Contents
www.frontiersin.org/articles/10.3389/fnins.2022.966019/full
Proteostasis unbalance in prion diseases: Mechanisms of neurodegeneration and therapeutic targets
- 1イタリア、ジェノバ、ジェノバ大学、内科(DiMI)、薬理学部門
- 2IRCCSOspedale Policlinico San Martino(イタリア、ジェノバ)
要旨
伝達性海綿状脳症(TSE)、すなわちプリオン病は、中枢神経系の進行性神経変性疾患であり、散発性、遺伝性、感染性の形態でヒトや動物に罹患する。アルツハイマー病や他の神経変性疾患と同様に、TSEの致死率を下げたり、罹患者の寿命を延ばしたりする試みは、これまで成功していない。一般に、神経細胞死は数十年にわたる経過の結果であると考えられているが、症状の発現は1年以内に致命的な結果をもたらすことを予期している。症状のない期間が長いこと自体が、効果的な神経保護療法を実施するための大きな障害となっている。
TSEの感染体であるプリオンは、プリオンタンパク質スクレイピー(PrPSc)と名付けられたプロテアーゼ耐性タンパク質で構成されている。PrPScのミスフォールディングは、生理的な対応物である細胞性プリオンタンパク質(PrPC)と異なり、すべてのTSEに共通する病原特性である。PrPScは細胞内で分解されにくく、可溶性の二量体やオリゴマーの形成を経てアミロイド様線維化を起こす。PrPScの除去不全は、アルツハイマー病、パーキンソン病、ハンチントン病など、プロテオスタシスの変化を特徴とする他の神経変性疾患と同様に、TSEをプロテオパチーとして定義する重要な病因事象である。
生理的な条件下では、ユビキチン・プロテアソーム・システムに代表されるタンパク質の品質管理とマクロオートファジーが、不適切に折り畳まれたタンパク質、冗長なタンパク質、凝集しやすいタンパク質を細胞質から除去する。これらの重要な恒常性維持経路の両方がTSE発症中に障害されるという証拠があるが、プロテオスタシスの変化がプリオンタンパク質のミスフォールディングを促進するのか、あるいはむしろPrPScプロテアーゼ耐性が細胞質タンパク質の品質管理を阻害するのかはまだ不明である。
本総説は、PrPCのミスフォールディングとプロテオスタシスの変化との因果関係に関する最新の進歩を批判的に分析し、ユビキチン・プロテアソームの能力を薬理学的に回復させ、オートファジーを刺激することで、PrPScの細胞内負荷を軽減し、プリオンに関連した神経変性の重症度を改善できる可能性について議論することを目的としている。
はじめに
環境変動に耐え、構造と機能を継続的に制御し続ける能力は、すべての生物にとって第一に必要なものであり、しばしばホメオスタシス(恒常性)と呼ばれる。単細胞生物でも多細胞生物でも、ホメオスタシスを維持するために非常に幅広いメカニズムが協力している。タンパク質の恒常性、すなわちプロテオスタシスは、細胞内タンパク質の数を細胞の必要量に適応させるだけでなく、それらの適切な折り畳み、輸送、リサイクルを制御するために、タンパク質の合成と分解のバランスをとる。これは、神経細胞の生存能力、シナプス機能、ひいては認知能力を長持ちさせるために、中枢神経系(CNS)内で特に関連している(Hetz, 2021)。
プロテオスタシスの欠陥は、いくつかの病態における中枢神経系の変化の基礎となっている。タンパク質のミスフォールディング、オリゴマー/フィブリルの蓄積、それらを排除できない細胞は、アルツハイマー病(AD)、パーキンソン病(PD)、前頭側頭型認知症(FTD)、ハンチントン病(HD)、筋萎縮性側索硬化症(ALS)、およびプリオン病としても知られる伝達性海綿状脳症(TSE)に共通する病理組織学的特徴である(Thellung et al. 2019;Le Guerroue and Youle, 2021)。
Walker and Levine III (2000)は、中枢神経系におけるタンパク質の立体構造の変化に関連するすべての疾患を意味的に統一するために、大脳プロテオパシーという用語を提案した。このような用語の統一は、病因、組織病理学、臨床像の点で異質な神経変性疾患を共通の基本的変化の下に包括するものであり、特に有用である。すべての神経変性疾患の中で最も多くみられるのは散発的発症であるが、遺伝性のプロテオパチーを研究することによって、発症機序の解明に大きな進展が見られる。
注目すべきは、疾患特異的なタンパク質をコードする遺伝子だけでなく、タンパク質の品質管理とターンオーバーのメカニズムに関与する遺伝子においても、病因となる変異が報告されていることである(Lehman, 2009)。
TSEは、プリオン病として普遍的に知られる、まれで致死的な神経変性疾患であり、ヒト、家畜および野生動物が罹患し、急激な神経機能低下を引き起こす。神経組織学的病変は、病型による違いはあるものの、脳実質の海綿状変性、神経細胞の減少、星状膠細胞症、アミロイド生成タンパク質であるプリオンタンパク質スクレイピー(PrPSc)の沈着で構成される。PrPScは、糖タンパク質である細胞性プリオンタンパク質(PrPC)の翻訳後の再配列に由来する(Prusiner and Dearmond, 1991)。クロイツフェルト・ヤコブ病(CJD)、ゲルストマン・シュトロイスラー・シャインカー病(GSS)、致死性家族性不眠症(FFI)、クル病などのヒトTSEは、散発性、家族性、異所性、感染性など病因が異なる。TSE特有の特徴は、PrPScのヒト間通過による伝播性である(Prusiner, 1998;Will and Ironside, 2017)。PrPCからPrPScへの移行は、PrPCをコードする遺伝子であるPRNP内の突然変異によって自然発生的に起こるか、あるいは好都合に起こるか、あるいは鋳型として働く外因性PrPScとの相互作用によって強制的に起こるかのいずれかである(Prusiner, 1982)。
PrPScの感染は、非経口的または経腸的経路をたどることがある(例えば、1990年代の「狂牛病」流行時に汚染された肉の摂取を介して;Diack et al., k14)。PrPScの形成につながるPrPCのミスフォールディングは、タンパク質の二次構造に関係し、βシート構造の含有量、疎水性、タンパク質分解に対する抵抗性を増加させる(Pan et al., n93)。PrPScの蓄積はアミロイド形成経路の活性化を引き起こし、オリゴマーやフィブリルの形成につながり、その蓄積は一般的に全てのプリオン病における組織学的病変と関連している(Corsaro et al., 2012)。
PrPScの蓄積と病態との直接的な因果関係についてはまだ議論の余地があるが(Parchi et al., 1996;Hill and Collinge, 2003)、PrPCのミスフォールディングは通常、タンパク質の品質管理システムによって阻止されており、その機能不全がすべてのTSEにおいて発症に関与しているという証拠が増えつつある。さらに、プロテオスタシスを制御する2つの主要な細胞機構、ユビキチン・プロテアソーム品質管理(UPS)とオートファジー(ALP)の障害は、タンパク質のミスフォールディングを特徴とする他の神経変性疾患との関連でも報告されている(Nixon, 2013;Ciechanover, 2015;Zheng et al.)すべてのプロテオパチーにおいて同定されたこれらの類似した発症機序は、これらすべての病態において効果的な神経保護介入を行うための共通標的の探索を促した(Kumar et al.)本総説では、プリオン病におけるプロテオスタシス不全の病原的役割と、UPSとオートファジーの薬理学的調節による神経細胞死への対抗の可能性に関する最新の知見に焦点を当てる。
タンパク質の品質管理
真核細胞では、新しく合成されたタンパク質の適切なフォールディングの制御と異常蓄積の防止は、小胞体(ER)でのリフォールディングの補助から、細胞質での分解や解離に至るまで、一連の相互に関連したメカニズムによって行われている。これらの経路はすべて、プロテオスタシスに重要な役割を果たすと同時に、相互に代償し合う複雑なネットワークで結ばれている(Ciechanover and Kwon, 2017)。タンパク質のフォールディングがうまくいかないと、疎水性のアミノ酸配列が露出することがよくある。シャペロンとの結合は、クライアントタンパク質の正しい構造の再構築を促進したり、ユビキチン・プロテアソームシステム(UPS)やオートファジー経路を介したタンパク質分解を促したりする。細胞性および病的プリオンタンパク質は、シャペロン支援リフォールディング(Tittelmeier et al., 2020)、ユビキチン化、プロテアソーム分解、リソソーム分解(Ciechanover, 2015)を受けるタンパク質品質管理機構の重要なクライアントであるという説得力のある証拠がある。また、これらの経路をより深く理解することで、プリオン病に対する新規で効果的な神経保護戦略につながることが期待される。
シャペロン支援によるリフォールディングとタンパク質分解
分子シャペロンは、進化的に保存されたタンパク質の大きなグループであり、タンパク質の品質管理に重要な役割を果たし、環境変化に対する細胞プロテオームの適応を可能にする。CNSにおけるその効力の喪失は、しばしば脳プロテオパチーと関連している(Ciechanover and Kwon, 2017)。
シャペロンは、タンパク質のミスフォールディングの特徴である、新生ポリペプチド中の露出した疎水性配列を認識し、アンフォールディング状態のタンパク質を一時的にブロックする複合体を形成することによって、タンパク質の適切なフォールディングを助ける。この複合体は、タンパク質の正しいリフォールディングを決定するか、ユビキチン-プロテアソーム系(UPS)やオートファジーによる分解を活性化する。ヒースショックタンパク質(HSP)は、熱、酸化、炎症などの様々な細胞ストレスによって活性化されるシャペロンであり、様々なサブグループ(それぞれの分子量に基づいて命名:Hsp90、HsP70、Hsp40など)がある。コ・シャペロンと呼ばれる補助タンパク質もまた、基質タンパク質との複合体にしばしば採用され、ユビキチン化やプロテアソームとの相互作用を誘導するアダプターとして働く(Abildgaard et al.)プリオンタンパク質のフォールディング制御は分子シャペロンの重要な仕事であり、現在、プリオン病の病態への関与や薬理学的調節による治療の可能性を明らかにするための研究が進められている(Tittelmeier et al.)
ユビキチン・プロテアソーム・システム
真核細胞が生存のためにUPSに依存しているのは、このタンパク質分解システムが、新しく形成される短命なタンパク質の質と量を継続的に制御しているからである。過剰に、あるいは間違って折り畳まれたタンパク質は、粗面小胞体からゴルジ体に輸送され、さらに処理されて細胞膜に挿入されたり分泌されたりする代わりに、細胞質に逆輸送され、複数のユビキチン鎖と結合し、26Sプロテアソームによって分解される(Pickart, 2001;Sloper-Mould et al., 2001)。基質のさらなるタンパク質分解処理に不可欠なポリユビキチン鎖の付加は、ユビキチン関連酵素E1、E2、E3によって触媒される、ATPを消費する3つのステップによって行われる。ユビキチンは一過性にE1(ユビキチン活性化酵素)に結合して活性化され、E2(ユビキチン結合酵素)に移行し、さらにE3(ユビキチンリガーゼ)に移行して基質にユビキチンを共有結合させる。このプロセスは、先に結合したユビキチンにさらにユニットを付加することによって数回繰り返され、26Sプロテアソームによって認識されるポリユビキチン鎖によって基質がタグ付けされる(Glickman and Ciechanover, 2002)。プロテアソームは、細胞質と核に存在する大きな26S、ATP依存性の酵素複合体で、複数のサブユニットからなる触媒(タンパク質分解)20Sユニットと制御19Sユニットからなる。20Sユニットの片側または両側にキャップを形成する制御性19Sユニットは、ベースとリッドを持ち、分解する基質を選択し、消化の準備をするゲートキーパーとして働く。大まかには、19Sユニットがユビキチン化タンパク質を認識し、ポリユビキチン鎖を除去し、タンパク質を解きほぐすことで、ユビキチン部分の再利用と基質の20Sタンパク質分解室への進入を可能にする(Sahu and Glickman, 2021)。触媒作用のある20Sサブユニットは、7つのサブユニットからなる4つのリングが積み重なった円筒形をしている。消化はβサブユニットによって行われるが、α構造はゲートの開閉を制御し、基質を取り込み、消化されたペプチドを排出する。
ユビキチンのライゲーションとプロテアソームの機能の極めて複雑な制御は、この総説の目的を超えているので、プリオン病におけるUPS障害と神経変性の発症との関連についての最新の知見に焦点を当てることにする。
オートファジー
オートファジーは、リソソーム依存的な、細胞質、損傷した小器官、潜在的に有害なタンパク質の生存を促進するタンパク質分解であり、細胞がいくつかのストレス状態に対処することを可能にする。オートファジーを通して、細胞は細胞質内の栄養素を再利用することで飢餓状態を生き延び、損傷したミトコンドリアを除去することでアポトーシスを回避し、異常タンパク質や凝集しやすいタンパク質を消化することでプロテオスタシスを制御することができる(Klionsky et al.)
オートファジーの種類は、基質の性質と基質がリソソームに運ばれるメカニズムに基づいて識別することができる。古典的にオートファジーと呼ばれるマクロオートファジーは、細胞質の大部分を二重膜のオートファゴソームに捕捉し、リソソームと融合して栄養素の消化と再利用を行うが、アグレファジーやシャペロン媒介オートファジー(CMA)と呼ばれるタンパク質を標的とした形態もあり、プロテオスタシスの制御において重要な役割を果たしている。
アグレファジーはUPSと協力し、プロテアソームの狭い孔に入り込めないような大きなタンパク質や凝集しやすいタンパク質の除去に主に関与している(Lamb et al., 2013;Kumar et al., 2022)。大まかに言えば、多量であったり、凝集構造であったり、あるいは特定のUPSステップの欠陥の結果として、プロテアソームによる分解を逃れたミスフォールドタンパク質は、細胞質に集まって封入体を形成し(Kopito, 2000)、オートファゴソームに取り込まれる(Fortun et al.)アグリファジーは極めて選択的なプロセスであり、分解が約束されたユビキチン化基質のクラスターが成長する食細胞に追い込まれ、リソソームと融合したオートファゴソームとして消化される(Bjorkoy et al., y05;Pankiv et al., v07)。おそらくこれらのアダプターの中で最もよく特徴付けられているのは、シーケストソーム1(SQSTM1)またはp62と名付けられた440アミノ酸からなるマルチドメインタンパク質であり、その活性はプロテオスタシスだけに関与しているのではなく、複数のシグナル伝達の交差点に位置している(Katsuragi et al.)P62の役割は、多くの神経変性疾患において広く研究されており、細胞質への蓄積は、今日では、オートファジーの活性化またはタンパク質品質管理システムの効率低下の信頼できる特徴であるとみなされている(Pankiv et al.)p62の塩基配列は詳細に定義され、タンパク質間相互作用を担う複数のドメインが同定された(Berkamp et al.)これらのドメインのうち3つは、UPSとアグレファジーの間でタンパク質分解の運命を支配するために重要であると考えられている:p62のオリゴマー化を可能にするPB1ドメイン、食細胞の膜に存在するLC3BIIと相互作用するLIRドメイン、ユビキチン化基質と結合するユビキチン結合ドメイン(UBA)である(Pankiv et al.)典型的なアグリファジーで起こるような、細胞質タンパク質の凝集は、オートファゴソームがクラスター化したミスフォールドタンパク質を除去する唯一のメカニズムではないという証拠が最近得られている。p62はそのUBAドメインを通して、ユビキチン化タンパク質をp62ボディと呼ばれる膜のない飛沫に封じ込め、徐々に合体してサイズを大きくする(Berkamp et al., p21)。凝集タンパク質から構成されるが、p62ボディは球形をしており、液液相分離で細胞質から分離する粘性を持つ(Brangwynne et al.)LIR配列を介して、p62は成長する食細胞の目印となるLC3-IIタンパク質と相互作用し、オートファゴソームへの食体の取り込みを促進する(Zaffagnini et al., i18;Simonsen and Wollert. 2022)。このプロセスは、オートファゴソームとリソソームの融合によって継続し、オートファゴソーム内に存在するユビキチン化基質の消化と再利用を可能にする(Klionsky et al.)
細胞質からのタンパク質の選択的除去は、シャペロン媒介オートファジー(CMA)と呼ばれるオートファジーの代替形態によって可能となる。この形態では、クライアント基質はシャペロンおよびコ・シャペロン複合体と結合し、リソソーム関連タンパク質2A(LAMP2A)によってリソソーム内に移動する(Bourdenx et al.)CMAの典型的な標的はアミノ酸配列KFERQを含み、ストレス条件下で過剰発現するHsp70ファミリーおよび熱ショックタンパク質70同族体(Hsc70)に属するシャペロンとの結合を必要とする。とはいえ、細胞周期の制御に関連する他のシャペロンも、例えばプリオンタンパク質のように、異なるペンタペプチド配列を認識するCMAを介して作用することが報告されている(Wang et al.)
プリオンタンパク質の品質管理
PrPCはグリコシルホスファチジルイノシトール部分を介して細胞膜外葉に固定された糖タンパク質である(Stahl et al., 1987)。PrPCが成熟する過程で、N-末端シグナルペプチドの除去に続いてジスルフィド橋が形成され、1本または2本のオリゴ糖鎖が付加される(Haraguchi et al.)GPIアンカーは22 AAペプチドの除去後、タンパク質のN末端に結合する(Stahl et al., 1992;Hegde and Rane, 2003)。成熟PrPCは細胞膜表面の外側に露出し、高いターンオーバーを特徴とし、エンドサイトーシス系を通って急速にリサイクルされることから(Shyng et al., g93)、UPS制御を受ける可能性が高い。PrPCの成熟は、膜貫通タンパク質の典型的なルートに従う。ERでシグナルペプチドとグリコシル化が取り除かれ、ゴルジ体に移動し、細胞膜の外葉に挿入される。
PrPCの生物学に関する重要な科学的疑問は、PrPCの適切なフォールディングを助けるだけでなく、PrPC-PrPSc変換に影響を与えたり、ミスフォールディング/凝集したプリオンを分解に導く分子パートナーに関係している。生理学的および病理学的状況下でプリオンタンパク質と相互作用することが報告されているいくつかのシャペロンのうち、熱ショックタンパク質、特にHsp70サブグループ(Hsp70s)に属するものは、ヒト、動物、実験的に感染させたげっ歯類や細胞におけるプリオン病と一貫した関連を示した。実際、散発性CJD患者と家族性CJD患者の脳を分析したところ、Hsp73の発現量は神経細胞の減少量に反比例することが示され、異なる脳領域におけるPrPScに対する脆弱性の違いは、シャペロンの神経保護活性に依存している可能性が示された(Kovacs et al., 2001)。同様の報告がスクレイピーに感染したヒツジでも得られており、そこでは熱ショックタンパク質の発現パターンがプリオンタンパク質の沈着、グリオーシス、スポンジオーシスと関連していた(Serrano et al., 2011)。スクレイピー株の脳内注射によって感染したマウスでは、リソソーム関連構造の近傍でHsp70の免疫反応性が検出され(Laszlo et al., o92)、Hsp70とユビキチン遺伝子の発現の純増が観察された(Kenward et al., d94)。慢性的にプリオンに感染した細胞株におけるHsp70の薬理学的誘導は、PrPScの細胞内蓄積を相殺し、他方、マウスにおけるHsp70の消失は、RMLプリオンの脳内注射後の神経変性の進行を加速する(Mays et al., s19)。その活性がプリオンタンパク質の翻訳後運命に関与するもう1つの分子シャペロンは、78KDaのグルコース制御タンパク質(GPR78)であり、結合免疫グロブリンタンパク質(Bip)としても知られ、主にERに常駐するHsP70のメンバーである。独立した研究者らは、Bipが生理的および疾患関連型のPrPと結合し、変異型PrPのプロテアソーム分解を誘導できることを実証している(Jin et al., n00;Park et al., k17)。重要なことに、BipはPrPScプロテアーゼ耐性を回復させ、持続感染細胞におけるPrPSc複製を阻害することができ、その発現はスクレイピー株の脳内感染後のマウスの生存期間を延長するのに必要である(Parkら、2017)。さらに、通常の状態では影響はないが、ヒースショック因子1などのストレス感受性タンパク質をノックアウトすると、スクレイピープリオンに感染したマウスの生存率が低下する(Steele et al., e08)。これらの報告を総合すると、Hspsに属するシャペロンの活性化は神経保護の試みであり、家族性TSEのほとんどが遅発性であることを説明できる可能性がある。
PrPCは一般的に半減期が約6時間と短いタンパク質であり(Taraboulos et al., s92)、新しく形成されたタンパク質の約10%は品質管理を通過せず、プロテアソームによって分解されると推定されている。実際、プロテアソーム活性を薬理学的に阻害すると、ユビキチン部位に富むプロテアーゼ抵抗性PrPの細胞質凝集体が蓄積する(Ma and Lindquist, 2001;Yedidia et al.プリオンタンパク質の変異はCJD、GSS、FFIの家族型と関連しており、不完全なプロセシングと変化したフォールディングとトポロジーを特徴とするPrP変異体を産生する。これらの変異型プリオンタンパク質の分解は、UPS、CMA、あるいは解離によるプロセシングを促進するために一時的にタンパク質と結合するシャペロンによって部分的に認められている(Jin et al., n00;Wang et al., g17;Thackray et al., y22)。PrP変異体を発現する細胞株で行われた試験管内試験研究では、UPSの薬理学的阻害が、凝集体に蓄積するデタージェント不溶性で部分的にプロテアーゼ耐性のPrPの細胞内蓄積を誘導することが示されている(Zanusso et al., o99;Jin et al., n00;MaとLindquist 2001;Mishra et al., a03)。さらに、ユビキチンリガーゼTRAF6によるPrPCのユビキチン化は、p62との相互作用とアグレソームの形成を促進する(Masperone et al., e22)。
疾患関連変異を持つPrPCやその変異体と同様に、PrPの病原体であるPrPScの存在は、防御戦略として、細胞にUPSの活性化を誘導し、凝集体の形成を促進する。Hommaは、感染モデルとして実験動物にプリオンを脳内および腹腔内に注射したところ、終末期の動物の脳でp62とオートファゴソーム関連LC3-IIの発現が増加することを観察した(Homma et al.)さらに、阻害剤MG-132を用いてプロテアソームを薬理学的に阻害すると、PrPSc持続感染神経芽腫細胞において、PrPScとp62の両方を含む核周囲凝集体の形成が促進された。重要なことは、PrPScとp62の共局在は、ユビキチンと結合するp62の能力に依存していることである。高レベルのp62はまた、変異型PrPCがUPS分解を免れ、PrPとp62を含む細胞質封入体を形成すると、アポトーシスへの細胞コミットメントを阻害する(Xu et al., u14)。PrPScの細胞内蓄積はオートファゴソーム-オートリソソソーム経路によって打ち消されると考えられているが、PrPScもシャペロンによって認識され、CMAの活性化によってリソソームで消化されるという証拠がある。これらのシャペロンの一つはポロ様キナーゼ3(PLK3)であり、その過剰発現はHsc70とLAMP2Aをアップレギュレートし、PrPScをリソソーム内腔に移動させる(Wang et al.)これらの証拠を総合すると、UPSシステムは、少なくとも一時的かつ部分的に、遺伝性・後天性TSEの発症中に、脳内の病的プリオンタンパク質の蓄積に対抗しうること、また、凝集したPrPScがUPSから逃れたり、過剰に凝集した場合に、選択的アグレファジーとCMAが補完的な消去システムとして機能しうることが示唆される。
進展する伝達性プロテオパシーの概念は、異常タンパク質、特にプリオンタンパク質の遠隔体内あるいは脳内への拡散を促進するメカニズムを明らかにするという重要な問題に直面した。軸索や細胞を介した輸送、感染した神経細胞から放出されるマクロベシクルを介した拡散など、いくつかのメカニズムが仮定されている(Glatzel and Aguzzi, 2000;Bellingham et al.)特に、マイクロベシクルを介した輸送は、短距離および長距離の細胞間情報伝達の極めて重要なメカニズムとして浮上している。多胞体(MVB)に含まれるマイクロベシクルであるエクソソームが細胞外腔に放出され、受容体を介したエンドサイトーシスや貪食によって隣接細胞に取り込まれることが報告されている。重要なのは、エクソソームの放出とオートファジーの活性化の両方が、細胞間コミュニケーションだけでなく、プロテオスタシス維持のためにも細胞によって利用されていることである(Xu et al.)
オートファジーとエクソソーム放出の協力関係は、不要なタンパク質の負担を減らすためにこれらのシステムを交互に利用する神経細胞にとって、特に重要である。MVBはオートファゴソームと融合し、オートファジーのフラックスに従ってエクソソームとそのカーゴを消化するか、あるいは細胞膜と融合してエクソソームを細胞外腔に放出する(Fader et al.)プリオン感染細胞はPrPScを含むエクソソームを放出し、このエクソソームはレシピエントマウスに病気を感染させる可能性がある(Fevrier et al., r04;Vella et al., a07)。これらの試験管内試験の結果は、TSEに罹患した脳におけるオートファジー関連構造の組織化学的解析と一致しており、PrPScに対して免疫反応性を示すMVBの存在を明らかにし、エクソソームを介したPrPScの脳内拡散を示唆している(Sikorska et al., a04;Liberski et al., i11;Guo et al., o16)。
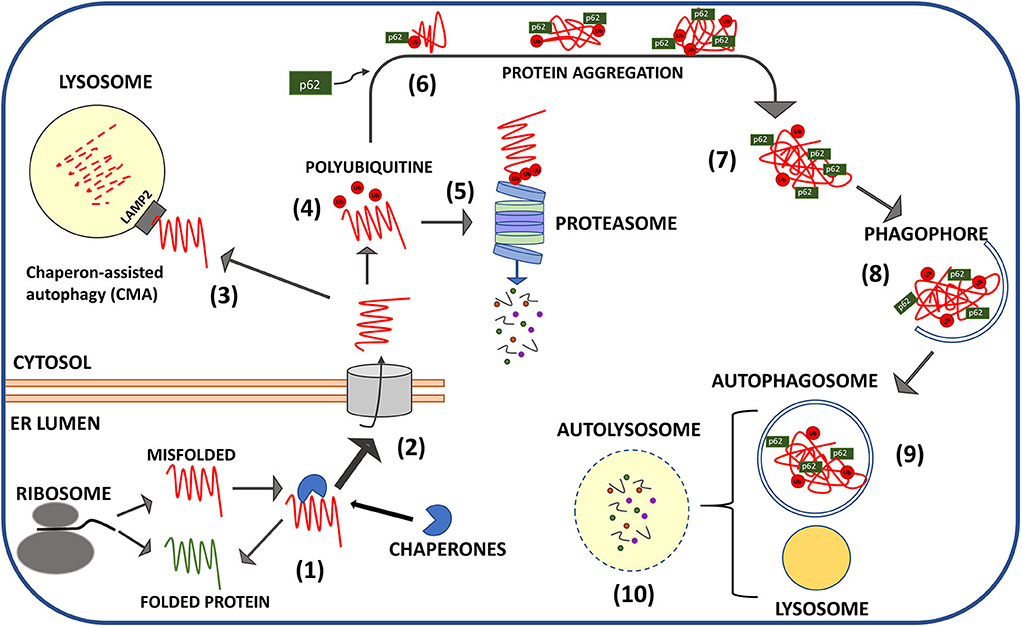
図1.PrPScの細胞内負担を軽減するためのエクソソーム放出とオートファジーの相互作用の模式図。PrPC(緑色)から神経毒性を持つPrPScアイソフォーム(赤色)への変換は、確率的事象によるPrPCリサイクル中、あるいは外因性PrPScとの相互作用後に起こる。細胞質PrPScを除去する神経細胞の戦略は、マクロオートファジーによるタンパク質分解、あるいは多胞体(MVB)に含まれるエクソソームへのPrPScの挿入である。MVBはPrPScを含むエクソソームを放出するか、オートファゴソーム、アンフィソーム(図には描かれていない)、オートリソソームを形成してオートファジーのサイクルに再び入る。
エクソソームを通して疾患関連タンパク質を取り込む神経細胞の能力は、現在広範な研究が行われており、タウオパチーやα-シヌクレインパチーを含む多くのタンパク質コンフォメーション疾患において「諸刃の剣」として説明されている。重要なことに、オートファジーの薬理学的調節は容易に達成可能であり、エクソソーム放出の間接的調節をもたらす(Abdulrahman et al., n18;Thellung et al., g19)。
最近、さまざまな神経変性疾患(特にADとPD)に由来する神経毒性タンパク質の凝集体も、患部から離れた神経細胞へ輸送されることが発見された。特に、b-アミロイドやα-シヌクレインのミスフォールド型は、おそらくエクソソームを介して細胞外環境に分泌され、それによって隣接する細胞の正常な生理機能にプリオンのような影響を及ぼす可能性がある。この仮説は、特発性ADやPDが中年期から晩年期になって初めて症状が現れるという時間経過のもっともらしい理由として提唱された。総説は吉田・長谷川(2022)を参照。
プロテオスタシス不全は、プリオン病や他の神経変性疾患に共通する発症特性である
神経細胞の可塑性と長期生存は、プロテオスタシスの擾乱を検知するセンサーと、シャペロン支援によるタンパク質のリフォールディングと分解、UPS、オートファジーなどの一連の対抗手段を実行に移すエフェクターの複雑なネットワークに依存している(Weibezahn et al., n05;True 2006;Overhoff et al., f21;Giandomenico et al., o22)。これらの制御システムの能力喪失は、生理的老化の際にも低いレベルで起こるが、多くの神経変性疾患では亢進し、ミスフォールディングタンパク質の蓄積、神経毒性オリゴマーの産生、中枢神経系内での拡散を促進する(Braak et al.)
病原性プリオンの発見は、伝染性の疾患関連形質としてのタンパク質フォールディングにかつてない関心を集めた。この観察により、神経科学者はタンパク質の凝集をダイナミックな現象としてとらえるようになった。異常なコンフォメーションをとる傾向があるタンパク質は、宿主細胞のタンパク質分解能力を引き継ぎ、ある状況下では脳内拡散を促進したり、他の生物に伝染したりする可能性がある(Harrison et al.)実際、Braak仮説は当初、パーキンソン病におけるレビー小体による隣接脳領域への進行性浸潤を説明するために提唱されたが、最近では、アルツハイマー病におけるジストロフィー神経細胞内の高リン酸化タウの進行や、感染性TSEにおける末梢から脳へのPrPScの求心的進行を説明するためにも応用されている(Braak and Braak, 1995;Braak et al.)さらに、もともとPrPScの特異的な性質として説明されていた、病的タンパク質がその空間的リフォールディングを駆動する生理的な相手をリクルートする能力は、最近、高リン酸化タウやα-シヌクレインの細胞内凝集を促進するメカニズムとして、アルツハイマー病やパーキンソン病の発症にも関与していると提唱されている(Caughey and Kraus, 2019;Duyckaerts et al.)
「毒性獲得」仮説によると、脳内のPrPScの蓄積に伴う神経解剖学的変化を説明するために、疾患の病因(感染物質からの伝播、PrPコード遺伝子の突然変異、確率的事象)にかかわらず、神経細胞死は、そのミスフォールディング後に起こるアミロイド生成過程で生成されるPrP可溶性オリゴマーの毒性活性に起因すると提唱された(Bucciantini et al. 2002;Chiovitti et al., 2007;Simoneau et al., 2007)。さまざまなヒト・プリオン病は、臨床症状や神経病理学、特に脳実質におけるPrPScの沈着に顕著な違いがあることが特徴である。PrPScを含むアミロイド斑は常に存在するわけではなく、変異型CJDや散発性CJDの症例ではごく一部に検出され、その超微形態は、馴染みのあるGSSに典型的な多中心型から、変異型CJDのほとんどの症例にみられる豊満型、あるいはkuruに典型的な単中心型まで様々である(Ghetti et al., 1995;Ironside and Bell, 1997;Sikorska et al.)このような不均一性にかかわらず、アミロイド斑を取り囲むユビキチンの存在と、海綿状領域におけるジストロフィー神経細胞との関連は、vCJD、GSS、kuru患者の脳の組織化学的および超微形態学的解析によって一貫して報告されており、プリオン病がアルツハイマー病や他の神経変性疾患と共有する重要な共通特徴である(Suenaga et al. 1990;Ironside et al., 1993;Lowe et al., 1993;Migheli et al., 1994;Sikorska et al., 2009;Zhou et al., 2015)。
ユビキチン化タンパク質を含むリソソームや、リボソームや他の細胞質成分を含む大きなオートファゴソームの特徴を持つ液胞構造の増加は、プリオン病に罹患した動物やヒトの脳や、実験的にプリオンに感染したげっ歯類の脳の死後分析でしばしば報告されている(Boellaard et al., d89、1991;Jeffrey et al., y92;Liberski et al., i92;Alves-Rodrigues et al., s98)。さらに、スクレイピー感染脳の脳組織を電子顕微鏡で観察したところ、未消化のプリオンタンパク質、ユビキチン、シャペロンを含む電子密度の高いリソソームの存在が観察され、タンパク質分解サイクルの障害がリソソームの崩壊と神経細胞死につながることが示唆された(Laszlo et al., o92)。
このような問題に関して、アルツハイマー病に関連した神経細胞死におけるオートファジー機能障害の発症結果に関する研究からも、関連する知見が得られている。アルツハイマー病に罹患した患者の大脳皮質ジストロフィー神経突起の免疫組織化学的解析では、年齢をマッチさせた健常人と比較して、電子密度の高いオートファゴソームが有意に増加していることが示されている(Boland et al.)この点に関して、β-アミロイドを含むプラークの細胞外沈着は、オートリソソームの酸性化の欠如と不完全なオートファジー・サイクルのために、Aβペプチドのオートファジー消化が損なわれた結果であることが最近証明された(Lee et al.)重要なことに、この変化はβアミロイドの細胞外沈着に大きく先行し、アミロイドプラーク形成の原因事象であると提唱された。
ヒトの広範なプロテオパチーにUPSとオートファジーの障害が関与していることを考えると(Bence et al., e01;Paul 2008)、プリオンタンパク質の細胞内凝集がプロテオスタシス破綻の結果なのか原因なのかを理解することは極めて重要であろう。このような重要な問題は現在のところ未解決であるが、タンパク質の凝集にオートファジーの障害が関与していることを示す証拠は、試験管内試験および前臨床試験から得ることができ、オートファジーを刺激することで凝集が抑制されるか、あるいは元に戻ることが実証されている。神経芽腫細胞をミスフォールディング(Corsaro et al., o06;Chiovitti et al., i07)、神経毒性(Thellung et al., g13、2017)の組換えPrPペプチドとインキュベートすると、プロテオスタシス機構が障害され、リソソームからのカテプシンDの漏出と、細胞死につながる正常なオートファジー流束の阻害につながることが示された(Thellung et al., g11、2018)。さらに、マウスにPrPScを脳内感染させると、空胞化やPrPScを含むアミロイド斑の沈着に興味を示す脳領域にユビキチンタグを付加したタンパク質が沈着することが報告された(Lowe et al., e90、1992;Lopez-Perez et al., z20)。これらの研究では、症状の重篤度とマウスの脳におけるユビキチン化タンパク質の存在との間に相関関係があることも報告されている。ユビキチン化タンパク質は、感染後数ヵ月以内にすでに検出可能であり、プロテアーゼ耐性PrPScの沈着と組織の変性に伴ってその量が増加した(Lowe et al., e92;Kenward et al., d94;Kang et al., g04)。ユビキチンタグ付きタンパク質の分布はまた、リソソーム酵素カテプシンDと有意に共局在し、電子顕微鏡分析によって明らかにされたように、小胞構造、おそらくリソソームかオートファゴソームに集中している(Lowe et al., e90、1992;Ironside et al., e93)。さらに、スクレイピー感染マウスでは、疾患の後期にp62の増加が観察され、オートファジー流束の進行の障害と、神経細胞のリソソームを介したプロテオスタシスの制御不全の可能性が示された(Homma et al.)
病的なプリオンタンパク質と特定のプロテオスタティック・エフェクターの障害との直接的な関連は、関連する論文によって報告されており、主にプリオン感染細胞株や動物におけるシャペロンのダウンレギュレーションと、凝集型PrPScを分解するためにプロテアソームが受け入れる能力の障害を含んでいる(図2)。
図2.ミスフォールドしたPrPのタンパク質品質管理機構の模式図。新生PrPCの構造異常(自然に起こるか、PRNPの突然変異によって好都合に起こる)は小胞体で感知され、適切な折り畳みが回復するまでPrPCをアンフォールディング状態でブロックするシャペロンのリクルートにつながる(1)。最終的にミスフォールディングしたPrPは、リソソームシャペロン会合オートファジー(CMA)(3)あるいはユビキチン化(赤い点:ユビキチン部位)(4)とプロテアソーム消化(5)のために細胞質(2)に移動する。ユビキチン化されたPrPの細胞質凝集体はプロテアソームから逃れ(6)、主にp62(7)のアダプタータンパク質の介在によって、より大きな封入体に集められる。P62は凝集体を、オートファゴソーム中のPrPクラスターを呑み込む新生食細胞に向かわせる(8)。オートファゴソームはリソソームと融合し、凝集体を消化し(9)、栄養素を再利用する(10)。
PrPの変異型を保有するトランスジェニックマウスやトランスフェクト細胞株で行われた研究では、PRNP変異が成熟PrPの構造、トポロジー、代謝に変化をもたらし、プロテアーゼ耐性PrPScへの自然変換を促進することが実証されている(Petersen et al., 1996;Capellari et al., 2000a,b;Chiesa et al., 2000;Stewart et al., 2001;Zaidi et al., 2005;Corsaro et al., 2011;Quaglio et al., 2011)。プリオンタンパク質の疾患関連変異がシャペロンをダウンレギュレートし、UPSを介したタンパク質分解を阻害することが証明されている(Zanusso et al., o99;Peters et al., s16)。細胞質での再トランスロケーションやユビキチン化を回避する能力は、PrP変異と常に関連しているわけではないが、身近なCJDに関連する変異プリオンタンパク質(例えば、V210IやM323R)の発現が、ERシャペロンBipのダウンレギュレーションとHrd1ユビキチンリガーゼの活性を誘導するという証拠がある(Peters et al.)PRNP遺伝子のアンバー変異(Y145stop)と関連したGSSの遺伝性型は、C末端のGPIを欠く切断型PrPを産生し、このPrPはN末端のシグナルペプチドを保持し、細胞膜に適切に露出しない。このような形態のPrPは無傷のUPSでは速やかに分解されるが、UPSが薬理学的に阻害されるとPK耐性タンパク質として細胞質に蓄積する(Zanusso et al., o99;Jin et al., n00)。特に重要なこととして、スクレイピーに感染したマウスの脳から抽出したPrPScと組み換えβ-リフォールドプリオンタンパク質の両方が、20Sコアを閉じたコンフォメーションで安定化させ、ユビキチン化基質による開口を防ぐために、26Sプロテアソームのタンパク質分解活性を阻害することが証明されている(Kristiansen et al.)逆に、プロテアソーム活性はフィブリル化PrPScでは阻害されず、PrPScやβ-リフォールド化リコンビナントPrPをそのオリゴマー凝集状態に対する抗体とインキュベートすると回復した(Kristiansen et al., 2007)。
UPS遮断は、主にプロテアソームのキモトリプシンやカスパーゼ様タンパク質分解活性に影響を与えるが、そうでなければ短命なタンパク質の細胞内蓄積を引き起こし、PrPScオリゴマーが神経毒性を発揮する可能性のあるメカニズムとして提唱されている(Amici et al.)ユビキチンの蓄積とプロテアソームの機能不全は、TSE以外にも、さまざまな病態における神経細胞の稀少化と関連するという確固とした科学的証拠がある。この減少は、加齢(Carrard et al., d02)、AD(Gregori et al., i97;Tseng et al., g08)、PD(Lindersson et al., n04)、ポリ(Q)リピート関連疾患(Verhoef et al., f02)で起こる。プロテアソーム活性がβ-アミロイドペプチドと高リン酸化タウの脳内蓄積を防ぐ可能性が、広範な研究から提唱されている。プロテアソームの20sタンパク質分解ユニットにAβが結合すると、その酵素活性が低下する(Gregori et al., i95、1997;Cecarini et al., i08)。主に細胞株やトランスジェニックマウス研究から得られたこの証拠と同様に、プロテアソーム活性の低下もアルツハイマー病患者の死後脳サンプルで確認された。プロテオソーム活性は、神経変性が顕著な海馬や皮質領域で特に低下していたが、通常組織の変性が検出されない小脳や後頭葉皮質では影響を受けなかった(Keller et al.)同様に、特発性パーキンソン病患者の黒質の自己光学的解析では、部分的に不活性なプロテアソームの存在が示され、α-シヌクレインやパーキンの変異がない場合でも、プロテアソームの障害がレビー小体の形成や黒質の神経変性に寄与していることが示された(Mcnaught et al.)さらに最近では、Aβ1-42、α-シヌクレイン、ポリQハンチンチンの可溶性オリゴマーが、そのαサブユニットとの特異的結合を介して、ゲートを閉じたコンフォメーションで安定化させるプロテアソーム活性を阻害できることが実証された(Thibaudeau et al.)これらの報告は、プリオン病で観察されるユビキチン化タンパク質の蓄積は、プロテアソームを介したタンパク質分解の障害を通じてPrPSc自身によって引き起こされる可能性があること、そしてこのメカニズムが、広範な神経変性疾患においてミスフォールドしたオリゴマーによって誘発される神経毒性の主要な原因である可能性を示している。
オートファジーの薬理学的増強がプリオン病治療の展望となる
1990年代半ばにvCJDが大流行し、プリオン生物学に対する科学界の関心が高まったことを除けば、ヒトTSEの発生率は非常に低く、最も頻度の高いsCJDは年間100万人あたり1~2人が罹患しており、治療法の追求に客観的な障害をもたらしている。採用可能な患者が少ないこと、診断後の神経学的および認知機能低下の進行が速いことが、実施された臨床試験の数が非常に少ない理由であり、これまでに試験されたすべての治療アプローチが有効性を示さないという苛立たしさも特徴である。注目すべきは、これらの臨床試験のほとんどが、前臨床試験で観察されたPrPC-PrPSc変換とPrPSc凝集を防ぐことを主目的として、構造的にも臨床的にも無関係な化合物を用いて実施されていることである(Forloni et al.)
しかしながら、プロテオスタシスの障害が、プリオン病と他のタンパク質のミスフォールディング疾患との間で共有される発症特性であるという証拠が増えてきていることから、中枢神経系の神経変性疾患すべてに、より有病率の高いものにも稀なものにも共通のアプローチを採用するよう、治療戦略を変更することの有用性が示唆されている(Butler et al., r06;Nixon. 2013;Engelender et al., r22)。α-シヌクレイン、Aβ、ポリ(Q)-ハンティンチンなどのアミロイド原性タンパク質に関連する神経細胞死を抑制し、生存期間を延長するためには、mTOR依存的・非依存的メカニズムによるオートファジーの薬理学的刺激が有益であるという考えが、重要な前臨床エビデンスによって裏付けられている。関与する分子経路は不均一であり、活性化されたmTORの減少、ミスフォールドしたオリゴマーの消化の増加、シトクロムCの拡散とアポトーシスの活性化を防ぐ損傷したミトコンドリアの除去を含む(Bove et al., e11;Bordi et al., i19)。
現在、ニューロンの自己防衛的なプロテオスタシス能力を高めることができる新しいアプローチが研究されている(Thellung et al.)これらの化合物の多くは、まだ特性化されていない抗プリオン活性を示し、オートファジーのフラックスを活性化する能力を証明し、プロテオスタシスの回復を目的とした中枢神経系のさまざまなプロテオパシーに対する共通の治療戦略の原理証明として提案されている(表1)。このような方向性を示す最初の証拠は、マクロライド誘導体の使用によってもたらされた。マクロライド誘導体は、もともと細胞周期の進行とタンパク質合成を阻害する能力を持つことから免疫抑制剤として使用されてきたが、mTORCのリン酸化を阻害してオートファジーを誘導することもできる(Rubinsztein et al.)これらのうち、シロリムス(ラパマイシン)とタクロリムス、およびそれらの誘導体(ラパログ)であるエベロリムスとテムシロリムスは、現在でも最も効果的で強力なオートファジー活性化剤である。ヒトに対するラパマイシンやラパログの抗プリオン、同情的、あるいは適応外使用はまだ報告されていないが、これらの薬剤が家族性プリオン病の治療において役割を果たす可能性があるという前臨床試験の証拠がある。特に、遺伝性のGSSに関連する変異型PrP(A116V)を発現するトランスジェニックマウスにおいて、ラパマイシンがアミロイド斑の沈着を減少させ、症状の発現を遅らせることが実証され(Cortes et al., s12)、細胞毒性を有する組換えPrP由来ペプチドでインキュベートした神経芽腫細胞においてオートファジーのフラックスを回復させた(Thellung et al., g18)。この証拠に従って、タクロリムスは持続感染細胞において新たにミスフォールドしたPrPSc分子の分解につながる持続的なオートファジー活性を誘導し、2種類のプリオン株を脳内接種したマウスの脳におけるPrPSc蓄積を打ち消す。したがって、mTORの薬理学的阻害は、非遺伝性のプリオン病の治療にも合理的に利用できる可能性がある(Nakagaki et al.)しかしながら、すべてのラパログを慢性的に使用した場合の副作用は、神経疾患に対するこのような治療の実現可能性に大きな不確実性をもたらすことを認めなければならない(Mandrioli et al.)
表1.オートファジー増強剤の前臨床抗プリオン活性。
mTORの阻害を介して作用するオートファジー刺激分子の抗プリオン活性の可能性が原理的に証明されたことで、オートファジーを刺激し、PrPScや脳プロテオパチーに関連する他の凝集しやすいポリペプチドの細胞内凝集を阻害する能力について、構造、起源、作用機序、本来の治療適応の可能性など、異種の多くの化合物のスクリーニングが促された。さらに、ラパマイシンアナログのように免疫系の機能を阻害することなく、オートファジーを刺激することができれば、慢性治療における毒性が穏やかになる可能性があり、非常に望ましい。実際、直接的なmTOR阻害以外にも、オートファジーを刺激してPrPScの細胞内蓄積を抑制するメカニズムが報告されている。
H1ヒスタミン受容体拮抗薬であるアステミゾールは、mTORシグナル伝達を阻害する能力を有し、持続感染した神経芽腫細胞株においてPrPScの複製を阻止し、RMLスクレイピープリオンの脳内接種を受けたマウスの生存期間を延長することができる(Karapetyan et al., n13;Lyu et al., u18)。
メトホルミンはII型糖尿病の第一選択薬であり、現在、独立した臨床適応に再利用されている(Wurth et al., h14)。メトホルミンは、AMP活性化プロテインキナーゼ(AMPK)の活性化を通じて血糖降下活性を発揮し(Zhou et al., u01)、同じメカニズムを通じてオートファジー促進活性を示す(Lu et al., u16)。AMPKはmTORを阻害することでオートファゴソームの形成を刺激し、永続感染する神経芽腫細胞株においてPrPScの量を減少させることが記載されている(Heiseke et al., e09;Howell et al., l17;Abdelaziz et al., z20)。
リチウムやバルプロ酸のような気分安定薬は、神経細胞のオートファジーを刺激し、神経毒性タンパク質に対する神経細胞の抵抗性を高める能力を有することが最近証明され、Aβペプチド、変異ハンチンチン、α-シヌクレインの細胞内蓄積と毒性を抑制する。神経保護活性の薬力学的基盤に関する最も詳細な特徴は、ホスホイノシトールのターンオーバーとIP3産生を阻害することによりオートファゴソームの形成を刺激するリチウムで得られている(Sarkar and Rubinsztein, 2006)。
もう一つの有望な化合物は、タンパク質フォールディングを安定化させる能力を持つことから、細菌、酵母、小型無脊椎動物によって生産される天然の二糖類トレハロースである(Wolkers et al.)トレハロースは、Aβ、タウ、ハンチンチン、α-シヌクレイン、SOD1など、様々な種類の凝集しやすいタンパク質によって誘導される神経細胞死に対して、神経保護活性を示す(Tanaka et al., 2004;Liu et al., 2005;Rodriguez-Navarro et al., 2010)。トレハロースは、オートファジー流束のmTOR非依存的活性化を通じて、アミロイド関連オリゴマーのクリアランスを刺激する(Tanaka et al., a04;Sarkar et al., r07;Mardones et al., s16)。PrPScのモデルとして持続感染神経芽腫細胞を用いると、トレハロースは細胞内に蓄積したプロテアーゼ耐性PrPScの量を減少させ、酸化的損傷から細胞を保護した(Beranger et al., r08;Aguib et al., b09)。最近では、トレハロースがアミロイド前駆体タンパク質のエンドソーム介在切断を変化させるAβペプチドの形成を阻害する可能性があることから、アミロイド斑の形成を減少させるトレハロースの有効性も提唱されている(Tien et al., n16;Liu et al., u20)。
タンパク質クリアランスを標的とした神経保護療法の現状と未来
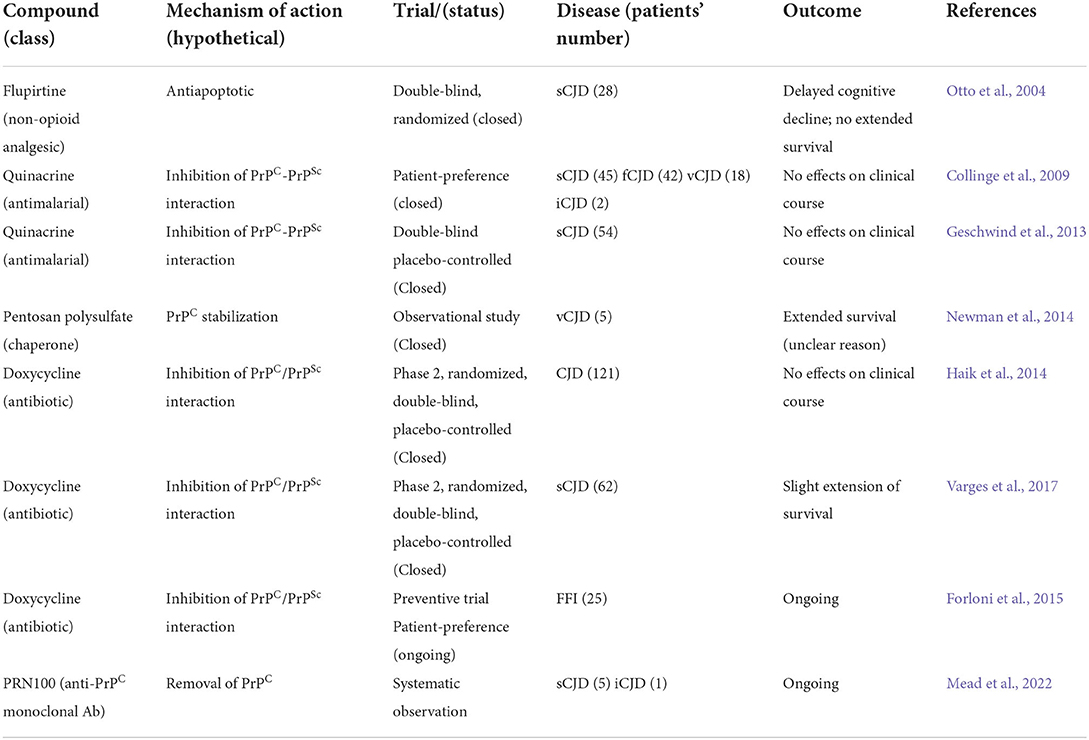
プリオン仮説の最初の定式化から40年が経過し、明らかに異端と思われた科学的仮説は、生物の生物学や、ヒトや動物の散発性、家族性、感染性の神経変性疾患の病理学にとって、大きな一里塚となった(Prusiner, 1982,1998)。ある状況下で異常なリフォールディングを起こす、脳内の病理学的PrPの生理学的対応物の発見は、神経科学者の間に特別な余波をもたらした。特に、凝集タンパク質の封入体、神経原線維変化、細胞外アミロイド斑への沈着を特徴とする多くの神経変性疾患の病因におけるタンパク質のミスフォールディングの役割に大きな関心が向けられた(Basler et al., r86;Scheckel and Aguzzi. 2018)。抗アポトーシス活性(Otto et al., 2004)により、CJD患者の認知機能低下を対照する非オピオイド薬フルピルチンの限定的な有効性に加え、ヒトTSEに対してこれまでに評価された治療アプローチのほとんどは、PrPScテンプレートとの相互作用を阻害することにより、PrPCのミスフォールディングを対照することに向けられていた。in silicoあるいは実験的に感染させたげっ歯類で得られた有望な結果に基づいて、PrPC-PrPSc転移の阻害剤として、慢性投与が倫理的と考えられる多くの薬剤が試験された。
PrPC-PrPSc転換を阻止し、PrPScの除去を促進するために追求された最初の戦略の一つは、PrPScとミスフォールドしたPrPCの結合能を持ち、シャペロンのような働きをする天然および合成ポリマーを用いることであった(照屋と堂浦、2022)。1990年代に英国で若者の間で発生したvCJDによる緊急事態に促され、スクレイピーに感染したマウスを陰イオン性ポリマーのペントサン・ポリサルフェートで治療したところ、感染後の生存期間が延長するという有望な結果が得られた(Doh-Ura et al.少数のvCJD患者がペントサンポリスルフェートで脳室内投与され、生存期間が有意に延長した(Newman et al., n14)。
抗マラリア薬であるキナクリンは、その平面的な立体構造によってPrPCとPrPScの界面に干渉し、TSEの細胞モデルや動物モデルで神経保護をもたらし(Forloni et al., i02;Villa et al., a11)、ヒトでのさまざまな臨床試験に影響を与えた。散発性、家族性、異所性、変異型CJDに罹患した150人以上の患者を対象に、2つの二重盲検無作為化試験が実施された。患者はキナクリンを経口投与され、生存期間、神経学的および認知機能の低下について評価された(Collinge et al.)同様の戦略を追求し、テトラサイクリン系抗生物質であるドキシサイクリンとミノサイクリンを用いた他の臨床試験では、試験管内試験で有効性が示され(Forloni et al., i02;GuとSingh 2004;De Luigi et al., i08;Corsaro et al., o09)、ドキシサイクリンはCJDとFFIに罹患した患者で試験された。しかし、有望な前臨床効果にもかかわらず、キナクリンまたはドキシサイクリンの投与はCJD患者の生存に有意な効果をもたらさなかった(Collinge et al., e09;Haik et al., k14;Varges et al., s17;Forloni et al., i19)。注目すべきは、まだ無症状の患者におけるFFIの予防的アプローチとしてドキシサイクリンを用いて実施された臨床試験の結果が近い将来に期待されていることである(Forloni et al., 2015)。
2018年には、ユニバーシティ・カレッジ・ロンドン病院で、CJDと診断された6人の患者(5人が散発性、1人が異所性)を対象に、PrPCに対するPRN100と名付けられたヒト化モノクローナル抗体を静脈内投与し、その忍容性と有効性を評価する実験的治療プログラムが開始された(Mead et al., d22)。前述のプログラムでは、PRN100による治療の忍容性は非常に良好であったが、安定した認知機能の改善や生存期間の延長を示したものはなかった(表2)。この有効性の欠如を説明する要因として考えられるのは、動物モデルとは異なり、ヒトのプリオン病は、PrPScの進行性蓄積が起こる非常に長い無症候期を特徴とすることである。したがって、診断と治療は、脳内のプリオン負荷と神経細胞死がすでにいかなる治療法にも感受性を越えているときに開始される(Forloni et al.)概念的に似ているが、アルツハイマー病やパーキンソン病における神経細胞死を遅らせることを目的とした疾患修飾治療アプローチは、高齢者の間で最も広範な神経変性疾患であり、Aβペプチド、高リン酸化タウタンパク質、α-シヌクレインの除去に向けられている(Engelender et al.)Aβ、タウ、α-シヌクレインに特異的に指向する抗体を用いて追求されたこれらの戦略は、これらの疾患の動物モデルにおいて良好な有効性を示し、Aβに対するヒトモノクローナル抗体(アデュカヌマブ)の米国食品医薬品局(FDA)による承認につながった(Cummings et al., s21)。残念なことに、ADに対する効果的な免疫療法を開発するための努力のほとんどは、効果的な結果をもたらさなかった。
プロテオスタシスのメカニズムの解明が進み、UPSとALPの機能不全とミスフォールディングタンパク質の蓄積との関係が証明されたことで、疾患特異的なタンパク質に作用するのではなく、神経細胞におけるタンパク質代謝制御の回復に焦点を当てた新たな治療戦略が開かれつつある、1995;CiechanoverとBrundin 2003;CiechanoverとKwon. 2017;Thellung et al., g19;Chen et al., n21;Leri et al., i21)。
表2.ヒト臨床試験。
著者寄稿
著者全員が原稿のデザイン、原稿作成、校閲、編集に貢献した。すべての著者が論文に貢献し、提出された原稿を承認した。
資金調達
本研究は、IRCCS Ospedale Policlinico San Martino Ricerca Corrente 2022助成金(TF)により行われた。
利益相反
著者らは、本研究が、潜在的な利益相反と解釈されうる商業的または金銭的関係がない状態で実施されたことを宣言する。
