおける脳腸軸に沿った炎症を標的とする様々な薬物療法について、初めて包括的なレビューを行った(表1と表2に要約)2)。これらの薬物は、炎症を緩和し、腸管バリアの完全性を維持し、運動性を促進し、健康なマイクロバイオームを促進することによって腸管機能を高める。重要なことは、これらの薬物療法はBBBを通過することによって直接的に、あるいは免疫、ホルモン、神経経路を調節することによって間接的に、神経学的転帰にも影響を及ぼすということである。
腸は最大の免疫貯蔵庫であり、身体の第二の「小さな脳」であるENS [348]を抱え、多様なマイクロバイオームを保持していることから、TBIの病因を理解する上で、腸が焦点となることが浮かび上がってくる。微生物異常、ENSの機能障害、EECの機能障害、ANSの調節障害、腸管免疫細胞の活性化の間の複雑な相互作用を解読することは、何十年もの間つかみどころのなかったTBIの治療法を開発する上で極めて重要である。そのためには、脳と腸にわたる免疫系活性化の時間的変化を理解すること、腸内微生物の代謝産物やホルモンの影響、急性・慢性TBIの病態生理学的過程における自律神経系と全身性炎症の役割を解明することが必要である。
略語
- TBI 外傷性脳損傷
- GI 胃腸系
- ENS 腸神経系
- ANS 自律神経系
- ICP 頭蓋内圧
- DAMP 損傷関連分子パターン
- ROS 活性酸素種
- BBB 血液脳関門
- AQP4 アクアポリン4
- ATP アデノシン三リン酸
- Bcl-2 B細胞リンパ腫2
- ICU 集中治療室
- HPA 視床下部-下垂体-副腎
- Ccr2 C-Cケモカイン受容体2型
- TNF-α 腫瘍壊死因子α
- IL-1β インターロイキン-1β
- TLR4 トール様受容体4
- EGC 腸グリア細胞
- Ach アセチルコリン
- α7nAChR α7 ニコチン性アセチルコリン受容体のサブタイプ
- EEC 腸内分泌細胞
- VNS 迷走神経/迷走神経刺激
- IBD 炎症性腸疾患
- IBS 過敏性腸症候群
- 中枢神経系
- GLP-1 グルカゴン様ペプチド1
- ChgA クロモグラニンA
- Lgr5 ロイシンリッチリピート含有Gタンパク質共役型受容体5
- Notch1 ノッチ受容体1
- Atoh1 アトナルbHLH転写因子1
- Neurog3 ニューロジェニン3
- TPH トリプトファン水酸化酵素
- SSRI 選択的セロトニン再取り込み阻害薬
- SNRI セロトニン・ノルエピネフリン再取り込み阻害薬
- ICC カハール間質細胞
- IP 腹腔内
- FGF 線維芽細胞増殖因子
- FGF-BP FGF結合タンパク質
- bFGF 塩基性FGF
- PI3K ホスホイノシチド3-キナーゼ
- PIP3 ホスファチジルイノシトール3,4,5三リン酸
- PIP2 ホスファチジルイノシトール3,4二リン酸
- Pkb/Akt プロテインキナーゼB
- IRI 虚血再灌流障害
- 脊髄損傷
- mTOR 哺乳類ラパマイシン標的タンパク質
- WD 体重減少
- ICH 脳内出血
- SMA 上腸間膜動脈
- BDNF 脳由来向神経性因子
- CD55 補体崩壊促進因子
- Nrf2 核赤血球2関連因子2
- ICAM-1 細胞間接着分子-1
- SAH くも膜下出血
- NE ノルエピネフリン
- HSP-70 熱ショックタンパク質70
- UCHL-1 ユビキチンカルボキシル末端ヒドロラーゼL1
- ZO-1 ゾヌラ・オクルーデンス-1
- vBNST 末端線条体腹側床核
- AD アルツハイマー病
- CD68 分化クラスタ68
- GFAP グリア線維酸性タンパク質
- VEGF 血管内皮増殖因子
- GSK-3beta グリコーゲン合成酵素キナーゼ-3β
- CD40 分化クラスタ40
- VCAM-1 血管細胞接着分子-1
- SCFA 短鎖脂肪酸
- Cb クロストリジウム・ブチリカム(Clostridium butyricum)
- LA 乳酸菌
資金提供
該当なし。