
Alternative Mitochondrial Electron Transfer for the Treatment of Neurodegenerative Diseases and Cancers: Methylene Blue Connects the Dots
2015年11月18日オンライン公開。doi: 10.1016/j.pneurobio.2015.10.005
PMCID: PMC4871783
NIHMSID: NIHMS739389
要旨
脳は、グルコースを唯一のエネルギー源とするエネルギー代謝に極めて高い要求を持っている。脳のエネルギー代謝とグルコース取り込みの低下は、アルツハイマー病、パーキンソン病、その他の神経変性疾患患者で認められ、神経変性疾患とエネルギー代謝の間に明確な関連性を示している。一方、神経膠芽腫を含むがんでは、グルコースの取り込みが亢進し、エネルギー代謝を好気的解糖に依存している。高効率の酸化的リン酸化経路から低効率の好気性解糖経路への切り替え(ワールブルグ効果)は、生合成と増殖のための高分子を供給する。
現在の研究では、100年来の薬物であるメチレンブルーが、複合体Iの存在下でNADHから電子を受け取り、それをシトクロムCに供与することで、別の電子伝達経路を提供することが示されている。メチレンブルーは試験管内試験で酸素消費量を増加させ、解糖を減少させ、グルコース取り込みを増加させる。
メチレンブルーは、急性投与によりラットのグルコース取り込みと局所脳血流を増加させる。さらに、メチレンブルーは、試験管内試験およびアルツハイマー病、パーキンソン病、ハンチントン病のげっ歯類モデルにおいて、神経細胞とアストロサイトをさまざまな障害から保護する。
神経膠芽腫細胞では、メチレンブルーはミトコンドリアの酸化的リン酸化を促進することによってワールブルグ効果を逆転させ、神経膠芽腫細胞周期をs期で停止させ、神経膠芽腫細胞の増殖を抑制する。従って、メチレンブルーはAMP活性化プロテインキナーゼを活性化し、下流のアセチル-コアカルボキシラーゼとサイクリン依存性キナーゼを阻害する。
まとめると、代替的ミトコンドリア電子伝達を介してミトコンドリアの酸化的リン酸化を促進することが、神経変性疾患に対する保護作用をもたらし、癌の増殖を抑制する可能性があるという概念を証明する証拠が蓄積されつつあるということである。
はじめに
がんと神経変性はしばしば、病因も治療法も正反対の2つの異なる病態疾患であると考えられている。がんが細胞死に対する抵抗性の亢進を特徴とするのに対し、神経変性疾患は進行性の早期神経細胞死を特徴とする(Plun-Favreau et al., 2010)。実際、アルツハイマー病(AD)のような神経変性疾患は、がんと逆相関することがわかっている(Bajaj et al.) 一方、がんと神経変性疾患は、共通の病因メカニズムや治療標的を共有している可能性があるという証拠が増えつつある。神経変性と癌の両方にとって、加齢は最も重要な危険因子である(Niccoli and Partridge, 2012)。食事制限は、寿命を延ばし、がんや神経変性疾患を含む加齢関連疾患を遅らせるための最も効果的な介入の一つであることが判明している(Hursting et al., 2013)(Graff et al., 2013; Hursting et al., 2010; Prolla and Mattson, 2001)。がんと神経変性疾患の両方の治療に有効な可能性のある薬剤は数多くある。例えば、T細胞リンパ腫治療用のレチノイドX受容体作動薬であるベキサロテンは、ADのマウスモデルにおいてAβ斑を減少させ、認知障害を減弱させることが示されている(Aicardi, 2013; Cramer et al.)
クレブスサイクル(Krebs and Johnson, 1937)の発見から、ミトコンドリアの酸化的リン酸化(Boyer et al., 1973; Gresser et al., 1982)に至るまで、生物学は1920年代から1980年代にかけて、細胞代謝に関する知識を大きく発展させてきた。過去30年間、分子生物学的アプローチは、細胞の代謝状態にあまり注意を払うことなく、生物学研究を支配してきた。分子生物学と現代医学の飛躍的な進歩にもかかわらず、神経変性疾患と癌は依然として最も壊滅的な疾患であり、効果的な治療介入が切実に求められている。最近では、転写が細胞代謝の決定に関与していることが認められ始めている(McKnight, 2010)。分子、細胞、代謝の統合的アプローチは、神経変性疾患や癌の理解を深め、これらの難病に対する新規治療法の発見につながる可能性がある。
代謝異常は多くの疾患の主要な特徴である。神経変性疾患は、主に中枢神経系に存在する神経細胞の異なるサブセットに影響を及ぼすと推定される疾患群であり、主にAD、パーキンソン病(PD)、ハンチントン病(HD)、筋萎縮性側索硬化症(ALS)などが含まれる。このような異質性にもかかわらず、多くの神経変性疾患は代謝機能障害を併発していることが多いことが、実験的研究と臨床研究の両方から指摘されている(Cai et al.) 同様に、がん細胞は、その発生経路の違いや腫瘍間のゲノム不安定性を考慮すると、遺伝的にも表現型的にも不均一である(Burrell et al., 2013)。それにもかかわらず、がん細胞は1920年代から代謝に特徴的な変化を示すことが知られていた(Warburg, 1956a, b)。エネルギー代謝のリプログラミングは、最近、新たながんの特徴として提唱されている(Hanahan and Weinberg, 2011)。そのため、代謝経路を標的とした新たな治療法が、がんや神経変性の治療のために模索されている。本総説では、神経変性疾患と癌における代謝の変化を明らかにした知見を要約し、100年来の医薬品であるメチレンブルーの代替ミトコンドリア電子伝達キャリアとしての新規機能をまとめ、代替ミトコンドリア電子伝達が神経変性疾患と癌の両方に共通する治療メカニズムであることを提案する。
脳のエネルギー代謝
脳の生体エネルギー
アインシュタインの有名な方程式E=MC2は、エネルギーと質量という2つの基本的な物理的実体の関係を単純化し、それらを光速と結びつけたものである。こうして、別個の存在として考えられていた質量とエネルギーは、交換可能であることが知られるようになった。アインシュタインの公式の意義は、物理学の枠を超えている。生物学では、エネルギーはバクテリアから人間に至るまで、すべての生物の属性である。質量とエネルギーの変換は、代謝と定義される生物学的プロセスの基本であり、生物はさまざまなメカニズムを通じてエネルギーを循環させ、必要な分子を生成し、生命に不可欠な機能を果たす。同化作用によって、複雑な化合物は、ATP加水分解によって提供されるエネルギー費用で、より単純な分子から生合成される。異化作用によって、複雑な栄養素は、ATP産生と連動したエネルギー放出プロセスによって、より単純な酸化分子へと分解される。生命とは、エネルギーと構造の相互作用である(Wallace, 2005)。代謝が進めば、生命も続く(図1)。
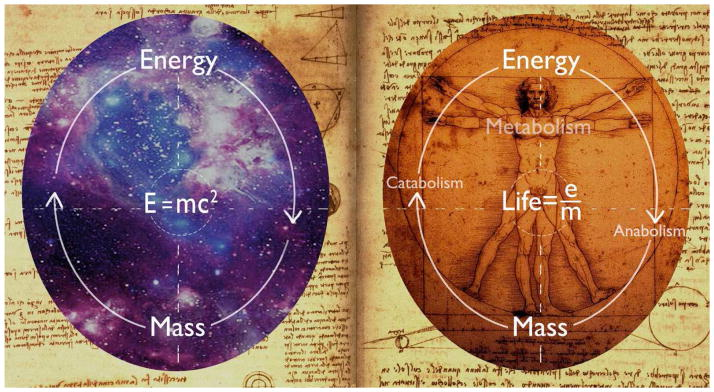
図1 エネルギーと質量:アインシュタイン方程式からエネルギー代謝へアインシュタインの有名な方程式は、質量とエネルギーの交換可能な関係を明らかにしている。アインシュタインの式の意義は、物理学の枠を超えている。生物学では、質量とエネルギーの変換は代謝として定義される基本的なプロセスである。同化作用によって、複雑な化合物は、ATP加水分解によるエネルギー消費によって、より単純な分子から生合成される。異化作用によって、複雑な栄養素は、ATP産生と結びついたエネルギー放出プロセスによって、より単純な酸化化合物に分解される。生命とは、エネルギーと構造の相互作用である。代謝が続けば、生命も続く。E:エネルギー。M:質量。C:光速。
哺乳類の脳は、神経細胞の活動に合わせて適切なエネルギー基質が供給されるよう、微細な調節機構を備えた高い代謝活動が特徴である。ヒトの脳は体重のわずか2%しかないが、心拍出量の15%を受け、酸素消費量のほぼ20%を占め、全身のグルコース利用量の約25%を消費する。人間の脳は、全身の中でエネルギー消費量が最も多い臓器なのだ。シナプス後電位と活動電位、神経伝達物質のリサイクルによって散逸する膜貫通静止電位の維持と回復が、脳における主なエネルギーコストである(Alle et al., 2009; Attwell and Laughlin, 2001; Howarth et al.) 最新の計算研究によると、大脳皮質では、シナプス後グルタミン酸受容体、活動電位、静止電位、シナプス前伝達物質放出、伝達物質リサイクリングが、それぞれエネルギー収支の50,21,20,5、4%を消費していることが示されている(Howarth et al.)
非常に高いエネルギー消費に加え、脳のエネルギー供給と消費は、神経血管と神経代謝のメカニズムによって緊密に連関している。脳のエネルギー貯蔵量は非常に限られているため、局所的な脳活動は、神経血管結合と呼ばれる脳血流(CBF)の増加と同時に対応しなければならない。脳血管系は、内皮、周皮細胞、アストロサイト、神経細胞(これらをまとめて神経血管ユニットと呼ぶ)の密接な関係によって特徴づけられる末梢とは異なる、多くのユニークな構造的・機能的特徴を持っている(Del Zoppo, 2013; Iadecola, 2004)。解剖学的に、神経血管の結合は、神経血管ユニットの相乗作用によって組織化されなければならない。したがって、脳活動によって誘発されるCBFの増加、すなわち機能的充血は、神経血管ユニットの構成要素に由来する多数の血管作動性物質の協調的な作用によって媒介されると考えられる(Girouard and Iadecola, 2006; Petzold and Murthy, 2011)。
正常な生理学的条件下では、成人の脳はエネルギー代謝の基質としてほとんどグルコースのみを使用し、グルコースは主にグルコーストランスポーター1(GLUT1)とGLUT3を介した促進拡散によって循環から脳へと送られる。GLUT1は最初にクローニングされたGLUTであり、2つのアイソフォームが脳で検出されている(Gerhart et al., t89;McCall et al., l96)。45kDaのアイソフォームはグリア細胞に局在し、高分子量(55kDa)のアイソフォームであるグリコシル化GLUT1は内皮の内腔膜と外腔膜の両方に存在することが見出されている(Birnbaum et al., m86)。さらに、55kDaのGLUT1は細胞内プールとして内皮細胞質に存在する(Duelli and Kuschinsky, 2001)。一方、GLUT3はほとんど神経細胞でのみ発現していることが判明している(Simpson et al., n07)。GLUT1とGLUT3はGLUT4に比べてインスリン感受性が低いため、脳はインスリンに鈍感な臓器と考えられている(Heidenrich et al., h89;Mueckler et al., r85)。脳における代謝速度の不均一性は、脳におけるGLUTの不均一な分布と関連している。代謝速度とGLUTの分布の相関から、GLUT1とGLUT3の密度が、異なる脳構造の局所的な代謝需要に寄与している可能性が示唆される(Duelli and Kuschinsky, 2001)。脳に運ばれたグルコースは、他の臓器と同様、ヘキソキナーゼによってリン酸化され、グルコース-6-リン酸を生成する。グルコース-6-リン酸は、解糖、ペントースリン酸経路、糖新生という異なる代謝経路を経て、さらに処理される。通常の状態では、グルコースは解糖、トリカルボン酸(TCA)サイクル、ミトコンドリアの酸化的リン酸化を経て、ほぼ完全にCO2と水に代謝される(Belanger et al.)
解糖は、進化的に保存されてきた、ほぼ普遍的なエネルギー生産経路であり、グルコースを嫌気発酵させて乳酸にし、3ATPを生産する。酸素の存在下では、解糖はTCAサイクルやミトコンドリアの酸化的リン酸化と緊密に結合し、分子間相互変換システムとして機能する。解糖の産物である乳酸は、ミトコンドリアのモノカルボン酸トランスポーター(MCT)を介してミトコンドリア内に移動し、そこでピルビン酸に酸化される。その後、ピルビン酸は二酸化炭素を放出しながらTCAサイクルによって一連の有機酸に変換される(Schurr, 2014)。さらに、TCAサイクルはNAD+をNADHに還元し、これをミトコンドリアの酸化的リン酸化に送り込み、究極の生化学的エネルギーであるATPを生成する。
ミトコンドリアの酸化的リン酸化には、ミトコンドリア呼吸鎖を介した電子伝達、ミトコンドリア膜を介したプロトンポンプ、ミトコンドリア膜電位の生成、そして最終的なATP合成が含まれる。ミトコンドリアは、炭水化物や脂肪由来の水素を酸化することにより、ATPの形でエネルギーを産生する。NADHまたはコハク酸に由来する電子は、電子伝達連鎖(ETC)複合体を順次通過し、放出されたエネルギーは、複合体I、III、IVを通して膜間空間にプロトンを送り込むのに使われ、ATP合成に連動するミトコンドリア膜電位を作り出す。プロトンがミトコンドリア内膜を流れ、複合体Vを通ってミトコンドリアマトリックスに戻ると、パイがADPと結合してATPが生成される(Wallace, 2005)。ミトコンドリアの酸化的リン酸化の副産物として、主に複合体IとIIIのETC複合体から漏れた電子は、直接O2に供与され、スーパーオキシドアニオンやその他の活性酸素種(ROS)を生成することができる(Birch-Machin, 2006; Murphy, 2009)。
ミトコンドリアの構造と機能は高度に保存されているが、異なる細胞や組織の間では、ミトコンドリアには質的にも量的にも多くの違いがある。加えて、ミトコンドリアは、融合と分裂の制御されたプロセスを通じて、ダイナミックに形や数が変化する。脳のミトコンドリアは、細胞や部位の不均一性と加齢によって複雑化し、末梢よりも速いペースでATPを生成できることを示した研究がある(Battino et al., o91;Kwong and Sohal 2000;Tesco et al., o07)。ミトコンドリア動態の乱れは、神経変性疾患や癌を含むヒトの様々な疾患に関わっていることが指摘されている(Chen and Chan, 2009; Grandemange et al.)
生体エネルギーを超えて
炭水化物由来のグルコースは、ミトコンドリアの酸化的リン酸化によるATP産生の主要な燃料であるだけでなく、タンパク質、脂質、核酸のde novo合成に不可欠な基質でもある(Bauer et al., 2005)。同様に、ミトコンドリアは細胞の生体エネルギーセンターとしての役割に加え、生合成ハブとして機能し、細胞増殖のための生合成需要を満たす高分子の合成に重要な役割を果たす(Jones and Thompson, 2009)。生体エネルギー経路では、解糖から生成されたピルビン酸がアセチル-CoAに変換され、TCAサイクルに供給されてエネルギー産生のために酸化される。生合成経路では、グルコース由来の炭素原子がミトコンドリアに入り、様々な段階でTCAサイクルから押し出され、脂質、ヌクレオチド、アミノ酸の生合成の前駆物質となる(Jones and Thompson, 2009)。
TCAサイクルでは、アセチル-CoAから誘導された炭素数2のアセチル基が炭素数4のオキサロ酢酸に転移し、炭素数6のクエン酸を形成し、さらにイソクエン酸に変換される。別の方法として、クエン酸は脂質生合成とタンパク質のアセチル化のために、ピルビン酸-クエン酸シャトルを介して細胞質に輸送される。細胞質では、クエン酸はオキサロ酢酸とアセチルCoAに切り戻される。クエン酸塩はさらにアセチル-CoAカルボキシラーゼによってマロニル-CoAに変換され、脂肪酸合成経路に供給される(Ahn and Metallo, 2015)。このように、代謝中間体アセチル-CoAは、生体エネルギーにおける重要な役割に加えて、脂肪酸合成のための中心的な生合成前駆体として機能している。一貫して、真核生物のミトコンドリアには、細胞質脂肪酸合成装置とは独立した、高度に保存された脂肪酸合成装置も存在するが、その最終産物や機能はほとんど不明である(Hiltunen et al.)
グルコース代謝産物もまた、アミノ酸のde novo生合成の基質となる。ピルビン酸、α-ケトグルタル酸、オキサロ酢酸は、アミノ基の付加によってアミノ酸に変換される。α-ケトグルタル酸は還元的アミノ化によってグルタミン酸に変換される。次に、グルタミン酸のアミノ基をピルビン酸とオキサロ酢酸に移し、それぞれアラニンとアスパラギン酸を生成することができる。グルタミン酸はプロリンやアルギニンの前駆体でもある。ペントースリン酸経路によるグルコース代謝は、ヌクレオチド生合成の重要な中間体であるリボース-5-リン酸を生成する(Munoz-Pinedo et al.) さらに、ペントースリン酸経路はNADPHを産生し、脂肪酸とヌクレオチドの生合成に還元当量を供給する(Jones and Thompson, 2009)。
ミトコンドリアの酸化的リン酸化は、脳における主要な代謝経路である。脳エネルギー代謝の中心的ドグマは、基礎状態でも活性化状態でも、脳内グルコースの利用がミトコンドリアの酸化的リン酸化を通じて神経細胞の活動を促進すると仮定していた(Simpson et al., 2007; Sokoloff et al., 1977)。しかし、脳のグルコース利用と酸素利用の間にアンカップリングがあることを示唆する証拠が増えつつある。グルコース利用の測定は、呼吸商を1として酸素消費量から計算すると、最大31mmol/100g体重/分であることがわかっている(Kety and Schmidt, 1948)(Pellerin and Magistretti, 2012)。したがって、ミトコンドリアの酸化的リン酸化によるATPの生体エネルギー産生に加えて、糖新生と脂肪新生の両方に関与する生合成経路が、酸素消費量を超える脳によるグルコース代謝に寄与している可能性がある。実際、成人ヒトの脳で消費されるグルコースのうち、好気的解糖が占める割合は10%程度であることがわかっている(Goyal et al.)
脳のエネルギー貯蔵量は非常に限られていると考えられているが、唯一最大のグルコース備蓄であるグリコーゲンは、脳内で主にアストロサイトの末端、小体、突起に局在していることが判明している(DiNuzzo et al.) 理論的には、解剖学的にも機能的にも神経細胞と密接な関係にあることから、アストロサイトは、通常時およびエネルギー需要が増大した場合や神経細胞が機能不全に陥った場合に、神経細胞の代謝のためにグリコーゲンを動員することができる。肝臓や骨格筋に比べて脳内のグリコーゲンの含量が比較的低いことを考えると、アストロサイトのグリコーゲンが低血糖時の脳の代替エネルギー供給源になるとは考えにくい。むしろ、アストロサイト・グリコーゲンは、通常時の脳代謝に不可欠な因子として機能している可能性が高い(DiNuzzo et al.) 糖新生とグリコーゲンの分解は、グリコーゲン合成酵素とグリコーゲンホスホリラーゼによって微調整されている(Obel et al.) アストロサイトのグリコーゲンがニューロン-グリア間の代謝結合にどのような役割を果たしているかについては、多くの議論があるが、この総説の範囲を超えている。重要なのは、アストロサイトのグリコーゲンが学習過程に関与していることである(Hertz and Gibbs, 2009)。さらに、アストロサイトとニューロン間のグリコーゲンのホメオスタシスが乱れると、神経変性が起こることが分かっている。最近の研究では、ニューロンには糖新生の酵素機構があり、ラフォラ病(進行性ミオクローヌスてんかん)に関連する遺伝子変異がニューロンのグリコーゲン合成を活性化し、アポトーシスを引き起こすことが示されている(Duran et al., 2012; Magistretti and Allaman, 2007; Vilchez et al.)
脂質代謝は、脳内に高濃度に存在し、脂質代謝酵素の変異が神経疾患の衰弱につながることから、特に注目されている。脂質は、β酸化によるエネルギー産生の基質として、また細胞膜の構造成分として、2つの主要な機能を果たす可能性がある。脳のエネルギー代謝において、脂肪酸よりもグルコースが優勢な基質として選択されるのは、β酸化のデメリットを避けるために進化的に有益なことなのかもしれないと推測された(Schonfeld and Reiser, 2013)。脂肪酸は脳でも利用されるが(Ebert et al., 2003; Kuge et al., 1995; Panov et al., 2014)、神経細胞には低レベルの脂肪酸異化酵素があり、一般に生体エネルギーのために脂肪酸β酸化に頼ることはないと考えられている(Cahoy et al., 2008; Lee and Wolfgang, 2012)(SchonfeldとReiser, 2013)。
潜在的なエネルギー基質の他に、脂質は細胞膜の構造成分であり、細胞内構造、膜輸送、膜内のタンパク質やサブコンパートメントの制御に関与していることがわかっている(Adibhatla and Hatcher, 2007; Liu and Zhang, 2014)。コレステロールは不可欠な生体膜成分である。脳は、全身のコレステロールの約25%を占める、コレステロールが非常に濃縮された臓器である(Dietschy and Turley, 2001)。脳内コレステロールの大部分は、脳の発達、シナプスと樹状突起の形成、軸索誘導に必要なミエリン鞘と細胞膜に存在する(Orth and Bellosta, 2012)。コレステロール合成は、発生段階の髄鞘形成時にオリゴデンドロサイトで最も高くなる(Dietschy and Turley, 2004)。成体脳では、BBBによって血液からのコレステロールの取り込みが妨げられるため、アストロサイトでのde novoコレステロール合成は低い割合で続いている(Nieweg et al., g09)(Vance, 2012)。一貫して、脂質合成経路はアストロサイトに非常に濃縮されている(Cahoy et al., y08)。
極めて重要な構造成分であることに加え、脳脂質は主に3つのメカニズムを通じて重要な細胞シグナル伝達機能を持つ。第一に、リゾリン脂質、エイコサノイド、エンドカンナビノイドなどの生理活性脂質は、Gタンパク質共役型膜受容体と相互作用し、下流のシグナル伝達を活性化することが知られている(Bieberich, 2012)。第二に、ホスファチジルイノシトールやセラミドなどの脳内生理活性脂質は、プロテインキナーゼ、ホスファターゼ、その他のシグナル伝達タンパク質と結合し、細胞内カスケードに影響を与える(Bieberich, 2012)。さらに、脳の生理活性脂質は、コレステロールやスフィンゴ脂質に富む脂質ラフトを通じて、細胞のシグナル伝達経路を制御している(Lingwood and Simons, 2010)。脂質ラフトは、生理学的にも病理学的にも、多くの脳機能に関与していることが示されている。例えば、脂質ラフトは神経伝達物質の受容体やトランスポーターの効力や有効性に影響を及ぼすことが示されている(Allen et al., 2007)。脂質L-カルニチンの主な役割は、β酸化サイクルのためにミトコンドリアへの長鎖脂肪酸の輸送を促進するようで、神経保護作用があることが指摘されている(Rau et al., 2012年;Wang et al., 2007)。脂肪生成は、成体神経幹の増殖を制御することが示されている(Folmes et al., s13;Knobloch et al., h13)。
アストロサイトとニューロンの代謝連関とアストロサイト・ニューロンの乳酸シャトリング
過去10年間、神経エネルギー学における私たちの知識は、「神経中心的」な見方から、ニューロン-アストロサイト結合の統合的な図式へと急速に発展してきた(Allaman et al.) 解剖学的に、ニューロンはBBBによって循環から隔てられており、血液由来の燃料に直接アクセスすることはできない。循環中のグルコースは、2つの内皮と2つのアストロサイトの計4つの細胞膜を通過してニューロンに到達する。そのため、ニューロンは自らの燃料供給を直接コントロールできない可能性がある。一方、アストロサイトは、その末端が脳毛細血管の99.7%を覆っているため、脳内へのグルコース流入を制御する上で解剖学的に有利である(Mathiisen et al.) したがって、アストロサイトは、神経細胞と血管系をつなぐ仲介役として機能している可能性がある。
解剖学的証拠と一致するように、ノックアウトマウスの研究では、グリア/内皮GLUT1が、神経細胞GLUT3と比較して、哺乳類の脳におけるグルコース輸送に主要な役割を果たしていることが示された。異種GLUT1ノックアウトマウスでは、脳内GLUT1発現が減少し、脳内グルコース取り込みの減少、発作、運動障害、低血糖、小頭症の協調運動障害や学習障害など、ヒトのGLUT1欠損症候群と類似した多くの特徴を示す(Wang et al., g06)。一方、GLUT3のハプロ欠損は、脳機能障害や脳内グルコース取り込み障害とは関連していない(Stuart et al., t11)。従って、アストロサイトは、脳のグルコース代謝において、ニューロンよりもさらに重要な役割を果たしている可能性がある。実際、海馬や小脳では、グルコースの輸送と代謝は、神経細胞区画よりもグリア区画の方が速いことがわかっている(Jakoby et al.) 脳スライスの生体内試験イメージングでは、アストロサイトの刺激がアストロサイト内カルシウムサージを引き起こし、それに続いて隣接する細動脈を拡張または収縮させることが示されている(Girouard et al.)
脳では、グルコース以外にも、乳酸、ピルビン酸、酢酸、グルタミン酸、グルタミンなど、さまざまな代謝中間体がエネルギー産生のために酸化される可能性がある(Belanger et al.) 重要なことは、ニューロンとアストロサイトは、生理的状態では異なる代謝プロフィールを示すことである。GLUT1とGLUT3の他に、解糖産物はモノカルボン酸トランスポーター(MCT)によってニューロン内外に輸送され、MCT1はBBBとアストロサイトに、MCT2はニューロンに存在する(Simpson et al., 2007)。さらに、アストロサイトはMCT4を発現している(Pierre and Pellerin, 2005)。MCT1-4は、ピルビン酸、乳酸、ケトン体のような内因性に生成されるモノカルボン酸を輸送することが示されている(Pierre and Pellerin, 2005)。解糖産物である乳酸は、長期記憶形成に関与していることが示されている。アストロサイトMCT4またはニューロンMCT2を標的とするアンチセンスオリゴヌクレオチドは、記憶障害を引き起こす。興味深いことに、外因性乳酸を添加すると、MCT4ノックアウトによる記憶定着の欠損のみが救済され、MCT2ノックアウトは救済されなかったことから、アストロサイトからニューロンへの乳酸シャトリングが示唆される(Bezzi and Volterra, 2011; Suzuki et al.) 神経細胞のグルコース由来のピルビン酸が神経細胞の主要な酸化燃料であることを示す証拠は数多くあるが(Patel et al., 2014)、脳における生体エネルギー代謝と生合成代謝の両方において、アストロサイトが重要な役割を果たしていることを示唆する証拠が増えつつある。さらに、多くの神経変性疾患において、アストロサイトの代謝機能障害が見つかっている(Stobart and Anderson, 2013)。
エネルギー代謝のマスターセンサーであり調節因子であるAMPK
エネルギー供給と消費を一致させることは、脳機能にとって不可欠である。ADPと無機リン酸から生成されるATPは、細胞内の普遍的なエネルギー通貨である。逆に、エネルギーが必要な細胞プロセスは、ATPが加水分解されてADPとリン酸になることで駆動する。したがって、ADP/ATP比は細胞のATP消費と合成の両方にとって重要なパラメーターとなる。さらに、ユビキタスに発現するアデニル酸キナーゼは、アデニンヌクレオチドの相互変換を触媒し、AMP/ATP比をエネルギー状態のもう一つの重要な指標とする(Hardie, 2011)。細胞レベルでは、ATP、ADP、AMPの相対レベルに反映されるエネルギー恒常性の維持は、エネルギー代謝にとって極めて重要である。最近の研究では、ATP、ADP、AMPの相対レベル(ADP/ATP比およびAMP/ATP比)が、AMP活性化プロテインキナーゼ(AMPK)シグナル伝達を通じて、エネルギー代謝の制御に重要な役割を果たしていることが示された(Gowans et al.
Ser/ThrキナーゼであるAMPKは、進化的に保存されたエネルギーセンサーであり、エネルギー代謝の調節因子である(Kahn et al., n05)。AMPKは、触媒αサブユニット(α1、α2)と制御βサブユニットおよびγサブユニット (β1、β2、γ1、γ2、γ3)からなるヘテロ三量体タンパク質である(Hardie, 2007)。ATP消費を増加させるか、細胞のATP産生を低下させるような出来事によって活性化される(Hardie, 2007; Long and Zierath, 2006)。βサブユニットのグリコーゲン結合ドメインにより、AMPKはグリコーゲンセンサーとしても機能する(McBride et al.) AMPKは、グルコースと脂質の代謝、細胞周期、タンパク質合成に多様な影響を及ぼす。活性化されたAMPKは、グルコースと脂肪酸の取り込みを増加させ、解糖と脂肪酸の酸化を促進し、ミトコンドリアの生合成を刺激することで、ATPを生成する異化経路を刺激する。同時に、AMPKの活性化は、ATPを消費する同化経路の速度を抑制するため、アデニル酸のエネルギーチャージが正しく回復する(Hardie, 2011)。AMPKの活性化は、グリコーゲン合成酵素とアセチル-CoAカルボキシラーゼ(ACC)をそれぞれリン酸化・不活性化することで、グリコーゲンと脂肪酸の合成を阻害する(Hardie, 2007)。さらに、AMPKはタンパク質合成と細胞増殖を抑制する(Horman et al., n02;Motoshima et al., a06)。栄養素が制限されている場合、AMPKは哺乳類ラパマイシン標的複合体I(mTORC1)の抑制を介して細胞増殖を阻害する(Gowans et al., s13)。さらに、AMPKの活性化はオートファジーを促進し、栄養不足に対応して必要不可欠な細胞活動を維持する(Kim et al., m11)。
哺乳類の成体脳では、AMPKは、異化活性が高いことを特徴とするニューロンで主に発現する(Culmsee et al., e01;Turnley et al., y99;Vingtdeux et al., x11)。グルコース代謝の高い成体脳では、AMPKの発現量が高いが、神経細胞のサブタイプによってAMPKの発現量が異なることが確認されている(Spasic et al., c09)。視床下部では、AMPKはエネルギー感知ニューロンで発現し、生体のエネルギーバランスのマスターレギュレーターとして機能している(Ronnett et al.) 低血糖(McCrimmon et al., n04)、甲状腺ホルモン(Ishii et al., i08)、グルココルチコイド(Shimizu et al., u08)、カンナビノイド(Kola et al., a05)、アディポネクチン(Kubota et al., a07)などのエネルギー不足シグナルは、視床下部のAMPK活性を増強し、摂食を増加させる。一方、高グルコース、レプチン、インスリン、およびレジスチンによって誘導されるエネルギー過剰シグナルは、視床下部AMPK活性を低下させることにより摂食行動を抑制する(Minokoshi et al., i04)。AMPKの活性化は、腫瘍抑制因子LKB-1複合体とカルシウム/カルモジュリン依存性プロテインキナーゼキナーゼβ(CaMKKβ)によるリン酸化を介して媒介される。LKB-1はユビキタスに発現しており、基礎的な構成的AMPK活性化を可能にするが、CaMKKβは主に神経細胞に発現していることが判明している(Spasic et al., 2009)。一方、アストロサイトは、その同化を促進する表現型と一致して、生理的条件下では最小限のAMPKしか発現していない(McCullough et al., 2005)。
神経変性疾患におけるエネルギー代謝
神経変性疾患は、中枢神経系におけるさまざまなニューロンサブセットの進行性喪失により、臨床症状や病態生理の点で異質である(Przedborski et al., 2003)。神経変性疾患の多くは加齢に関連しており、進行性の運動機能障害や認知機能障害を呈し、生活の質の低下、医療費の増大、そして最終的には死亡に至る。世界的に高齢化人口が増加するにつれて、神経変性疾患は社会的にも経済的にも大きな負担となっている。残念なことに、神経変性に対する有効な治療法を発見するための努力は実を結んでおらず、神経変性疾患の治療法は今のところ開発されていない。
何十年もの間、神経変性疾患の研究は、神経変性は主として神経細胞の欠陥によって引き起こされるという神経中心的な見解に支配されてきた。しかし、血管系やアストログリアも多くの神経変性疾患の発症や進行に関与している可能性を示す証拠が増えつつある(Maragakis and Rothstein, 2006; Zacchigna et al., 2008)。神経変性疾患の異質性は、共通の生化学的特徴を妨げるものではない。神経変性疾患における神経血管ユニットとその共通の生化学的特徴を統合的に捉えることは、今後の神経変性研究に光を当て、最終的に有効な治療法の発見につながるかもしれない。加齢と加齢に伴う神経変性疾患の両方において、ミトコンドリア機能障害が原因的役割を果たすことを示す証拠が蓄積されつつある(Bratic and Larsson, 2013; Chen and Chan, 2009)。実際、原因因子はミトコンドリアの機能不全を超えて、エネルギー代謝に関与している可能性が高い。
ADにおける代謝変化
ADは最も一般的な神経変性疾患であり、神経乳頭や脳血管におけるアミロイドβペストや、神経細胞における高リン酸化神経原線維変化(Girouard and Iadecola, 2006)によって病理学的に特徴づけられる。したがって、AD研究は「アミロイドカスケード仮説」と「タウともつれ仮説」によって支配されてきた(Korczyn, 2008; Mudher and Lovestone, 2002)。しかし、Aβとタウに焦点を当てた数十年にわたる研究は、ADの有効な治療法の特定には至っていない。アミロイドβ叢を減少させるとされる治療薬はすべて、臨床試験で有効性を示すことができなかった(Karran and Hardy, 2014; Karran et al., 2011)。アミロイド沈着が認知機能と強い相関がないという事実は、アミロイド仮説に反するものである。さらに、アミロイドカスケードとタウのリン酸化亢進は、多くの脳損傷によって誘導されることが明らかになっている。実験モデルでは、虚血性脳卒中がβアミロイド前駆体タンパク質の過剰発現を誘導し(Nihashi et al., i01;Shi et al., i00)、βセクレターゼの発現と活性を増加させ(Tesco et al., o07;Wen et al., n04a)、部位特異的なリン酸化亢進を誘導し(Wen et al., n04c)、AD様のタウオパシーを引き起こす(Wen et al., n04b)。一貫して、虚血性脳卒中後のヒト海馬では、Aβ1-40とAβ1-42の蓄積が見られた(Qi et al., 2007)。アミロイドβとタウの病態も、ヒトの外傷性脳損傷後に観察されている(Chen et al., n09;Johnson et al., n12b)。これらの証拠は、アミロイドやタウのカスケードがADの神経変性の原因ではないことを示唆している。したがって、アミロイドβとタウのカスケード以外の代替仮説を探る必要がある。
メタボリックシンドロームは、高齢者の認知機能障害に関与し、ADと関連していることがわかっている(Razay et al., y07;Yaffe et al., e04)。代謝低下は神経変性の強固なバイオマーカーとして浮上してきた(Johnson et al., n12a)。グルコース代謝の低下は、以前からAD脳に関与している(Iadecola, 2015)。脳のエネルギー代謝とグルコース利用における障害は、認知機能障害の初期段階を伴う、あるいはそれに先立つ、ごく初期の異常のひとつであることがわかっている(Hoyer, 2004; Nordberg et al., 2010; Sims et al., 1980)。脳内グルコース輸送と利用における低下は、ADの遺伝的リスクを持つ認知正常者やアルツハイマー病患者の初期状態で認められた(Iadecola, 2015; Nordberg et al., 2010)。AD脳におけるグルコース代謝の障害には、多くの基礎的メカニズムがあると考えられる。内皮およびアストロサイトに局在するGLUT1は、哺乳類の脳におけるグルコース輸送に主要な役割を果たしている。AD脳ではGLUT1の減少が認められている(Kalaria and Harik, 1989; Mooradian et al., 1997)。ごく最近の研究では、GLUT1の欠損がADマウスの脳の病態生理を悪化させ、AD病態と相乗的に作用して認知症の進行を促進する可能性が示された(Winkler et al., r15)。逆に、GLUT1を回復させると、ADマウスの脳におけるAβレベルが減少した(Winkler et al., 2015)。さらに、ケトジェネティック食や経鼻インスリンによるグルコース取り込み促進などの代替エネルギー基質が有益な効果を示している(Mamelak, 2012; Stafstrom and Rho, 2012; Yarchoan and Arnold, 2014)。
ADにおける代謝の変化には、血管のメカニズムも関与している可能性がある。脳血管系へのアミロイド沈着を特徴とする脳アミロイド血管症は、高齢者では一般的であり、AD患者ではさらに一般的である(Castellani et al.) 従って、ADにおいて脳血管の機能が著しく変化することは驚くべきことではない。皮肉なことに、脳血管障害の存在はAD診断の唯一の基準と考えられており、ADの発症と進行に対する血管因子の寄与については長い間議論されてきた(Iadecola, 2004; McKhann et al.) 剖検と長期観察研究の両方から、ADは病理学的に非常に不均一であり、その多くが梗塞を含む混合病態を示すという証拠が増えつつある(Schneider et al., 2009)。実際、ADと血管性痴呆の共存は混合型痴呆と呼ばれ、最も一般的な痴呆である(Langa et al., 2004)。脳では、神経細胞、アストロサイト、血管系が脳の恒常性を維持する神経血管ユニットを構成している。神経-血管結合の変化は脳血管障害のみならず、ADを含む神経変性疾患においても認められている。脳アミロイド血管障害では、血管反応性の障害、平滑筋細胞の減少、血管の完全性の破壊を特徴とする進行性の神経血管ユニット機能障害が引き起こされる(Zipfel et al., l09)。AD脳のアミロイド斑の主成分であるアミロイドβは、試験管内試験でも生体内試験でも血管作動性である(Crawford et al., 1998; Niwa et al., 2000)。機能的充血は、動物モデルでもアルツハイマー病患者でも障害されていることがわかっている(Iadecola, 2004; Petzold and Murthy, 2011)。したがって、神経-血管結合の障害は、他の細胞機構とともに、ADにおける神経変性の引き金となり、促進する可能性がある。
アミロイドβプラークや神経原線維変化以外にも、ADの脳には脂肪細胞内封入体が多く見られる(Foley, 2010)。ADのリスクは、コレステロール代謝の重要な制御因子であるアポリポ蛋白E(APOE)の異なるアイソフォームの遺伝に影響され、ε4対立遺伝子がADの最も強い遺伝的危険因子である(Bertram and Tanzi, 2008; Corder et al. 多くの脂質がAD発症に関与していることはよく知られている(Di Paolo and Kim, 2011)。研究では、脳内コレステロールレベルの変化が、APPプロセシングとAβ産生に影響することが示されている(Lim et al., m14)。また、脂質ラフトが神経毒性タンパク質を産生するプラットフォームとして機能し、AD発症に関与しているという証拠もある(Allen et al., n07;Ehehalt et al., t03)。
ミトコンドリアは、エネルギー産生、細胞増殖、アポトーシスなどの細胞機能において極めて重要な役割を果たしている(Birch-Machin, 2006; Wallace, 2005)。脳では、ミトコンドリアはシナプス伝達、脳機能、認知を制御している(Picard and McEwen, 2014)。ADにおける代謝機能障害が確認されていることから、ミトコンドリアの欠損がADの発症と進行に関与していても不思議ではないかもしれない。AD脳ではミトコンドリアの形態、生化学、遺伝学的異常が見つかっているが、ADにおけるミトコンドリアの作用についてはまだ議論の余地がある(Mancuso et al., 2008; Swerdlow et al.) ADに関連する酸化ストレスは、ミトコンドリアETC複合体の変化と関連している(Morais and De Strooper, 2010)。AβはETC複合体のアセンブリーや酵素活性を変化させることが示されている(Moran et al.) それゆえ、AD患者とトランスジェニックADマウスの両方において、Aβがミトコンドリアに入り込む可能性があることが示されている(Chen and Yan, 2006)。さらに、AD脳で検出された神経原線維のもつれは、ETC複合体の活性を阻害し、活性酸素産生を増加させ、ミトコンドリア膜電位を低下させることが示されている(Moran et al.) 逆に、AD患者由来のmtDNAを再増殖させた内因性mtDNAを枯渇させた細胞であるADトランスモコンドリア・サイブリッドは、Aβの過剰産生を示したが、AD患者では原因となるmtDNAの栄養化は発見されていない(Mancuso et al., 2009)。したがって、Aβ、神経原線維のもつれ、ミトコンドリアの欠損は、相乗的にADの病態生理を開始し、悪循環の中でADの進行を促進する可能性がある。
AMPKがマスターエネルギーセンサーおよび調節因子であることを考慮すると、AMPKシグナル伝達の機能的欠陥が、AD脳におけるエネルギー代謝の欠乏に関与している可能性がある。一貫して、AD患者およびADマウスモデルの脳では、AMPKリン酸化の増加が認められている(Ma et al.) AMPKが多くの部位でタウのリン酸化を制御しているという証拠がある(Thornton et al., n11)。正常な脳では、リン酸化AMPKは主にニューロンの核に局在している。一方、AD脳では、タウのリン酸化が亢進している神経細胞で活性化AMPKの細胞質集積が見つかっており、このことは、AMPKの細胞質移動と活性化が、タウのリン酸化亢進ともつれ形成に先行している可能性を示している(Vingtdeux et al., x11)。逆説的だが、AMPK活性化物質である5-アミノイミダゾール-4-カルボキサミド-1-d-リボフラノシド(AICAR)は、タウのリン酸化を抑制することが示されている。さらに、一次ニューロンにおいて、レプチンがAMPKの活性化を通じてタウのリン酸化を低下させることが示唆されている(Greco et al., 2009a;Greco et al., 2009b)。同様に、アミロイド形成におけるAMPKの逆説的作用も指摘されている。Aβ42オリゴマーはAMPKを活性化することが示されている(Mairet-Coello et al., o13)。一方、AMPKがアミロイド生成を抑制することも、研究によって証明されている(Salminen et al., n11)。AMPKの遺伝学的および薬理学的活性化は、試験管内試験と生体内試験の両方でAβ蓄積を減少させた(Vingtdeux et al., x10;Won et al., n10)。これらを総合すると、タウオパチーおよびアミロイド生成に対するAMPKの効果は、文脈依存的である可能性がある。アミロイドβは特定の条件下でAMPKを活性化し、タウタンパク質をさらに直接リン酸化する可能性がある一方で、AMPKの活性化は他の場合においてアミロイド形成とタウ凝集を抑制する可能性がある(Salminen et al.)
PDにおける代謝変化
PDは、黒質におけるドーパミン作動性ニューロンの進行性死滅を病理学的に特徴とする、2番目に多い神経変性疾患である。PDの典型的な症状には、動作の緩慢さ(ブラジキネジア)、筋肉のこわばり(硬直)、振戦、平衡障害などがある。病因論的には、PDは黒質におけるドーパミン作動性ニューロンの著しい喪失と、それに続く線条体におけるドーパミンの枯渇によるものである。現在までのところ、パーキンソン病患者には、特に初期の段階では対症療法しかない。PDを治癒させたり、進行を止めたりする治療法は発見されていない。
グルコース代謝の調節異常は散発性パーキンソン病患者の初期症状として認められている。FDG-PET研究では、PD脳におけるグルコース代謝の有意な低下が示されている(Borghammer, 2012; Borghammer et al., 2010; Borghammer et al., 2009; Dunn et al., 2014)。興味深いことに、グルコース代謝低下は、認知症の有無にかかわらずパーキンソン病患者の大脳皮質で広く認められた(Edison et al., n13)。
黒質におけるドーパミン作動性ニューロンの変性の根底にある病態生理学はまだ明らかではない。PDの脳では、ミトコンドリア機能障害と酸化ストレスが一貫して観察されている。PDとミトコンドリア呼吸鎖機能障害、特にミトコンドリアETC複合体Iの欠損との関連を支持する薬理学的および遺伝学的証拠が増えつつある(Franco-Iborra et al.) ミトコンドリア複合体I阻害剤である1-メチル-4-フェニル-1,2,3,4-テトラヒドロピリジン(MPTP)への事故暴露は、急性かつ不可逆的なPD症候群を引き起こすことが知られている(Calne and Langston, 1983; Langston and Ballard, 1983)。その後、散発性パーキンソン病患者の脳でもミトコンドリア複合体I阻害が確認された(Schapira et al., a90)。さらに、農薬ロテノンによるミトコンドリアETC複合体Iの慢性的な全身的阻害が、散発性PDと関連していることが判明している(Betarbet et al., t00)。興味深いことに、複合体Iの欠損は死後の黒質だけでなく大脳皮質でも見つかっており(Schapira et al., a90)、これはパーキンソン病患者で観察される皮質のグルコース代謝低下と一致している。実際、PDの病態には、SNc以外のいくつかの脳領域と、ドーパミン以外の多くの神経伝達物質が関与していることがわかっている(Lang and Obeso, 2004a,b)。現在では、MPTPやロテノンを用いたPDモデルがPD研究に広く用いられている(Beal, 2010)。
PD症例の大半は散発性だが、家族性PDで確認されたミトコンドリア機能障害には、多くの遺伝子の変異が直接的または間接的に関係していることが判明している。常染色体劣性パーキンソン病では、パーキンとPINK1の変異が同定されている(Pickrell and Youle, 2015)。PINK1とパーキンの機能は、ミトコンドリアの動態を制御することによる健全なミトコンドリアの維持と、機能不全に陥ったミトコンドリアを排除するオートファジーであることが指摘されている(Moran et al.) 最近では、抗酸化物質であり転写調節因子でもあるDJ-1タンパク質が、PINKsやパーキンと協力してミトコンドリア機能を制御していることが示唆されている(Irrcher et al., 2010)。
PDの病態生理学におけるエネルギー代謝の欠乏は、AMPKシグナル伝達の潜在的な機能障害と関連している可能性がある。AMPKシグナル伝達の活性化は、MPTP、MPP+、6-OHDAを用いたPDモデルにおいて、試験管内試験と生体内試験の両方で証明されている(Choi et al., i10;Kim et al., m13)。さらに、化合物CによるAMPKの阻害は、MPP+による細胞死を引き起こした(Choi et al., i10)。一方、AMPKの薬理学的および遺伝学的活性化は、PDモデルにおいて保護作用をもたらすことが示されている(Choi et al., i10;Ng et al., g12)。
HDとFRDAにおける代謝変化
ADやPD以外にも、ミトコンドリアや主要なエネルギー代謝シグナルの機能不全は、他の神経変性疾患でも観察されている。HDは、変異型Huntingin遺伝子の結果として知られており、線条体の中棘GABAニューロンの優先的な喪失が特徴である(Damiano et al., 2010)。変異型ハンチンチンがミトコンドリアに直接結合し、ミトコンドリア機能を変化させることを示す証拠が増えている(Choo et al., 2004;Orr et al., 2008)。HD患者では、尾状核のミトコンドリアETC複合体II、III、IVの減少が見られた(Damiano et al., o10)。さらに、HD患者およびHDモデルマウスの脳では、AMPKの亢進が認められている(Ju et al., u11)。一貫して、AMPK活性化物質であるAICARは、神経細胞死を誘発し、HDマウスの寿命を縮めた(Ju et al., u11)。
フリードライヒ失調症(FRDA)は、フラタキシン欠損による最も一般的な早期発症の遺伝性失調症である。フラタキシン遺伝子(FXN)のGAAトリヌクレオチド拡張により、フラタキシンの転写が欠損し、フラタキシンの発現量が減少する(Gonzalez-Cabo and Palau, 2013; Kaplan, 1999)。フラタキシンは鉄代謝に関連し、ヘム生合成と鉄-硫黄クラスターの形成に重要な役割を果たすことが知られている(Gonzalez-Cabo et al., o05;Karthikeyan et al., n03)。FRDA患者では、ミトコンドリアETC複合体I、II、IIIおよびアコニターゼの鉄-硫黄クラスター含有サブユニットの欠損が同定されている。さらに、FRDA患者では、ミトコンドリアの酸化的リン酸化の重大な欠損とミトコンドリア内鉄の上昇が観察されている。一貫して、ミトコンドリアATP産生の最大速度の低下がFRDA患者の筋肉で認められた(Lodi et al., 1999)。従って、ミトコンドリア呼吸は、FRDAの新規治療法を発見するための主な標的となっている。
エネルギー代謝と癌
ワールブルグ効果を再検討する
100年前の1924年、オットー・ワールブルクは、腫瘍組織が酸素の豊富な環境で活発にグルコースを代謝し、過剰な乳酸を産生する一方で、呼吸数は比較的に低いという観察結果から、癌は解糖の増加と呼吸障害によって引き起こされる可能性があると提唱した(Koppenol et al.) このユニークなカナー代謝は、後にワールブルグ効果として知られるようになり、その後多くの癌研究者によって実証された。ワールブルグ効果は、ATPを大量に必要とする高度に増殖したがん細胞が、18倍も効率の低いATP産生経路を使用するというパラドックス現象を提示した。ワールブルグ効果の根底にあるメカニズムや意義がわからないまま、癌の起源に関するワールブルグの仮説は、癌研究において限定的な影響しか与えず、前千年紀の終わり頃になっても、代謝シグネチャーは癌の特徴として認識されていなかった(Hanahan and Weinberg, 2000)。
新ミレニアムの初めに、ワールブルグ効果はがん研究者の注目を再び集めた。生物学の発展、エネルギー代謝と遺伝学の知識の蓄積により、がん細胞の代謝スイッチの根底にある細胞や分子のメカニズムがさらに詳しく解明されるようになった。現在では、がん細胞における好気的解糖によるATP産生の効率低下は、グルコーストランスポーターのアップレギュレーションによって補われることがわかっている。加えて、増殖性の高いがん細胞では、ミトコンドリアの酸化的リン酸化が欠損している必要はない(DeBerardinis et al., 2008a; Moreno-Sanchez et al., 2007)。エネルギー代謝経路が生体エネルギーと生合成の両方の機能を果たすことを考えると、好気性解糖の増加機能は、主にがん細胞の異常な増殖と成長速度を支えているのかもしれない(DeBerardinis, 2008; DeBerardinis et al.)
抗増殖シグナルに対する不感受性と増殖シグナルの自己充足は、がんの特徴の2つであり(Hanahan and Weinberg, 2000)、がん細胞は増殖速度が速いだけでなく、自己複製に必要なタンパク質、脂質、核酸を大量に必要とする。この課題に対処するため、がん細胞は十分な栄養を取り込み、適切な代謝を確立して高分子を生産し、がんバイオマスを構築する能力を獲得しなければならない。脂質とヌクレオチドの生合成は、炭素源としてグルコースを使用し、TCAサイクル中間体を消費し、還元力としてNADPHを必要とする(Deberardinis et al.)がん細胞は、TCAサイクルやミトコンドリアの酸化的リン酸化からグルコース代謝を転換し、ピルビン酸-クエン酸シャトルを介してアセチルCoAを生成し、脂肪酸合成やタンパク質のアセチル化を行う(Kamphorst et al.) さらにがん細胞は、解糖からペントースリン酸経路に炭素を移動させ、ヌクレオチド生合成のためのリボース-5-リン酸を生成する(Deberardinis et al.) このように、ミトコンドリアの酸化的リン酸化による効率的なエネルギー生産と、好気的解糖による効率的なエネルギー生産とのトレードオフは、最終的にガンのバイオマス増加に寄与する(Deberardinis et al., 2008b; Jones and Thompson, 2009)。現在、エネルギー代謝の転換が癌に広く見られることが証明され、エネルギー代謝のリプログラミングが癌の新たな特徴として認識されている(Hanahan and Weinberg, 2011)。
がん細胞の発がんと好気性解糖作用
ワールブルグが癌発生説を提唱して間もなく、癌におけるミトコンドリアの欠陥に関する彼の主張は、他の研究者によって否定された(Chance and Castor, 1952; Weinhouse, 1956)。実際、ほとんどのがんにおいて、ミトコンドリアの酸化的リン酸化能は欠損していない(DeBerardinis et al., s08a; Moreno-Sanchez et al., z07; Ward and Thompson, 2012a)。一貫して、私たちは、膠芽腫細胞において、酸化的リン酸化の増加をあまり伴わない解糖系フラックスの劇的な増加を観察した(Poteet et al.) したがって、がん細胞は依然としてミトコンドリアの酸化的リン酸化によってかなりの割合のATPを産生しており、がんにおけるワールブルグ効果はミトコンドリアの損傷によるものではない。遺伝学と分子生物学の技術的進歩により、がん細胞の代謝スイッチの根底にあるメカニズムを調整し、ワールブルグ効果と腫瘍形成の関連性を再評価することが可能になった。
過去30年以上にわたる分子生物学の進歩により、がん抑制遺伝子とがん遺伝子が、がん増殖を維持するだけでなく、がん細胞の成長と増殖に伴う生合成の課題に対応するために、がんのエネルギー代謝を再プログラムする多くの細胞シグナル経路をコードしていることが理解されるようになった(Hanahan and Weinberg, 2011; Vander Heiden et al.) PI3K/Akt/mTORC1やHIFを含む多くのシグナル伝達経路が、細胞生合成のためのがん代謝リプログラミングに関与している(Jones and Thompson, 2009; Ward and Thompson, 2012a,b)。さらに、癌遺伝子と癌抑制因子は、これらの代謝シグナルネットワークの重要な構成要素であることが判明している。PI3K/Aktの活性化は、重要な癌抑制因子であるPTENによって厳密に制御されている。PTENの欠損やPI3K変異は、生合成のリプログラミングを引き起こすヒトのがんで最も一般的な遺伝子型である(Jones and Thompson, 2009)。H-Ras、c-Myc、srcなど多くの癌遺伝子は、癌細胞において同化代謝を促進することが知られている(Dang, 2011; Jones and Thompson, 2009)。これらを総合すると、癌遺伝子の変異、癌抑制遺伝子の欠損、多くの異常シグナル伝達が、癌が発生し増殖するための代謝再プログラミングに関与している。さらに、細胞内シグナル伝達と代謝の関係は双方向的である可能性がある(Metallo and Vander Heiden, 2010; Ward and Thompson, 2012b)。生合成活性に対する細胞内代謝産物のフィードバック制御は、翻訳後修飾と遺伝子発現の両方のレベルで見出されている(Metallo and Vander Heiden, 2010; Ward and Thompson, 2012b)。
MB、1世紀前の薬剤の新たな応用
拡大し続ける疾患領域に対して幅広い医療応用が可能な1世紀前の薬物 1800年代後半、繊維産業の繁栄により染料需要が劇的に増加し、合成染料研究が急速に発展した(Oz et al.) 1876年、アニリンベースの染料であるメチレンブルー(MB)が、Heinrich Caroによって綿の染色用に合成された(Schirmer et al.) MBは繊維産業の基準を満たすことはできなかったが、その生物学的応用はすぐに発見された。100年以上もの間、MB構造に由来する医薬品が拡大し続け、その広範な医学的応用が、拡大し続ける様々な疾患の治療に役立てられてきたことを考えると、MBはおそらく、最も重要な医薬リード構造であろう(Ohlow and Moosmann, 2011)。
MBは、フェノチアジンおよびその誘導体の最初のリード化合物である。1883年、ドイツの化学者Heinrich August Bernthsenがフェノチアジンを合成した。1891年、ポール・グットマン(Paul Guttmann)とポール・エーリック(Paul Ehrlich)によってMBの原虫染色とマラリアに対する効果が開発され、近代化学療法の基礎となった(Kaufmann, 2008; Parascandola, 1981)。1890年代には、精神科患者のコンプライアンスを監視するためにMBが投与され、その結果、抗精神病作用が発見され、後に1951年に最初の合成抗精神病薬であるクロルプロマジンが発見された(Ohlow and Moosmann, 2011; Schirmer et al.) 20世紀初頭には、メチレンブルー誘導体であるフェノチアジンの抗真菌、殺虫、駆虫活性が発見された(Ohlow and Moosmann, 2011)。1930年代以降、MBはメトヘモグロビン血症(Wendel, 1939)やシアン中毒(Alston, 2014)の第一選択薬として静脈内投与されてきた。1990年代以降、MBはイホスファミド誘発脳症の治療に導入された(Kupfer et al., r94;Pelgrims et al., s00;Zulian et al., n95)。さらに、MBはがん診断のためのトレーサーや、がん治療のための光増感剤としても使用されている(Chen et al., 2007)。
1940年代から、フェノチアジンはポリエチレンオイルの酸化変化を防ぐ酸化防止剤として使用されてきた(Murphy et al., 1950)。1990年代から、フェノチアジンおよびその誘導体の神経保護作用が研究されてきた(Yu et al., u92a;Yu et al., u92b)。フェノチアジン誘導体の保護作用は、虚血性脳卒中(Yu et al., u92a)やPD(Mocko et al., o10)のモデルで実証されている。一方、MBの保護作用が発見されたのは最近のことである。
MBは半世紀以上前から電子伝達物質として知られており、単離されたミトコンドリアにおけるシトクロムcの還元を促進する作用が知られている(Weinstein et al., n64)。MBが、他のフェノチアジン誘導体とは異なるメカニズムで神経保護作用を持つ可能性を示す証拠が増えつつある(Poteet et al.) 私たちの最近の構造活性相関研究により、MBの細胞保護作用は、ミトコンドリアの代替電子キャリアとしての作用と、ミトコンドリアにおける再生成可能な抗酸化剤としての作用において、他のフェノチアジン誘導体とは異なることが示された(Poteet et al.)
5.2. 代替ミトコンドリア電子伝達キャリアとしてのMB
MBは、濃紺の酸化状態(MB)と無色の還元状態(ロイコメチレンブルー)の間で平衡状態に存在する非常にユニークな酸化還元特性を持ち、異なる条件下でプロオキシダントとアンチオキシダントの両方を作る。MBのユニークな酸化還元特性は、イホスファミド誘発脳症のようないくつかの疾患に対する治療効果に直接寄与している可能性がある。イホスファミド誘発脳症の正確な病態生理学的メカニズムはわかっていないが、イホスファミドの代謝産物であるクロロエチルアミンによるミトコンドリア呼吸鎖の阻害が原因であるという証拠がある(Kupfer et al., r96;Pelgrims et al., s00)。メトヘモグロビン血症の治療には、赤血球のメトヘモグロビン還元酵素によってMBをロイコメチレンブルーに還元し、メトヘモグロビンをヘモグロビンに還元する(Clifton and Leikin, 2003)。
MBの電子キャリアとしての能力は、長い間認識されてきた。MBはNADH、NADPH、FADH2から直接電子を受け取る。(Atamna et al., 2008; Buchholz et al., 2008; Dixon, 1971; May et al., 2004; Wen et al., 2011b)。MBは、嫌気的・好気的条件下で、ある種の酵素からシトクロムcへの電子の流れを仲介することができる(McCord and Fridovich, 1970)。したがって、MBは損傷を受けたミトコンドリアの呼吸複合体に代わる電子伝達キャリアとして働くことができるかもしれない。この考え方は、MBが網膜と線条体におけるロテノン誘発神経障害を抑制するという事実からも支持される(Rojas et al., s09a;Rojas et al., s09b;Zhang et al., g06)。一貫して、単離されたミトコンドリアを用いると、MBはミトコンドリアETC複合体I、I-III活性を増強するが、複合体II-III活性は増強しないことが証明されている(Atamna et al., 2008; Poteet et al., 2012; Wen et al., 2011a)。さらに重要なことは、MBによって誘発されるミトコンドリア複合体IとI-III活性の上昇は、複合体IとIIIの阻害に対して感受性がないことである(Atamna et al., a08;Poteet et al., t12;Wen et al., n11b)。さらに、還元型MB(MBH2)は、ミトコンドリア内の酸素存在下で、シトクロムcに電子を供給できることが判明している(Atamna et al., 2008; Wen et al., 2011b)。これらを総合すると、MBがミトコンドリアの代替電子伝達物質として機能する可能性があるとの見方が強まっている。その酸化還元サイクル(MB-MBH2-MB)において、複合体IとIIIの阻害にもかかわらず、NADHからの電子は代替経路でシトクロムcに送られる(Atamna et al., a08;Bruchey and Gonzalez-Lima 2008;Wen et al., n11b)(Poteet et al., t12;Rojas et al., s12)(図2)。
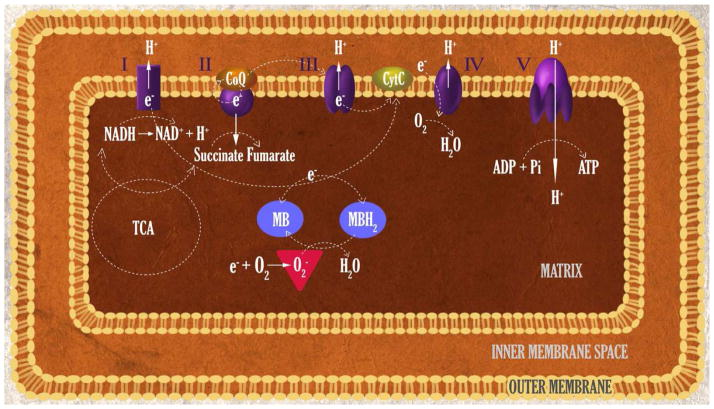
図2
代替ミトコンドリア電子伝達キャリアおよび再生可能な抗酸化剤としてのメチレンブルー(MB)の機能。MBは複合体Iの存在下でNADHから電子を受け取り、酸化還元サイクル(MB- MBH2-MB)の際に、複合体IとIIIが阻害されているにもかかわらず、別の経路でチトクロムcに電子が供給される。このような明確な酸化還元特性により、MBはミトコンドリアにおいて、従来のフリーラジカル消去物質とは異なる再生可能な抗酸化物質として機能する。
ミトコンドリアのETC複合体IVであるシトクロムcオキシダーゼは、ミトコンドリア呼吸の終末酵素であり、ミトコンドリア内膜を横切ってプロトンを送り出し、酸素を水に還元する(Wallace, 2005)。MBは試験管内試験でも生体内試験でもシトクロムcオキシダーゼ活性を増強することが証明されている(Atamna et al., 2008; Callaway et al., 2004; Gonzalez-Lima and Bruchey, 2004; Poteet et al.) MBとシトクロムcオキシダーゼとの直接的な相互作用が、MBの記憶増強作用の根底にある可能性が提唱された(Rojas et al.) 一方、MBによるシトクロムcオキシダーゼ活性の増加は、少なくとも部分的には、電子輸送を促進し、シトクロムcの還元を増加させる代替電子キャリアとしてのMBの作用による二次的なものかもしれない(Atamna et al., a08;Poteet et al., t12)。私たちの最近の研究では、フェノチアジン誘導体である2-クロロフェノチアジンが、HT-22細胞において、酸素消費速度を増加させることなく、グルタミン酸誘発酸化ストレスに対して同様の神経保護作用を有することが示された。興味深いことに、2-クロロフェノチアジンはシトクロムcオキシダーゼの発現と活性には影響を及ぼさないことから、MBの代替ミトコンドリア電子輸送としての作用が、シトクロムcオキシダーゼに対する作用に直接寄与している可能性が示唆された(Poteet et al.)
ミトコンドリアの電子輸送に対するMBの作用は、ミトコンドリアの酸化的リン酸化に対するMBの作用と一致している。酸素消費に対するMBの作用は、1930年代にさかのぼり、好気的解糖を持つ正常組織や腫瘍の酸素消費を増加させることが明らかにされている(Barron, 1930)。Rihaらは、光ファイバー酸素センサーを用いて、ロイコメチレンブルーが試験管内試験のラット脳ホモジネート中の酸素濃度を低下させることを発見した(Riha et al.) また、生体内試験では、MB処理24時間後のラットの脳ホモジネートにおいて、酸素濃度が有意に低下することが判明した(Riha et al., 2005)。クラーク酸素電極を用いて、Atamnaらは、MBが線維芽細胞の酸素消費量を増加させることを発見した(Atamna et al., 2008)。Seahorse細胞外フラックスアナライザーとルテニウム蛍光寿命イメージング顕微鏡を用いて、MBがニューロン細胞とアストロサイトの両方で酸素消費速度を増加させ、細胞外酸性化速度を減少させることを発見した(Poteet et al.) マルチメトリック・ニューロイメージングと血管血中酸素濃度測定を用いると、さらに、MBの急性投与によってラットの脳内酸素代謝率(CMRO2)が上昇することが観察された(Lin et al.) ミトコンドリアの電子輸送に対するMBの刺激作用は、グルコース代謝に対するMBの作用と一致している。線維芽細胞において、MBは2-デオキシグルコースの取り込みを刺激することが示されている(Louters et al., s06;Roelofs et al., s06)。MRIとPETを用いて、私たちは、MBの急性投与がラットのグルコース取り込みを有意に促進し、局所CBFを増加させることを証明した(Lin et al.)
ミトコンドリアの酸化的リン酸化は、脳のエネルギー供給と消費との間に緊密な連関を持つ、脳における支配的な代謝経路である。従って、MBは脳代謝および血行動態改善剤として、生理学的条件下で脳機能を改善する可能性がある。神経系に対するMBの効果は、1880年代後半に研究された。ポール・エーリックが、MBが生きた神経組織に選択的に親和性を持つことを発見し、エーリックの言葉を借りれば神経向性であると期待された(Parascandola, 1981)。実際、ラットにおける薬物動態学的研究により、MBはBBBを通過し、循環中の濃度より10倍高い濃度で脳に到達することが示されている(Peter et al., 2000)。1970年代以降、MBはげっ歯類の様々な実験的記憶課題を改善することが分かっている(総説(Rojas et al.) 加えて、加齢と加齢に関連した神経変性疾患の両方において、脳代謝機能障害が原因となっている可能性があることから、MBは神経変性疾患の治療薬として、将来的に理想的な候補となる可能性が示唆された。
MBは、神経疾患治療の臨床試験で失敗した従来の抗酸化剤とは異なる。第一に、MBの作用は、ミトコンドリアの酸化的リン酸化を維持するだけでなく、スーパーオキシドや活性酸素種の産生を抑制する(Atamna et al., a08;Poteet et al., t12;Wen et al., n11b)。第二に、独特の酸化還元特性により、MBはミトコンドリア内で再生可能な抗酸化物質となり、従来のフリーラジカル消去物質とは一線を画す(Poteet et al.) 加えて、ミトコンドリアの代替電子伝達作用により、MBは脳代謝を促進するだけでなく、ワールブルグ効果を切り替え、がんの増殖を抑制することもできるかもしれない。
MBによる神経変性疾患の治療
MBとA. 治療薬の開発は何十年にもわたるが、AD治療薬として臨床応用が承認されているのは、メマンチンと4種類のコリンエステラーゼ阻害薬のみである。しかし、承認されたこれらの薬剤の効果は極めて限定的であり、治療上有用かどうかについては議論がある(Schneider et al.) MBは、その合成から1世紀以上が経過し、最近になって科学的関心が高まっている。2008年のアルツハイマー病協会の年次総会で、ADの可能性が高い患者332人の認知機能障害に対するMB治療を試験した臨床第II相試験から有望な結果が出た。1年間にわたり、MB治療は認知機能を有意に改善し、プラセボを投与された患者と比較して認知機能低下率が81%減少した(Gura, 2008; Oz et al.) 薬剤の製剤、潜在的なタウ溶解メカニズム、MBが尿を青くすることによる盲検化の問題、臨床試験から得られた査読論文の欠如などに関する懸念が提起されている(Gravitz, 2011; Oz et al.) それにもかかわらず、MBの臨床試験は、実験モデルと臨床研究の両方において、ADに対するMBの効果を探求する世界的な関心を引き起こした。MBは現在、ADと前頭側頭型痴呆を対象とした第III相臨床試験が世界的に進行中である。
ADにおけるMB治療の第II相臨床試験は、MBの前臨床動物モデルでの公表試験を行うことなく、試験管内でMBのタウ溶解特性が早期に確認されたことに基づくものであった。MBは、臨床的には達成できないかもしれない高濃度で、リピートドメインを介してタウ-タウ結合相互作用をブロックする(Wischik et al., k96)。同様に、MBは50μMの濃度で、トランスジェニックADマウス由来の器官型スライスにおいて、タウのリン酸化を減少させることが証明されている(Jinwal et al., l09)。後期には、MBの慢性投与が生体内試験でアミロイド沈着と神経原線維のもつれを減少させることが示されている(Hochgrafe et al., e15;Hosokawa et al., a12;Melis et al., s15)。これと同様に、MB含有飼料を8カ月間与えると、Aβプラークが有意に減少し、5 X FADマウスの水迷路と橋渡し歩行の成績が改善することがわかった(図3)。
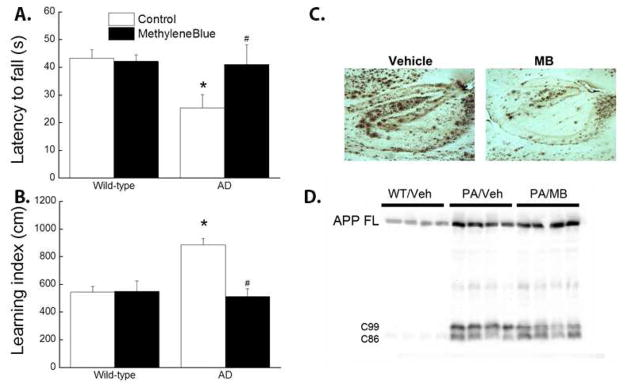
図3
メチレンブルー給餌により、5 X FADマウスの水迷路および橋上歩行の成績が改善した。野生型および5 X FAD雌性マウスを8週齢でMBまたはビヒクルを含む飼料に切り替え、8カ月間継続摂取させた。10カ月齢になったマウスを、前述(Shetty et al.) MB群では対照群に比べ、橋渡し歩行試験における平均転倒潜時が有意に短縮した(A)。水迷路試験では、MB群では学習指数が有意に改善した(B)。すべてのマウスを犠牲にし、脳のAβプラークに対するMBの効果を測定した。MB摂取によりAβプラークが減少することが、6E10免疫組織化学(C)およびウェスタンブロット分析(D)により証明された。# p<0.05 vs 対照AD。* p<0.05対対照野生型。
ADの認知機能低下に対するMBの有益な効果は、タウオパチーに対する作用とは無関係かもしれないという証拠が増えつつある。実際、MBは正常なげっ歯類において、神経代謝メカニズムを通じて記憶機能を高める可能性がある(Rojas et al., s12)。MBの抗タウ凝集作用には、生体内では到達できないかもしれない高濃度が必要である。ヒト変異型タウを発現するゼブラフィッシュでは、MBはタウのリン酸化と凝集を抑制できなかった(van Bebber et al.) MBの慢性投与は、ADマウスモデル(3 x Tg-AD)において、タウ病理に影響を与えることなく、脳のAβレベルを低下させ、学習と記憶を改善した(Medina et al., 2011)。これらの研究は、タウ凝集やAβプラークに対する阻害作用以外にも、MBの有益な作用の根底にある新たなメカニズムを論じている。興味深いことに、ADマウスにMBを慢性投与すると、タウオパチーやアミロイド斑が減少するが、これにはオートファジーの亢進が伴うことが判明した(Hochgrafe et al.) オートファジー活性の上昇は、タウオパシーを含む神経細胞内のタンパク質凝集を防ぐことが指摘されている(Lee et al., e13)。実際、MBは試験管内試験でも生体内試験でも、mTORシグナル伝達の阻害を通じてオートファジーを誘導し、タウオパチーを抑制することが実証されている(Congdon et al.) AMPKは、細胞エネルギー恒常性のマスターレギュレーターであり、オートファジーにおいて重要な役割を果たしている(Hardie, 2011)。MBはミトコンドリア呼吸を増加させ、AMPKシグナルを増強し、オートファジーをさらに活性化する可能性がある(Xie et al., e13)。これらの研究を総合すると、MBが代替電子伝達物質としてAD治療に有効である可能性が示された。
MBとPD 脳内ドーパミンを回復させるドパミン前駆体であるレボドパは、依然としてPDに対する唯一で最も有効な治療法である。しかし、レボドパは疾患の進行に影響を与えず、治療開始後5〜10年以内にほとんどの患者でジスキネジアが治療を複雑にする(Lang and Obeso, 2004a)。PDの進行性経過を変える治療法は今のところ開発されていない。
PDの病因にミトコンドリアETC複合体の機能障害と酸化ストレスが関与していることを示す証拠は数多くある(Abou-Sleiman et al., n06;Sayre et al., e08)。上述したように、ロテノンやMPTPなどのミトコンドリア複合体I阻害剤が散発性PDの病因に関与していることが指摘されている。代替ミトコンドリア電子キャリアとしてのMBのユニークな作用は、ETC複合体IとIIIをバイパスし、ロテノンとMPTPの毒性からMBを保護する。単離されたミトコンドリアでは、MBはロテノンとアンチマイシンによるミトコンドリア複合体IとIIIの阻害にもかかわらず、呼吸を維持する(Poteet et al., 2012; Wen et al., 2011b)。細胞培養では、MBは神経細胞HT-22細胞と初代ラット網膜神経節細胞をロテノン障害から保護する(Daudt et al., 2012; Poteet et al., 2012; Wen et al., 2011b)。げっ歯類では、MBは網膜と線条体において、それぞれロテノンの硝子体内投与または脳内投与によって誘発される神経変性を予防することが示されている(Rojas et al., s09a; Rojas et al., s09b; Zhang et al., g06)。MPTPとロテノンを用いたPDの実験動物モデルは、新規治療法の開発にとって貴重なツールである。MPTPやロテノンの全身投与は、線虫(Braungart et al., t04)、マウス、ラット、霊長類(Beal 2001)においてドーパミン作動性神経変性を引き起こす。MBは、PDの線虫モデルにおいて保護的であることが示されている(Mocko et al., o10)。重要なことに、私たちの研究では、ラットPDモデルにおいて、MBがロテノンによる黒質ドーパミン作動性ニューロンの減少を抑制し、運動機能を改善することが示された(Wen et al.) 一方、ドパミン作動薬はPDの運動機能障害を改善するのみである。従って、MBはレボドパに比べ、症状を改善するだけでなく、ドパミン作動性ニューロンの変性を予防し、病気の進行を遅らせる優れた治療法かもしれない。
MBとHD HDは、典型的な散発的、急速、不随意的な四肢運動、四肢のこわばり、精神障害、認知機能障害などを伴う壊滅的な神経変性疾患である(Kumar et al.) HDにおける現在の治療法は、主に症状をコントロールすることで患者の生活の質を改善することを目的としており、疾患修飾的な治療法はない。変異型ハンチンチンはミトコンドリア呼吸を障害し、ATP産生を低下させるため、酸化ストレス、興奮毒性、アポトーシスを促進することが指摘されている(Kumar et al.) その結果、ミトコンドリア呼吸はHDの治療標的となりうる。ミトコンドリアETC複合体の構成成分であるコエンザイムQ10は、HDマウスモデルにおいて、ミトコンドリア活性を増強し、運動障害の発現を遅延させ、体重減少を減少させ、脳萎縮を予防し、生存期間を延長することが示されている(Ferrante et al., e02)。現在、コエンザイムQ10はHDを対象とした第II相臨床試験中である(Kumar et al., 2015)。
ミトコンドリア呼吸を促進する代替ミトコンドリア電子キャリアとしてのMBのユニークな機能は、HDの治療効果を高める可能性がある。MB投与は、ショウジョウバエとR6/2マウスのHDモデルにおいて、行動表現型を減少させ、病気の進行を遅らせることが判明している(Sontag et al.) ミスフォールドしたハンチンチンの凝集は、HD発症の主な原因と考えられてきた(Kim and Kim, 2014)。そして、HDに対するMBの保護作用は、ハンチンチン変異体の凝集に対するMBの作用の二次的なものであると提唱された(Sontag et al., g12)。MBは確かに、ゼブラフィッシュにおいて10μMと100μMの用量で、ポリQ拡張ハンチンチンの凝集と沈着を阻止した。しかし、これらの濃度ではハンチンチンの凝集をほぼ完全に抑制しても、MBの保護効果は観察されなかった(van Bebber et al.) 同様に、MBは一次ニューロン培養において、1,10,100μMの濃度で急性処理すると変異型ハンチンチンの凝集をブロックした(Sontag et al.) 一方、低用量のMB(100 nM)投与では、変異型ハンチンチンのオリゴマー形成を減少させるのに10日かかった。興味深いことに、100 nMのMBは、変異型ハンチンチンを発現している一次ニューロンの生存率を3日間で25%増加させることがわかった(van Bebber et al.) 従って、MBの抗凝集作用は、おそらく代替ミトコンドリア電子伝達体としての機能による保護作用の二次的なものであろう。
6.4 MBとFRAD
FRADは、後根神経節、脊髄の皮質脊髄路と脊髄小脳路、歯状核の感覚ニューロンの変性による進行性の運動失調と構音障害を特徴とする衰弱性の進行性神経変性疾患である。FRADはまた、筋力低下、糖尿病、心筋症、最終的な心不全などの非神経学的特徴も伴う。現在の治療法は、心不全、不整脈、糖尿病に対する標準的な治療に限られている(Pandolfo, 2009)。この10年間、コエンザイムQ10とその類似体であるイデベノンがFRADの治療薬として広く研究されてきた。これまでのところ、FRADに対するコエンザイムQ10とイデベノンの有効性は、まだ解明されていない(Parkinson et al., n13)。補酵素Q10はミトコンドリアETCの必須補酵素であり、その欠乏はFRADを含むいくつかの神経変性疾患に関与している(Molyneux et al., x08)。補酵素Q10は、複合体IまたはIIから複合体IIIに電子を移動させる。さらに、還元型コエンザイムQ10は抗酸化物質でもある(Sohal, 2004)。内因性のミトコンドリアETC成分として、ミトコンドリア呼吸に対するコエンザイムQ10の効果は、複合体IIとIIIの活性によって大きく制限される。実際、コエンザイムQ10は、単離されたミトコンドリアと神経芽細胞腫細胞における基礎酸素消費率には影響を及ぼさなかった(Fink et al.)
MBはコエンザイムQ10とは異なり、ミトコンドリアETC複合体IからIIIの阻害をバイパスし、ミトコンドリア呼吸を増加させるというユニークな作用を持つ。MBは、FRAD線維芽細胞培養において、L-ブチオニン(S,R)-スルホキシミン(BSO)誘発酸化ストレスに対して保護的であることが示されている(Richardson et al., n13)。ごく最近の研究では、MBがフラタキシンを欠失させたショウジョウバエのFRDAモデルにおいて心臓の欠損を救うことが示された。MB誘導体のさらなる分析から、イデベノンではなく、電子キャリア特性を持つ化合物のみが心臓機能不全を予防できることが示された(Tricoire et al., e14)。したがって、代替的なミトコンドリアの電子伝達機構は、MBをFRAD治療のためのより良い治療戦略とする可能性がある。
がん治療のためのMB
がん治療のための代謝シグナルと酵素の標的化
ワールブルグ効果への新たな関心から、代謝は抗癌剤探索の新たなホットターゲットとなっている(Vander Heiden et al., 2009)。がん遺伝子とがん抑制遺伝子は、がん細胞の代謝再プログラミングに関与する多くの細胞シグナル経路を制御している。同定された代謝シグナル伝達経路上の抗がん剤治療標的が探索されている。PI3K/Akt/mTORシグナル伝達は、癌細胞の生存、増殖、代謝に関与する成長因子受容体チロシンキナーゼの下流にある重要な経路として、長い間認識されてきた(Cantley, 2002; Shaw and Cantley, 2006)。それに応じて、PI3K/Akt/mTORシグナル伝達経路の阻害剤は、がん治療薬として広く利用されてきた(Engelman, 2009)。がん細胞がシグナル伝達の冗長性やクロストークを利用して、同化の課題に取り組むことが懸念されている(Logue and Morrison, 2012)。実際、がん代謝に関する理解は進んでいるものの、代謝シグナル制御に関する知識は不完全である。がんにおける代謝ネットワークの高い可塑性を明らかにするためには、さらなる研究が必要である。それにもかかわらず、シグナル伝達ネットワークは複雑であるため、がん治療において単一のシグナル成分を標的とすることには反対である。
がん細胞の特徴的な異化代謝と代謝依存性は、がん治療のために代謝酵素を標的とすることへの関心を高めている(Vander Heiden, 2011)。主要な代謝酵素に対する多くの阻害剤が、潜在的な抗がん治療薬として開発されている。標的となる酵素は、グルコース取り込み、ペントース経路、解糖、TCAサイクル、脂質合成の広い範囲に分布しており、GLUT、ヘキソキナーゼ、ホスホフルクトースキナーゼ2、ピルビン酸キナーゼMなどが含まれる、ピルビン酸キナーゼM2(PKM2)、ピルビン酸デヒドロゲナーゼ、ピルビン酸デヒドロゲナーゼキナーゼ、乳酸デヒドロゲナーゼ、MCT、グルタミナーゼ、ATPクエン酸リアーゼ、モノアシルグリセロールリパーゼ、脂肪酸合成酵素などである(Jones and Schulze, 2012; Vander Heiden, 2011)。これらの酵素を標的とすることで、がん治療に十分な治療効果が得られるかどうかは不明である(Vander Heiden, 2011)。そして、これらの阻害剤の臨床的有効性は、今後さらに明らかにされる予定である。代謝酵素を標的とした抗がん剤開発には、2つの大きな障害がある。第一に、合成経路の冗長性により、特定の代謝酵素を阻害する薬剤のがん治療効果が損なわれる可能性がある。幸いなことに、遺伝子変異によって、がん細胞は特定の代謝経路を冗長性の少ないものにするという証拠が増えつつある(Vander Heiden, 2011)。それにもかかわらず、腫瘍間のゲノムの不安定性は、がん細胞の突然変異率の上昇につながり、薬剤耐性をさらに発達させる可能性がある(Burrell et al.) 第二に、異化経路と同化経路の両方を含む代謝は、正常細胞の重要な機能である。がん細胞における特異的代謝酵素の同定は、正常組織への副作用を最小限に抑えた薬剤開発にとって極めて重要である。従って、正常な増殖細胞の代謝酵素を阻害するという受け入れがたい副作用は、抗がん剤開発におけるもう一つの大きな課題となるだろう(Vander Heiden, 2011)。
MBはがん治療のための代替ミトコンドリア電子伝達キャリアとして機能する
細胞代謝に対するMBの効果は、1920年代にはすでに研究されていた。MBは血液細胞において糖の分解を促進し、乳酸の生成を減少させることが発見され、MB投与が細胞の糖代謝を嫌気的経路から酸化的経路へとシフトさせることが示された(Harrop and Barron, 1928)。MBはさらに、ヒトおよび動物の腫瘍において、酸素消費率を19.2〜116%増加させることがわかった(Barron, 1930)。しかし、ワールブルグ効果のメカニズムや意義が理解されないまま、MBのがん代謝に対する劇的な効果は、がん研究者の注目を集めることはなかった。
MBは生体内試験で特定の腫瘍に選択的に蓄積されることが分かっている(福井ら、1983;ギルら、1984;ギルとストラウス、1984)。興味深いことに、MBは脳や神経組織にも高い親和性を示し、全身あるいは静脈内投与される(Kristiansen, 1989; Oz et al., 2011; Parascandola, 1981; Peter et al., 2000)。MBが神経細胞や癌組織に選択的に作用するメカニズムは明らかではないが、両組織とも代謝率が高いことから、細胞代謝がこれらの組織へのMBの蓄積に関与している可能性がある。一貫して、MBはミトコンドリアに積極的に結合し、ミトコンドリア膜電位によって増強される形でミトコンドリアマトリックスに入り込むことがわかっている(Gabrielli et al., i04)。神経組織やがん組織に対するMBの高い親和性から、臨床研究や組織化学研究の両方で、神経細胞構造やがん組織の染色に応用されている(Oz et al., z11)。MBによるリンパ節郭清は、乳がんや直腸がん患者にも導入されている(Chen et al., 2007; Markl et al., 2007; Resch and Langner, 2013; Thevarajah et al., 2005)。
光増感剤には電子移動反応が関与し、生体分子にラジカル誘起の損傷を引き起こすことが指摘されている(Cappugi et al., 2001)。MBの可逆的な電子移動というユニークな酸化還元作用は、光増感剤としての作用をもたらすと考えられる。実際、MBは光増感剤として、基底細胞癌、メラノーマ、カポジ肉腫など、様々な癌治療に用いられてきた(Tardivo et al., 2005)。
ミトコンドリアETCは、電子伝達とミトコンドリア内膜を横切るプロトンポンプを結合し、ATP産生の原動力となる同化経路の最下流に位置する。ETC複合体を標的とすることは、がん治療の可能な戦略として提案されている(Rohlena et al.) 実際、ETC複合体の構成成分を阻害する抗がん剤もある。例えば、第一選択の抗乳がん剤であるタモキシフェンは、ミトコンドリア複合体Iを阻害する作用がある(He et al., 2015)。タモキシフェンの抗がん作用におけるミトコンドリアETC複合体I阻害の役割については、議論の余地がある。それにもかかわらず、がん治療のためにミトコンドリアETCを阻害するという戦略は、正常細胞におけるATP産生におけるミトコンドリアETCの不可欠な役割を考えると、受け入れがたい副作用をもたらす可能性がある。一方、私たちの最近の研究では、代替ミトコンドリア電子伝達アプローチによるミトコンドリア呼吸の増強が、ワールブルグ効果を逆転させ、がん増殖を抑制する可能性が示された(Poteet et al.)
ワールブルグ効果は、がん細胞がミトコンドリア呼吸速度を比較的に低下させ、主に同化作用を示すという、がんの代謝的特徴を示している。興味深いことに、酸化的リン酸化を増加させると、試験管内試験および生体内試験でのがん増殖が抑制されることが示されている(Schulz et al., 2006)。私たちの最近の研究では、MBが代替ミトコンドリア電子キャリアとしてワールブルグ効果を逆転させ、同化作用を減衰させ、神経膠芽腫細胞の増殖を抑制するという概念実証がなされた(Poteet et al., 2013)。膠芽腫細胞において、MBは酸素消費率を高め、乳酸産生と細胞外酸性化率を低下させ、NADPHを減少させ、増殖を抑制する(Poteet et al.) これらの結果は、MBが代替ミトコンドリア電子伝達体として、ミトコンドリアの電子伝達を促進し、酸化的リン酸化を増加させ、解糖系フラックスと代謝中間体を減少させ、したがって、がん細胞増殖のためのビルディングレンガを使い果たすことを示している。一貫して、MBはがん細胞をS期で停止させ、サイクリンA2、B1、D1の発現を減少させる(Poteet et al.) 私たちはさらに、MBがAMPKシグナル経路を活性化する能力を持つことを示したが、これはおそらくその生体エネルギー作用による二次的なものであろう(Poteet et al.) このように、ミトコンドリアの電子伝達を直接標的とすることは、生合成活性に対する細胞内代謝産物のフィードバックの可能性を考えると、さらなる利点をもたらすかもしれない(Metallo and Vander Heiden, 2010; Ward and Thompson, 2012b)。
代替ミトコンドリア電子伝達は、他の代謝標的化戦略と比較して多くの利点を持つ、抗がん剤開発のための新しいメカニズムを提供する。第一に、異化代謝が全ての癌の特徴であることから、代謝適応を標的とすることは多くの癌に応用できる。第二に、代謝シグナル伝達経路と同化経路が重複しているため、がんの同化を抑制するために細胞シグナル伝達や代謝酵素を阻害しても効果がない可能性がある。一方、ミトコンドリアETCはミトコンドリアの酸化的リン酸化に不可欠である。従って、ミトコンドリアの電子伝達経路を代替するアプローチは、ミトコンドリア呼吸を増強し、がん治療により効果的な方法を提供する可能性がある。第三に、代謝シグナル伝達経路と同化経路の冗長性から、がん治療効果を高めるために複数の代謝シグナル伝達経路や酵素を標的とすることが求められるが、どの代謝シグナル伝達経路や酵素もがん細胞に特有ではないため、重篤な副作用を引き起こす可能性が高い。代替的ミトコンドリア電子伝達戦略は、細胞毒性が低いという点で従来の化学療法剤とは異なる。ミトコンドリア電子伝達の増加は、解糖およびその他の同化経路を相互に減少させ、ミトコンドリアの役割を生合成ハブから生体エネルギーハブに切り替え、その結果、癌の成長と増殖を阻害する可能性がある。従って、ミトコンドリア電子伝達の代替戦略は、重篤な副作用の少ない、より安全な抗癌剤治療アプローチとなりうる。
結論
神経変性疾患とがんは、世界的に最も主要な死因のひとつであり、公衆衛生にとって最悪の脅威となっている(Fitzmaurice et al.) 少子化と平均寿命の伸びにより、世界の人口高齢化のスピードは今後数十年で加速する(Lutz et al., 2008)。間違いなく、主要な老化関連疾患である神経変性疾患とがんの世界的な負担は、今後1世紀にわたって大幅に増加し続けるだろう。本総説で繰り返し述べたように、神経変性疾患とがんは、代謝の再プログラミングなど、共通のメカニズム的基盤を共有している可能性がある。従って、エネルギー代謝を標的とすることは、神経変性疾患と癌の両方の治療に一石二鳥の戦略を提供する可能性がある。脳の生体エネルギー代謝を強化することで、脳の機能を高め、神経変性疾患の進行を遅らせたり、予防したりすることができる一方、がんの代謝表現型を生合成的なものから生体エネルギー代謝的なものに切り替えることで、がんバイオマスを構築するためのレンガを使い果たし、がんの増殖を抑制することができるかもしれない(図4)。総説で述べたように、MBは代替ミトコンドリア電子伝達担体として機能し、様々な実験モデルにおいて神経変性疾患と神経膠芽腫の両方に有効であることから、代替ミトコンドリア電子伝達が、がんや多くの神経変性疾患に対する共通の新規治療メカニズムである可能性が示された。
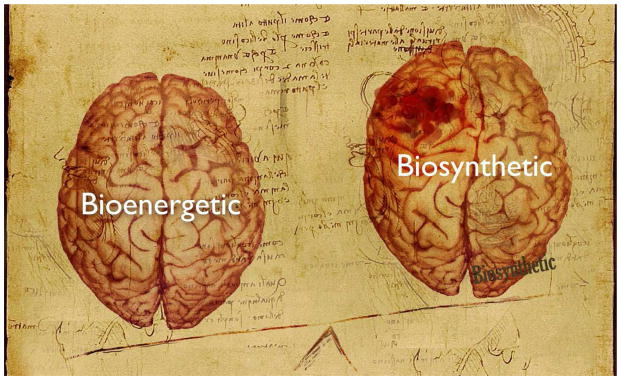
図4 神経変性疾患や脳腫瘍の治療のために、エネルギー代謝を再プログラムする。代謝のリプログラミングは、神経変性疾患と癌の共通のメカニズム基盤として機能する可能性がある。従って、エネルギー代謝を標的とすることで、神経変性疾患と癌の両方を治療する一石二鳥の戦略を提供できる可能性がある。脳の生体エネルギー代謝を強化すれば、脳機能が向上し、神経変性疾患の進行を遅らせたり、予防したりできるかもしれない。一方、がんの代謝表現型を生合成型から生体エネルギー代謝型に切り替えれば、がんバイオマスのレンガを使い果たし、がんの増殖を抑制できるかもしれない。
MBは1世紀以上の歴史を持つ薬物であり、よく知られた薬物動態、低毒性でBBBを通過して脳内に蓄積する能力、神経細胞組織と癌組織の両方に対する高い親和性など、神経変性疾患や癌の治療に望ましい特性を数多く持っている(Kupfer et al., r94;Peter et al., r00)。神経変性疾患や癌に対するMBの効果については、さらなる研究が必要である。しかしながら、神経変性疾患の病因病態が不均質であることを考えると、MBがすべての神経変性疾患の特効薬になるとは考えられない。実際、筋萎縮性側索硬化症(ALS)のSOD1 G93AモデルマウスとTDP-43モデルマウスでは、MB投与による神経保護効果は認められなかった(Audet et al., 2012; Lougheed and Turnbull, 2011)。同様に、代替ミトコンドリア電子伝達戦略も、がん治療には限界があるかもしれない。AMPKは、状況に応じて、がんにおいて抗腫瘍性と促進性の両方の役割を発揮する可能性がある。したがって、MBによって誘導されるAMPKの活性化は、代謝ストレスに対処する柔軟性をがん細胞に与えることで、化学療法中のがん細胞の生存に有利に働く可能性がある(Faubert et al.)
ハイライト
- エネルギー代謝のリプログラミングは、神経変性疾患やがんに共通する特徴である。
- メチレンブルーは、代替ミトコンドリア電子伝達キャリアとして機能し、生体エネルギーを増強し、生合成を抑制する。
- メチレンブルーは、パーキンソン病、アルツハイマー病、ハンチントン病、フリードライヒ失調症のげっ歯類モデルにおいて保護効果を示す。
- メチレンブルーはワールブルグ効果を逆転させ、癌の増殖を抑制する。
- 代替ミトコンドリア電子伝達は、癌や神経変性疾患に対する共通の新規治療メカニズムを提供する可能性がある。
謝辞
本研究の一部は、米国国立衛生研究所(National Institutes of Health)の助成金R01NS054651(SY)、R01NS088596(SY)、米国心臓協会(American Heart Association)の助成金SDG16960084(RL)により行われた。
略語リスト
- MB メチレンブルー
- AD アルツハイマー病
- PD パーキンソン病
- ALS 筋萎縮性側索硬化症
- HD ハンチントン病
- CBF 脳血流
- GLUT グルコーストランスポーター
- TCA トリカルボン酸
- ETC電子伝達鎖
- NADH ジヒドロニコチンアミドアデニンジヌクレオチド
- NADPH ニコチンアミドアデニンジヌクレオチドリン酸水素
- MCT モノカルボン酸トランスポーター
- AMPK AMP活性化プロテインキナーゼ
- ACC アセチル-CoAカルボキシラーゼ
- CaMKKβ カルシウム/カルモジュリン依存性プロテインキナーゼキナーゼβ
- APOE アポリポ蛋白質E
- MPTP 1-メチル-4-フェニル-1,2,3,4-テトラヒドロピリジン
- FRDA フリードライヒ失調症
- FXN フラタキシン
- mTORC1 哺乳類ラパマイシン複合体I標的タンパク質