

コンテンツ
rwmalonemd.substack.com/p/what-is-adulteration-of-pseudo-mrna
2023/10/24

最近、シミアンウイルス40(SV40)由来のDNA配列を含むDNA断片がmRNAワクチンに混入していることに関して、COVIDの”mRNAワクチン”の話に詳しい関係者や関係者の間で多くの議論が交わされている。

これは何を意味するのか?そして、なぜ気にする必要があるのか?
これは中の野球の、「ソーシャルメディアの専門家/理論家」が宣伝するフリンジ陰謀論や、酸化グラフェンや生きたヒドラ、ワクチンに含まれるヘビ毒に関する恐怖政治が増幅された論争、あるいは脂質-擬似RNAナノ粒子が実は24世紀のスター・トレックSFのナノボットであり、私たちの脳をすべてプログラムし直すという論争と同じような、急須の内輪揉めに過ぎないのだろうか?
このDNA汚染/混入の問題は本物なのか?
デイビッド・シュパイヒャー博士、ケビン・マッカーナン博士、そして同僚たちは、実際に、配列および分子生物学的解析手法の実世界での応用における、真に真剣な科学的・技術的専門家である。彼らはそれを生業としている。それがたまたま、彼らが報告している特定の技術分野なのである。
彼らはフリンジの”熱の沼“の陰謀論者(スティーブ・バノンの用語)ではない。
Dr. David J. Speicher, University of Guelph Department of Pathobiology, 50 Stone Rd E, Guelph, ON, N1G 2W1, speicher@uoguelph.ca ,ORCID 0000-0002-1745-3263
Speicherらが観察し、下記リンク先の科学原稿で報告していることは、FDAや世界の規制当局が、その最も重要な仕事-医師や医療従事者が販売・使用することを許可した医薬品の純度や不純物の混入のなさを保証すること-に重大な失敗をしていることを明確に示している。
少なくとも、ピーター・マークス博士の「真の信者」指導の下、FDA/CBERワクチン部門に蔓延しているように見える故意の盲目さが改めて示された。ピーター・マークス博士は、ワクチンの専門家でも免疫学者でも分子生物学者でもなく、非ウイルス性脂質ナノ粒子をベースとしたポリヌクレオチド送達を理解する者でもなく、むしろ臨床血液・腫瘍学者であり、ワクチン(そして現在は抗がん剤)開発における「ワープ・スピード作戦」アプローチの最初の発案者であり、継続的な支持者である。つまり、数十年にわたる生物学的製剤や医薬品の開発、製造、販売承認、市販後調査から学んだ通常の手順や教訓をほとんどすべて回避するということである。
悪く言えば、この新情報によって、米国をはじめとする西側行政国家の医薬品規制当局と製薬業界との腐敗した癒着を示す「決定的証拠」が出現したことになる。
これらのデータに対する私の個人的な評価によれば、この汚染は、米国連邦法で厳しく禁止されている医薬品の「不純物混入」の正式な基準を満たしているようだ。医薬品、医療機器、食品の「不純物混入」の防止は、FDAの中心的な任務の一つであり、基本的には、FDAが最初に設立された中心的な理由である。
未解決の重要な問題のひとつは、なぜこのようなことが起こったのかということだ。
この不純物はFDA、EMA、ポール・エーリック研究所、カナダ保健省などに知られ、一般には隠されていたのだろうか?もし知られていなかったとしたら、この不純物はどのようにして事実上すべての西側諸国公認の政府規制専門家による発見を逃れたのだろうか?
以下は、このツイートのスクリーンショットと、リンク 今回の騒動の発端となったプレプリント原稿へのである。

速報-27本のバイアル瓶の最新調査。すべてのバイアルは蛍光測定法でガイドラインを桁違いに超えている。qCRを用いた単回投与では、すべてのバイアルがガイドラインを下回っている。何本も接種すればオーバーする。
XBB1.5にはまだ問題がある。
要旨
背景 SARS-CoV-2ワクチン用のヌクレオシド修飾RNA(modRNA)を作製するために用いられる試験管内試験転写(IVT)反応は、現在、DNA鋳型から転写するRNAポリメラーゼに依存している。オリジナルのファイザー無作為化臨床試験(RCT)で使用されたmodRNAの生成には、PCRで生成されたDNA鋳型が使用された(工程1)。数十億のワクチン用量を生成するために、このDNAを細菌のプラスミドベクターにクローニングし、大腸菌で増幅してから直鎖化した(工程2)。モデルナは、臨床試験用ワクチンと臨床試験後使用ワクチンの両方に、同様のプラスミドベースのプロセスを使用していたようだ。最近、DNAシークエンシング研究により、ファイザー・バイオエヌテックワクチンとモデルナmodRNAワクチンの両方において、このプラスミドDNAが有意なレベルで存在することが明らかになった。これらの研究は限られたロットを調査したものであり、国際的に観察された残存DNAのばらつきに疑問が残る。
方法Using previously published primer and probe sequences, quantitative polymerase chain reaction (qPCR) and Qubit® fluorometry was performed on an additional 27 mRNA vials obtained in Canada and drawn from 12 unique lots (5 lots of モデルナ child/adult monovalent, 1 lot of モデルナ adult bivalent BA.4/5、モデルナ小児・成人2価BA.1 1ロット、モデルナXBB.1.5 1価1ロット、ファイザー成人1価3ロット、ファイザー成人2価BA.4/5 1ロット)。ワクチン有害事象報告システム(VAERS)データベースは、試験された各ロットの報告された有害事象(AE)の数と分類について照会した。ファイザー社製COVID-19ワクチンの既研究バイアル1本の内容物を、DNA断片のサイズ分布を決定するためにオックスフォード・ナノポアシーケンシングで調査した。このサンプルはまた、残存DNAが脂質ナノ粒子(LNP)内にパッケージされているためDNaseIに耐性があるのか、あるいはDNAがLNPの外側に存在しDNaseIに不安定なのかを決定するために使用された。
結果プラスミド複製起点(ori)およびスパイク配列の定量サイクル(Cq)値(1:10希釈)は、ファイザーではそれぞれ18.44~24.87および18.03~23.83、モデルナではそれぞれ22.52~24.53および25.24~30.10であった。これらの値は、qPCRで測定したオリおよびスパイクのそれぞれについて、0.28~4.27ng/doseおよび0.22~2.43ng/dose(ファイザー)、0.01~0.34ng/doseおよび0.25~0.78ng/dose(モデルナ)に相当し、Qubit®蛍光光度計で測定したファイザーおよびモデルナのそれぞれについて、1,896~3,720ng/doseおよび3,270~5,100ng/doseに相当する。SV40プロモーター-エンハンサー-オリは、Cqスコアが16.64~22.59のファイザーバイアルでのみ検出された。探索的分析において、1回あたりのDNA量と重篤な有害事象(SAE)の頻度との用量反応関係の予備的証拠を発見した。この関係はファイザー社とモデルナ社で異なっていた。サイズ分布解析の結果、DNA断片の平均長は214塩基対(bp)、最大長は3.5 kbであった。プラスミドDNAはLNPの内部にあり、ヌクレアーゼから保護されていると考えられる。
結論 これらのデータは、これらのワクチン中に1回投与あたり数十億から数千億のDNA分子が存在することを実証している。フルオロメトリーを用いると、全てのワクチンはFDAとWHOが設定した残留DNA量のガイドラインである10ng/doseを188~509倍上回っている。しかし、qPCRによる残留DNA量はすべてのワクチンでこのガイドラインを下回っており、定量的ガイドラインを解釈する際の方法論の明確さと一貫性の重要性が強調された。qPCRによる残存DNA量とSAEの用量反応効果に関する予備的な証拠は、確認とさらなる調査の必要性を示している。私たちの知見は、ワクチンの安全性に関する既存の懸念を拡大し、LNPを用いた効率的なトランスフェクションが導入される前に考案されたガイドラインの妥当性に疑問を投げかけるものである。 いくつかの明らかな限界はあるが、私たちの研究を法医学的条件下で再現し、高効率DNAトランスフェクションおよび累積投与を考慮したガイドラインの改訂を強く要望する。
原稿の全文はこちらのリンクから見ることができる。
この発見の背後にある科学を理解する
発見され実証されたことの技術的側面や意味を理解するためには、分子生物学の基本をある程度理解しておく必要がある。私は、大学で分子生物学を学んだことのない人たちにも、必要な背景を説明できるよう最善を尽くすつもりだ。私はこのテーマに少し近づきすぎており、時には背景知識を前提としすぎていることを認める。もしそうなら、申し訳ない。リチャード・ファインマン教授の言葉にあるように、「簡単な言葉で説明できないなら、あなたはそれを理解していない」のである。私は彼の基準に沿うよう努力するつもりだ。
生物学の「セントラル・ドグマ」から始めなければならない。DNAはRNAを作り、RNAはタンパク質を作るもちろん逆転写酵素のことは知っている。
純粋なRNAを大量に製造したいのであれば、基本的には大量のDNAからスタートし、タンパク質酵素(私のオリジナルの方法ではバクテリオファージT7 RNAポリメラーゼ、これは今でも使われている)とRNA化学サブユニット、そしてエネルギー源(ATP)を使ってDNAからRNAを作る必要がある。次に、DNAを小さな断片に分解し、大きなRNAはそのまま残す必要がある。そして、大きなRNAから小さなDNA断片を精製する必要がある。私のオリジナル・プロセスでは、この精製は一種のフィルター(ゲルクロマトグラフィー)を使って行われ、分解された小さなDNA断片と未使用の小さな化学サブユニットを、大きなRNA分子よりも速く通過させる。そして、最初に出てきた小さなもの(DNA断片と未使用の化学物質)を捨て、後から出てきた大きなもの(基本的に水に溶けた純粋なRNA)を保存する。
意味があるのか?
そして、マイナスに帯電した精製RNAを水に入れたら、それを多かれ少なかれ濃縮し、自己組織化したプラスに帯電した脂肪のような他のものと派手な方法で混ぜて脂質ナノ粒子を作り、ガラス瓶に保管し、人に注射する。これが擬似mRNAワクチンの製造工程である。
何が問題なのか?
この場合、少なくとも2つのことが間違っているように見える。ひとつは、RNAの製造に使われるDNAが関係していることである<生物学のセントラル・ドグマを思い出してほしい>。そしてもうひとつは、DNAの分解と精製プロセスである。
DNAの製造には2種類の方法がある。ポリメラーゼ連鎖反応は、より大きな直鎖状DNA断片(精度にやや問題がある)を作ることができ、それを使ってRNAを製造した。これは、世界的な投与をサポートするために必要なレベルでの大量生産をサポートするには、あまりにも困難で、高価で、時間がかかることなどが判明した。そこでファイザー/バイオエンテックとモデルナは、私が使用した、バクテリア(大腸菌の特別な実験用菌株で、腸内に一般的に存在するバクテリア)を使用して生成される環状「プラスミド」DNAに依存するオリジナルの方法に戻ったようだ。

プラスミドは、細菌ウイルスの最も純粋な形のようなものだと考えることができる。バクテリアに感染するもっとウイルスに似たもの(バクテリオファージと呼ばれる)は他にもあるが、プラスミドは文字通り純粋なDNAとしてバクテリアに感染することができる環状DNAであり、バクテリアに自分自身や他のプラスミドをあるバクテリアから別のバクテリアに移すよう指示することができる。
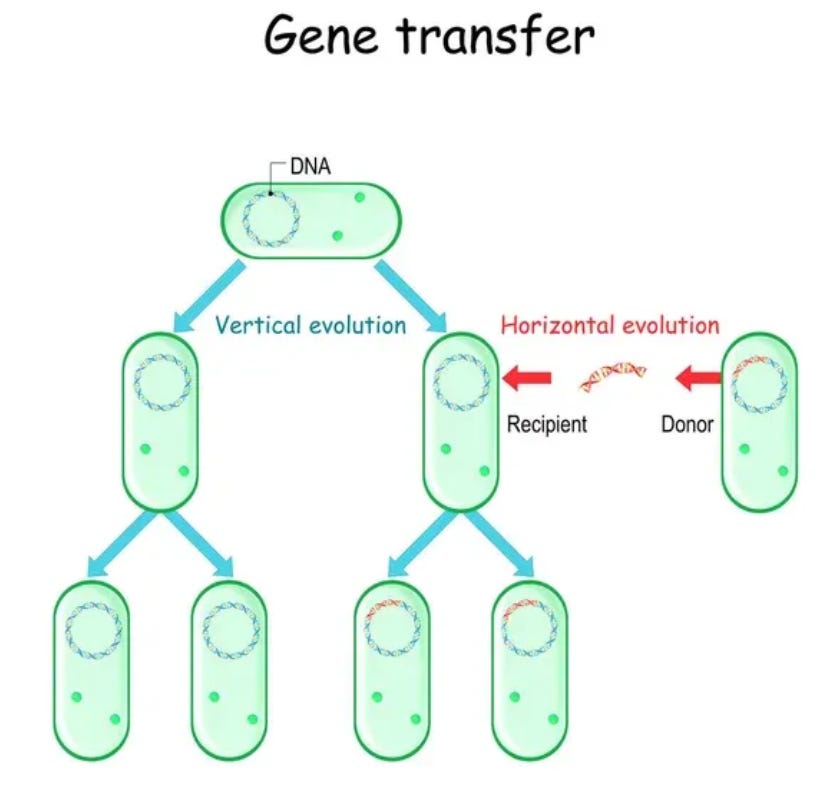
これらのプラスミドは小さな寄生DNAサークルのようなもので、抗生物質への暴露など特定の条件下で細菌宿主の生存を助けることが多い。このような淘汰圧のもとでは、プラスミドは生存や繁殖に有利に働くため、細菌によって維持される。もしプラスミドが利点をもたらさない場合、他の類似した細菌がプラスミドを持つ細菌に勝ってしまうだろう。<本題に関連する話なので我慢してほしい>。
大腸菌の培養で、できる限り多くのプラスミドDNAを増殖させ、回収(つまり製造)したいのであれば、工学的に設計できる最も小さく、最も削ぎ落とされたプラスミドを使いたい。なぜなら、プラスミド中の余分なDNA配列は、結果として得られる細菌培養物中の1リットルあたりのプラスミド生産量を少なくするという代償を伴うからだ。したがって、プラスミド複製、抗生物質選択(この場合はカナマイシンかネオマイシン)、最終的なRNA製造に必要のないDNA配列はプラスミドに加えたくない。お分かりいただけただろうか?
ではなぜ、DNAテンプレートからRNAを大量に合成するプラスミドベースの製造プロセスを開発・展開する企業が、意図した目的には必要のない配列をプラスミドに含めるのだろうか?なぜシミアン・ウイルス40(SV40)のような既知のがん原性(つまり、がんを引き起こす)DNAウイルスから取り出した配列を加えるのか?

Speicherらによって文書化されたプラスミドDNA断片汚染(上記)で同定されたこれらの特異的なSV40配列は、分子生物学者が使用するために数十年前に開発された特定のタイプの人工細菌プラスミドで一般的に使用されていることが判明した。これは確立された。「コモン・コア」組換えDNA技術である。
細菌プラスミドは、細菌と動物細胞の両方において複製し、RNA(およびタンパク質)を産生するよう設計することが可能であり、また長い間そうされてきた。このようなプラスミドは、業界では「シャトルベクター」と呼ばれている。このプラスミドは、実験用の大腸菌株を使って大量に製造・精製され、動物細胞に移植(「トランスフェクション」)される。
では、SV-40の塩基配列は、プラスミドの中で一体何をしているのだろうか?その唯一の目的は、精製され、市販の酵素ベースの製造プロセスを使って「試験管の中で」大量のRNAを生産するために使われることなのだろうか? いい質問だ。
私は推測や仮説を立てることはできるが、その疑問に答えるのは製薬会社と政府規制当局の仕事だと思う。そして、SV-40やその他のプラスミドDNA配列の小断片(抗生物質耐性遺伝子断片を含む)が、世界史上最も効率的な生体内非ウイルス送達技術を用いて患者の体内に送達される際に、起こりうるリスクについて正式な評価を受けることはおろか、一般に開示されることもなかったのはなぜなのか、その理由を明らかにすることである。
考えられるリスクを想像できるだろうか?
短く言えば、イエス、できる。何らかの形で、最低限これらの断片はDNAを取り込む人間の細胞の遺伝子発現に影響を与える可能性が高い。一つの可能な影響は、癌の発生に関与するかもしれない―これは分子生物学者や癌研究者が「変換」と呼ぶものである(強調している)。このようなリスクは、これが進行し、人々に(彼らの知らない間に)注入される前に調査されるべきだったか?もちろん、そうすべきだった。また、これがすべて関係者に開示されるべきだったことも自明である。もしFDA、EMA、ポール・エーリッヒ研究所、ヘルス・カナダなどが知らされていなかった場合、それは詐欺である。知らされていて何もしなかった場合、それは刑事過失である<私の意見だが、私は医師であり、弁護士ではない>。
ただし、Pfizer/BioNTechとModernaのプラスミドにおけるSV40の配列に関する重要な注意点がある。それは、SV40が固形腫瘍(肉腫)の発展を促進する主要なメカニズムは、ウイルスが産生する「Large T抗原」タンパク質であるという点である。このタンパク質のDNA配列は、いずれのプラスミドにも存在していない。
この全てについて、事実検証者のプロパガンダ、曖昧さ、何それ主義が嵐のように吹き荒れると予想するが、中核の事実は明白である。
というわけである。
バチン。

ファインマン教授、私はあなたの期待に応えられたか、あるいはそれ以上であったかと思う。
それなら、不純物がどう関係するんだ?(ティナ・ターナーに謝罪する)
「不純物」は法的規制用語であり、米国連邦規則集(CFR)において広く使用されている。例えば、21 U.S. Code § 351 – Adulterated drugs and devices.を参照のこと。
Webstersウェブスターによれば
ADULTERATE(不純物)の意味は、異物または劣った物質や要素を加えることによって、腐敗させる、堕落させる、不純物にすることである。
米国議会によると、不純物は定義されている(CFRTitle 21,CHAPTER 9,SUBCHAPTER V§ 351)以下のように:
医薬品または器具は、不純物とみなされる。
(a)毒物、不衛生な成分などである場合、製造において適切な管理がなされていない場合。
(b)医薬品であって、その製造、加工、包装又は保管に使用される方法又は設備若しくは管理が、当該医薬品が安全性に関して本章の要件を満たし、かつ、当該医薬品が有すると称し又は表示される同一性及び強度を有し、品質及び純度の特性を満たすことを保証するための現行の適正製造規範に適合しないか、又は適合するように運営若しくは管理されていない場合。
(b)公式コンペンディウムと異なる強度、品質または純度
公式大要にその名称が認められている医薬品であると称し、又はそのように表示され、かつ、その強度が当該大要に定められている基準と異なる場合、又はその品質若しくは純度が当該大要に定められている基準を下回る場合。ただし、当該大要に試験または測定方法が規定されていない場合、または規定されている試験または測定方法が、長官の判断により、当該判定を行うには不十分である場合は、この限りではない、 ただし、当該大要に試験または分析方法が規定されていない場合、または規定されている試験または分析方法が当該判定を行うには不十分であると長官が判断した場合には、長官は、当該大要の改訂を担当する適切な機関にその事実を通知し、当該機関が合理的な期間内に、本項の目的には十分であると長官が判断する試験または分析方法を規定しなかった場合には、長官は、強度、品質または純度に関する当該判定を行うための適切な試験または分析方法を規定する規則を公布するものとする。
(c)コンペンディウムで医薬品が認識されていない場合の強度等の不当表示
本節(b)の規定に該当せず、かつ、その強度が、当該医薬品が有すると表示され、又は表示されている強度と異なり、又はその純度若しくは品質が、当該医薬品が有すると表示され、若しくは表示されている純度若しくは品質を下回る場合。
(d)他の物質との混合または置換
医薬品であり、かつ、(1)その品質または強度を低下させるように他の物質が混合され、または包装された場合、または(2)その全部または一部が代替された場合。
不純物混入の法的救済措置は?
FDA文書CPG Sec.420.100 Adulteration of Drugs Under Section 501(b) and 501(c)を参照のこと。*セクション501(b)に基づく粗悪医薬品の直接参照押収権限*.
食品医薬品化粧品法(FDA)第501条(b)は、公定薬物(すなわち、公定書の中でその名称が認められている医薬品であると称され、またはそのように表示されている医薬品)が、品質、強度または純度に関する公定書の基準に適合しない場合、粗悪品であるとみなしている。501(b)に基づきこのような適合性を判断する際には、一般的な試験または測定法が使用される。この基準は、個々のモノグラフおよびUSP/NFの一般通知のセクションの一部に記載されている。規格および試験方法は、力価、無菌性、*溶出性*、重量変動および含量均一性等の特性について確立されている。公的医薬品が、強度、品質、純度に関する1つ以上の規格に適合していないが、ラベルに 規格とどのように異なるかを明確に記載している場合、その医薬品は第501条(b)に基づき粗悪品とはみなされない。
方針:公的な医薬品は、その品質、強さ、純度について、公式の方法で試験したときに、公式の基準に適合しない場合、そのような基準との相違が医薬品のラベルに明白に記載されていない限り、不純物となる。公式大要で認められていない医薬品は、その強度が、科学的に健全な方法で試験された場合に、その医薬品が有すると称する、または表示される強度と異なる場合、またはその純度もしくは品質が、その医薬品が有すると称する、または表示される純度もしくは品質を下回る場合、粗悪品である*。
規制措置の指針: 規制措置の勧告は、上記の不純物混入の事例において考慮される。選択される規制措置は、各事例の状況による。
健康被害がある場合、特に殺菌剤であることが判明した医薬品や、力価試験や溶出試験に不合格となった治療域の狭い医薬品については、回収を第一選択とすべきである。しかしながら、医薬品品質管理局(OPQO)内の適切な部署が、そのような欠陥製品について会社に助言し、会社が回収に応じない場合には、押収を検討すべきである。
セクション501(c)に基づく粗悪品を告発する押収勧告は、医薬品評価研究センター(HFD-300)(CDER)のコンプライアンス部に提出されなければならない
OPQO内の適切な部門事務所は、以下の状況下では、CDERの審査なしに、501条(b)に基づく粗悪品を理由とする押収勧告を、州際通商への導入または導入のための引渡しが文書化されている場合に限り、OPQOプログラム・ディレクターおよび執行部に直接提出する権限を有する:
1.原薬または最終製剤の公式試料が、原薬の分析法をそのまま用いて分析され、原薬分析および確認分析の両方において不合格であることが判明した場合。
2.分析機関は、提出された覚書において、修正されていない公式の方法が使用されたことを証明する。注:公式コンペンディウムで規定されている以上の公差を適用する必要はない。3.無菌製品については、コンペンディショナル無菌試験が修正されずに利用され、製品が無菌であることが要求されるものであり、関連するすべての検査室管理(陽性および陰性を含む)が満足のいくものであれば、確認分析は必要ない。分析施設が公式のコンペンデンシャル分析法から逸脱した場合、その逸脱の詳細な説明と正当な理由をCDERに提出し、審査を受けることができる。このような場合、CDERは逸脱のみを審査し、規制措置の選択やその他の文書については審査しない。押収処分の場合、告発は以下のように作成することができる:当該医薬品が、合衆国法律集第 21 編第 351 条(b)の意味において、州際通商に導入された時および州際通商中に混入され、州際通商における出荷後に販売のために保管されている間に混入されたこと。351(b)の意味において、公式大要(United States Pharmacopeia)にその名称が認められている医薬品であると称し、そのように表示され、その強度が公式の(テストの種類を挿入する)テストに不合格であるため、その品質および純度が当該大要に規定されている基準とは異なり、その基準を下回っている。
または
当該医薬品は、米国連邦法典第351条(c)の意味において、州際通商に導入された際、および州際通商において出荷された後、販売のために保管されている間に粗悪品であり、米国連邦法典第351条(b)の規定に従わない医薬品であり、その強度は、(例. 例えば、ラベルに記載されている(INSERT NAME OF INGREDIENT)の含有量より少ない場合)。501(b)および501(c)の下で発見された不純物の種類は、製造者が現行の適正製造規範を遵守していないことを含む、より広範な問題を示している可能性があり、これに対処すべきであることを念頭に置くべきである。
第一に、上述したように、私の知る限り(上記で検討したデータと事実が真実であると仮定して)、Speicherらによってテストされた製品は、粗悪品の正式な規制基準を満たしているというのが、私の専門家としての見解である。
第二に、私の知る限り(上記で検討したデータと事実が真実であると仮定して)、Speicherらによって試験された製品は、健康被害の重大なリスクを示すものであるというのが、私の専門家としての見解である。さらに、CDC VAERS、VSAFE、ファイザー・ファーマコビジランス、および世界中の安全性データベースによって蓄積された安全性データは、これらの製品が健康被害の重大なリスクを示す合理的な証拠があることを示し、CFRTitle 21,CHAPTER 9,SUBCHAPTER V§ 351および FDAガイダンスCPG Sec.*セクション501(b)*に基づく粗悪医薬品の直接参照押収権限、これらの製品は即時リコールの対象となるべきである。
第三に、最後に、これらの製品がスポンサーによって回収されない場合(EUAの特別なケースでは、スポンサーは FDAであることに留意されたい )、米国50州それぞれの検事総長によって差し押さえが強く検討されるべきであるというのが私の専門的見解である。
2023年10月23日(月)署名

余談だが、コロナ危機の陰謀を読んだり聞いたりすることでドーパミンのヒットを求める人々にとって、高校レベルの教育を受けた落ちぶれた賞金稼ぎ、経済学の博士号を持つ高齢の看護師、科学的訓練を受けたことのないカイロプラクター、独立ジャーナリストを装った関連科学的背景のない既知のサイバーストーカー、あるいは元製薬業界のマーケティング専門家が、これまでに大量生産され、何十億人もの人間に注射によって投与された最も複雑な医薬品の組成、リスク、作用機序の詳細な分析について信頼できる情報源であると正直に考えているのだろうか?
What is Adulteration of pseudo-mRNA vaccines, and why should you care?