
GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7089874/
オンラインで公開2020年3月17日
要旨
アルツハイマー病は、高齢者に存在する認知症の最も一般的な形態である;その病因は、遺伝的要因と環境要因が含まれる。近年の疫学研究では、アルツハイマー病患者のかなりの数が後に発作を起こすことがあるため、アルツハイマー病と慢性てんかんとの間に相関関係があることが示されている。アルツハイマー病の発作の病態生理は完全には解明されていないが、アミロイドβペプチド(アミロイドβ)の蓄積とタウタンパク質の過リン酸化に関連したいくつかの分子機構の結果である可能性があり、それは興奮性と抑制性の神経伝達物質の放出と回収の不均衡、神経細胞の細胞骨格の構造変化、シナプスの損失、神経炎症を誘発する可能性がある。これらの変化は、慢性的な状態では、神経変性過程を促進し、アルツハイマー病で観察される認知機能の低下に影響を与える、非同期放電とてんかん発生の再発の開発を支持する可能性がある。また、側頭葉てんかん患者の脳組織の病理組織学的研究では、アミロイドβの沈着や神経原線維のもつれにタウタンパク質が蓄積していることが明らかになっており、それに伴ってグリコーゲン合成酵素キナーゼ-3β(GSK3β)活性が上昇し、タウを中心とした微小管関連タンパク質(MAP)の翻訳後修飾に変化が生じている可能性がある。本研究では、TLEやアルツハイマー病の発症におけるGSK3βとタウの病理学的側面の理解に焦点を当てている。
キーワード:タウタンパク質、GSK3β、てんかん、神経変性、海馬硬化症
序論
アルツハイマー病は、進行性の記憶喪失、行動の変化、認知機能の低下を特徴とする。病理組織学的には、アミロイドβペプチド(アミロイドβ)と微小管関連タンパク質(MAP)タウによって形成された神経原線維性のもつれ(NFT)によって形成された神経斑(NP)の存在によって定義される。これらの病変は、ミクログリアおよびアストロサイトの活性化を生じ、シナプスの喪失および神経細胞の死につながる(Meraz-Ríos et al 2013)。アミロイドβはFrizzled受容体と相互作用し、Wnt/β-カテニンシグナル伝達経路を障害し、主要なタウ蛋白質キナーゼであるグリコーゲン合成酵素キナーゼ-3β(GSK3β)の活性を増加させることが提案されている。タウの高リン酸化は、細胞骨格微小管(MT)の不安定化を促進し、軸索輸送異常や神経細胞死を引き起こす(Inestrosa and Toledo, 2008)。アミロイドβとタウタンパク質は、興奮毒性や興奮性亢進と関連している(Holth et al 2013)。タウと興奮性の関係は、神経回路および神経変性プロセスの変化に起因する可能性があるアルツハイマー病患者で観察される非誘発性発作の発生率の増加の観点から重要である(PandisおよびScarmeas 2012)。このように、発作やネットワークの過興奮性は、認知機能障害の発症を加速させる一因になる可能性があると考えられる。さらに、アルツハイマー病で影響を受ける神経回路には、てんかん誘発性が高いことが示されている脳領域が関与しており、最も一般的なタイプのてんかんである側頭葉てんかん(TLE)と強く関連している。TLE患者の病理組織学的研究では、高リン酸化タウタンパク質凝集体の存在が示されている(Sen et al 2007;Thom et al 2011;Tai et al 2016)。本レビューは、アルツハイマー病およびTLEの発症におけるタウおよびGSK3βタンパク質の役割を評価することを目的としている。
タウタンパク質
タウは主に軸索に位置するII型MAPである。中枢神経系(中枢神経系)におけるMTの組み立ておよび安定化を阻害する熱安定性タンパク質である。ヒトのタウは、染色体17q21上に位置するMAPT遺伝子によってコードされ、16個のエクソンからなる(Andreadis et al 1992; Andreadis 2005; 図1A)。このタンパク質の6つの異なるアイソフォームが、成人ヒトの脳で発現している。各アイソフォームは、3つまたは4つの微小管結合リピート(3R/4R)を含み、1つまたは2つのN末端インサートの存在または不在を示す(Buée et al 2000;Martin et al 2011;図1B)。
図1 タウタンパク質
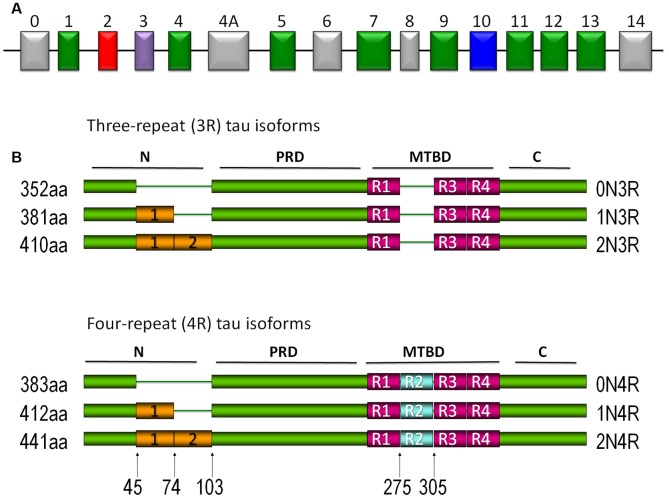
(A) タウ遺伝子。中枢神経系(CNS)ではエクソン2,3,10が交互にスプライシングされている。エクソン9〜12はそれぞれ微小管結合ドメイン(MBD)を含む。エクソン4aと6は末梢神経系のアイソフォームで発現しているが、エクソン8はどのアイソフォームでも報告されていない。B)タウタンパク質の異なるアイソフォームが中枢神経系で発現している。異なるアイソフォームの発現は発生によって制御されている。3つの繰り返しドメインを持つアイソフォームは胎児期に優先的に発現するが、成体期には6つのアイソフォームが存在することが特徴である。微小管(MT)に結合する繰り返しドメインはR1,R2,R3,R4と指定されている。もう一つの特徴は、タンパク質のアミノ末端に位置する1つ(1N)または2つ(2N)の挿入物の有無(0N)である。
正常な状態では、タウは、ダイニンおよびキネシンなどの運動タンパク質と相互作用し、逆行性および前行性輸送に参加し(Dixit et al 2008)胚発生、長期増強(LTP; Ahmed et al 2014)および長期うつ病(LTD; Kimura et al 2014; Regan et al 2015)。病理的条件下では、ペアらせん状フィラメント(PHFs; Goedert, 1999)として知られる不溶性構造に自己組織化する。タウの2つの翻訳後修飾がPHFに存在する:高リン酸化および切断(Flament et al 1990; Alonso et al 1996; Hasegawa et al 1998)。
過リン酸化はタウ微小管の結合を阻害し、その結果、細胞骨格の安定性が変化し(Evans et al 2000)、その後の軸索輸送の喪失、および他のシグナル伝達関連機能の低下をもたらす(Mandelkow et al 1995);また、フィラメントにおけるタウ病理学的凝集の引き金となる主要なイベントと考えられている(Grundke-Iqbal et al 1986; Wood et al 1986; Alonso et al 1996)。
タウとてんかん
近年、タウタンパク質は神経細胞の同期性や興奮性の乱れに関与していることが知られており、てんかんとの関連性も示唆されている。具体的な病理学的メカニズムはまだ明らかにされていないが、これらの主張を裏付ける様々な報告がある。
いくつかのタウ病理モデルでは、結合性が劇的に変化し、ガンマ・シータ振動の強いアンカップリングを引き起こすことが示されている。しかし、てんかん様活性の徴候は登録されていなかった(Ahnaou et al 2017)。この考えに挑戦して、ヒトアミロイド前駆体タンパク質(hAPP)のトランスジェニックマウスモデルは、アミロイドβの過剰産生とその結果としての自然発作の発生を提示した。このモデルにタウ遺伝子をノックアウトしたものを加えると、タウのレベルが低下し、N-メチル-D-アスパラギン酸受容体(NMDAR)の機能障害が防止され、LTPが低下し、認知機能の低下が改善され、海馬のてんかん活動が低下することが明らかになった(Roberson et al 2007,2011)。
タウと興奮性亢進との関係を評価したもう一つの興味深い研究は、TLEモデルであるKcna1-/-マウスである。このマウスはKv1.1のαサブユニットに対するヌル対立遺伝子を持っており、生後3週目の自然発作の発生を条件とする電圧依存性カリウムチャネルである。このモデルでのタウ遺伝子ノックアウトは、自然発作の発生と頻度を減少させ、特に海馬のCA3錐体領域での生存を促進した(Holth et al 2013)。別の同様のモデルでは、アミロイドβ誘発性の過興奮性が、樹状突起の興奮性とシナプス可塑性の調節に重要な樹状突起カリウムチャネルであるKv4.2のレベルの低下と関連していることが実証された。興味深いことに、これらの現象の両方の十分な量が、タウノックアウトマウスで劇的に減少したことから、タウが神経細胞の興奮性を調節する上で直接的な役割を持っている可能性が示唆された(Hall et al 2015)。同様の結果は、ペンチレネテトラゾール(PTZ)で処理したタウ-/-マウスモデルでも観察された。この場合、タウ発現の減少が発作の重症度を保護することが観察された(DeVos et al 2013)。これは、アルツハイマー病患者の内因性タウレベルが発作発症のリスクに影響を与える可能性があることを考えると興味深い。
脳脊髄液中のタウの高い基底レベルを持っていること自体は発作を引き起こすことはないが、これらの患者は、例えば、虚血性脳卒中や外傷性脳損傷の後に、傷害後に発作を発症する可能性が高くなる(Camilo and Goldstein, 2004; Kwan, 2010)。このため、将来的には、タウ遺伝子の多型(Myers et al 2007;Kauwe et al 2008)または脳脊髄液中のタウのレベル(Cruchaga et al 2013)を同定することにより、遺伝子解析によりハイリスク患者を同定し、この集団において予防的な抗てんかん治療を行うことが有用であるかどうかを判断することが重要になるかもしれない。
高リン酸化タウ凝集体およびNFTは、いくつかのてんかん患者において観察されている(Sen et al 2007;Thom et al 2011;Tai et al 2016)。さらに、難治性てんかん患者における側頭葉切除に関する研究では、高リン酸化タウの存在および前タングルおよびNFTへの蓄積が認知機能の低下と相関しているようであることが示された(Tai et al 2016)。証拠は、てんかん患者における発作の長期化または再発が認知障害を引き起こすか、または悪化させる可能性があることを示唆している(Holmes, 2015; Kneynsberg et al 2017)。病理学的タウは、興奮毒性(Roberson et al 2007)てんかん(DeVos et al 2013)および認知障害、特に記憶(Holmes 2015)と相関している。しかしながら、現在までのところ、高リン酸化タウがどのようにして高興奮性を誘導するかを説明する具体的なメカニズムは、活発な研究の領域である。
てんかん発症は、興奮性神経伝達物質と抑制性神経伝達物質の間の不均衡と関連している。てんかんの主な興奮性神経伝達物質はグルタミン酸であり、このグルタミン酸はNMDARの過剰刺激により興奮性を促進する。さらに、NMDARの活性化は、タウのリン酸化を促進するもう一つのメカニズムとして提案されている。この過程を経て、タウはNMDARの活性化、シナプス可塑性、神経毒性を調節している可能性がある(Mondragón-Rodríguez er al)。
これらのタウの潜在的な役割は、神経病理学との直接的な関係を確立し、海馬ニューロンネットワークや神経変性プロセスの不均衡にリンクして発作を伴うTLEのような他の疾患の知識を確立するための研究の新しい分野を開いた。このように、タウがてんかん発症においてどのように極めて重要な役割を果たしているかの正確なメカニズムを明らかにし、それによって診断、治療、予後の価値のある潜在的な介入を行うために、さらなる研究が行われなければならない。
GSK3
GSK3はプロリン指向型キナーゼであり、19番染色体にグリコーゲン合成酵素キナーゼ-3α(GSK3β)3番染色体にグリコーゲン合成酵素-3β(GSK3β)の2つのアイソフォームがある。GSK3βは主に中枢神経系に発現し、通常は軸索に位置し、タウタンパク質をリン酸化する主要なキナーゼである(Muyllaert et al 2008)。GSK3βはセリン9のリン酸化により不活性化され、チロシン216のリン酸化により活性化される。GSK3βの不活性化は、PI3K/AktとWnt/β-カテニン経路によって制御されている。アルツハイマー病では、両方とも変化しており、GSK3βの活性の増加に有利である(Hooper et al 2008)。
GSK3βの過剰発現または過剰活性は、タウのリン酸化を増加させ、タウがMTから分解することを可能にし、軸索輸送の変化、海馬神経変性(Avila et al 2006; Muyllaert et al 2008)および学習障害(Gómez De Barreda et al 2010)につながる。GSK3は42の部位でタウをリン酸化することができる(図2;Hanger et al 2009;Martin et al 2013)。また、GSK3活性は、アルツハイマー病脳におけるNFTの量と相関している(Leroy et al 2002)。また、GSK3キナーゼのアルツハイマー病におけるもう一つの意義は、アミロイドβペプチドが細胞内アポトーシスの経路を制御することであると考えられている(Takashima et al 1996)。GSK3βは、プロアポトーシスタンパク質であるBax活性を促進し、細胞が毒性のある障害に対して感受性を持つことを回避する転写因子を阻害する。これらの因子の一つは、細胞ストレスに対する応答を調節するヒートショック転写因子1(HSF1)である(Mines et al 2011)。
図2 タウ蛋白質上のリン酸化部位
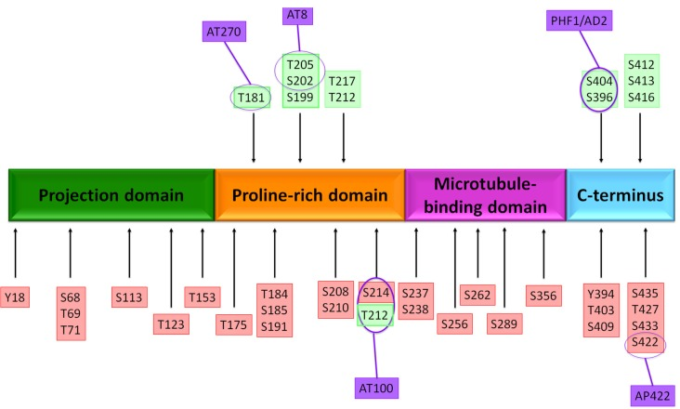
ネイティブのタウタンパク質は、セリン残基またはスレオニン残基でリン酸化されており、85の仮定的リン酸化部位が含まれている。いくつかのアミノ酸がアルツハイマー病 (アルツハイマー病) 脳 (赤枠) と正常と アルツハイマー病 の両方の脳 (緑枠) でリン酸化されている。抗体エピトープの局在を示す(紫丸)。
最近の研究では、GSK3βが記憶障害や学習障害に関連していることが示されており、GSK3βキナーゼの活性化や過剰発現はNMDA受容体の調節を介してLTPを低下させ、LTDを増加させることが示されている(Peineau et al 2007; Salcedo-Tello et al 2011)。リチウムによるGSK3βの阻害は、神経変性から細胞を保護し、脾臓摘出ラットモデルではホスホタウレベルと空間学習・記憶障害を有意に減少させる(Tan er al)。
GSK3βとてんかん
GSK3βは、シナプス可塑性の調節やニューロンの細胞骨格動態の制御に生理的な役割を果たしていることから、興奮性の亢進やてんかん発症と関連している可能性があると考えられている。ラットモデルでは、カイニン酸(KA)で誘発された発作や海馬の神経細胞損傷は、GSK3β活性を刺激し、Wnt/β-カテニン経路を阻害する可能性がある。これらのラットにGSK3β阻害剤であるリチウムを投与すると、海馬の神経細胞の損傷は抑制されたが、KAによる発作は抑制されなかった(Busceti et al 2007)。神経新生障害はTLEの後期に頻繁に観察され、Wnt/β-カテニン経路の阻害と関連している。GSK3β阻害剤は、この経路を回復させ、神経新生を回復させる(Huang et al 2015);さらに、GSK3β活性は、PI3k/Akt経路によっても制御される。この経路は、KAおよび電気痙攣性てんかんモデルにおいて損なわれる結果となり、また、発作前のGSK3β活性化、神経細胞損傷およびタウリン酸化を促進し得る(Crespo-Biel et al 2007;Gangarossa et al 2015)。
上記の情報に沿って、他の研究では、KA注入後48時間後のウエスタンブロットアッセイでBcl2/Bax発現のプロアポトーシス傾向が証明されている;Bcl2ファミリータンパク質はGSK3βの重要な下流標的であるように思われる(Linseman et al 2004)。これに関連して、Bcl2発現の減少は、神経変性プロセスの別の結果である可能性がある(Bhowmik et al 2015)。これらの知見はいずれも、興奮性インシュルト後のGSK3βの変化が、神経変性だけでなく、正常な神経細胞生存シグナル伝達経路の破綻にも重要な役割を果たすことを示唆している。
GSK3β自体がてんかんを引き起こすことは実証されていないが、脳の興奮性が正常で健康な状態から「プロてんかん」状態に変化した後であっても、GSK3βの阻害が抗てんかん活性をもたらすことを示す証拠が存在する(Aourz et al 2019)。さらに、GSK3βの変化は発作感受性を増加させ、GSK3β活性の増加は他のてんかんモデルにおいても証明されている(Lohi et al 2005;Tripathi et al 2010;Lee et al 2012)。
他のてんかんモデル(Lohi et al 2005; Tripathi et al 2010; Lee et al 2012)では、GSK3β活性の増加が確認されているにもかかわらず、KAの添加は、他のてんかんモデル(Lohi et al 2005; Tripathi et al 2010; Lee et al 2012 2012)GSK3βの構成的に活性な形態を過剰発現したマウスの海馬スライスへのKAの添加は、おそらくグルタミン酸α-アミノ-3-ヒドロキシ-5-メチル-4-イソオキサゾールプロピオン酸(AMPA)受容体のGluA1サブユニットの減少したリン酸化によって媒介される、誘発されたてんかんの進行の減少をもたらした(Urbanska et al 2019)。この結果は、GSK3β阻害に起因する有益な効果のために矛盾しているように思われるが、最近の研究では、状態てんかんの重症度に関連する海馬損傷に対するGSK3β活性の変調に対して脳が限定的な耐性を有していることが暴露された(Engel et al 2018)。
TLEを有利にするGSK3βおよびそのてんかん発生における役割の別の強力な仮説は、NMDARおよびその刺激との密接な関係であり、これは疾患の急性段階で興奮性を誘導する可能性がある(Liu et al 2017)。
上記のすべてのエビデンスは、てんかん予防におけるGSK3βのホメオスタシスの重要性を示している。興奮障害性の障害が最初にGSK3βを混乱させることがあっても、それが起こると、GSK3βは積極的に発作感受性を高める役割を果たす(図3)。GSK3βの変化が正常な生存シグナル伝達経路の破綻と興奮性の亢進を促進し、これらが一緒になっててんかん発症や神経変性を引き起こす可能性があることは間違いない。
図3 グリコーゲン合成酵素キナーゼ-3β(GSK3βとてんかん)
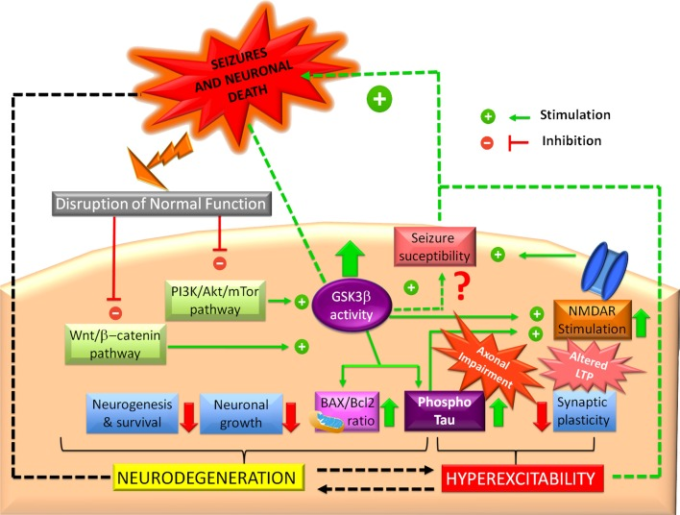
PI3K/AktおよびWnt/b-カテニンシグナル伝達経路の障害は、GSK3βの活性を増大させ、N-メチル-D-アスパラギン酸受容体(NMDAR)の過剰刺激を促進し、興奮性の亢進やシナプス可塑性の変化を引き起こす可能性がある。GSK3βの活性が上昇することで、海馬の痙攣が抑制されるという研究もあるが、おそらくこのメカニズムがGSK3βと海馬の痙攣との主な関連性を示していると思われる。また、GSK3βはタウの高リン酸化を促進し、軸索輸送障害や海馬の神経細胞死を促進している可能性もある。高リン酸化されたタウは興奮性の亢進や神経変性に積極的に関与している。また、GSK3βはミトコンドリアのプロアポトーシス(BAX/Bcl2比が高い)を促進する。この意味で、GSK3βはまた、神経原性構造の一つである海馬のシナプス平衡の回復を妨げる神経原性機能不全の引き金になる可能性もある。
セラピューティクス
前述のように、タウとGSK3βは、アルツハイマー病やTLEを含むいくつかの神経疾患と関連しており、その両方が治療標的となる可能性がある。
興味深いことに、様々な抗てんかん薬(AED)の有効性は、てんかん実験モデルおよびアルツハイマー病患者において試験されており、認知障害予防において有望な機能を示している(Sánchez et al 2018)。タウの高リン酸化はNFT形成の主なメカニズムであるため、タウの高リン酸化に関与するCDK5やGSK3βなどの異なるタウキナーゼを阻害することで、アルツハイマー病(Morris et al 2011)やてんかん(Sen et al 2007;Thom et al 2011;Tai et al 2016)で観察されるそれらの凝集を減少させる可能性が示唆されている(Xie et al 2017;Holzer et al 2018)。
健康なボランティアにAEDを使用し、異なる神経心理学的変数を測定した場合、研究者は、一般に、カルバマゼピン、フェニトインおよびバルプロ酸塩のようなAEDは、軽度の認知作用を有することを見出した(Meador et al 1995;Martin et al 2001;Salinsky et al 2002)。対照的に、オクスカルバゼピン、ガバペンチン、カルバマゼピンの認知効果を健康なボランティアを対象に評価したところ、その結果は、長期記憶には何の影響もなく、集中注意課題と手書き速度において個人の方が優れたパフォーマンスを示した(Curran and Java, 1993; Meador et al 1999)。いくつかの要因(神経病理、遺伝、発作の影響、心理社会的背景など)が複雑に相互作用しているため、高齢の患者は一般的にいくつかのAEDの否定的な認知効果に対してより敏感であることは言及しておく価値がある。
アルツハイマー病患者の発作予防におけるAEDの有効性が検証されている。しかし、高齢者におけるAEDの認知作用に関する報告は比較的限られており、特にアルツハイマー病患者の高齢者ではその効果が確認されていない。古いAEDの多くは、気分、行動、記憶などの認知機能に悪影響を及ぼす(Craig and Tallis, 1994; Meinhold er al)。 対照的に、新しいAEDは有害性が低いようで、トピラマートのような新しい薬の中には言語記憶や言語記憶を損なうものがあるにもかかわらず、わずかな認知的利益をもたらす可能性がある(Fritz et al 2005)。ラモトリギンやギャバペンチンのような他の様々な新しいAEDは、記憶を損なうことはなく、記憶、さらには気分にわずかな有益な効果をもたらす可能性がある(Tekin et al 1998; Meador et al 1999)。レベチラセタムは認知的効果を実証しており、日常臨床では特に注意力と流暢な口調で測定することができる(Cumbo and Ligori, 2010)。
AED治療の主な目的は患者の生活の質を改善することであり、発作の量を減らすことが主な目的であるにもかかわらず、今回のレビューは、認知のような他の治療変数を考慮した場合に、医師と患者が治療を決定する際に役立つ追加的な情報を提供する可能性がある。
これまでの臨床における最も進んだプロテインキナーゼ阻害戦略は、GSK3βを標的としたものであり、チデグルシブのような新規薬剤の開発につながっている(Hooper et al 2008;Medina 2018)。このチアジアゾリジノン(TDZD)化合物による治療は、アミロイド沈着の減少、タウリン酸化のレベルの低下、およびトランスジェニックマウスモデル(APPsw-tauvlw;Serenó et al 2009)における記憶障害の予防をもたらした。リチウムやバルプロ酸は、抗けいれん作用を有し、てんかんや他の精神疾患にも使用されている(Jope and Roh, 2006)。さらに、これらの阻害剤とGSK3βの選択的阻害剤であるTDZD-8は、GSK3βの不活性化を導くセリン9のリン酸化を促進する(Zhang er al)。 阻害戦略は、タウの高リン酸化を減少させ、神経細胞の死を防ぎ、状態てんかんの設定での海馬における神経新生過程の変化を抑制する(Busceti et al 2007;Bhowmik et al 2015;Huang et al 2015;Urbanska et al 2019)。メマンチンおよびイフェンドロピルは、NMDARアンタゴニストであり、GSK3βの不活性化およびタウリン酸化の減少に伝導することが実証されている(Liu et al 2017)。最近では、インディルビンやBIO-アセトキシムなどの強力かつ選択的なGSK3β阻害剤の使用により、抗けいれん作用が示されている(Aourz et al 2019)。本報告では、PTZ処理したゼブラフィッシュ幼虫を両化合物で処理したところ、抗痙攣活性を示し、てんかん状放電が減少した。同様に、インディルビンおよびBIO-アセトキシムは、6-Hz難治性発作マウスモデルにおいて抗痙攣活性を示した(Aourz et al 2019)。
アセチル化は、障害されたタウ機能につながり、その病理学的凝集を促進する別の翻訳後修飾である;したがって、タウアセチル化阻害剤は、アルツハイマー病および他のタウ症のための潜在的な治療戦略として提案されている(Medina et al 2016)。サルサレートは、アセチルトランスフェラーゼp300誘導タウアセチル化を阻害する非ステロイド性抗炎症剤である;この薬剤は、タウ誘導記憶欠損を救済し、前頭側頭型認知症(FTD)のマウスモデルにおいて海馬萎縮を予防することが示されている(Min et al 2015)。Leuco-methylthioninium bis-hydromethanesulfonate(LMTM)は現在第III相臨床試験中であり、タウの凝集を阻害する効果が示されている(Bulic et al 2013)。受動的および能動的なタウ免疫療法は、タウ症および関連疾患に存在するモノマー、オリゴマー、プレフィラメント、顆粒、フィブリル、または不溶性凝集体などの有害なタウ種を除去するために設計された有望な戦略である。免疫療法の主な目的は、タウ凝集を減少させ、神経変性を予防し、認知機能を改善することである(Bittar et al 2018; Medina 2018; Novak et al 2018)。
これらの治療戦略のすべてが主にアルツハイマー病を標的としているにもかかわらず、将来的には、それらがTLEを含む他のタウ症の治療に十分に役立つ可能性があることは明らかである。したがって、これらの疾患の将来において、タウおよびGSK3βをベースとした戦略の潜在的な治療効果を調達し、奨励することが重要である。
結論
TLEは、海馬硬化症と慢性発作を特徴とする一般的な疾患であり、しばしば薬理学的治療に抵抗性を示し、障害を伴う併存疾患や認知機能の低下と関連している。疫学研究では、慢性てんかんではアルツハイマー病を含むいくつかの認知症の有病率が高いことが示されている。これらの病態はタウタンパク質の過リン酸化に収束し、明らかにアポトーシス、異常な神経新生、シナプス可塑性の障害、海馬での過興奮性を促進することができるGSK3βの活性の増加によって媒介されている。これらの変化はアルツハイマー病ではよく知られているが、TLEではまだ完全には解明されていない(図4)。このため、病態や構造異常におけるタウとGSK3βの役割を明らかにし、新規タウ免疫療法の有効性を探るためには、さらなる研究が必要である。また、アルツハイマー病やTLEは散発性・多因子性疾患であることから、TLEやアルツハイマー病の発症の遺伝的危険因子となりうるGSK3βやタウ(MAPT)をコードする遺伝子の異なる変化を研究することは興味深いことである。
図4 神経変性過程におけるタウとGSK3βの役割を模式的に示したもの
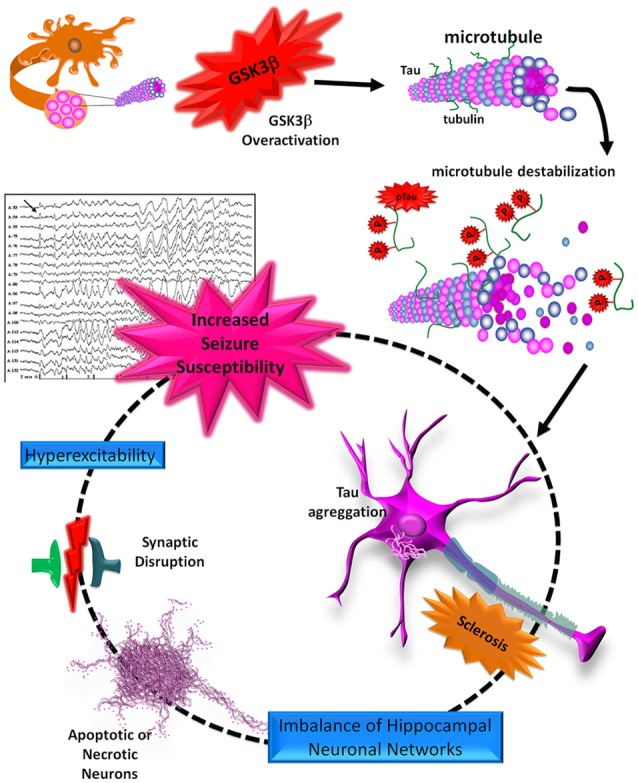
タウタンパク質はGSK3βの活性が増加することで高リン酸化され、pTauはMTとの結合を失い、凝集して神経原線維のもつれ(NFT)を形成する。これらの凝集体は、神経斑(NP)のように、海馬のニューロンネットワークの不均衡やシナプス機能不全を促進し、その結果、アポトーシスや壊死を介してニューロンの死を促進する可能性がある。最後に、これらの変化は、増加した発作感受性を有利に、海馬における興奮性と興奮毒性のプロセスにつながる。