
コンテンツ
Toward Therapy of Human Prion Diseases
www.annualreviews.org/doi/10.1146/annurev-pharmtox-010617-052745
58巻:331-351(巻頭言掲載日2018年1月)
初出は2017年9月27日付のReview in Advance
アドリアーノ・アグッツィ、アスビン・K.K.ラッカラジュ、カール・フロンツェク
チューリッヒ大学神経病理学研究所、CH-8091チューリッヒ、スイス
要旨
クロイツフェルト・ヤコブ病やその他の伝達性海綿状脳症の原因としてプリオンが発見されてから30年が経過したが、いまだに有効な治療法は見つかっていない。数多くの薬理学的介入が病気の進行の様々な段階を標的として試みられてきたが、病気の経過を有意に改善したものはない。プリオンがどのように脳に損傷を与えるかについてのメカニズム的な理解がまだ不十分であり、このため有効な薬理学的標的が乏しいのである。この総説では、プリオンの複製を阻害し、そのクリアランスを促進する試みについて述べる。また、プリオンの生物学的知見が深まるにつれて、新たな標的を同定する可能性が出てくるかもしれない。
キーワード
プリオン,神経変性,治療薬,発光性ポリチオフェン結合体,免疫療法,ユビキチン・プロテアソーム系
プリオンとプリオン病
プリオン病、すなわち伝達性海綿状脳症(TSE)は、細胞性プリオンタンパク質PrPCのミスフォールディングバージョンであるプロテイナーゼK耐性型プリオンタンパク質PrPScの秩序だった凝集によって引き起こされる。同様のメカニズムが他の多くの神経変性疾患や全身疾患にも見られることから、後者を引き起こすタンパク質凝集体はプリオノイドと呼ばれるようになった(1,2)。われわれの定義では、プリオノイドは分子レベルではプリオンと同様に作用するが、個人から個人へ伝播することは(まだ)証明されていない。プリオン病の場合、凝集は自己持続的であるため、個体間で感染することになる。これらの疾患には、クロイツフェルト・ヤコブ病(CJD)、クル病、致死性家族性不眠症、ヒトの遺伝性TSEがある(7)。神経病理学的には、非特異的徴候(アストログリオーシス、神経細胞消失、アミロイド沈着)は、時に変性小器官を含む神経細胞内空胞や神経細胞内空胞という海綿状変化を伴う(8)。神経細胞の海綿状変性はプリオン病に極めて特異的であり、通常、確定診断が可能である。
細胞内プリオンタンパク質の存在は、プリオンのde novo生成に必要なだけでなく、宿主生物がプリオンに関連した神経毒性を経験するためにも必要である(9)。PrPCを除去したマウスはプリオンに曝されても発症しない。PrPCをコードするPrnp遺伝子の対立遺伝子を1つしか持たないプリオン感染マウスは、野生型マウスよりはるかに遅れて発症することから、PrPCの利用可能性が発症率を制限しているようである。
プリオン病はまれな病気で、年間100万人あたり1.5〜2人の患者が報告されている。しかし、必ず致死的であり、現在のところ有効な治療法はない。潜在的な抗プリオン療法が同定されれば、プリオノイドによって誘導されるミスフォールディング障害の治療にも道が開ける可能性がある。ここでは、成功する可能性のある治療薬と、それを待ち受ける課題に焦点を当てる。
プリオン転換をターゲットに
細胞内のPrPCは細胞膜上に発現し、そこで洗浄剤耐性の膜ドメインに選別される(10)。ある種の突然変異が存在すると、PrPCはよく理解されていないメカニズムで病的な、そして最終的には感染性のコンフォメーションをとるようになり、疾病につながる(11)。秩序化されたPrPSc凝集体は、さらなるプリオンの核形成の種となる。PrPScは大きな不溶性の凝集体やプラークから小さなオリゴマーまで、様々な構造をとることができる(6)。
ポリアニオン性化合物やアミロイドトロピック色素は、試験管内試験でPrPCから PrPScへの変換を阻害することができる(12,13)が、毒性、薬物動態の乏しさ、有効性の低さなどのために、治療薬にはなり得なかった(14,15)。プリオンに感染した神経芽腫細胞を分岐ポリアミンで処理すると、PrPScがクリアランスされた(16)。この化合物は酸性pHでプロトン化され、エンドソームやリソソームでのプリオン変換に作用すると考えられる(16)。しかし、これらの化合物はいずれも生体内では有益な効果を示さなかった。
デンドリマーは、主に修飾可能な末端基を持つ分岐ポリアミンからなる合成分子である(17)。リン酸デンドリマーは試験管内試験で有効な抗プリオン剤であり、PrPScを有意に除去したが、それ以上の開発は行われなかった(18)。ペントサン・ポリサルフェートはプリオン感染マウスの生存期間を延長し、PrPCから PrPScへの変換を阻害すると考えられたが、プリオン感染ヒトでは再現性のある効果は得られなかった(19,20)。アマンタジンは、もともとインフルエンザウイルスに対する予防薬として使用されていたが、様々な報告においてCJDの臨床経過を改善したことが示唆され、最初の症状発生から数年後まで生存したという逸話がある(21)。しかし、CJD患者におけるアマンタジンの有益な効果を再現できなかったという報告もある(22,23)。別の抗ウイルス薬であるアシクロビルはCJD患者2人では無効であり(24,25)、インターフェロンも患者2人のケースシリーズでは無効であった(26)。フルピルチンは一般に鎮痛薬として使用されるアミノピリジンであるが、プラセボ対照二重盲検試験で使用され、生存期間は変わらないにもかかわらず、散発性CJD(sCJD)と遺伝性CJDにおける認知障害の改善が示唆された(27)。
キナクリンは1930年代に抗マラリア薬として導入された抗寄生虫薬であり、新しい千年紀の変わり目に変異型CJD(vCJD)が流行した際、効率的な抗プリオン化合物がなかったため、研究者たちは臨床研究のために患者を集めざるを得なかった(28)。30人のsCJD患者と2人のvCJD患者を対象としたキナクリンの公開同情使用試験では、生存期間の有意な延長や機能障害の改善はみられず、脳病理に対する有益な効果も示されなかった(29)。キナクリンの前向き患者選択試験であるPRION-1でも、登録された107人の患者において、有意な生存期間の延長や認知障害の改善はみられなかった(30)。試験責任者らは、脳脊髄液中の薬物濃度が低かったことが、臨床エンドポイントに達しなかった原因かもしれないという仮説を立てたが、キナクリンの適用は全体的に許容できる安全性プロファイルを示した(30)。プラセボ群がないため、コンパッショネート使用試験では分子の小さな効果を検出することができないので、その後、sCJD患者を対象としたキナクリンの無作為二重盲検プラセボ対照試験が行われた(31)。最終的に51人の患者が機能解析と生存率解析に組み入れられた。キナクリン投与群の患者は治療初期には機能的スコアでわずかに良好であったが、キナクリン投与による生存利益は観察されず、プリオン病治療薬としては除外された(31)。
われわれは、活性酸素種の発生がプリオン誘発神経毒性の下流エフェクターであることを報告しており、アセチル化ヒドロキシチロソールのような抗酸化物質の投与は、プリオン病マウスの生存期間を効果的に延長した。ある症例報告では、CJD患者にビタミンEとαリポ酸を含む複合的な抗酸化物質を投与したところ、神経疾患に対する有益な効果が示唆されたが、その患者は発症から22カ月後に病死した(32)。
ドキシサイクリンのパイロット的な同情的使用試験では、年齢、性別、PRNP遺伝子のコドン129多型とは無関係に、患者の生存に有益な効果が示された(33)。初の多施設共同前向きプラセボ対照無作為化第II相試験では、ドキシサイクリンはプラセボと比較して最初の中間解析で優越性を示さず、試験は中止された(34)。後者の研究は、ドキシサイクリンがプリオン病患者の生存期間を延長しないというクラス1の証拠を提供したが、その後に発表された症例報告では、ドキシサイクリンで治療された可変性プロテアーゼ感受性プリオン病に罹患した患者の生存期間が異常に長かった(5年以上)ことが示唆されている(35)。これらの報告は、抗プリオン薬の効率の悪さが証明されているにもかかわらず、抗プリオン薬を処方する臨床医の無力さを示している。化合物B、IND24、anle138bは、ヒトプリオンに対して効果を示さなかった他の化合物の一つである(34)。
これらの残念な結果は、プリオン治療薬を同定する戦略の実行可能性に疑問を投げかけるものである。特に、細胞培養モデルが生体内における有効性の予測に乏しいことが明らかになってきた。そのもっともらしい理由は、不死化し、継続的に増殖する細胞においてプリオン感染性を維持することは一般に困難であるという事実にある。このような培養細胞がプリオンに慢性的に感染し続けるためには、少なくとも細胞分裂と同じ速さで複製が行われなければならない:負の差異が生じれば、時間の経過とともに感染性が失われるのは必然である。さらに、プリオンの複製は感染細胞にフィットネス・コストを課し、その結果、非感染細胞(不均一に感染した培養細胞や後天的に耐性を獲得した細胞)が系を過剰増殖させるかもしれない(36)。このような特徴から、感染細胞培養系は本質的に不安定な系であると予測される。この予測は、慢性的に感染した細胞株のほとんどが、時間の経過とともに自然に低い感染力価に引き寄せられるという観察によって実験的に検証されている(Aguzzi研究室のメンバーによる未発表の観察結果)。したがって、N2A細胞に関する重要な問題は、いかに治癒させるかではなく、むしろいかに感染状態を維持させるかである、と主張することができる。このように解釈すれば、培養細胞を治癒させることのできる薬剤が、脳内常在細胞のターンオーバー速度がはるかに遅く、自然抵抗性がほとんど発達する機会のない生体内で試験されると、ほとんど常に全く効果がないことが証明されるのは驚くべきことではない(表1)。
略語:6-OHDA、6-ヒドロキシドーパミン;CJD、クロイツフェルト・ヤコブ病;COCS、小脳器官型培養スライス;DMSO、ジメチルスルホキシド;EEG、脳波;gCJD、遺伝性CJD;gPrD、遺伝性プリオン病;GSS、ゲルストマン・シュトロイスラー・シャインカー症候群;i.c.、脳内;iCJD、異所性CJD;i.p.、腹腔内;LCP(発光性共役ポリチオフェン);ns(特定せず);MPA(ミスフォールド・プロテイン・アッセイ);PrPSc(プロテイナーゼK耐性プリオンタンパク質);RMLX(ロッキー・マウンテン研究所プリオン株のx継代);SCEPA(スクレイピー細胞エンドポイント・アッセイ);sCJD(散発性CJD);TMAO(トリメチルアミンN-オキシド);vCJD(変異型CJD)。 |
表1 プリオン病標的化合物
小脳器官型培養スライス(COCS)は、抗プリオン化合物を試験するための、より現実的なシステムであるように思われ、実際、生体内試験での抗プリオン効力の予測において、N2A細胞よりもはるかに優れた実績を上げている(37–42)。しかし、N2A細胞に対するCOCSの主な欠点は、その生産に手間がかかることであり、ハイスループット・スクリーニングには適さない。代わりに、COCSは試験管内試験で同定された化合物の中間的な検証のための二次スクリーニングとして最適である。
効果的な治療法を考案する上で重要なことは、病的タンパク質凝集体のフランジビリティー(壊れやすさ)を考慮することである。プリオン凝集体のフランジ性は、プリオンの感染性を決定する上で最も重要なパラメーターである。第一原理からのこの予測は、動物モデルでほぼ確認された(40)。大きな凝集体を多数の小さなオリゴマーに変換するβシート破壊化合物を使うことで、より多くのプロパゴンを不注意に作り出すかもしれない(43)。従って、効果的な治療法は、PrPSc凝集体を分解することではなく、むしろ凝集体を高安定化することを目指すべきである(図1)。発光性共役ポリチオフェン(LCP)は、このようなプリオンの超安定化剤として作用するようである。LCPはPrPScのクロスβスパインに結合する(44,45)。LCPはPrPSc凝集体を高感度で検出することができ、その発光スペクトルは異なるアミロイド(45)やプリオン株(46)を区別することができる。
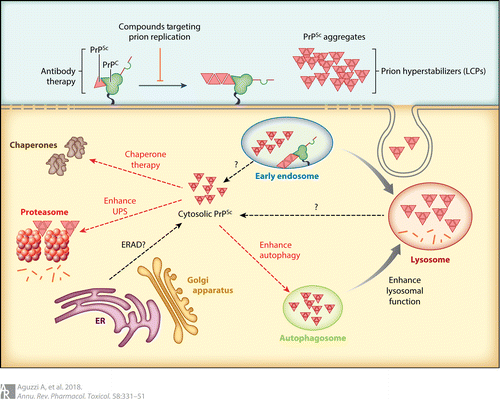
図1プリオンプロパゴンに遭遇すると、細胞内のPrPCは変換され、PrPScに取り込まれる。PrPCから PrPScへの変換はおそらく細胞膜上で始まり、エンドサイトーシス経路全体を通して続く。PrP凝集体は細胞質でも観察されており、ERAD、欠陥のあるエンドソームやリソソームからの漏出、あるいはその両方が原因である可能性がある。治療的介入の可能性としては、プリオンクリアランス(抗体療法)とプリオンの複製(凝集体の過安定化を含む)がある。細胞内標的としては、オートファジーやリソソーム機能のエンハンサー、UPSやケミカルシャペロンのモジュレーターなどがある。略語ERAD、小胞体関連タンパク質分解、PrPC、細胞性プリオンタンパク質、PrPSc、プロテイナーゼK耐性プリオンタンパク質、UPS、ユビキチン-プロテアソーム系。
LCPは感染マウスの脳から採取したプリオン凝集体を含むサンプル中のプリオン感染性を低下させた(47)。LCPとアミロイド線維との結合は原子レベルで分解され、協力的な静電相互作用に依存していることがわかった。しかしながら、フィブリルの安定性の代用であるプロテイナーゼKによる消化はLCPによって促進され、同じプリオン調製物の感染性は用量依存的に低下した(47)。これらの観察結果は、LCPがPrPSc凝集体を超安定化することにより、プリオンの感染性を実際に低下させるという仮説と一致している。LCPとアミロイドとの相互作用の構造的解明により、より結合力の強い新しいLCPを設計することができた。これらはプリオン感染マウスの生存期間の延長に高い効果を示した(48)。
細胞経路を標的としたプリオン治療
ユビキチン・プロテアソームシステム(UPS)は、ミスフォールドや損傷を受けたタンパク質を分解することにより、細胞内の品質管理を維持している(49)。初期の研究では、タンパク質の凝集体中にユビキチンが存在することが明らかにされた(50,51)。プリオン感染マウスの脳におけるユビキチン化タンパク質のレベルの上昇は、UPSの機能不全と関連しており(52)、これが神経毒性の一因となっている可能性がある。PrPScは20Sプロテアソームサブユニットの外葉に結合し、その機能を損なう可能性がある(53)。他の研究では、プリオンオリゴマーが触媒Bサブユニットを阻害する、あるいは基質がタンパク質分解コアに入るのを妨げると仮定している(54)。これらの仮説は、プリオン感染におけるプロテアソームの失敗を説明できるかもしれないが(52,55)、小胞体内腔や細胞外腔に存在するPrPが、細胞質でプロテアソームと遭遇するようになるとは考えにくい。PrPが顕著な小胞体関連タンパク質分解(ERAD)を受けるという考えはもっともらしいが(56,57)、反論もある(45)。
19Sプロテアソームサブユニットに結合している脱ユビキチナーゼであるユビキチンカルボキシ末端ヒドロラーゼ14(USP14)を阻害すると、凝集しやすいタンパク質がクリアランスされる(58)。USP14を標的とする低分子は、TDP43、タウ、アタキシンなどの神経変性疾患に関連するタンパク質の分解を促進する。神経変性疾患におけるミスフォールディングタンパク質を標的とした、まだ未解明の戦略として、低分子化合物の開発が挙げられる。低分子化合物は、内在性のE3ユビキチンリガーゼを基質に誘導する。PROTAC(タンパク質分解標的化キメラ分子)は、特定のユビキチンリガーゼを認識するペプチドと、標的タンパク質を認識する低分子が化学的に結合したものである(59)。標的タンパク質に結合すると、基質とユビキチンリガーゼの間に空間的な近接性が生じ、ポリユビキチン化と標的基質の分解が促進される。研究者たちは、がん治療のためにPROTACの特性を明らかにするための広範な研究を行い(60)、PROTACをミスフォールディングしたプリオンの標的に使える可能性があることを突き止めた。新しい戦略では、シャペロンタンパク質と低分子化合物を組み合わせる。低分子は基質へのガイドとして働き、シャペロンはミスフォールドしたタンパク質に関与してプロテアソーム分解を受けやすくする(61)。このような戦略は、脊髄性球筋萎縮症や筋萎縮性側索硬化症で実施された(62)。
アンフォールド・タンパク質反応を標的とする
タンパク質のミスフォールディング障害に共通する事象は、ERストレスとも呼ばれるアンフォールデッドタンパク質応答(UPR)のアップレギュレーションである(63)。全細胞タンパク質の30%以上が、修飾され最終目的地へ運ばれる前にERを通過する。ERは、タンパク質が合成され、折り畳まれ、翻訳後修飾される一連の複雑な細胞内プロセスを制御している(64)。ERの機能障害は、ミスフォールディングタンパク質の蓄積やカルシウムホメオスタシスの変化を引き起こし、ストレスの誘発につながる可能性がある。
UPRは、全体的な翻訳を停止させることで細胞のプロテオスタシスを回復させ、それによって小胞体内のミスフォールドタンパク質の負荷を軽減することができる(65)。またUPRは、小胞体内のミスフォールドタンパク質を修復するために、シャペロンやタンパク質のフォールディングを補助する他のタンパク質の合成を促進する(66)。ミスフォールディングしたERタンパク質は細胞質に再転移し、そこでERAD経路によって分解される(67)。UPRの主要な伝達因子は、プロテインキナーゼRNA様ERキナーゼ(PERK)、イノシトール要求性酵素-1(IRE1)、活性化転写因子-6(ATF6)である。PERKは真核生物の翻訳開始因子(eIF2α)のリン酸化による翻訳の減衰に不可欠な膜貫通タンパク質であり、IRE1とATF6は主にタンパク質のフォールディングに必要なシャペロンの合成に関与している(68)。
プリオンに感染したヒトとマウスでは、小胞体シャペロンGRP94、GRP78、GRP54の上昇が観察された(69)。精製PrPScにさらされた細胞は、CJD患者で同定されたシャペロンの発現上昇とともに、UPRの活性化とERからのカルシウム放出を示した(70)。さらに、UPSとERストレスの間には複雑な相互作用が存在し、プロテアソーム機能の阻害がUPRを引き起こすと広く信じられている(71)。
プリオン感染マウスは、PERKの持続的な活性化とeIF2αのリン酸化を示し、その結果、eIF2αのリン酸化を介してグローバルなタンパク質翻訳がダウンレギュレーションされ(72,73)、シナプスタンパク質の減少と神経細胞死を引き起こす。eIF2α特異的リン酸化酵素GADD34を過剰発現させると、少なくともしばらくの間、シナプス欠損と神経細胞死が回復する(73)。PERKを薬理学的に阻害すると翻訳が回復し、ある程度の神経保護が得られる(73)。eIF2αの下流の翻訳阻害をターゲットとする統合ストレス応答阻害剤Bもプリオン病態を改善することが示された(74)。
PERK阻害剤とは対照的に、グアナベンズとその誘導体であるセフィン1は、GADD34を阻害し、eIF2αのリン酸化状態を高めることによって、筋萎縮性側索硬化症モデルマウスにおける神経変性を予防した(75)。この長期にわたる翻訳停止は、新しいプロパゴンの合成を防ぎ、それによって神経保護をもたらすと考えられる。グアナベンズはプリオンクリアランスを促進することが以前に示されている(76)が、重篤な副作用のために、これまでのところその使用は制限されている。この問題は、UPRに基づく治療法の基本的な難問を示している:翻訳の一般的な制御機構に干渉する過程は、必然的に有害で予期せぬ結果を伴う(図1)。
リソソーム分解とオートファジーを標的とする
PrPCから PrPScへの変換は、細胞膜で起こり(77)、エンドソームや多胞体のリサイクルを含むエンドサイトーシス経路で起こる(78,79)。エンドサイトーシスコンパートメントにおけるミスフォールディングプリオンの蓄積は、小胞コンパートメントの組成とその機能を変化させる可能性がある。リソソームは細胞内PrPCの主要な分解部位であり、PrPScはリソソームに蓄積することがある(80)。細胞培養では、PrPScはリソソームによって除去される。しかし、エンドリソソーム機構に生じる他の欠陥やPrPScの過剰負荷によって、最終的にリソソームは機能しなくなる可能性がある。実際、プリオンに感染すると膜結合型rab7のレベルが低下し、リソソームの成熟とタンパク質の分解能力に影響を及ぼす(81)。
PrPScがリソソームへ運ばれ分解されるもう一つの重要な経路はオートファジーである(82)。オートファジーでは、細胞質成分が二重膜構造のオートファゴソームに取り込まれ、そのオートファゴソームはリソソームと融合し、内容物を放出し分解する。プリオン感染マウスの神経細胞、プリオン感染細胞培養物、遺伝的プリオンモデルにおいて、巨大な多胞体とオートファジー小胞(AV)が観察される(83)。オートファジーは、凝集体をすくい上げ、分解することによって、保護的な役割を果たしているのかもしれない。研究者たちは当初、プリオン病で観察される海綿状液胞はAVであると信じていたが、これらの液胞はAVの膜特性を持たず、オートファジーのマーカーも示さない。薬理学的あるいはsiRNAによってオートファジーを阻害すると、細胞がPrPScを分解する能力が阻害される(84)。したがって、オートファジーとリソソーム分解の促進剤はプリオンに対する治療薬となりうる(85,86)。リチウムはオートファジーを誘導することにより、培養細胞株におけるPrPScのクリアランスを促進し(87)、細胞内のPrPCレベルをわずかに低下させることが示されている。同じくオートファジーを促進するラパマイシンとタクロリムスも同様の結果を示した(88,89)。
トレハロースは、タンパク質の変性を防ぐことによって環境ストレス条件から保護するために、菌類や植物によって合成されるα結合二糖である。細胞培養では、トレハロースはオートファジーを誘導し、ミスフォールドしたタンパク質のクリアランスを改善する可能性がある(90)。プリオンに感染した細胞培養物からのPrPScは、トレハロースで処理することにより速やかに除去された(91)。同様に、もう一つのオートファジーを促進する化合物であるイマチニブは、細胞培養中のPrPScレベルを減少させた(92)。
シャペロン療法
分子シャペロンは他のタンパク質と相互作用し、それらが安定したコンフォメーションを獲得するのを助ける。分子シャペロンは、ミスフォールディングや凝集を防ぐ重要な品質管理システムである。酵母では、熱ショックタンパク質104(Hsp104)解離酵素が、酵母プリオンΨであるSup35の細胞質凝集体を可溶化することができる(93)。研究者らは、Hsp110/70/40の三者が哺乳類の最小分解酵素であることを同定した(94)。Hsp70を単独でアップレギュレーションすると、モデル系で神経保護が得られた(95)。しかし、プリオン病に対してこの3要素を治療的に利用できるかどうかは不明である。
ケミカル・シャペロンはタンパク質に結合し、タンパク質をリフォールディングして安定した構造にすることで機能を回復させる低分子化合物である。その非特異的な作用様式と低い親和性にもかかわらず、タンパク質の凝集体を除去するその能力は、治療薬として魅力的である。メチルアミンとグリセロールは、細胞培養モデルにおいてPrPCから PrPScへの変換を阻害するのに有効であった(96)。アントラサイクリン、ポルフィリン、ジアゾ染料も試験管内試験ではプリオンの複製を阻害するのに有効であった(97)。
プリオン病に対する積極的免疫療法
様々なタンパク質ミスフォールディング障害において、免疫戦略が有望視されている(98)。プリオンに対する積極的な免疫は、細胞性プリオンタンパク質PrPCの体内における広範な発現が自己寛容をもたらすために妨げられている。主要組織適合性複合体クラスII結合ポケットの既知の溝に適合するように設計された小さなプリオン断片による免疫は抗PrPC免疫を誘発し、その抗体はプリオン感染腫瘍移植におけるプロテイナーゼK耐性PrPScレベルを低下させた(99)。組換えプリオンタンパク質をマウスに積極的に免疫すると、免疫原が予防的に投与された場合にはプリオン病が遅延し、動物がすでに感染していた場合には、その程度は低かった(100)。経口投与されたプリオンによって誘発された臨床疾患は、マウス(101,102)およびシカ(103)へのワクチン接種によって抑制された。しかし、別の報告では、シカやヘラジカのプリオン病である慢性消耗病に罹患しているシカに予防的プリオン接種を行っても、発病しやすさの違いを示すことはできなかった(104)。組換えプリオンタンパク質断片による免疫と腹腔内プリオン接種により、緩やかな発病遅延が達成された(105,106)。DNAとタンパク質を組み合わせたワクチン接種法を用いて自己寛容を破壊する試みは、さまざまな結果をもたらした(107,108)。
免疫系は自己抗原に対して寛容であるため、免疫から得られる抗体はしばしば有効な治療に必要な親和性を欠く。PrPCの自己寛容の分子的所在を調べたある研究では、少量の神経細胞外PrPCでも効率的な免疫反応が消失することが見いだされた(109)。フロイントアジュバントによって発症遅延が達成されたことから、免疫系の非特異的活性化による効果が示唆された(110)。別の研究では、自然免疫を刺激することが示唆されているCpGオリゴデオキシヌクレオチド(CpG-ODN)を反復投与することで、プリオンに対するその場限りの免疫刺激による強い神経保護効果が示唆された(111)。しかし、CpG-ODNを慢性的に投与すると、リンパ濾胞破壊、肝毒性、出血性腹水などの重篤な免疫抑制を引き起こすことが示された(112)。さらに、マウスを反復免疫すると、プリオンクリアランスの減少、濾胞樹状細胞(FDC)ネットワークの縮小、あるいはその両方によって、末梢で誘発されるプリオン病に対する感受性が高まることから、個々の免疫状態(例えば、活性化亢進や低下)が、プリオン病に対する脆弱性の素因となっている可能性が示唆された(113)。
プリオンタンパク質に対する受動免疫療法
プリオン免疫療法の最初の概念実証は、無細胞の精製プリオンをPrP特異的抗血清で暴露することによるプリオン感染性の低下を示した(114)。モノクローナル抗PrP抗体6H4、SAF32、SAF61、あるいはPrP特異的抗体D13、D18、R1、R2のFab断片を、慢性的にプリオンに感染したN2A神経芽腫細胞に投与すると、試験管内試験でPrPScレベルを減少させる受動的プリオン免疫療法が示された(115–118)(試験した抗体の全リストは表2を参照)。D13を二価抗体(D13-IgG)として投与すると、広範な神経細胞アポトーシスが観察され、PrPCの架橋を介して神経細胞死が起こることが示唆されたが、この所見はD18のホロIgG分子では見られなかった(119)。D18の一本鎖断片をアデノ随伴ウイルス9ベクターに組み込み、Rocky Mountain Laboratory感染マウスの脳に導入したところ、接種したマウスの生存期間の延長が観察された(120)。D13の毒性作用は2番目の研究で再現された(41)。
| クローン | エピトープ領域 | プリオン接種 | 投与経路 | 毒性と保護の評価 | 結果 | 参考文献 |
|---|---|---|---|---|---|---|
| 6H4 | α1 | RML5, i.p. | μ鎖のトランスジェニック発現 | マウスバイオアッセイ | 遺伝子導入動物が病気に倒れることはなかった。 | 121 |
| D13 | CC2 | NA | 定位注射 | 組織学 | 1μgで無害、2μgで有毒 | 119 |
| 定位注射 | 組織学 | 2μgまでは無害 | 126 | |||
| 定位注射 | 組織学、MEMRI | 6μgで毒性 | 41 | |||
| 定位注射 | 組織学、MEMRI | 2μgで無害、6μgで毒性;推定安全上限量:3.7~5.4μg | 127 | |||
| D18 | HC-α1終了 | NA | 定位注射 | 組織学 | 2μgで無毒 | 119 |
| NA | 定位注射 | 組織学 | 2μgまでは無害 | 126 | ||
| RML (ns), i.p. | AAV9 | サバイバル | AAV9媒介抗体を接種した動物における疾患の遅延 | 120 | ||
| 8B4 | CC1とORの間 | 139A, i.p. | i.p. | サバイバル | 疾患の大幅な遅延 | 122 |
| 8H4 | α2 | |||||
| 8F9 | α3とGPIの始まり | |||||
| ICSM18 | α1-3 | RML (ns) | i.p. | 生存期間、PrPScレベル | ICSM18をi.p.接種したマウスの生存期間は延長したが、i.c.接種したマウスの生存期間は延長しなかった。 | 125 |
| NA | 定位注射 | 組織学 | 2μgまでは無害 | 126 | ||
| NA | 定位注射 | 組織学、MEMRI | 2μgで無害、6μgで毒性;推定安全上限量:3.1μg | 127 | ||
| ICSM35 | OR-CC2終了 | RML (ns) | i.p. | 生存期間、PrPScレベル | ICSM18をi.p.接種したマウスの生存期間は延長したが、i.c.接種したマウスの生存期間は延長しなかった。 | 125 |
| NA | 定位注射 | 組織学 | 2μgまでは無害 | 126 | ||
| 31C6 | α1 | 帯広、チャンドラー | 脳室内 | 生存期間、組織型、PrPScレベル | 接種直後で臨床症状があるときに投与した場合、生存期間が延長した。 | 124 |
| チャンドラー | 静脈内 | プリオン感染、31C6処理マウスの生存期間延長 | 123 | |||
| 4H11 | または | 6PB1 | 脳室内 | 生存率、組織学、行動 | プリオン感染マウスでは4H11の生存期間は延長せず、4H11による行動障害と神経細胞喪失が誘発された | 132 |
| POM1 | α1-3 | NA | 定位注射 | 組織学、MEMRI | 重篤な神経毒性 | 41 |
| COCS | 生化学、マイクロアレイ | 真性プリオンに類似した毒性経路の誘導 | 38 , 39 | |||
| COCS、i.c. | 生存期間、組織型、PrPScレベル | 感染力は発生しない | 129 | |||
| POM2 | または | NA | 定位注射 | 組織学、MEMRI | 2μgまでのscFvPOM2で無毒(12μgのホロIgGにモル等価) | 41 |
| RML6 | COCS | 組織学、PrPScレベル | プリオン感染COCSにおけるPOM2による神経保護作用 | 38 , 39 |
略語:α1-3、PrPのα-ヘリックス1-3;AAV9、アデノ随伴ウイルス9;CC1/2、PrPの荷電クラスター1/2;COCS、小脳器官型培養スライス;GPI、グリコシルホスファチジルイノシトール;HC、PrPの疎水性コア;i.c.、脳内;i.p.、MEMRI:マンガン増強磁気共鳴画像法;NA:該当せず;ns:特定せず;OR:PrPのオクタペプチド反復;PrP:プリオンタンパク質;PrPSc:プリオンタンパク質のプロテイナーゼK耐性型;RMLX:Rocky Mountain Laboratoryプリオン株のx番目の継代。
表2 生体外および生体内のPrPに対する治療用抗体
抗PrP抗体6H4のIgMaμ鎖のトランスジェニック過剰発現は、プリオン感染マウスにおけるプリオン感染性とPrPScのレベルを減少させた(121)。また、モノクローナル抗PrP抗体8B4,8H4,8F9の末梢注射は、PrP-α1らせん標的抗体31C6の注射と同様に、臨床的疾患の発症を減少させた(122)。31C6は、脳室内投与ではあるが、臨床症状がすでに現れているような遅い時期に投与すると、プリオン病に対する予防効果があると報告されている(124)。
抗PrP抗体ICSM18とICSM35の安全性プロファイルについては、大いに議論のあるところである(125)。ある報告では、マウスに2μgの抗体を定位注射した後、両化合物に薬物関連毒性は認められなかった(126)。しかし、ICSM18を用いた用量漸増試験では、安全量とされる2μgの抗体で薬物起因性の神経毒性作用がみられ、ICSM18の臨床試験への適性に懸念が生じた(127)。注目すべきは、ICSM18と同様のエピトープに対するモノクローナル抗体であるPOM1が、ex vivoおよび生体内試験で重篤な神経毒性を示したことである(39,41,128,129)。
市販のプールされた免疫グロブリンから得られた変異型プリオン断片PrPA117V106-126を認識するヒト自己抗体は、ミクログリアが変異型断片を取り込むことにより、試験管内試験でのPrPA117V106-126誘発神経細胞死を防御すると提唱された(130,131)。しかし、PrPA117V106-126は自然界には存在しないので、このような推測はありえない。
プリオンタンパク質のオクタペプチド反復ドメイン(OR)のエピトープを標的とするモノクローナル抗体によって、プリオンチャレンジ後の神経保護が得られた。N2A細胞では、mab110とSAF34の2種類の抗OR抗体によってPrPScの形成が阻害された。さらに最近では、ORを標的とする高親和性モノクローナル抗体であるPOM2が、COCSにおいてプリオン感染からの保護をもたらした(図2)(41)。プリオンの力価は影響を受けなかったが、プリオンの複製下流の細胞障害事象は選択的に抑制された(39)。しかし、興味深いことに、抗PrPC-OR抗体4H11は、細胞培養実験ではPrPScを除去することが示されていたが、マウスではプリオン病を改善しなかった(132)。それどころか、4H11を注射した動物は行動障害を示し、神経細胞の減少とアストログリオーシスが亢進した(132)。抗OR抗体POM2の脳内注射が毒性を示さなかったのに対して、4H11抗体投与が防御を与えなかった理由を理解するためには、4H11と他の抗OR抗体との違いを理解することが重要であろう(127)。
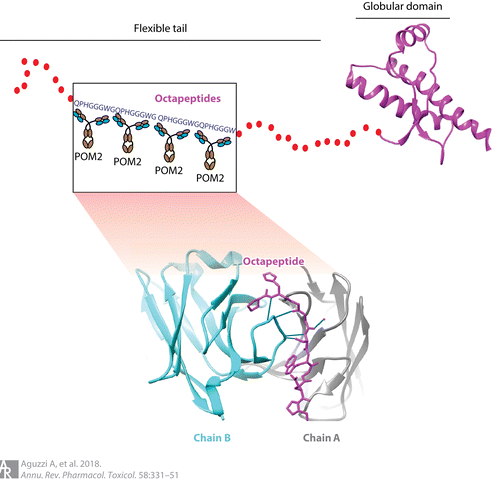
図2 免疫療法はプリオン病に対する魅力的な治療戦略として急速に発展している。モノクローナル抗体POM2は、PrPCのオクタペプチド反復領域にある変性エピトープに結合し、プリオンが誘発する神経変性を防御する。赤い点はプリオンタンパク質のN末端を示し、フレキシブルテールとも呼ばれる。PrPCの秩序化された球状ドメインはマゼンタ色で示されている。POM2抗体のF(ab)1フラグメント(シアンとグレー)とPrPC上の同族エピトープ(紫)との相互作用を挿入図で可視化した。青い線は相互作用を示す。略号PrPC、細胞性プリオンタンパク質。
4H11は、PrPCと PrPScの間の病的変換の間のミスフォールドした中間体であると仮定されたマウス人工PrP二量体に対して上昇させた(132)が、POM2はPrPCノックアウトマウスの免疫から上昇させた(133)。この2つのOR結合抗体の相違を説明する一つの可能性は、非天然タンパク質で免疫したことによる4H11のオフターゲット効果かもしれない。
プリオンの末梢複製と神経侵入を標的とする
B細胞の成熟を遺伝的に阻害すると、末梢プリオン接種後のプリオン病の発症が抑制された(134)。これらの所見から、B細胞の薬理学的切除(例えば、抗CD20抗体リツキシマブ)によって、曝露後の予防が可能であると推測される。プリオンの初期蓄積は神経浸潤に先立ち二次リンパ系臓器で起こるが(135)、他のプリオン株、いわゆる向神経性プリオンは末梢での複製を必要とせず、主として中枢神経系に浸潤する(136)。初期の研究から、プリオンの神経浸潤には成熟したPrPC発現FDCが必要であることが主張されている:分化したB細胞を切除すると、B細胞から分泌されるFDC成熟シグナルの欠如により、末梢でのスクレイピーの発症が阻止され(134)、成熟FDC上のPrPCの発現または成熟FDCのいずれかを欠損したマウスは、末梢で発症したプリオン病に屈しなかった(137)。FDCは発生と維持のためにB細胞からのリンパ毒素と腫瘍壊死因子(TNF)に依存しているので、プリオンの複製を標的とする機会を提供する(138)。リンパ毒素β受容体とヒト免疫グロブリン(LTβR-Ig)からなるハイブリッドタンパク質の投与は、リンパ毒素α/β経路の阻害を通じてFDCを脱分化させ、LTβR-Igを発症後期に投与した場合でも、末梢接種時にプリオン病の発症を遅延させた(139)。ヒト免疫グロブリンIgG1のFc部分に結合した可溶性ヒトTNFレセプターの単回注射によるFDCの脱分化もまた、末梢投与されたプリオンに対する疾患感受性を低下させた(141)。
FDCはFcγ受容体に結合することで免疫複合体を捕捉する。また、補体レセプターCD21/CD35を介してオプソニン化抗原とも結合する。補体因子C3またはその受容体CD21/CD35の薬理学的および遺伝学的切除は、末梢プリオン接種マウスにおけるインキュベーション時間を延長させた(142,143)。したがって、PrPScを介した補体の活性化は、FDC結合PrPScを増加させ、プリオンの複製を促進する可能性がある。膜結合型免疫グロブリンも分泌型免疫グロブリンもプリオンの神経侵入を変化させなかった(143)。様々なFcγレセプターの欠失変異体はプリオンのインキュベーション時間に影響を及ぼさなかったので、PrPScに結合した循環免疫複合体はプリオンの病態に関与していない可能性がある(143)。
交感神経系(SNS)は二次リンパ系器官を支配しており、末梢プリオン接種後、脾神経は早期に複製される部位であり(144)、交感神経および感覚神経節にもプリオンが蓄積することから(145)、プリオンの発症にSNSが関与しているという実験的証拠が指摘されている。6-ヒドロキシドーパミンまたは抗神経成長因子抗体の注射による一過性のSNSの薬理学的切除は、末梢接種後のスクレイピー発症の遅延につながった(146)。
経口プリオン暴露後にプリオンを腸関連リンパ組織へ運搬する役割を担う細胞を扱った研究では、特殊な上皮細胞であるマイクロフォールド細胞(M細胞)(147)を、核因子κBリガンドの受容体活性化因子に対するモノクローナル抗体の適用によって枯渇させた(148)。M細胞の枯渇は、FDCの状態を変化させることなくプリオンのFDCへの取り込みを減少させ、経口プリオン暴露後の発病を予防した(148)。
ヒトのプリオン病に対する治療法
現在までのところ、プリオン病に対する臨床試験は成功していない。プリオン病の有病率が低いため、大規模な患者群を対象とした二重盲検無作為化プラセボ対照多施設共同試験を実施することは、研究者にとって本質的に限界がある。希少疾患は産業界からの資金提供を受けにくく、実際、システマティック・レビューによると、CJDの臨床試験7件中1件しか産業界からのスポンサーがいなかったが、これとは対照的に、全体平均では4件中3件が産業界からのスポンサーであった(149,150)。
プリオン病に特異的な疾患評価尺度がないため、初期の臨床研究は、プリオン病の表現型に対応するように特別に設計されていない認知テスト用バッテリーを用いたり(27)、生存期間をアウトカム指標として用いたりして行われた(151)。限られたサンプルサイズと不均一なエンドポイントにより、治療介入は症例報告として発表されることになる。しかし、症例報告は本質的に出版バイアスによる欠陥がある:例外的な治療の成功は、治療の失敗よりも発表される可能性が高い。プリオン病臨床試験におけるエンドポイントベースの主要アウトカム(すなわち生存期間)を神経心理学的、精神医学的、その他の機能的ランキングシステムに拡張することで、将来の臨床試験の検出力計算が改善される可能性がある(152)。
プリオン病における緩和ケア
現在、プリオン病に対する有効な治療法がないため、すべての医療ケアは基本的に支持的で緩和的なものである。主に、患者の安全を守るための看護が行われる(歩行器や車椅子による歩行補助、終末期には病床での定期的な皮膚や口のケア、食事摂取の補助など)(153)。特に、発熱(体温の幅広い変化)は、放っておくと興奮を高める可能性のある一般的な症状であることが示唆され、扇風機の使用やぬるめのスポンジ浴によって緩和される可能性がある(154)。注意深く対処する必要のあるさらなる苦痛な症状は、ミオクロニー・ピクピク、感覚過敏の亢進、息切れ、失禁、便秘である(154)。
農業および人体医学における厳格な予防措置により、vCJDの発生率はほぼ完全に消滅した(155)。現行の世界保健機関(WHO)の「伝達性海綿状脳症における組織感染性の分布に関する表」(156)では、vCJD以外の患者からの血液や尿を正式に感染性とみなしていないが、sCJD患者からの尿の検出可能な感染性や、ヒト遺伝性プリオン病患者からの血液の霊長類への感染性を示唆する報告もある(157,158)。従って、vCJD以外のプリオン病患者を扱うすべての職員は、適切な保護具を着用し、さまざまなヒト組織の相対的な感染性について知識を得るなど、予防的行動をとらなければならない(159)。また、CJD患者は緩和ケアと精神的ケアの両方を必要としているため、集学的なガイドラインを作成することで、洗練された治療スキームを開発し、患者のケアを向上させることができる(160)。
ディスクロージャー・ステートメント
アドリアーノ・アグッツィは、プリオン病を含む難治性疾患に対するヒト抗体の開発に専念するマビロン社の創設者であり、取締役である。著者らは、本総説の客観性に影響を及ぼすと思われる、その他の所属、会員資格、資金提供、金銭的保有について承知していない。
謝辞
A.A.は、欧州研究評議会(European Research Council)の助成金およびスイス国立科学財団(Swiss National Science Foundation)、チューリッヒ大学(University of Zurich)の臨床研究優先プログラム「Small RNAs」および「Human Hemato-Lymphatic Diseases」、SystemsX.ch.の助成金を受けている。A.K.K.L.はシナプシス財団からの助成金を受けている。K.F.はTheodor und Ida Herzog-Egli Stiftungより助成を受けた。