
コンテンツ
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7242821/
要旨
アルツハイマー病では一酸化窒素/環状グアノシン一リン酸(cGMP)シグナル伝達が障害され、cGMPを分解するホスホジエステラーゼ5(PDE5)がアップレギュレーションされる。シルデナフィルはPDE5を阻害し、cGMPレベルを上昇させる。
これまでの知見を統合すると、シルデナフィルのほとんどの用量(特に低用量)は、プロテインキナーゼG介在性環状アデノシン一リン酸(cAMP)応答エレメント結合タンパク質(CREB)のリン酸化および/またはサーチュイン-1の活性化およびPGC1αの脱アセチル化を介して、ペルオキシソーム増殖因子活性化レセプター-γコアクチベーター1α(PGC1α)を活性化している可能性が高いことが明らかになった。
PGC1αシグナル伝達を介して、低用量シルデナフィルはβ-セクレターゼ1の発現とアミロイドβ(アミロイドβ)の生成を抑制し、抗酸化酵素をアップレギュレートし、ミトコンドリアの生合成を誘導する可能性が高い。さらに、シルデナフィルは、神経のアポトーシスや炎症を抑制しながら、脳の灌流、インスリン感受性、長期増強、神経新生を増加させるはずである。
アルツハイマー病におけるシルデナフィルのシステマティックレビューが行われた。試験管内試験では、シルデナフィルは神経ミトコンドリアをアミロイドβおよび高度な糖化最終生成物から保護した。
トランスジェニックADマウスでは、シルデナフィルはCREBリン酸化と記憶の障害を救済し、脳由来の神経栄養因子をアップレギュレートし、反応性アストロサイトとミクログリアを減少させ、インターロイキン-1β、インターロイキン-6,腫瘍壊死因子-αを減少させ、神経アポトーシスを減少させ、神経新生を増加させ、タウの高リン酸化を減少させることが明らかになった。
アミロイドβレベルを試験したすべての研究は、最高用量を使用した2つの研究を除いて有意な改善を報告し、PGC1αシグナル伝達に対するcGMP誘導ホスホジエステラーゼ2(PDE2)活性化およびcAMP枯渇の用量制限効果と一致した。
アルツハイマー病患者において,シルデナフィルの単回投与は,自発神経活動を低下させ,脳血流を増加させ,脳内酸素代謝量を増加させた。アルツハイマー病患者を対象としたシルデナフィル(理想的にはPDE2阻害薬との併用が望ましい)の無作為化比較試験が必要であると考えられた。
キーワード
アルツハイマー病、サイクリックGMP、ミトコンドリア生合成、ペルオキシソーム増殖因子活性化受容体γ-アクチベーター1α、クエン酸シルデナフィル
はじめに
アルツハイマー病は世界的に認知症の主要な原因となっており、アルツハイマー病患者とその家族は、この壊滅的な疾患の進行を予防し、遅らせるための新しい治療法を緊急に必要としている。アルツハイマー病の特徴としては、アミロイドβ(Aβ)ペプチドの分泌と神経斑への沈着、タウタンパク質の高リン酸化と神経原線維のもつれ形成、金属イオンの不均一性[1-9]、酸化ストレス、脂質、核酸、タンパク質の損傷[10-13]などが挙げられる。
細胞周期再突入異常[14-26]、神経炎症・微生物異常[27-33]、インスリン抵抗性[34,35]、脳血管機能障害[36-38]、シナプス機能障害[39,40]、神経細胞喪失、小胞体ストレス[41-44]、ミトコンドリア機能障害[45-48]などがある。
* *
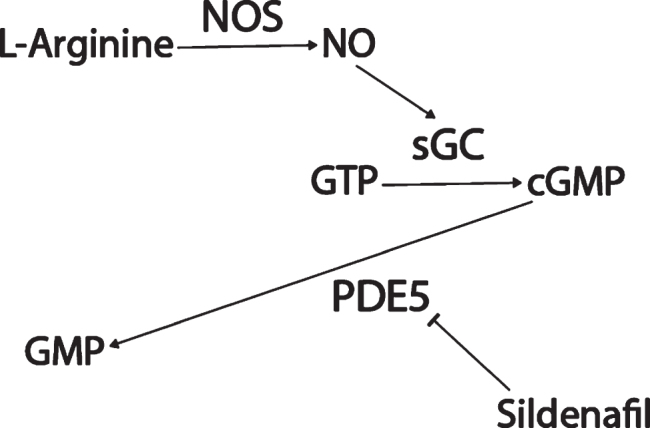
シルデナフィル(バイアグラ)は、ホスホジエステラーゼ5(PDE5)を阻害する勃起不全や肺動脈性肺高血圧症の治療に用いられる薬剤である(図1)。PDE5は環状グアノシン一リン酸(cGMP)を分解する。cGMPの上流では、通常、アミノ酸のL-アルギニンは、一酸化窒素合成酵素(NOS)という3種類の酵素によって一酸化窒素(NO)に変換される。
NOは細胞伝染性の小さな気体分子であり、細胞膜を越えて拡散し、可溶性グアニルシクラーゼ(sGC)を活性化し、sGCはグアニル三リン酸(GTP)をcGMPに変換する[49]。しかし、アルツハイマー病では、NOS/NO/cGMP経路の活性が著しく損なわれている。
NOS活性は、アルツハイマー病患者の上前頭前庭および海馬において、年齢をマッチさせた対照群と比較して有意に低下している[50]が、異常なニューロンNOS(nNOS)タンパク質の発現は、アルツハイマー病患者の脳の等皮質錐体ニューロンのサブ集団で観察されており[51,52]、アストロサイト内皮NOS(eNOS)および誘導性NOS(iNOS)発現の強度は、アルツハイマー病患者の深部皮質層で増加していた[52,53]。
NO誘発性可溶性sGC(基底性sGCまたは微粒子グアニルシクラーゼではなく)活性は、対照群と比較してアルツハイマー病患者の上側頭皮質で50%減少することが判明した[54]。cGMPレベルは対照群と比較してアルツハイマー病患者の脳脊髄液(脳脊髄液)で有意に低いことが判明し、cGMPレベルの低下は脳脊髄液 アミロイドβ42レベル[55]、併存するうつ病[56]、およびMini-Mental State Examination(MMSE)で測定される認知機能の低下と相関した[55,57]。
図1 シルデナフィルの作用機序
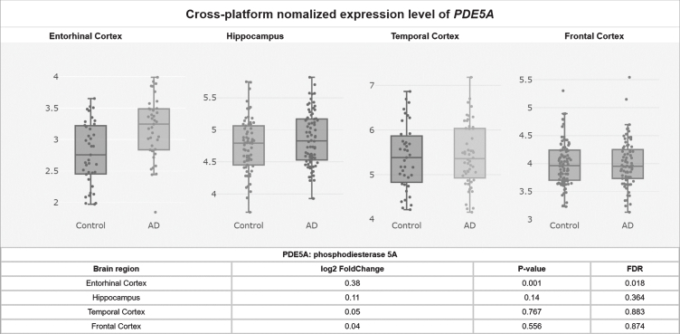
このcGMP枯渇に潜在的に寄与していると考えられる、PDE5タンパク質レベルは、アルツハイマー病患者の側頭皮質で有意にアップレギュレーションされていることが発見されており[55]、mRNAデータセットのメタ解析(p = 0.001,FDR = 0.018; 図2)によると、PDE5 mRNAレベルは対照群と比較してアルツハイマー病患者の側頭皮質で有意に上昇している[58]。
しかし、ヒト中枢神経系における PDE5 mRNA の発現は末梢組織と比較して低いことが報告されており [59-61]、ある研究では、放射性 in situ ハイブリダイゼーション組織化学を用いた高齢者や アルツハイマー病 患者の脳内では PDE5 mRNA に特異的なハイブリダイゼーションシグナルは観察されず、PDE5 阻害が アルツハイマー病 に関連しているかどうかについては懐疑的である [62-64]。
しかし、それにもかかわらず、証拠の重みは、PDE5が正常なヒトの脳で(末梢組織よりは比較的少ないものの)発現していることを示唆しており[58-61]、さらに、PDE5はアルツハイマー病の側頭葉と側頭葉でアップレギュレートされていることを示唆しており[55, 58]、PDE5がアルツハイマー病における重要な治療標的である可能性があるという考えを支持している。
図2 アルツハイマー病内耳皮質でアップレギュレートされたPDE5A mRNAレベル
cGMP/PGC1αシグナル伝達
このマルチメカニズムのNOS/NO/cGMPシグナル伝達機能不全は、複数の理由からアルツハイマー病における重要な治療標的である。一つの理由は、cGMPがペルオキシソーム増殖因子活性化受容体-γコアクチベーター1α(PGC1α)の発現および活性を増加させることに関与しているからである。
PGC1αの過剰発現(または低用量NO)は、アミロイドβ生成の律速酵素であるβ-セクレターゼ1(BACE1)の発現を抑制する[65]ことから、PGC1α活性がアミロイドβ生成を抑制していることが示唆される。さらに、PGC1αは、ミトコンドリア生合成、酸化的呼吸[66,67]、脂肪酸β酸化[68]、抗酸化防御[69]のマスター転写調節因子である。
PGC1αは、フォークヘッドボックスO3a(Foxo3a)に結合し、Sirtuin-1(SIRT1)によって脱アセチル化されると、ミトコンドリアマンガンスーパーオキシドジスムターゼ(MnSOD)を含む複数の抗酸化遺伝子をアップレギュレートする[69]。
しかしながら、PGC1αタンパク質レベルは対照と比較してアルツハイマー病患者の海馬において有意に低く[48]、ミトコンドリア生合成およびMnSOD発現はそこで障害される[45,48]。シルデナフィルは、アルツハイマー病における海馬のPGC1α抑制を逆転させる可能性がある。
10μMのシルデナフィルを試験管内試験で24時間,0.3mg/kgのシルデナフィルを生体内試験で投与すると、腎近位尿細管細胞およびマウス腎皮質でそれぞれcGMPが増加し、PGC1α発現およびミトコンドリア生合成が誘導されることが示されている[70]。
さらに、シルデナフィルはラット肝臓(1.48mg/kgシルデナフィル)およびヒト血液(100mgシルデナフィル1回投与)において、SODおよびカタラーゼ活性をアップレギュレートすることが示されている[71, 72]。
シルデナフィルとcGMPによるPGC1αの多段階調節
NOS/NO/sGC/cGMPシグナル伝達は、腎近位尿細管細胞[70]だけでなく、褐色脂肪細胞、U937単細胞、HeLa子宮頸がん由来細胞、白色3T3-L1脂肪細胞[73,74]、およびおそらくニューロン[75]を含む多様な細胞型においてPGC1αをアップレギュレートする。マウスでは、皮質下の脳組織が低酸素に反応し、PGC1αのアップレギュレーション(ミトコンドリアの生合成と同様に)がnNOSの発現に依存する方法で行われた[75]。
* *
しかし、シルデナフィルとcGMPがPGC1αを活性化するという主張に対する重要な注意点は、NOS/NO/cGMPはPGC1αシグナル伝達の多相レオスタットであり、その結果はcGMP濃度、持続時間、およびホスホジエステラーゼ3(PDE3)およびホスホジエステラーゼ2(PDE2)活性によって媒介されるcAMPシグナル伝達とのクロストークに依存しているようであるということである。
低濃度のcGMPはPDE3の活性部位と競合し[76]、それによりPDE3がcAMPを分解するのを阻害し、cAMPレベルを上昇させる。対照的に、高濃度のcGMPはPDE2をアロステリックに活性化し、それによってcAMP分解を促進する[76,77]。
これを反映して、低用量シルデナフィル(1μMまで)はPDE3を阻害し、cAMPの蓄積をもたらす[76]一方、高用量シルデナフィル(10および100μM)はPDE2を活性化し、cAMP分解をもたらし[76]、cAMP/EPAC/アデノシン一リン酸活性化プロテインキナーゼ(AMPK)経路を抑制する[76, 78, 79]。
この理由から、シルデナフィルの過剰な高用量投与は、非アルコール性肝性ステアトーシスおよび肥満の前臨床モデルにおいて、低用量投与と比較して治療効果が劣る結果となった[76]。この注意点はアルツハイマー病にも関連しているかもしれない。
cAMPレベルに対するシルデナフィルおよびcGMPシグナル伝達の用量依存的効果は、cAMPがアルツハイマー病における強固なPGC1α発現およびミトコンドリアの生合成に必要であるため、PGC1αの調節に影響を与えるはずである[48, 67]。
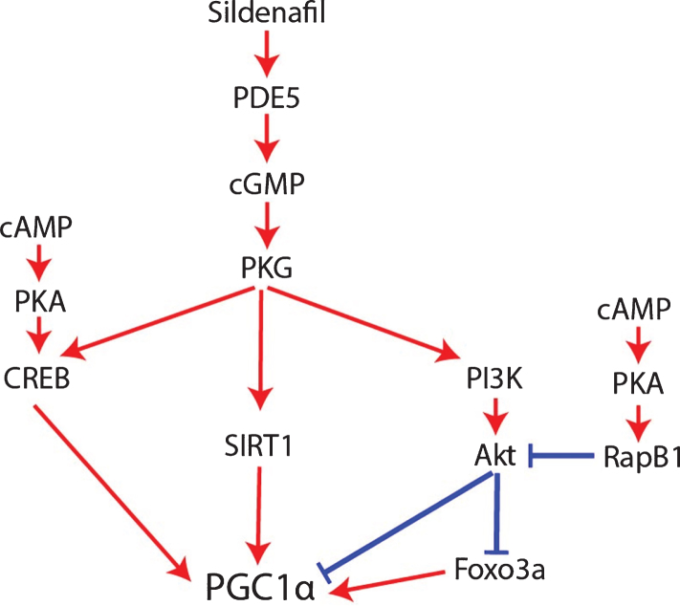
これに加えて、NO/sGC/cGMPシグナルはPGC1α活性を典型的に促進するが、同時にプロテインキナーゼG(PKG)/ホスホイノシチド3-キナーゼ(PI3K)/Aktシグナル伝達カスケードを活性化し、PGC1α活性を低下させる[80-83];cGMPによるPGC1αのこの負の制御に対抗して、cAMPはPKAおよびRap1bを介してAktを阻害する(図4)[84]。
言い換えれば、cGMP自体は、異なるメカニズムでPGC1αシグナルを同時に増加させたり減少させたりしており、おそらくPGC1αが全体的に活性化されているか阻害されているかは、cAMP経路とのクロストークによって決定されると思われる。
したがって、高用量のシルデナフィルはPDE2を活性化し、cAMPレベルを低下させるので、高用量のシルデナフィルは、PGC1αの活性化、ミトコンドリア生合成の誘導、抗酸化遺伝子のアップレギュレーション、およびBACE1のダウンレギュレーションにおいて、低用量のシルデナフィルよりも効果が低いと予想される[76]。
図4 シルデナフィルとcGMPがPGC1αをどのように調節するか
cGMPレベルが低く、高くても中等度ではない場合はPGC1αを活性化する?
PGC1αに対するシルデナフィルとcGMPの用量依存的効果と一致するように、10μMの8-Br-cGMP処理を24時間行うと、腎近位尿細管細胞においてPGC1αのmRNA発現とミトコンドリア生合成がアップレギュレートされた[70]。
対照的に、内皮細胞では、100μMの8-Br-cGMPを6~24時間処理すると、PGC1αがダウンレギュレーションされた[80, 83]。さらに、12時間未満のNOドナーでの処理はPGC1α発現を減少させたが、24時間以上の処理はPGC1α発現を増加させた[83]。
NOをトリガーとしたPGC1αの発現低下とその結果としての活性酸素種(ROS)産生は、内皮細胞の遊走に必要であり、その効果はNO活性化されたPI3K/Aktシグナル伝達とFoxo3aのAktリン酸化阻害によって媒介されていた[80]。
* *
しかし、驚くべきことに、100μMよりもはるかに高い用量のcGMPは、低用量と同様にPGC1αをアップレギュレートする:例えば、PGC1αは、1 mMの8-Br-cGMPで4日間処理した褐色脂肪細胞でアップレギュレートされた[73]。
ヒト単球性U937細胞、ラットL6筋管およびラットPC12神経分泌細胞において、3 mM 8-Br-cGMPを6日間毎日投与すると、PGC1αだけでなく、Nrf1およびTfam、ミトコンドリアタンパク質Cox IVおよびシトクロムC、およびミトコンドリアDNA含量がアップレギュレートされた[66]。さらに、これら3つの細胞型において、3mM 8-Br-cGMPを6日間投与すると、酸化的リン酸化共役酸素消費量の増加をもたらした[66]。
* *
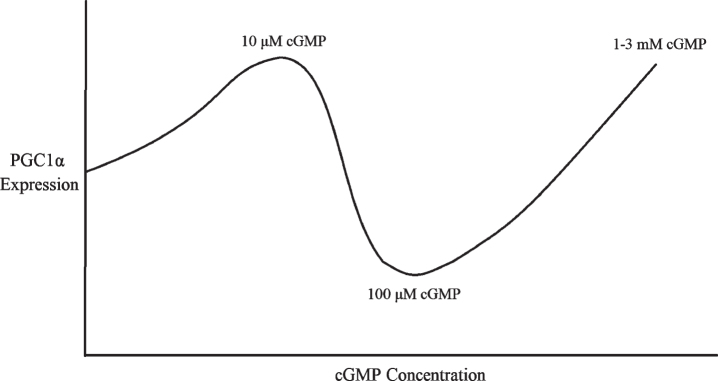
したがって、cGMP濃度とPGC1α発現との間にはU字型の用量反応曲線があるようであり、PGC1αは100μM cGMPでは6~24時間[80, 83]まではダウンレギュレートされるが、10μM cGMPまたは1~3mM cGMPでは24時間以上はアップレギュレートされる(図3)[66, 70, 73]。
これは、100μMのcGMPがPDE2を活性化し、cAMPを低下させるのに十分なシルデナフィルの10-100μMの用量に相当するためかもしれない[76]が、はるかに高い用量のcGMPおよびシルデナフィルは、それにもかかわらず、ミトコンドリアの生合成およびPGC1αの転写を刺激する[66]。
73] なぜならば、cAMP応答要素結合タンパク質(CREB)リン酸化およびSIRT1活性化(次のサブセクションを参照)を介して、おそらくcAMP応答要素結合タンパク質(CREB)リン酸化およびSIRT1活性化を介して、cAMP枯渇を克服して、cAMPとは無関係にロバストなPGC1α活性を誘導するのに十分な超骨格的cGMPシグナル伝達があるからである。
もしそうであれば、これは前例のないことではないだろう:線維芽細胞では、PDE2の過剰発現は、cAMPレベルを低下させ、筋線維芽細胞の親線維性表現型への移行を誘導するのに十分であったが、この変化は、cGMP上昇剤によって逆転された。
したがって、最適なPGC1α発現、ミトコンドリア生合成、およびBACE1ダウンレギュレーションのためには、十分に低用量または高用量のシルデナフィルを使用するか、またはPDE2阻害剤とシルデナフィルを併用しなければならないようである。
図3 cGMP濃度に対するPGC1α発現の多相用量応答
この複雑ではあるが臨床的に関連性のある用量特異的効果に加えて、NO/cGMPがPGC1αを典型的にアップレギュレートするメカニズムは未だ解明されていない[67]。ここでは、CREBまたはSIRT1のいずれかが関与する2つの可能性のあるメカニズムについて説明する。
シルデナフィルとcGMPがPGC1αを活性化するメカニズム
長期増強中の海馬や他の神経組織では、cGMP/PKGとcAMP/PKA経路の両方がCREBのリン酸化に寄与しており[85-90]、PKA媒介のCREBリン酸化はPGC1αの転写を促進する[67]ので、cGMP/PKG/CREBシグナル伝達はPGC1αの発現も促進する可能性がある。
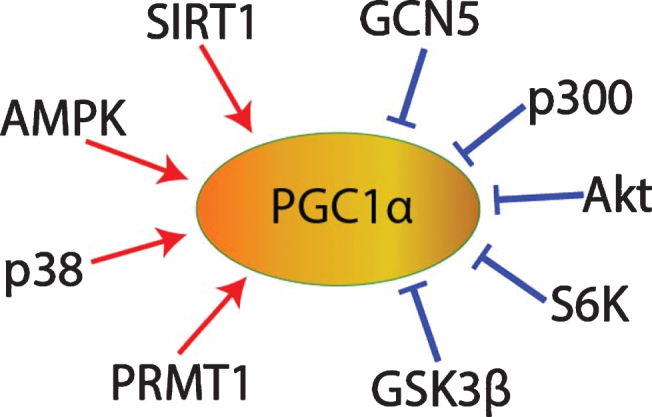
さらに、cGMPはPGC1αの翻訳後制御にも寄与している可能性がある。転写および翻訳されると、PGC1αタンパク質の安定性、細胞内局在性、および共活性化活性は、複数の翻訳後修飾によって制御される。例えば、PGC1αは、Akt[81,82]、S6キナーゼ[91]、またはグリコーゲン合成酵素キナーゼ3β(GSK3β)[74,92]によるリン酸化を介して、またはアミノ酸合成5の一般制御(GCN5)[81,93-95]またはp300[96]によるアセチル化を介して阻害され得る。
逆に、PGC1αは、アデノシン一リン酸活性化プロテインキナーゼ(AMPK)[97-101]またはp38マイトジェン活性化プロテインキナーゼ(MAPK)[101,102]によるリン酸化、プロテインアルギニンメチルトランスフェラーゼ1(PRMT1)[103]によるメチル化、またはSIRT1(図5)[69,93,94,96,101,104-107]による脱アセチル化を介して活性化され得る。
PGC1αのSIRT1介在性活性化を考慮すると、興味深いことに、シルデナフィルは心臓、心筋細胞[108]、血清、皮下脂肪組織[109]でSIRT1をアップレギュレートすることが示されている。cGMP類似体は、白色脂肪組織[110,111]および骨芽細胞[112]でSIRT1発現をアップレギュレートした。
骨芽細胞特異的PKGIIを過剰発現させたマウスは、SIRT1発現の増加を示した[112]。PKG阻害剤KT-5823は、SIRT1活性化因子であるレスベラトロールによって誘導される脊髄性アロディニーアの緩和をブロックした[113,114]。
低酸素心筋細胞において、1μMシルデナフィル処理はPGC1αタンパク質のアセチル化を減少させた[115,116]。したがって、シルデナフィルがcGMP/PKG/SIRT1シグナリングを介してPGC1αの脱アセチル化を促進する可能性がある(図5)。
図5 PGC1αの翻訳後制御
アルツハイマー病におけるシルデナフィルの効果
ほとんどの用量のシルデナフィル/cGMPはPGC1αを活性化し、PGC1αシグナルはミトコンドリア生合成を誘導し[67,70]、抗酸化酵素をアップレギュレートし[69]、BACE1発現を抑制する[65]ので、シルデナフィルはアルツハイマー病患者に有意な利益をもたらすはずである。
PGC1α特異的な利点に加えて、シルデナフィルはcGMPを介して平滑筋弛緩および血管拡張を促進する[117]が、低灌流はアルツハイマー病患者の脳における重要な障害でもあるので、アルツハイマー病患者に追加の利点を提供する可能性がある[37, 38, 118]。
シルデナフィルは低酸素ニューロンのアポトーシスを抑制し、神経新生を促進するので、アルツハイマー病ニューロンの喪失を遅らせ、新しいニューロンの補充を促進する可能性がある[117, 119]。さらに、シルデナフィルは患者のインスリン感受性と内皮炎症を改善することから、シルデナフィルもインスリン感受性を促進し、アルツハイマー病脳の炎症を抑制する可能性がある[124-126]。
cGMP/PKGシグナリングはCREBリン酸化を介して長期増強を媒介するので[85-90, 117]、シルデナフィルはアルツハイマー病に関連する学習および記憶障害を改善するはずである。
したがって、理論的には、ほとんどの用量のシルデナフィルは、過剰なアミロイドβ生成、ミトコンドリア生合成の障害、酸化ストレス、神経炎症、低灌流、インスリン抵抗性、神経細胞の喪失、不十分な神経新生、および記憶障害を含むアルツハイマー病の複数の特徴を改善するはずである。
シルデナフィルとアルツハイマー病の併存疾患と危険因子
また、多くのアルツハイマー病患者がこれらの疾患の1つ以上に罹患していることから、一般的なアルツハイマー病の併存疾患および/または危険因子となるII型糖尿病、心血管疾患、うつ病などの疾患に対するシルデナフィルの効果を考慮することも重要である。
* *
うつ病におけるシルデナフィルの効果については、NOS/NO/sGC/cGMPとセロトニンシグナル伝達は互いに対立する傾向がある。cGMPは脳血管拡張を誘発するが[127]、セロトニンは脳血管収縮を誘発する[128,129]。
NOS/NO/sGC/cGMP/PKGシグナリングはセロトニントランスポーター(SERT)を活性化し、セロトニンの再取り込みを誘導する[130-132]。このため、シルデナフィルは選択的セロトニン再取り込み阻害薬(SSRI)抗うつ薬の効果を低下させると予想される。
しかし、そのような報告はないようであり、シルデナフィルはSSRIを服用している患者の勃起不全の治療に安全かつ成功裏に使用されている[133]。また、シルデナフィル自体がマウスにおいて抗うつ作用を示すことが示されている[134]。
* *
シルデナフィルのⅡ型糖尿病への効果については、無作為化二重盲検プラセボ対照試験において、シルデナフィルを1日3回25mgを3ヶ月間投与することで、プレ糖尿病患者のインスリン感受性が向上することが示されており、アルツハイマー病およびⅡ型糖尿病患者に有効である可能性が示唆されている[125]。
シルデナフィルと心血管疾患の影響については、初期の懸念にもかかわらず[135]、シルデナフィルの使用は心筋梗塞や心臓突然死のリスクに寄与していないようである[136]。実際、心筋梗塞を起こした患者の勃起不全をPDE5阻害薬(アルプロスタジルは除く)で治療した場合、死亡リスクおよび心不全による入院リスクの低下と相関があった(n = 43,145)[137]。
前臨床試験では、高コレステロール血症のマウスモデルにおいて、シルデナフィルは大動脈の動脈硬化性プラークを40%減少させた[138]。さらに、シルデナフィルは心肥大を減少させる[139]。したがって、心血管疾患を併存するアルツハイマー病患者におけるシルデナフィル治療は、安全であり、潜在的に有益であると期待される。
材料および方法
シルデナフィルとアルツハイマー病の研究における現在の進展を明らかにするため、PubMedで “sildenafil Alzheimer’s “を検索した。前臨床研究と臨床研究の両方をレビューした。シルデナフィルの患者への効果やアルツハイマー病の前臨床モデルに関係のない研究(例えば、長期増強におけるcGMPとアミロイドβの相互作用に関する研究)は破棄した。臨床的関連性を確保するために、シルデナフィルの誘導体に関する研究も破棄された。
結果
方法の項にあるように、2つの試験管内試験、10のげっ歯類試験、1つのシステマティックレビュー、および2つの患者を対象としたパイロット試験が含まれている。全体として、すべての研究でアルツハイマー病におけるシルデナフィルの使用が支持された(結果の要約は表1を参照)。
表1 文献レビュー結果
表1
文献レビューの結果
| 投与量 | 人口 | 結果 | 調査 |
| 10-100 μ Mシルデナフィル | Aβに曝さHT-22海馬神経細胞 25から35 | シルデナフィルは、ミトコンドリアのCa救出2 +過負荷および障害Aβに起因する 35から 25まで開放することによりATP感受性K +チャネル | [ 140 ] |
| 20 μ Mシルデナフィル | 高度な糖化最終産物に曝露されたHT-22海馬神経細胞 | シルデナフィルは、 HO1のアップレギュレーションを介してミトコンドリア伝染性遷移の細孔開口とアポトーシスを減少させた | [ 141 ] |
| 可変、3 mg / kg | スコポラミン誘発コリン作動性機能障害マウス | シルデナフィルは記憶を救った | [ 143 ] |
| 可変、主に3mg / kg /日の腹腔内シルデナフィル | APP / PS1マウス | シルデナフィルは、長期増強、CREBリン酸化、記憶、およびAβレベルの低下を助けた | [ 144 ] |
| 6 mg / kg、3か月間毎日腹腔内 | APP / PS1マウス | シルデナフィルは、記憶力、アミロイド斑負荷、炎症、および神経新生を改善した | [ 145 ] |
| 2mg / kgシルデナフィルを1日2回4ヶ月間 | APP / PS1マウス | シルデナフィルは、記憶とアミロイドの病状を救い、PDE5をダウンレギュレートし、NOS、NO、およびcGMPレベルを増加させた | [ 146 ] |
| シルデナフィルを0.9%生理食塩水に1.0mg / mlの濃度で溶解した。この溶液の10.0mg / kgを,0.1ml / 10gの注射量で腹腔内投与した。 | APP / PS1マウス | シルデナフィルは記憶力を改善し、Aβ、IL-1β、IL-6,TNF-αのレベルを低下させ、p-CREBを増加させた | [ 147 ] |
| 水中で10週間毎日15mg / kgのシルデナフィル | J20マウス | 改良されたメモリ、タウ過剰リン酸化およびAktおよびGSK3βリン酸化ではなく、前頭前皮質Aβシルデナフィル 42のレベル | [ 61 ] |
| 15 mg / kg毎日シルデナフィル、腹腔内 | Tg2576ADマウス | シルデナフィルは記憶を改善し、前頭葉アミロイド病理ではなくタウを改善し、GSK3βを阻害し、CDK5 p25 / 35比を低下させ、BDNFとアークをアップレギュレートした | [ 148 ] |
| 変数 | 老齢マウス | シルデナフィルは空間記憶の保持を改善したが、獲得は改善しなかった | [ 64 ] |
| 3mg / kgの腹腔内シルデナフィルを3週間毎日 | 老齢マウス | シルデナフィルは、二本鎖DNA切断、アポトーシス促進性カスパーゼ-3およびBaxを減少させ、抗アポトーシス性Bcl2およびBDNFをアップレギュレートした。 | [ 149 ] |
| 7.5mg / kgシルデナフィルを1日1回4週間腹腔内投与 | SAMP8マウス | シルデナフィルはアミロイドとタウの病理、記憶、神経膠症を改善した | [ 145,150 ] |
| 7.5mg / kgシルデナフィルを1日1回4週間腹腔内投与 | SAMP8マウス | シルデナフィルは、海馬のJNK蛍光体活性化、タウリン酸化、および記憶障害を減少させた | [ 151 ] |
| 50 mgシルデナフィル、単回投与 | アルツハイマー病患者10人 | シルデナフィルは右海馬の自発的神経活動を減少させた | [ 152 ] |
| 50 mgシルデナフィル、単回投与 | 14人のアルツハイマー病患者 | シルデナフィルは、12人の患者で酸素と脳血流の脳代謝率を増加させ、8人の患者で脳血管反応性を減少させた | [ 127 ] |
HT-22海馬神経細胞をアミロイドβ25-35で処理した場合、ミトコンドリアカルシウム過負荷が生じ、ATP枯渇、活性酸素の発生、伝染性遷移孔の開孔、カスパーゼ9の活性化、細胞死と関連していた。シルデナフィルは、ATP感受性K+チャネルの開通を促進することで、これらの影響を防止した[140]。
培養HT-22海馬神経細胞において、高度な糖化最終生成物[141](アルツハイマー病の危険因子[142])への曝露は、ミトコンドリアの活性酸素の発生、細胞内ATPの枯渇、ミトコンドリアの伝染性遷移孔の開通、シトクロムCの放出、カスパーゼ-3の活性化、アポトーシスを誘導した。
シルデナフィルを投与すると、ヘムオキシゲナーゼ1(HO1)が増加し、ミトコンドリアを伝染性遷移孔の開放とチトクロムCの放出から保護し、カスパーゼ-3の活性化とアポトーシスを減少させた[141]。HO1の発現は、シルデナフィルによるミトコンドリアの還元能と伝染性遷移孔の開口の保護に必要であった[141]ことから、シルデナフィルがHO1のアップレギュレーションを介してミトコンドリアを保護したことが示唆された。
* *
スコポラミン注射によるアルツハイマー病を模倣したコリン作動性機能障害を有するマウスでは、シルデナフィルは迷路のパフォーマンスを回復させ、3mg/kgは1.5や4.5mg/kgの投与量よりも効果的であることが明らかになった[143]。
* *
トランスジェニックアミロイド前駆体タンパク質(APP)/プレセニリン1(PS1)ADマウスから海馬スライスでは、50 nMのシルデナフィルは、APP/PS1遺伝子型によって引き起こされるシャッファー側副経路における破傷風誘発性長期増強の障害を救済した[144]。
生体内試験でのシルデナフィル治療でも同様の結果が得られた。APP/PS1マウスでは、3mg/kgのシルデナフィルを一度に投与すると、文脈的恐怖記憶が回復した。3mg/kgのシルデナフィルを2~3週間毎日腹腔内投与することで、橈骨腕水迷路試験における空間作業記憶の欠損が部分的に改善された。
同様の効果は、治療中止後9~12週間後に3mg/kgのシルデナフィルを毎日投与した場合にも認められ、治療中止後も長期的な効果が持続することが示唆された。シルデナフィルはまた、モリス水迷路とプローブ試験で長期的な空間参照記憶を回復させた。
シルデナフィル投与により、破傷風によって誘発されたCA1海馬のCREBリン酸化が正常レベルに回復した。毎日3mg/kgのシルデナフィルを3週間投与することで、大脳皮質サンプルのアミロイドβ40およびアミロイドβ42レベルを低下させるのに十分であった[144]。
* *
APP/PS1マウスにシルデナフィル6mg/kgを毎日3ヶ月間腹腔内投与したところ、行動試験(営巣行動、Y迷路での腕の進入、Morris水迷路の脱出潜時および経路長)において有意な改善が認められた。
同様に、炎症性ミクログリアとアストロサイトマーカーの免疫反応性イオン化カルシウム結合アダプター分子1(Iba1)とグリア線維性酸性タンパク質(GFAP)歯状回のNeuN陽性ニューロンとダブルコルチン(DCX)陽性細胞によって示される神経新生、およびアミロイドプラーク負担[145]と同様に。
* *
APP/PS1 ADマウスでは、2mg/kgのシルデナフィルを1日2回4ヶ月間投与したところ、Y-迷路試験での自発的な交替と電気刺激からの逃避によって示されるように認知が回復し、皮質および海馬のアミロイドβPP、アミロイドβ40,アミロイドβ42レベルの低下、PDE5発現の低下、およびnNOS、eNOS、iNOS、NO、およびcGMPレベルの増加によって示されるようにアミロイド病理が減少した[146]。
* *
APP/PS1マウスにおけるシルデナフィルの腹腔内投与は、新規物体認識選好性、海馬における前炎症性サイトカインであるインターロイキン-1β(IL-1β)インターロイキン-6(IL-6)腫瘍壊死因子-α(TNF-α)の発現低下、海馬可溶性アミロイドβ40およびアミロイドβ42発現の低下、およびCREBリン酸化の増加によって示されるように記憶力を改善することが明らかになった[147]。
* *
最近の系統的レビューでは、Morris水迷路とT迷路において、シルデナフィルは高齢マウスの空間記憶保持を改善したが、獲得は改善しなかったことが明らかになった[64]。興味深いことに、このレビューでは、PDE5 mRNAプローブの特異的なハイブリダイゼーションの欠如に基づいて、PDE5がアルツハイマー病に重要な脳構造に位置していないことが報告されている[62-64]が、指摘したように、他のグループは、それぞれアルツハイマー病患者の側頭葉[58]または側頭葉[55]でPDE5 mRNAまたはタンパク質の発現の増加を発見しており、PDE5 mRNAの発現は、末梢組織よりも低いレベルではあるが、正常なヒトの脳で複数のグループによって発見されている[58-61]。
* *
J20 ADマウスでは、15 mg/kgのシルデナフィルを毎日10週間飲水させると、モリス水迷路でのパフォーマンスが改善し、タウの高リン酸化が減少し、AktとGSK3βのリン酸化が増加したが、前頭前野のアミロイドβ42レベルは変化しなかった[61]。
* *
Tg2576 ADマウスでは、15 mg/kg/日の腹腔内シルデナフィルは、有意にモリス水迷路と恐怖条件付けタスク、減少した海馬タウ高リン酸化、GSK3β活性、およびCDK5のp25/p35比で示されるように学習と記憶障害を救済した。
シルデナフィルは恐怖条件付け訓練後に海馬のp-CREBとc-Fosをアップレギュレートし、脳由来神経栄養因子(BDNF)と活性調節された細胞骨格関連タンパク質(Arc)(記憶のエンコードに関与する即時応答遺伝子)の海馬発現を増加させた[148]。しかし、シルデナフィルは前頭皮質の総アミロイドβ42レベルに影響を与えなかった[148]。
* *
高齢のマウスでは、シルデナフィルを毎日3mg/kg腹腔内に3週間投与すると、CA1海馬における末端デオキシウリジン三リン酸ニックエンドラベリング(TUNEL)アッセイで可視化されたように、二本鎖DNA切断とアポトーシス細胞が減少した。
プロアポトーシスタンパク質であるカスパーゼ-3とB細胞リンパ腫2-関連X(Bax)をダウンレギュレートし、抗アポトーシスB細胞リンパ腫タンパク質-2(Bcl2)とBDNFをアップレギュレートし、アミロイドβPP発現をダウンレギュレートし、アミロイドβ42/アミロイドβ40比の年齢に関連したシフトを抑制した[149]。
* *
老化が加速し、散発的な アルツハイマー病 を発症したマウスの SAMP8(senescence accelerated mouse-prone 8)モデルでは、7. 5 mg/kgのシルデナフィルを4週間投与すると、モリス水迷路と受動的回避テスト[145, 150, 151]、タウ高リン酸化[145, 151]、GFAPのダウンレギュレーションによって示される炎症、およびBACE1,カテプシンB、および海馬アミロイドβ42のダウンレギュレーションによって示されるアミロイド病理学によって示されるように認知能力が改善された[150]。シルデナフィルはまた、Aktを活性化し、GSK3β、カルパイン、サイクリン依存性キナーゼ5(CDK5)[145,150]、およびc-Jun N末端キナーゼ(JNK)[151]を阻害した。
アルツハイマー病患者におけるシルデナフィル
アルツハイマー病患者10名において、シルデナフィル50mgの単回投与は、アルツハイマー病患者の海馬および海馬傍神経において異常に増加することが示されているパラメータである血中酸素濃度依存性信号の機能的磁気共鳴イメージングで記録された低周波揺らぎの分画振幅によって示されるように、右海馬における自発的神経活動を有意に減少させた[152]。
アルツハイマー病患者12人の高齢者では、シルデナフィル50mgの単回投与で、脳内酸素代謝率と脳血流が有意に増加した[127]。8人のアルツハイマー病患者では、脳血管反応性を低下させた[127]。
考察
序文で予測されたように、これらのパラメータを試験した前臨床研究では、シルデナフィルがCREBリン酸化、長期増強、および学習と記憶を回復させ[61,143-148,150,151]、神経新生を増加させ[145]、および神経炎症を減少させることが明らかになった[145,147,150]。
さらに、これらの研究は一貫してシルデナフィルがタウ高リン酸化および関連するパラメータを減少させることを見いだした[61,145,148,150,151]。これは、アルツハイマー病海馬ニューロンにおけるMnSODのダウンレギュレーションがタウ高リン酸化に寄与しているため[45,153]、および低用量シルデナフィルはPGC1α活性化を介してMnSODをアップレギュレーションするようであるため、その一端であるかもしれない[69-72]。
比較的高用量の15mg/kg投与では、主に高用量シルデナフィルがcGMP/PKG/PI3K/Akt経路を活性化し、タウキナーゼGSK3βのSer9リン酸化阻害の増加をもたらしたため、タウの高リン酸化が減少したようである[61, 80, 148]。
* *
しかし、シルデナフィルのアミロイドβレベルへの影響については矛盾が報告されている:ほとんどの研究でシルデナフィルがアミロイドβレベルを低下させることが示されているが[144-147, 150]、最高用量(15mg/kg)を用いた2つの研究では、前頭皮質のアミロイドβレベルには影響がないことが報告されている[61, 148]。
低用量のシルデナフィルとcGMPはPGC1αを活性化し、BACE1発現を抑制するようであるが[65, 70, 76]、高用量のシルデナフィルと100μMのcGMPはcAMPとPDE2シグナルとのクロストークによりPGC1αを阻害し、BACE1発現を抑制しないようである。
言い換えれば、15mg/kgのシルデナフィルはPDE2を活性化し、cAMPを減少させ[76]、cAMPシグナル化されたPGC1αの活性化とBACE1の抑制を阻害するので、15mg/kgのシルデナフィル試験ではアミロイドβレベルが有意に減少していない可能性がある[61, 148]。
もう一つの興味深い観察は、15mg/kgの投与量でのみAktの活性化および/またはGSK3βの阻害が報告されていることである[61, 148]。これは、15mg/kgの投与量がAktを活性化し[61, 148]、結果としてPGC1αの発現およびその抗アミロイド性を抑制した可能性を示唆している[80-82]。
* *
興味深いことに、試験管内試験での研究では、シルデナフィルはATP感受性K +チャネルまたはHO1のアップレギュレーションを介してそれぞれアミロイドβまたはAGEsからミトコンドリアを保護していることが明らかになっている[140, 141]。
* *
患者において、特に有望な知見は、シルデナフィル50mgを投与すると脳内酸素代謝量が増加したことである[127]。この効果は脳血流の増加[127]によって完全に説明されているか、あるいはPGC1αによって制御されたミトコンドリアの生合成によっても部分的に媒介されている可能性がある。
しかし、検討された研究では、PGC1αのmRNA、タンパク質、アセチル化レベル、ミトコンドリア生合成の他のマーカーを測定したものはなく、この可能性を評価することは不可能であった。また、これらの研究では、SIRT1活性やSIRT1/PGC1α経路に対するシルデナフィルの影響も検討されていない。
また、これらの研究では、インスリン抵抗性のマーカーや抗酸化酵素発現に対するシルデナフィルの影響も検討されていない。トランスジェニックADマウスを用いた将来の前臨床研究では、シルデナフィル誘発のアミロイドβ抑制、ミトコンドリア生合成、抗酸化酵素発現におけるSIRT1およびPGC1α活性の役割、ならびにインスリン抵抗性に対するシルデナフィルの仮説的効果を評価するために、これらの点に直接取り組むべきである。
今後の研究では、シルデナフィルとPDE2阻害剤を併用して、治療期間を通してシルデナフィルの最大有効量を継続的に増加させる可能性も評価すべきである。これにより、PDE2活性化およびcAMP枯渇に対するシルデナフィルの用量制限効果を回避することができ[76]、cAMPおよびcGMPシグナル伝達が同時に確実に行われるため、最大のPGC1α活性、ミトコンドリア生合成、抗酸化酵素発現、およびBACE1抑制が可能になるであろう[48, 65-67, 69, 70, 73-75, 96, 108, 109, 112, 115, 116, 154-156]。
この役割のための最良の候補は、プロペントフィリンであり、強力で広範なスペクトルのPDE阻害剤であり、カフェインのようなメチルキサンチン誘導体であり、cGMP刺激PDE2活性およびPDE4の阻害に特に効果的である[157]。
プロペントフィリンは、主に300 mgを食事の前に1日1時間を3回服用することがテストされており、5つの第III相臨床試験[158-164]で軽度から中等度のアルツハイマー病患者で安全かつ効果的であることが判明しているため、他のPDE2阻害剤よりも優れているであろう。興味深いことに、ヘックマンらによる最近のレビューは、前臨床証拠に基づいて、PDE2,PDE4,およびPDE5の阻害は、アルツハイマー病 [165]の治療のための最も有望な保持するように見えた、とシルデナフィルとプロペントフィリンは、一緒に投与すると、強力に同時にこれらの3つの治療標的を阻害するだろうと見解を述べた[157]。
最終的には、アルツハイマー病患者を対象としたシルデナフィルの無作為化対照試験を実施し、高齢の対照群と比較してこの集団におけるシルデナフィルの長期投与の臨床的有用性を評価すべきである。このRCTでは50mg/日の用量を使用すべきである[127]。
アウトカム指標として、RCTではMMSEとアルツハイマー病評価尺度-認知サブスケールでの認知、老年期うつ病尺度での併存うつ病をテストすべきである[56]。2-(1-6-[(2-[F-18]フルオロエチル)(メチル)アミノ]-2-ナフチルエチリデン)マロノニトリルポジトロン断層撮影(FDDNP-PET)によるアミロイドとタウの病理結合[166, 167]、MRIによる脳血流[127]、MRIによる脳内酸素代謝率[127]。
168,169]、18F-フルオロデオキシグルコースポジトロン断層撮影(FDG-PET)によるグルコースの脳代謝率[34,35,170-174]、脳脊髄液 IL-1β、IL-6,およびTNF-αによる炎症[124-126]。
147,175-182]、脳脊髄液 cGMPによるNOS/NO/sGC/cGMP/PGC1α経路の機能不全[55,56]、および尿中8-オキソ-2′-デオキシグアノシンを間接的なバイオマーカーとする抗酸化酵素活性[45,69,71,72,183]。