
Contents
Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer’s Disease?
www.ncbi.nlm.nih.gov/pmc/articles/PMC6436584/
2018年8月12日オンライン公開
概要
アルツハイマー病(AD)は、脳の特定部位における広範な神経変性と炎症が特徴で、重篤な認知障害に結びつくる。アルツハイマー病は、脳の特定の部位に広範な神経変性と炎症が生じ、重篤な認知障害を引き起こすことが特徴である。このため、最初の分子的な引き金から、検出可能な症状が現れるまでのタイムラグが著しく長くなっている。注目すべきは、過去10年間の多くの研究により、軽度認知障害の初期段階において、神経細胞の障害とは対照的に見える事象が発生する可能性があることが明らかにされたことだ。これらは、in vitroや動物モデルで再現され、シナプス要素の増加、シナプスおよび代謝活動の増加、神経栄養環境の強化、グリア細胞の反応性と炎症の変化などが含まれている。これらは、疾患の進行を遅らせるか、あるいは長期的には有害な結果をもたらす代償反応であると解釈されている。このため、これらのメカニズムは、これまで過小評価されていた新たな介入の機会を提供するものである。その重要性は、特にその初期に現れることにある。新しい薬理学的標的を同定するために、この段階をよりよく特徴づける努力をすることは、AD研究の将来の進歩のための刺激的な新しい道である。
キーワード:βアミロイド、認知障害、補償、グリア反応性、神経変性
1. はじめに
アルツハイマー病(AD)は、広範な神経細胞の損傷と死によって特徴づけられる神経変性疾患であり、その結果、認知機能が徐々に低下し、すべての基本的技能の自立を完全に失うに至るとされている。ADには、早期に発症するまれな家族性と、主に高齢者に発症する一般的な散発性のものがある。世界人口の平均年齢が上昇していることから、現代社会にとって大きな緊急課題となっている。ADの病態には、分子生物学的な観点から、βアミロイドペプチド(Aβ)の蓄積とタウタンパク質のリン酸化亢進の2つの要因が大きく関わっているとされている。Aβは、オリゴマー(oAβ)からプロトフィブリル、フィブリルへと徐々に分子量の大きい分子種に凝集しやすく、病態のエフェクターとしての関連性は様々である。oAβは、神経細胞およびグリア細胞の表面にある様々な受容体と特異的に相互作用し、シグナル伝達カスケードを引き起こし、その結果、遺伝子転写/タンパク質発現の細胞内プロファイルを変更する6,7)。7, 8, 9 一般に、Aβ毒性に関する研究は、シナプス活性、神経細胞代謝、神経栄養因子の著しい低下、グリア活性化/炎症の増加など、主にその有害な作用に焦点を当てている。しかし、過去20年間の文献をより注意深く見ていくと、これらの減少の前に一過性の逆説的な上昇があるという二相性の傾向があることが指摘されている。10, 11これらは、AD患者において記述され、動物およびin vitroモデルにおいて再現され、最初の損傷に対する代償反応と解釈されている。代償はおそらく中枢神経系(CNS)の回復力に寄与し、より大量のAβに「耐える」能力を与え、症状の出現を遅らせる。このことは、Aβの病理学的レベルと実際の認知障害の程度との間に一定の相関がないことと一致する12。
このように、ADの後期介入に焦点を当てた戦略はすべて失敗しているので、初期の代償メカニズムに焦点を当てることは、この分野の今後の研究においてより注目に値する別の治療戦略であると言える。ここでは、ADの逆説的代償反応について、その主要な生物学的プロセスを個別にレビューしながら、最新の概要を紹介する。
2. シナプス要素
14, 15, 16, 17 したがって、疾患の進行に伴うシナプスの構造および機能の変化に着目することは、治療法の開発にとって極めて重要である。シナプスを構成する成分の発現が、初期には一過性に増加し、その後、神経変性の進行に伴って減少するという二相性の傾向を示すことが初めて観察されたのは、今から数十年前のことだ。ヒトやトランスジェニック動物モデルを用いた多くの研究により、グルタミン酸作動性、コリン作動性、GABA作動性のシナプス前ブトン密度の「予期せぬ」あるいは「逆説的」な増加が報告され始めた18, 19, 20, 21, 22。さらに、海馬の5-HT1A受容体密度は、MCIの初期には代償的に増加したが、ADの後期には劇的に減少した23。PETで評価した受容体密度の代償的増加は、実験モデルのラットの海馬で、セロトニン濃度の減少を誘発した結果、観察されている24。
その後、死後ADや痴呆性高齢者脳のさまざまな領域で、後期に減少する多くのシナプス成分について、同じ二相性発現パターンが示された。例えば、シナプス前タンパク質であるシナプトフィジン(SYP)、シナプトプレビン、SNAP-25、rab 3A、シナプス後タンパク質であるシンタキシン、ドレブリン、PSD-95などである。25、26、27、28、29 この観察はAD動物モデル30、31、32およびin vitroモデルで再現された33。34, 35 初期には、やはり軽度の認知症状と同時に、これらの多くが一過性に増加することが分かっている。26, 29, 36 中でも、シナプス小胞成分SYPは、シナプスの状態を評価する特徴的なマーカーとして知られている。一般に、SYPの増加はシナプス機能の改善と関連している。また、3xTg-ADの動物脳では、シナプス成分が最初に失われた後でも、SYPの領域および時間選択的な上昇37が現れ、この代償的な上昇は認知機能の部分的回復に関係し、その後、症状の悪化とともにSYPが失われた。我々の最近の研究では、ラットの海馬器官型スライスを亜致死濃度のoAβ42に暴露して得た遅発性神経細胞障害モデルにおいて、SYPレベルと小胞リサイクルにおける初期の代償的増加を示した。35 シナプス後密度タンパク質PSD-95についても同様の傾向が認められた。
これらの結果から、ここで議論したように、シナプス結合の減少を補うために、主要なプレーヤーを過剰発現させるという複合的な試みが存在することが確認された。
3. 神経活性化
Aβ毒性によってシナプスに引き起こされる代償的な構造変化は、機能的磁気共鳴画像法(fMRI)研究によって見られるように、神経活動の強化に相関を見出すことができる38。特に、Aβの集積が検出された脳領域でのみ、神経活動の亢進が見られることが、Aβの集積の有無にかかわらず、MCI患者と健常者を比較したfMRI研究によって示されている39, 40, 41, 42, 43。神経細胞の過活動の存在下では、認知機能が一時的に改善するが、この効果は神経細胞の損傷の程度が閾値を下回った場合にのみ顕著であった。このことは、ApoE 4対立遺伝子を持つ非健常高齢者が、代償機構を活性化することによって、マッチング3の患者と同等の記憶・学習能力を達成したという観察からも裏付けられている45。
47 亜致死濃度のAβを用い、神経細胞障害をゆっくりと進行させる動物実験やin vitroの研究に基づいて、シナプス可塑性や記憶の初期の改善は、神経伝達物質の放出速度の増加によってもたらされると提唱された。しかし、時間の経過とともに、これは興奮毒性障害とこれらの機能の低下の原因となるであろう48。
予期せぬことに、神経細胞の活性化だけに関係しないいくつかの分子も、疾患の進行に伴い二相性の発現パターンを示すようになった。例えば、発症前のADマウスモデルでは、一酸化窒素(NO)合成酵素のアップレギュレーションとNOの動員による代償機構が作用する。その結果、NOシグナルがカルシウム応答を増大させ、シナプス可塑性を救済することになる。同様に、コリン作動性ニューロンに選択的に発現するChol-1αガングリオシドが、AD患者の前頭葉でコリン作動性伝達を維持するための代償事象として増加している50。
ADの保護戦略としての神経活性化を伴う代償メカニズムの真の意義は、まだかなり議論の多い問題である。神経活動の改善は機能の回復を示す一方で、過活性化は長期的には興奮毒性につながり、神経細胞障害を悪化させる可能性がある。
4. 脳代謝活動
最近の研究によると、AD初期の一過性の代償反応として、[18F]-fluoro-deoxyglucose PET(FDG-PET)スキャンによって測定される脳のブドウ糖代謝の増加がある。51、52、53、54 しかし、ブドウ糖消費速度とAβ沈着の程度との真の相関については、矛盾が生じた。しかし、グルコース消費量とAβ沈着の度合いとの真の相関については、矛盾が生じた。Aβ病変のレベルが異なるMCI被験者グループのFDG-PET分析では、皮質の選択的領域における代謝率の増加は、Aβレベルの低い被験者に限られることが明らかになった。51 別の研究では、脳代謝は、MCIではAβ負荷と直接相関するが、ADでは逆相関することが示された54。ダウン症患者においても、グルコース代謝の二相性パターンが報告されており、認知症に先行する段階では代謝亢進が特徴的で、疾患が進行するにつれて徐々に減少していた55。対照的に、認知障害のある異種集団を登録した最近の研究では、このような正の相関は確認されていない。動物モデルで得られた結果は、臨床データと一致しているように思われる。ADのTg2576マウスでは、生後7ヶ月で脳代謝率のピークがあり、徐々に減少して生後19ヶ月で野生型レベルに達することが報告されている57。
MCIにおける代謝亢進は、ADの確立された危険因子であるインスリン抵抗性(IR)58とも関連していた。59 IRは、MCI安定被験者とは対照的に、ADへ進行するMCIにおける代謝亢進と、ADにおける代謝亢進と、領域特異的な形で関連していた。IRの役割は現在のところ不明であるが、MCI進行者に選択的に観察されるこのような二相性の傾向は、AD発症時にIRによって引き起こされる一過性の代償効果を示唆するものである。
興味深いことに、グルコース代謝亢進は、認知症状が現れる前のAβに対する耐性の程度に対する教育の影響の根底にあることも示された52。実際、教育水準が高いほど、AD患者における認知機能低下の速度が遅れる60。教育水準は必ずしも考慮されていないため、この知見は研究間の食い違いを説明するのにふさわしいと考えられる。
5. 神経栄養環境
ニューロトロフィンは、脳由来神経栄養因子(BDNF)、神経成長因子(NGF)、ニューロトロフィン(NT)3および4/5からなる小ペプチドのファミリーで、それぞれが選択的な受容体を介して作用する。ニューロトロフィンは、CNSの発達と恒常性、そしてシナプスのモデリングと機能に影響を与える。61 したがって、神経栄養環境のバランスが崩れると、必然的に神経細胞の機能と生存に打撃を与えることになる。MCIから早期および進行ADまで、さまざまな病期の患者の脳や血漿、動物モデルやin vitro試験におけるBDNFの発現を調べる多くの研究が行われているが、その結果は極めて多様である(62, 63に広範なレビューあり)。前述したように、ADの病態の程度と認知症状の程度との間に相関がないことはよく知られているが、このような比較は困難であり、病態の進行段階に由来するものと思われる。特に、代償反応は一過性であるため、同定が困難である可能性がある。しかしながら、BDNFの二相性発現の傾向を見分けることは可能であり、ADの初期に代償性の増加が現れ、その後、進行したADでは減少する62。興味深いことに、BDNFの発現を誘導することができる経口投与可能な神経栄養化合物P021の早期投与は、異なる老化およびADの動物モデルにおいて認知機能障害を回復させた65。これは、代償期の神経栄養環境をサポートすることが新たな戦略として期待できる結果である。
NGFとそのプロフォームの発現が増加したことが報告された。しかし、これは、BDNFレベルがすでに低下している進行したADの後期の出来事であった。66, 67, 68, 69, 70 したがって、これは、代償的生存の試み以外のメカニズムに関与している。
6. 酸化ストレス応答
活性酸素種(ROS)の産生と除去の不均衡に由来する酸化ストレスは、ADの初期の事象である。71, 72 脳はその高い酸素消費量により、特に高いROS濃度に曝される。加齢に伴い、酸化ストレスに対抗する能力が変化すると、脳はさらに活性酸素を蓄積しやすくなり、神経細胞の損傷や死が促進される73。興味深いことに、MCIでは、Mn-SODおよびグルタチオン還元酵素(GSSG-R)タンパク質の発現が代償的に増加することが報告されている77。AD患者においても、グルタチオンペルオキシダーゼ、GSSG-R、カタラーゼのタンパク質およびmRNAレベルが、過酸化脂質の増加を特徴とする選択的領域において、対照群と比較して増加した78, 79, 80 これらの結果は、活性酸素レベルの増加に対する代償的局所上昇を示唆しており、それはADを通じて持続し、抗酸化機能の喪失に先行する可能さえも含んでいる。また、抗酸化物質であるヘムオキシゲナーゼ1の発現は、ADと同様にMCIにおいても側頭葉およびHCで増加し、このことは認知能力と負の相関があった81。一般的な代償反応が実際に酸化ストレスに対抗することができるという証拠は、AD患者を対象とした研究から得られている。病態の異なる段階での評価により、酸化的損傷は初期段階で最も高く、A沈着が進行し、同時に病勢が進行するにつれて減少することが示された。注目すべきは、この相関関係がApoE4キャリアでより顕著に見られることである82。
83 したがって、銅結合タンパク質であるセルロプラスミンは、脳内への銅の侵入と鉄の酸化還元状態に関与しており、AD の脆弱な脳領域で選択的に増加していたことから、局所的に増加した酸化ストレスに対する代償反応であることが示唆された84。
最後に、ライソゾームの活性化とオートファジーは、ADの初期に代償的に誘導される抗酸化保護機構として浮上した。85, 86 しかし、疾患の進行の後半に、負荷がライソゾームのクリアランス能力を超えると、これらはうまくいかなくなる87。
7. 神経原性
成体神経新生は、HCの歯状回(DG)および哺乳類脳の脳室下帯(SVZ)において成体を通じて継続される。加齢や疾患は脳のニューロン新生の潜在能力を変化させ、一般的に低下する傾向にある。90 神経新生の障害は、患者やAD動物モデルで観察されるように、代償的な増加によって打ち消される。同様に、老化した脳では、ニューロン機能の低下は、ニューロン新生が維持されている個体で補われる91。
AD患者の死後脳研究では、DGおよびCA1における海馬のニューロン新生の増加が認められた92。さらに詳細な研究では、ニューロン新生の変化は、ニューロン新生プロセスの特定の段階および選択的ニューロン新生ニッチにおいて、ADの進行段階にも依存して異なることが示された。特に、DGの前駆幹細胞はADの初期段階で減少し、この影響は通過増幅細胞の代償的な増加によって対照をなす。93 したがって、神経新生の増加は、疾患のさまざまな段階においてではあるが、動物モデルのAD進行中に一貫して出現した。3ヶ月齢のAPPswe/PS1dE9マウスのDGとSVZの両方で、増殖の亢進と未熟な神経細胞マーカーの発現の増加が見られた。これらの変化は、アミロイドの沈着や神経細胞の消失に先行しており、初期の神経機能障害によって引き起こされたと思われる初期の代償反応を示している。94 ADの異なる遺伝子モデルマウスに関するさらなる研究では、若い動物では神経新生が代償的に増加し、その後、高齢になって海馬成人の神経新生が減少することが確認されている95、96、97 一方、同様の増加は、すでに記憶障害とAβ沈着を起こした高齢動物でも報告された98。前駆細胞の増殖の増加が、必ずしも成熟した機能的なネットワーク統合型ニューロンへの新細胞の実際の発達に続くとは限らないことは興味深い。93, 99 全体として、これらのデータは、代償性神経新生の活性化の時間/領域パターンは一定ではないものの、その促進は疾患の進行に対する重要な防御機構である可能性を示すものである。したがって、アセチルコリンエステラーゼ阻害剤ガランタミン、NMDA拮抗剤メマンチン、エストロゲン、アロプレグネノロン、ドーパミンD2/3受容体作動薬プラミペキソールなど、神経新生をアップレギュレートすることが知られている薬剤やホルモンが、in vitroおよびin vivo試験から大きな抗神経変性能を有すると考えられている100, 101, 102, 103, 104。
注目すべきは、他の反応でも証明されているように、補償の増強における環境の充実の寄与は、神経発生においても証明され、新しく生成されたニューロンの生存と成熟の減少を救うことができることである105。
8. グリア細胞
グリア細胞は、神経細胞を構造的に支持するだけでなく、神経細胞の発達と機能において積極的な役割を担っていることは、長い間、確立されてきた。シナプスレベルでは、神経細胞とグリア細胞は密接に相互作用し、グリアはシナプス伝達を直接調節している106, 107。
ミクログリアは、疾患進行の初期と後期において、2つの活性化段階を示す。MCIとADの患者を14ヶ月にわたって追跡調査した結果、MCIでは、抗炎症性で保護的なミクログリア表現型の初期ピークが実際に出現していることが示された。しかし、アミロイドの負荷が増加し、おそらくそのクリアランス能力を超えると、二次的な活性化が始まり、ミクログリアは炎症性表現型に切り替わる。108 アミロイド濃度の上昇にさらされた初代ミクログリアでは、膜貫通型受容体TREM2の過剰発現が抗炎症表現型に有利であることが分かっている。このことは、TREM-2を介した保護は、特定の疾患段階においてのみ、おそらくミクログリアの機能状態に依存した意義をもたらすことを示唆している。
ミクログリアで起こることは、時にアストロサイトでも再現され、両方の細胞タイプがADのモデルにおいて神経栄養環境を改善する。例えば、トランスジェニックマウスのプラーク周辺ではアストロサイトおよびミクログリアのBDNF産生が有意に増加し111 、致死濃度以下のオリゴマーAβ42に暴露したラットの初代ミクログリア細胞では選択的に増加した112 。
10 アストロサイトの反応性表現型の出現は、ADマウスモデルにおいてもAD患者においても、Aβ沈着が出現する前に起こる比較的早いイベントである114。このようなアストログリオシスは代償的なイベントであり、実際、この活性化を抑制すると、APP/PS1マウスにおいてジストロフィック神経突起の数が増え115、プラーク形成を加速した116。
一方、トランスジェニックマウスでは、Aβ沈着の結果としてのみ、GFAPレベルの上昇を伴う肥大化したアストロサイトが出現し、疾患後期のプラークを取り囲むだけであると報告している117, 118 興味深いことに、これらの反応の相違は、地域特異的であると思われた。117, 119, 120著者らによると、ECで観察された低栄養は、この領域がAD病理学に対してより脆弱であることに寄与している可能性があり、反応性グリオシスの欠如がネガティブな転帰と相関していることが確認された。別の研究では、認知症を伴うAD病態(AD-D)または伴わないAD-Nの被験者の死後EC領域におけるアストログリア反応性を分析した121。どちらも正常脳と比較してI/II層でGFAP+アストロサイト数が増加することが特徴的であった。しかし、AD-Nでは、これらのアストロサイトは、グリア反応性を示す太く長い突起を示し、グルタミン酸トランスポーター-1(GLT-1)の発現も亢進していた。これらのアストロサイトはCA1へのシナプス伝達の維持に重要な役割を果たしており、AD-N群におけるこれらの選択的な反応性は、シナプス損傷に対する代償機構であることが示唆された121。
注目すべきは、老齢3xTg-ADマウスおよびAD患者におけるアストロサイトの代償には、膜Kir6.2チャネルも関与しており、その機能によりグルタミン酸の取り込みが促進されることである122。
最後に、アストログリアがAβ沈着後/疾患発症後期に発現するという仮説に沿って、in vitroでのAβへの曝露、またはTgCNRD8 AD動物におけるAβ沈着の存在は、解糖系酵素6-phosphofructo-2-kinase/fructose-2,6-biphosphatase(PFKFB3)の発現を通じたアストロサイト生体エネルギーの変化を引き起こした。これにより、酸化的リン酸化過程が損なわれている状況下で、代替エネルギー源としての乳酸の神経細胞への供給が増加する。特に、PFKFB3を阻害すると、アストロサイトはAβの毒性に対して脆弱になる118。
9. 結論
ADの代償機構は、長年にわたってよく知られている。代償機構は、脳機能と恒常性に関わる幅広い生物学的プロセスから構成され、本疾患に罹患した脳領域において、早期に、一過性に、そして選択的に出現するものである。シナプス構造の強化は、シナプス活性のアップレギュレーションにつながり、また神経新生も、破壊されたネットワークに新しいニューロンを送り込むことに貢献する。同時に、脳の代謝が増加し、抗酸化作用が増強される。この間、グリア細胞は活性化され、これらすべての反応の実行に寄与している。神経細胞にとって有害な状態に対抗するための試みである可能性が高いが、補償の最終的な結果が本当にポジティブなものとなり得るかどうかは、まだ明らかではない。一方では、代償反応は病態の進行に対する回復力を促進することに成功したことを示唆するデータもあるようだ。一方、神経活動やオートファジーのようなプロセスが逆説的に増加することで、神経細胞の損傷が悪化する可能性もある。神経細胞の障害に対する抵抗力の異なる閾値の存在は、認知的予備能と表現されている。123 これは、ADの初期脳機能低下に対する個人の補償を活性化する能力を決定する上で主要な役割を担っている(図(Figure1).1)。したがって、代償作用の増強が有効な戦略である可能性が高く、認知的予備能-代償-回復力という軸の中心にある細胞メカニズムに注目することが必要であると考えることができる。つまり、代償現象の有益な部分を利用し、その一方でネガティブな部分を抑制することで、ADの疾患修飾、あるいは予防につながる新たな根拠を提供することができる。
図1 ADの神経保護戦略としての補償
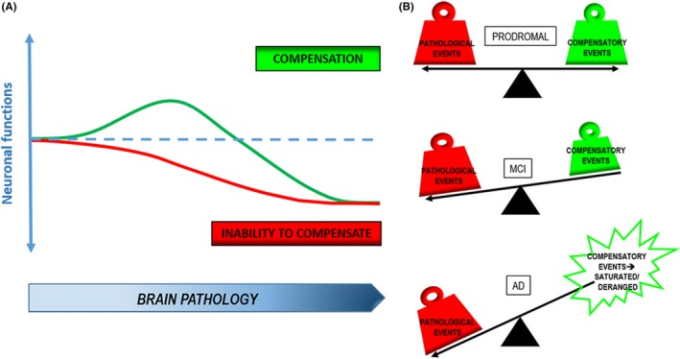
A, Aβ/tauによる脳病変が増加すると、神経機能の低下を打ち消すために代償現象が起こり、脳病変に対する耐性が増加する。代償反応の導入ができない場合、より早い段階で機能が低下する。B, 代償事象は、疾患の前駆期には病理学的事象と均衡を保っているが、MCIでは病理学の過負荷により機能しなくなり、重度のADではもはや病理学と効率的に対比できなくなるか、有害なものとさえなっている