
Does Glyphosate Acting as a Glycine Analogue Contribute To ALS?
要旨
筋萎縮性側索硬化症(ALS)は、グリシンを多く含む領域のいくつかのタンパク質変異を伴う致死的な神経変性疾患であり、治療法は限られている。全症例の90~95%は家族性ではなく、グリホサートに曝露された労働者のリスクが有意に高いことが疫学的研究で示されている。
本論文では、ラウンドアップ®の有効成分であるグリホサートが、主にタンパク質合成時にグリシンと誤って置換され、ミネラルのホメオスタシスを破壊し、バイオシスの状態を作り出すことにより、ALSの一翼を担っていることを提案する。
ALSのマウスモデルでは、症状が出る前の腸内環境異常のプロファイルが明らかになっている。この腸内生物異常状態は、最初は腸内代謝に障害を与え、最終的には一連の中間段階を経て、ALSで見られる運動ニューロンの軸索損傷に至るまでの一連のイベントのカスケードを開始する。
腸内代謝異常の副産物であるリポ多糖類は、ALS患者では統計的に高い値を示している。本論文では、グリホサートがどのようにしてグリシン置換によるミトコンドリアストレスや酸化損傷などの有害作用を発揮するのかを明らかにした。
さらに、グリホサートのミネラルキレート特性は、マンガン、銅、亜鉛のバランスを崩し、シナプス内でグルタミン酸の毒性を誘発し、その結果、損傷を受けた骨格筋に供給されている運動ニューロンの軸索でダイバック現象を引き起こすことになる。
はじめに
2016年9月5日にALSの合併症のために亡くなったドウェイン・グラベリン博士を偲んで
筋萎縮性側索硬化症(ALS)は、筋肉の萎縮とともに運動ニューロンが徐々に失われていくことを特徴とする致死的な神経変性疾患である。ALSは成人に発症し、通常、最初に手足の脱力感が現れ、麻痺や呼吸不全による死亡へと進み、診断から5年以内に発症することが多い[1,2]。生涯のリスクは400人に1人から1000人に1人であり、最も一般的な運動ニューロン疾患である[3-5]。現在のところ、治療法や治療法は知られていない。
最近のエビデンスでは、この疾患の初期段階では腸内環境異常が重要な役割を果たしていることが指摘されている。2015年に発表されたWuらの論文では、ALSマウスモデルにおける腸内微生物と関連代謝物の変化を調べ、特に疾患発現前の障害を標的とした研究が行われた[6]。このALSマウスモデルでは、残基93のグリシンがアラニンに置換されたヒトスーパーオキシドジスムターゼ1(SOD1)の変異体が発現している。この研究により、ヒトG93A SOD1を発現した若いマウスは、損傷したタイトジャンクション構造を示し、腸管透過性が増加したことが明らかになった。
これは、タイトジャンクションタンパク質であるゾヌリン・オクルーデンス-1およびE-カドヘリンの発現低下と関連しており、腸管バリア機能の障害につながっていた。パネス細胞もまた、抗菌ペプチドであるディフェンシン5αの発現が低下した異常な細胞であった。ブチリブリオ・フィブリオ・フィブリソルベンスやフェルミカスのような酪酸産生腸内微生物のレベルの低下、炎症性サイトカインIL-17の発現の増加、オートファゴリソザイム1のレベルの低下(誤って折り畳まれたタンパク質を除去する能力の低下につながる)は、マイクロバイオームの障害のさらなるマーカーであった。これらの特徴はすべて、ALSの症状が現れる前の生後2ヶ月のマウスに現れた。
ALSのうち、既知の遺伝的欠陥と関連があるのは約5~10%の症例(家族性ALS)のみであり、残りは特発性または散発性のALSである。家族性のALSの約20%はSOD1の変異と関連している[7]。家族性疾患と特発性疾患の間には、病理学的および臨床的に顕著な類似性があり、研究者は、マウスSOD1モデルが家族性疾患だけでなく散発性疾患の代表である可能性があると考えている。
SOD1の主な機能は、ミトコンドリアの酸化的リン酸化の副産物として生成されるスーパーオキシドを水や過酸化水素に変換することである。しかし、変異型SOD1の劇症的な影響は、この機能の喪失によるものではなく、ほとんどの変異型は完全な活性を示すか、あるいは活性レベルが向上している[3]。
ヒトにおけるSOD1の変異は180以上確認されているが、3つの変異がトランスジェニックマウスモデルで広範囲に特徴づけられており、そのすべてが元のタンパク質の保存されたグリシン残基の置換を伴うものである(SOD1G85R、SOD1G37R、およびSOD1G93A)。
多くの場合、変異体タンパク質は、内因性SOD1のレベルよりも数倍高いレベルでマウスゲノムに発現する[8]。fALS患者において、アルギニンを保存性の高いグリシン(G10R)に置換すると、タンパク質の二次構造が強く不安定化し、細胞内凝集体が形成されることが明らかになった[9]。
ミトコンドリア機能の低下はALS、特にチトクロームc酸化酵素(CcO)に関連しており[10,11]、変異型SOD1はCcO活性を抑制することが示されている[12]。興味深いことに、CcOとSOD1の両方が銅を触媒としている。G93A-SOD1マウスを用いた研究では、変異マウスは複合体IVに特異的なミトコンドリア呼吸の減少を示し、この欠損はチトクロームc酸化の障害と関連しており、自覚症状が現れるずっと前から発生していたことが示された[13]。
ミトコンドリア内膜上のCcOの量が減少し、これはカルジオリピンを含む脂質の過酸化の増加と相関していた。一つの説明として、変異型SOD1の過剰発現は、銅のCcOに対するバイオアベイラビリティーの低下をもたらし、複合体IVの酸化的リン酸化の障害と活性酸素種の過剰発現の両方をもたらしたと考えられている。
もう一つ提案されている理論は、変異型SOD1は、触媒作用を持つ銅とペルオキシナイトライトや過酸化水素との相互作用を促進する構造変化を誘導し、有毒なフリーラジカルを発生させ、細胞のタンパク質や脂質を損傷させるというものである[12]。
驚くべきことに、銅の協調結合に必要なすべての重要な残基を欠いたSOD1の変異体は、他のヒトSOD1変異体を発現したマウスの疾患表現型と区別がつかないALS様疾患をマウスに誘導する[14]。この病気の症状は、誤って折り畳まれた高分子量のSOD1凝集体の蓄積として現れる。この特徴は、グリシン置換マウスモデルでも観察されている。このことは、銅の欠乏または銅結合の障害が、SOD1およびCcOの両方の誤った銅欠乏バージョンと一緒に遊離銅による酸化ストレスおよび窒素ストレスを引き起こし、病気のプロセスの要因である可能性を示唆している。
Beckmanらの研究では、ヒトSODとスーパーオキシドジスムターゼ(CCS)のためのヒト銅シャペロンの両方を過剰に発現させたマウスを調べた[15]。これらのマウスはALSで加速度的に死亡するが、これはおそらくCCSがマウスの銅供給量を隔離し、脊髄に銅欠乏を誘発したためであろう。
銅の枯渇はSOD1とCcOの両方の機能に影響を与えた。2016年の研究では、SOD1への銅供給とCcOへの銅供給の障害がこの病気の強い要因であることをほぼ決定的に示している[16]。これらの著者らは、ジメチルスルホキシドに溶かして子犬の首に滴下した銅キレート剤CuATSMが皮膚に吸収され、中枢神経系への生物学的に利用可能な銅の供給源となることを示した。
この治療法は、SOD1の変異とCCSの過剰発現の両方に悩まされた疾患マウスの運動性を数時間以内に大幅に向上させる効果があった。一般的なモデルとして、変異型SOD1はタンパク質のミスフォールディングと酸化ストレスの両方を誘導し、おそらく遊離銅を介してミトコンドリアの機能不全を引き起こし、軸索に沿った輸送障害が神経炎症と運動ニューロンのアポトーシスにつながるということがわかってきている[17]。
ALSにおけるミスフォールドされたタンパク質の蓄積はSOD1に限定されない。2つの核内RNA/DNA結合タンパク質、TAR(トランザクティブ・レスポンス)DNA結合タンパク質43(TDP-43)とFUS(FUsed in Sarcoma)は、いずれもALSと遺伝的に関連していることがわかっている[18]。
TDP-43は、家族性、散発性を問わず、ALSの包接体ではほぼ常にユビキチンとの凝集体で見られる[3,19]。実際、驚くべきことに、TDP-43またはFUSの病理学的形態は、内因性野生型スーパーオキシドジスムターゼの細胞障害性ミスフォールディングをプリオンのような方法で播種することがわかっている[18]。
これらの包接体は免疫炎症反応を誘発し、ミトコンドリアや他の細胞プロセスにさらなる損傷を与える。ユビキチン陽性細胞内包体は、ユビキチン-プロテアソームシステムの障害を受けているという事実[20]。
本論文では、研究文献を深く広く検索することで、4つの異なるトピックに関連する論文を見つけ出した。
- (1)ALS の疾患メカニズム、
- (2)細胞外マトリックスや糖タンパク質におけるグリコサミノグリカンの合成に関与する代謝経路、
- (3)グリホサートが ALS の原因因子であることを示す疫学的証拠とグリホサートの毒性メカニズムに関する論文、
- (4)上記のすべてに関連するタンパク質におけるグリシンの多くの役割であること、
である。私たちはまず、ALS のマウスモデルにおける疾患過程における腸内環境異常の早期関与を示した Wu ら[6] の新しい論文に刺激を受けた。
私たちの最初のステップは、ALSに関連する遺伝的変異を持つほとんどのタンパク質を列挙し、これらのタンパク質のグリシンの置換やグリシンに富んだ領域内での変異がALSと関連しているかどうかを特別に探すことであった。
私たちは特に、フコースが枯渇した免疫グロブリンとALSとの関連性に注目し、フコースの合成、輸送、糖タンパク質への付着に関与するすべての酵素や代謝経路を詳細に記述した論文を徹底的に検索した。また、ALSにおけるミネラルアンバランスの役割や、フコースの前駆体であるフルクトースに関連する複雑な代謝経路についても調査した。我々の方法の新規性は、様々な分野の研究文献を共通のストーリーラインに統合したことである。
この論文の残りの部分では、ALSの発症過程のギャップを埋め、腸内細菌叢の異常がどのようにして最終的に運動ニューロンの劣化につながるのかを、いくつかの段階を経て示していく。腸内細菌叢の崩壊は、ALSの症状が表に出る前から始まり、代謝障害、特に果糖代謝の障害、ステアトーシス、骨格筋細胞の損傷、そして最終的には脊柱の運動ニューロンの劣化にまで及んでいる。普及している除草剤ラウンドアップ®の有効成分であるグリホサートがALSに関連して観察される障害の多くを引き起こす可能性があるという理論の文脈で、このカスケード全体について説明する。
この障害の多くは、グリホサートがグリシンアナログとして作用し、タンパク質合成の際にグリシンの代わりになることに起因すると考えられている[21]。グリホサートが鉱物の恒常性を破壊することもまた、特にマンガン、銅、亜鉛の役割を果たしている可能性が高いと考えられる[22-25]。
グリシンアナログとして作用するグリホサート
グリホサート製剤は、加工食品産業の主要作物、特にトウモロコシ、大豆、キャノーラ、テンサイなど、グリホサート耐性になるように遺伝子操作された作物の間で生育する雑草を制御するために広く使用されている。米国では、雑草の間でグリホサート耐性が広く出現したことに伴い、過去 20 年間でグリホサートの使用量が劇的に(100 倍に)増加している[26,27]。一般的にグリホサートは人間にはほとんど毒性がないと考えられているが、世界保健機関(WHO)のIARCは2015年にグリホサートを「発がん性物質の可能性が高い」と表示した[28]。
信頼できる試験では、人間がグリホサートに曝露されていることが確認されている。Krügerらは数百人のヒトの尿サンプル中のグリホサートのレベルを測定し、主に有機食品を摂取している人と比較して、従来の食事を摂取している人の方が統計的に有意に高いレベルであることを発見した(p < 0.0002) [29]。さらに、慢性疾患のある人は、健康な人に比べて有意に高いレベルを示した(p < 0.03)。
最近の証拠は、グリホサートは、障害されたマイクロバイオームによって部分的に媒介されている陰湿な毒性効果を持っていることを示唆している[27,30]。植物における重要な障害経路であるシキメート経路は、多くの腸内微生物にも見られ、この経路は宿主に必須の芳香族アミノ酸やその他の栄養素を供給する。この経路の閉塞は、腸内微生物のバランスを崩し、果糖代謝の障害につながる。我々はここで示すように、これは人間の生理に深刻な影響を予測している。
グリホサートのより不穏な特徴は、グリシンのアミノ酸アナログとしての役割に由来する。Samsel and Seneff[21]では、グリホサートがタンパク質合成中に誤ってグリシンの代わりになることが提案されている。実際には、これがシキメート経路のEPSPSを混乱させる働きをしているのだろう。ストレス状態の間に生物によって産生される他の天然アミノ酸類似体との前例の証拠に基づいて、強力なケースが作られた。
実際、シアノバクテリアが産生する非コードアミノ酸β-メチルアミノ-L-アラニン(BMAA)は、L-セリンの代わりにタンパク質に取り込まれ、ALS様の状態を引き起こしている[31,32]。
グアムでのALSのパンデミックはソテツの種子にBMAAが混入していることが原因とされている[31]が、日本の紀伊半島ではALSの発生率が非常に高いことが原因と考えられている[32]。腸内細菌叢に存在するシアノバクテリアが合成するBMAAは、ALSやパーキンソン認知症、馬の運動ニューロン病などに関与していることが知られている[33]。
根圏とは、植物の根が関与する土壌の領域であり、特に栄養素の吸収に重要な役割を果たしていることを説明するために使用される用語である[34]。興味深いことに、グリホサート処理後の根圏における細菌遺伝子発現の変化を調べた研究では、タンパク質生合成とタンパク質分解の両方に関与するタンパク質の顕著なアップレギュレーションが示された[35]。このことは、グリホサートがタンパク質の誤形成を引き起こし、誤形成されたタンパク質の合成と分解のサイクルを繰り返すことでエネルギーの浪費を誘発したことを示唆している。
Swansonら[36]は、主要作物へのグリホサート使用量の劇的な増加に歩調を合わせて、米国では多くの衰弱性疾患が驚くほど増加していることを示している。SamselとSeneff[21]では、Swansonらの論文で同定された疾患の多くは、グリシンが高度に保存されており、重要な機能的な役割を果たしている特定のタンパク質におけるグリシンのためのグリホサート置換を通して説明することができるかが示された。
我々は、ALSに至る病態はゆっくりと始まるが、肝機能と腸管上皮バリアが深刻な障害を受けると加速するという仮説を立てた。腸管バリア機能が障害されると、グリホサートが全身循環に到達するようになる。腸内での果糖代謝の障害により、過剰な果糖が蓄積する可能性がある。
肝臓が果糖を除去することができないため、特に運動量が多く、脂肪組織が少ない状況下では、筋肉細胞にストレスがかかる。プロテアソームによってクリアできない筋肉内のグリホサートが埋め込まれたタンパク質の高いタンパク質のターンオーバーと蓄積は、最終的には筋肉のシグナル伝達に関与するタンパク質に対する自己免疫反応につながる。
これは、病気になった骨格筋とシナプスを形成する運動ニューロンにストレスを与える。軸索末端から始まるダイバックは、最終的には制御している運動ニューロンの細胞アポトーシスを引き起こし、運動ニューロンタンパク質を標的とした自己免疫反応のさらなる進展と関連していることが多い。
農薬とALSの関連性の証拠
一般人口におけるグリホサート曝露の最も重要な原因は食品であることが疑われるが、農業従事者は職業性グリホサート曝露のリスクがはるかに高い。グリホサートに関する具体的なデータを得ることは難しいが 2014年の韓国の研究では、農薬曝露(OR = 1.44)と農業職業(OR = 1.42)の両方でALSへのリスクの増加が示されている[37]。
ミシガン州で実施されたALSと診断された150人の患者を対象とした調査では、さらに強い関連性が見出された。累積的な農薬曝露はALSと高い有意な関連性を示した(OR = 5.09,p = 0.002)[38]。ミシガン州では遺伝子組み換えラウンドアップレディ大豆が大量に生産されていたため、韓国に比べてグリホサートがはるかに高いレベルで使用されていた可能性がある。
大豆は2012年のミシガン州最大の輸出品目であり、その価値は8億ドル以上であった。オーストラリアを拠点とする研究では、工業用除草剤および農薬への曝露について、用量反応関係があり、ORは5.58であることが明らかになった[39]。ブルターニュの研究では、「農業活動」とALSの関連性についてオッズ比2.919(p = 0.01)が得られており、農業に従事している人の間ではALSのバルバー型が優勢であった[40]。
グリホサートは、L型アミノ酸トランスポーターによる活性輸送を介して、鼻腔内の上皮粘膜バリアを容易に通過することが最近明らかにされている[41]。
これらの著者は、”グリホサートが脳に入るためのこの追加の経路は、経口または静脈内暴露に基づいて以前に予想されていたよりもはるかに高い脳濃度をもたらす可能性があり、また、報告されている神経毒性の発生を説明する可能性がある “と書いている。
レジスタンス運動は、L型アミノ酸を含む筋肉細胞内のアミノ酸トランスポーターの発現を増加させる(p < 0.05)[42]。これは、運動が筋肉のターンオーバーを増加させるため、タンパク質合成を必要とすることから予想される。
しかし、このことは、運動量が多く身体的に健康な人ほど、L型アミノ酸トランスポーターを介した輸送を介してグリホサートを筋細胞に蓄積する可能性が高いことを示唆しており、身体的に健康な人の間で観察されたALSのリスクの増加を説明するのに役立つかもしれない[43,44]。グリホサート残基はニワトリ[45]やウシ[29]の筋肉から検出されている。
表1:筋萎縮性側索硬化症に関与している機能障害を持ついくつかのタンパク質で、必須グリシンが高度に保存されているもの
タンパク質 グリシン残基 参考文献
- SOD1 G10,G37,G85,G93 C Ricci er al 2010[9]
- L Bruijn er al)。 1996[8]
- TDP-43 C末端グリシンリッチ領域 GS Pesiridis er al 2009[63]
- RNA結合に重要なFUSグリシンリッチ領域 SK Dhar er al 2014[61]
- SLC351 G180,G198,G277 Zhang er al 2012[80]
- 複合体I Gx(x)GxxGモチーフ ME Bakerら[66]
- CcO G283 L サロモンソンら 2004 [87]
- ユビキチンC末端ダブルグリシンA Zuin et al 2014[46]
- ミオシンG699 NM。キノセ et al 1996[175]
- キネシンG292 BJ Grant et al 2007[176]
ALS関連タンパク質における保存グリシンの役割
私たちは以前、SOD1 内の複数のグリシン置換が ALS マウスのモデルとなりうることを指摘した。実際、私たちは、表1に示すように、SOD1 の他に、ALS の病態に関与する高度に保存されたグリシンを持ついくつかのタンパク質を同定した。最も興味深いのは、ユビキチン自体が、タンパク質の分解をシグナルする複雑なユビキチン鎖を構築するために、高度に保存されたカルボキシ末端の二重グリシン対に決定的に依存しているという事実である[46]。グリホサートをこれらの必須グリシンのいずれかに置換すると、誤って折り畳まれたタンパク質のリサイクルプロセスが損なわれると予想される。このことは、ALSの特徴である誤って折り畳まれたタンパク質の蓄積を容易に説明することができるだろう。
ALSには、SOD1,TDP-43,FUSなど複数のタンパク質の凝集が関連している。これら3つのタンパク質はすべて保存性の高いグリシンを含んでおり、ALSに関連した変異の多くはグリシンを多く含む領域に集中しており[47,48]、保存性の高いグリシンのために他のアミノ酸の置換を伴うことが多い。このことから、これらのグリシンは、タンパク質のミスフォールディングからの保護を介して、ALSに対する保護を行っている可能性があることが示唆されている。
アシルホスファターゼは、生命の3つのドメインにまたがるグリシン残基の大部分が高度に保存されている小さな酵素である[49]。ヒト筋肉アシルホスファターゼの6つの異なるグリシン残基を他のアミノ酸で系統的に置換する実験では、G15A置換のみが酵素活性の劇的な低下を引き起こすことが明らかになった。
しかし、テストした他のすべての置換は、タンパク質の凝集傾向を著しく増加させる結果となった。著者らは、これらの他のグリシンが高度に保存されている可能性が高い理由は、タンパク質の凝集から保護するためであると結論づけた。グリシン残基は、側鎖の欠如に起因する、コンフォメーション空間のより広い領域を占有することができる。グリシン残基が無秩序な構造からβ鎖、凝集に向けた重要なステップに変換するとき、これは、実質的なエントロピーペナルティにつながる。グリホサートをこれらのグリシンのいずれかに置換すると、タンパク質の凝集も促進することが予測される。
ある論文では、SOD1を150のミスセンス変異について解析し、全体的に、変異によって正味電荷が非常に有意に減少し、凝集傾向が増加することを示した[50]。安定性とネット電荷の変化の程度は、ALS患者の生存期間と逆相関していた。著者は「プロテオームの枯渇」という用語を用いて、病気と結びついた根本的な病理学のために提案した理論を説明している。誤って折り畳まれたSOD1分子を交換するために高いターンオーバー率が必要とされることは、すでに高エネルギーを必要とする細胞タイプである運動ニューロンに高いエネルギー需要をもたらす。この著者は次のように書いている:「この解析は、ALSのようなタンパク質のミスフォールディング疾患が、ミスフォールディングされたタンパク質種の特定の分子毒性によって必ずしも引き起こされるのではなく、タンパク質のターンオーバーの上昇による全身的な疲労によって引き起こされる可能性があることを示している。このメカニズムは、このような悪質なタンパク質状態の同定がこれまでのところ成功していない理由を説明することができる。” SOD1の保存されたグリシンのいずれかのグリホサート置換は、負の電荷の増加と凝集傾向の増加の両方をもたらすだろう。このことは、野生型SOD1でさえもALSの病理学にリンクさせることができることを説明することができる[51,52]。
興味深いことに、TDP-43とFUSの両方は、「RNA結合タンパク質」として知られている幅広いクラスのタンパク質のメンバーである[53]。FUSはRNAからタンパク質合成への翻訳の活性化に関与している[54]。TDP-43はタンパク質の品質管理を維持し、タンパク質の折り畳みストレスに応答し、誤って折り畳まれたタンパク質のレベルを調節する[55]。オプティニューリンはもう一つのRNA結合タンパク質であり、これもまたALSと関連している[56]。オプティニューリンの機能が損なわれると、受容体間相互作用キナーゼ1(RIPK1)依存性のシグナル伝達により、進行性の脱髄と軸索変性を引き起こす。RIPK1活性化は変異型SOD1によっても誘導される[56]。特にオリゴデンドロサイトが標的となり、オプティニューリンの障害はオリゴデンドロサイトによるミエリン鞘へのミエリンの供給障害をもたらす。
RNA結合タンパク質は、通常は可逆的なプロセスでメッセンジャーRNAとともにタンパク質の凝集を「ストレス顆粒」(SG)に誘導することでストレスに反応する[53]。細胞質内のSGはRNAの代謝や恒常性に大きく関与している[53,57-59]。SGは、ホメオスタシスや温度の変化、感染や化学物質への曝露などの外部要因、ミトコンドリアや酸化ストレスなどの内部要因など、特定の細胞ストレス要因の部位に蓄積することで機能すると考えられている。かつては病理学的プロセスで役割を果たすと考えられていたが、現在ではSGが生理的プロセスとして保護的な役割を果たすことが認識されている。RNA結合タンパク質は標的mRNAをリクルートしてSGに丸め込み、細胞ストレスが収まると分解される[60]。これは、選択されたタンパク質の合成を一時的に停止させる方法である可能性がある。しかし、病的なALSタンパク質はSGの機能を劇的に破壊し、それによって神経細胞の損失をさらに永続させる能力を持っているという証拠がある。ALSや他の多くの神経疾患では、ストレス顆粒がタンパク質包接体として蓄積することで、最終的に細胞機能が障害される。
RNA結合タンパク質が、そのグリシンを豊富に含むドメインを介して特異的に凝集することは驚くべきことである[53,61]。FUSのエクソン6にはアミノ酸残基222から231までの10個のグリシンが伸長しており、fALSの複数の症例はこれらのグリシンのいくつかの欠失と関連している[62]。TDP-43のグリシンに富むC末端領域は特に凝集しやすい。図1に示すように、このグリシンに富む領域における複数のミスセンス変異がALSと関連している[63]。これらのタンパク質はいずれもSOD1のミスフォールディングに応答してアップレギュレーションされることが予想される。細胞毒性顆粒関連RNA結合タンパク質(TIA-1)もまた、グリシンに富んだ領域を持つRNA結合タンパク質である[53]。
SOD1のG93を代替アミノ酸で置き換えようとする進化的圧力があるように思われる。挑発的に言えば、これはグリシンのグリホサート破壊に対する脆弱性などの環境要因が突然変異率に影響を与えうることを意味している可能性がある。これまでに、セリン、バリン、アスパラギン、アラニン、システイン、アルギニンの6つのアミノ酸置換が確認されている[64]。興味深いことに、一般的な突然変異であるG93Sは、親に比べて子孫においてより病原性の高い表現型を持っている。9組の親子ペアで平均してみると、平均発症年齢は親では64.4歳であったのに対し、子孫では44.8歳であった。グリホサートのような相乗的な環境因子の増加が、この観察結果を説明する可能性がある。
短鎖脱水素酵素/還元酵素(SDR)スーパーファミリーは、これまでに知られている最大のタンパク質スーパーファミリーの一つである。これらの酵素は、ニコチンアミド・アデニン・ジヌクレオチド(P)(H)-(NAD(P)(H)-)依存性反応を触媒し、その基質はポリオール、レチノイド、ステロイド、脂肪酸誘導体、異種生物学的物質など多様である[65]。このファミリーのメンバーはヒトゲノム上に少なくとも73人いる。これらのメンバーは、高度に保存されたグリシンに富んだ構造モチーフであるGx(x)GxxGを、複合体IのNADH:ユビキノン酸化還元サブユニットと共有している[66,67]。高度に保存されたGxGxxGモチーフは、様々な種のNAD(P) H:キノン酸化還元酵素に見られる[68]。共通の祖先から子孫を継いでいない酵素はこのモチーフを共有しているため、このモチーフは基本的なものであると考えられている。ALS患者のリンパ球では、コンプレックスIの活性が低下していることが示されている[69]。
ヘキソサミン生合成経路は、基質としてフルクトース-6-リン酸とグルタミンから始まり、最終生成物はウリジン二リン酸N-アセチルグルコサミン(UDP-GlcNAc)であり、これはヌクレオチド糖であり、グリコシル化タンパク質のヘパラン硫酸およびコンドロイチン硫酸鎖に組み込まれる他の多くの誘導体糖の前駆体である。
図1:TDP-43の複数のALS関連遺伝子変異は、C末端のグリシンに富んだ領域に集中している
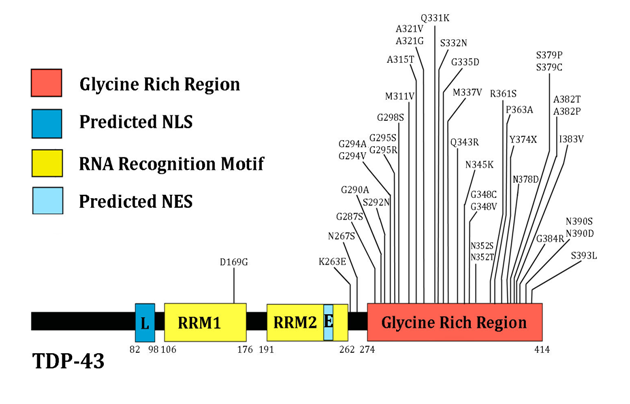
NLS:核局在化シグナル;NES:核輸出シグナル;RRM:RNA認識モチーフ。図はLagier-Tourenneら[48]より引用。
フコースやキシロースなど、これらの誘導体糖を産生する酵素の多くは、SDRファミリーのNADPH依存性メンバーであり、したがって、これらの酵素はNADPH結合に必須のGx(x)GxxGモチーフを持っている[70]。
ヘリコバクター・ピロリのUDP-GlcNac脱水酵素であるFloA1のGxxGxxGモチーフのG20A突然変異は、活性の完全な喪失をもたらし、ヌクレオチド結合部位の構造変化が関与していることを示唆している[71]。ナマコのコンドロイチン硫酸鎖の研究では、フコースの分岐が分解に対する耐性を誘導することが示された[72]。
これらの酵素の欠乏は、腸粘膜ライニングを維持する能力の障害だけでなく、未代謝のフルクトース-6-リン酸およびグルタミンの過剰量の蓄積をもたらす可能性がある。
より一般的には、GxxGモチーフは、酵素のこの広いが重要なクラスのメンバーであるオキシドレダクターゼにおけるFADおよびNAD(P)の両方への結合を安定化するために重要である[70]。UDP-GlcNAcとその誘導体であるフコースとキシロースの両方は、グリコサミノグリカン、プロテオグリカン、グリコ脂質の必須構成要素である[73]。SDRファミリーの重要なメンバーはUDP-キシロース合成酵素(UXS)であり、UDP-グルクロン酸(UDP-GlcA)デカルボキシラーゼおよびUDP-GlcAカルボキシラーゼとしても知られており、UDP-グルクロン酸の脱炭酸を触媒してUDP-キシロースを生成する[74,75]。
キシロシル転移酵素は、プロテオグリカンコアタンパク質中の選択されたセリン残基へのUDP-キシロースからのキシロースの転移を触媒する酵素である[76]。これは、哺乳類グリコサミノグリカン合成における初期および速度制限ステップである。したがって、UDP-キシロースの安定した供給は、腸内の健康な粘膜ライニングを維持するために絶対に不可欠である。
キシロース合成およびキシロース輸送の両方が必須のグリシン残基に依存しているだけでなく、キシロースのためのグリオシル化されたタンパク質の付着部位は、しばしばグリシンが濃縮されている。コンドロイチン硫酸またはヘパラン硫酸鎖の合成を開始するキシロース糖の付着部位はセリン残基であるが、この残基は常にグリシン残基に高度に富む短いペプチド配列に埋め込まれている[77]。
キシロシルトランスフェラーゼのために知られている最良のアクセプタータンパク質は、α-トリプシン阻害剤であるビクニンである。この場合のコンドロイチン硫酸付着部位は、セリン残基の前の保存されたグリシン残基と、セリン残基の後の3個のグリシン残基の配列を含む。50種類の異なるプロテオグリカンの比較は、’a’が任意の酸性アミノ酸を表すa-a-a-Gly-Ser-Gly-A-Glyの一貫したパターンを明らかにした。
したがって、これらのグリシン残基のいずれかに対してグリホサートが置換されると、キシロースの付着が破壊され、したがってコンドロイチン硫酸またはヘパラン硫酸の合成が開始されることが予想される。これは、腸内粘膜ライニングの維持を大きく阻害する可能性が高い。
プロテオグリカンの合成に関与するトランスポータータンパク質もまた、グリホサートによって破壊される可能性が高い。
プロテオグリカン合成に関与する様々なヌクレオチド糖のためのいくつかの異なるトランスポーターが存在し、それらはタンパク質クラスであるSLC35に属している[78,79]。腸内の哺乳類細胞は、グリホサート曝露によるこれらのトランスポーターのグリシンの破壊により、ゴルジ体装置にフコースとキシロースを輸入する能力が損なわれる可能性がある。
GDP-フコーストランスポーターSLC351は、決定的にグリカンのフコシル化を制御し[80]、欠陥のあるバージョンは深刻な障害にリンクされている[78]。このトランスポーターの膜貫通らせん5,6および8にある3つの高度に保存されたグリシン残基、Gly180,Gly198,およびGly277は、その活性において重要な役割を果たしている。
Gly180をチロシンに置換するとタンパク質の活性が著しく低下し、Gly277をイソロイシンに置換すると細胞表面のフコシル化が完全に消失することが明らかになった。グリホサートは、大腸菌におけるフコース代謝に関与する4つの酵素の発現を(少なくともファクター2の)アップレギュレートすることに注意することは興味深いことである。
L-フコース-1-リン酸アルドラーゼ、L-フコースイソメラーゼ、L-フクロキナーゼ、フコースペルメラーゼ[81]である。これは、フコースを分岐糖鎖に取り込む能力が抑制されていることを反映していると考えられる。
ALS患者は、高度に糖化されたタンパク質であるT細胞発現免疫グロブリンGs(IgG)の血清レベルが不足している[82]。より具体的には、ALS患者におけるIgG N-グリカンのフコシル化グリカンの血清レベルは、シアル化グリカンのレベルと比較して有意に低下していることが明らかになった[83]。フコシル化不足のIgGもまた、ALS患者の運動野に発現している。
ALS患者血清由来のフコース欠損糖鎖A2BG2は、抗体依存性の細胞毒性を増強し、G93A-SOD1マウスにおけるCD16および活性化ミクログリアの過剰発現をもたらす[82]。A2BG2はALSに特異的であるだけでなく、その産生量は病気の進行と相関している[84]。癌治療薬研究の活発な分野には、腫瘍細胞が生存に決定的に依存するタンパク質に対する特異的な抗体の産生が含まれている。
腫瘍細胞への毒性を高めるために、極端にフコシル化されていない抗体を生産するための特別な技術が開発されている[85]。IgGにおけるコアフコシル化の欠如は、抗体依存性の細胞毒性を50~100倍に増加させる[86]。したがって、ALS患者によって産生されるフコシル化不足の抗体は、自己免疫疾患において特にウイルス性であることが予測できる。
シトクロムc酸化酵素(CcO)は、複合体IVとしても知られており、呼吸電子輸送鎖の最後の酵素である。CcO活性の低下がALS患者の脊髄で検出されている[10,11]。CcOは酸素の水への還元を触媒する酵素として、また膜貫通プロトンポンプとしても機能する。プロトンをゲートするセグメントは、酸素が触媒部位に送られるチャネルと重なっている。
このチャネルの狭い部分の単一のグリシンをバリンで置き換えると、触媒部位への酸素のアクセスが完全にブロックされ、小さなガス分子に不浸透性の部位の周りのコンパートメントの形成をもたらす。この破滅的な結果は、野生型のCcOよりも数桁遅いO2を結合することである[87]。これは、スーパーオキシドの漏出や、近隣の脆弱な分子への酸化的損傷につながることが予測できる。
図2:病気につながるフルクトースとグリホサートによる破壊が関与する代謝経路の模式図
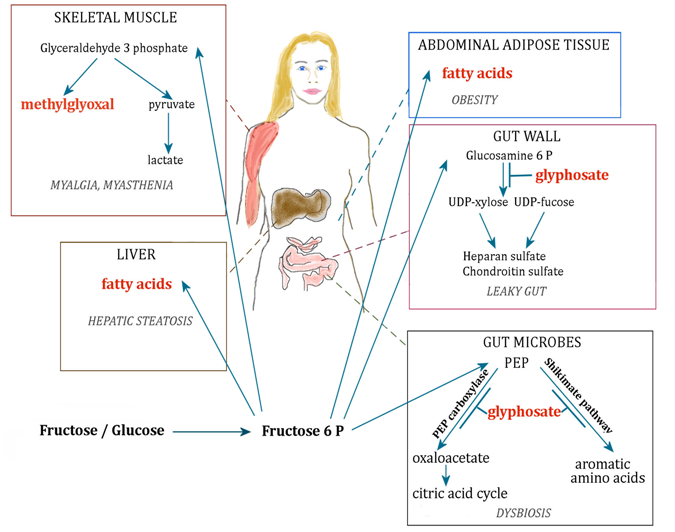
PEP:ホスホエノールピルビン酸。フルクトース6 P:フルクトース6リン酸塩。グルコサミン6 P:グルコサミン6リン酸塩。
表2:ポリGGGCCから合成されたジペプチド配列中のグリシンと対をなすアミノ酸、およびそれらの関連する非コード化アナログ
ハンチントン病に連結されたCAG CAGリピートもまた、完全性のために、表に含まれる。アゼ(Aze)。アゼチジン-2-カルボン酸。DON:6-ジアゾ-5-オキソ-L-ノルロイシン。
パターンペアリングされたアミノ酸アナログ参考文献
GGG GCC Gly-Ala L-アラニン R S Roy er al)。
GGG CCG Gly-Pro Aze K Bessonov er al)。
GGC CGG Gly-Arg L-カナバニン J Krakauer er al)。
CAG CAG Gln-Gln-Gln DON CC Clarkら[215]。
代謝撹乱
腸の不調
ALSは罹患率が高く、治療の予後が非常に悪い衰弱性の病気である。したがって、最終的にALSを発症させる要因を早期に発見することが、この病気を克服するための最善の道であると考えられる。したがって、ALSの症状が表出する前の代謝病理を明らかにすることに研究の努力を傾けるべきである。図2に図示したように、我々はALSの2つの重要な危険因子は、加工食品からの過剰な食事性果糖とグリホサートへの慢性的な曝露であるという仮説を立てている。変異型SOD1マウスの論文[6]で証明されているように、病気のプロセスは腸から始まる可能性が高い。
腸内微生物分布のアンバランス、特に大腸での酪酸産生種の欠乏は、腸管透過性の上昇と肝臓ストレスにつながるが、これはおそらくフルクトースの微生物クリアランスの障害が原因の一部である。フルクトース代謝の負担は、その後、肝性ステアトーシスに直接つながる肝臓に主に落ちる。肝臓が故障し始めると、より多くのフルクトースは、一般的な循環に入るために肝臓の障壁を越えて壊れる。その結果、骨格筋は果糖とメチルグリオキサール[88]のような高度に糖化する誘導体への過剰な曝露に屈する。
時間の経過とともに、骨格筋は、特に神経伝達物質の放出を通じて十分な興奮性刺激を産生するための運動ニューロンへの負担を増大させるグリホサートへの同時曝露によって、損傷を増大させることになる。最終的には、運動ニューロンが広範囲に損傷を受け、ALSの明白な症状が現れる。ミネラルの不均衡は衰えを加速させる。
大腸環境は大部分が嫌気性であり、正常な腸内細菌叢は、食物源から得られる腔内複合炭水化物や、ヒトのゴブレット細胞が産生するムチンから得られる炭水化物を発酵させることで繁栄する[89,90]。大腸菌叢は酪酸、コハク酸、プロピオン酸を含む短鎖脂肪酸(SCFAs)を産生し、これらは大腸上皮細胞の重要なエネルギー源である。特に酪酸は、ヒストン脱アセチラーゼ活性[91]、大腸炎や大腸癌からの保護[90]を含む顕著な有益な生物学的効果を持っている。
酪酸産生細菌は、ALSマウスモデルでは明らかなALS症状が現れるずっと前に減少している[6]。N-アセチルトリプトファンは、ALSのマウスモデルにおいて治療効果が期待できることが示されている[92]。トリプトファンは、細菌間の細胞間シグナル伝達を媒介する定足数感知分子インドールの前駆体である。インドールは、タイトジャンクション、アドヒデンスジャンクション、アクチン細胞骨格、ムチン産生に関わる遺伝子の発現を誘導することで、腸管上皮細胞のバリア機能を高める[93,94]。2015,Parkerは、グリホサートがシキメート経路を破壊した結果、トリプトファンの供給量が減少することで、腸管透過性の障害を誘導する可能性があることを提案した[95]。
乳児が最初に固形食を摂取すると、腸は複雑な炭水化物を分解することに長けた種であるバクテロイデス・ザタイオタミクロンの急速な成長を伴う深遠な変化を受ける[96]。この種は、順番に、腸ゴブレット細胞によるフコシル化および硫酸化グリカン(ムチン)の合成を誘導する。バクテリオデス種は、腸壁からこれらのムチンを剥離し、それ自体を消費するだけでなく、他の近縁生物への栄養源として供給することに長けている。
このような宿主と共生関係は、生涯を通じて健全な微生物の分布につながっている。無菌マウスを用いた実験では、離乳後のフコシル化糖鎖発現はB. thetaiotaomicronによるコロニー化に依存することが示されている[97]。特にB. thetaiotaomicronはヒト消化管遠位部の支配的な細菌であり、酪酸合成微生物にアセテートを供給する重要な役割を果たしている[98]。
バクテロイデス種は、ヒトの腸内で最も豊富なグラム陰性細菌であり、糞便1グラムあたり1010個以上の細菌密度に達している[99]。彼らは、植物と宿主生物の両方から多糖類を分解する驚異的な能力を持っている。B.thetaiotaomicron、そして実際には、ほぼすべてのバクテロイデス種は、UDP-キシロース合成酵素の2つの高度に保存された明確に異なる形態を持っている[100]。それは、彼らがUDP-キシロースを合成する理由として研究者にパズルを提示してきた、なぜなら彼らはそれを何のために使用するそれが不明なので。細菌の細胞壁にはキシロースは存在しないし、キシロシルトランスフェラーゼ酵素も持っていない。
我々は、細菌の主な目的は、宿主が細菌のための健康な粘膜環境を維持することができるように、膜タンパク質のグリコシル化を開始するために、宿主にこの重要な栄養素を供給することを提案する。重要なことに、哺乳類の細胞は、培地にキシロシルトランスフェラーゼを分泌する – 糖タンパク質中のセリン残基にキシロースを結びつける酵素。哺乳類細胞の培養では、酵素に供給するためにキシロースを輸出していなかったため、この事実はまた、このことを調査した研究者に謎を投げかけた。我々は、B. thetaiotaomicronと宿主の粘膜細胞は、大腸の厚いムチン壁を生成するために協力しているという仮説を立てた。
最近の興味深い研究では、B. thetaiotaomicronのような重要な常在微生物が、腸内の病原体に対する炎症反応によって誘導される免疫攻撃からどのようにして自分自身を守ることができるのかについての重要な洞察を提供している[101]。B. thetaiotaomicronは、脂質-Aホスファターゼ酵素の活性によって宿主免疫細胞によって産生されるカチオン性抗菌ペプチドとの結合に抵抗する。
このホスファターゼ酵素は、4つのペプチドの高度に保存された配列を含む脂質ホスファターゼの幅広いクラスのメンバーである。PSGH[102]。機能に対するこのペプチド配列の意義はまだ明らかではないが、グリホサートをグリシン残基で置換すると酵素の挙動が乱れ、炎症反応後の腸内のB. thetaiotaomicronの個体数の減少につながる可能性があることが予測できる。グリホサートは抗菌剤として特許を取得しており、ヒトの腸内に生息する多数の微生物種に悪影響を与える[30]。特にバクテロイデスの増殖速度はグリホサートによって著しく低下する[103]。
食物繊維がALSに対する保護効果があることは明らかにされていないが[104]、小麦、オーツ麦、大麦、豆類などの重要な食物繊維源は、収穫直前に乾燥剤としてグリホサートを散布するという慣行が広く行われているため、グリホサートで高度に汚染されている可能性が高いことを念頭に置いておかなければならない[105]。したがって、繊維の潜在的な利点は、グリホサートへの曝露量の増加によって相殺される可能性がある。
果糖の過剰負荷
脂肪組織はフルクトースの主要な代謝物質であり、おそらく肝臓と腸だけがそれを上回っている[106]。唯一の炭水化物としてのフルクトースの存在下でのマウス線維芽細胞は容易に脂肪細胞に分化し、フルクトースが脂肪原性であることを示唆している[107]。痩せた体躯の人は脂肪組織が非常に少ないため、この経路を介して過剰な果糖をクリアする能力が著しく制限される。
不思議なことに、糖尿病はALSに対して防御的であり、これは、インスリンベースのグルコース取り込みが障害されているときに、筋肉細胞が糖化糖にさらされる量が減少していることに起因していると考えられる[108]。逸話として、イタリアのプロサッカー選手は、フルクトース1,6ビスフォスフェートを日常的に摂取していたが、45歳でALSのバルバー型の早期発症を発症した[109]。
グリホサートは、サトウキビ作物の熟度調整剤プレハーベストとして頻繁に使用されており、サトウキビ中の糖の貯蔵量を増加させる。これは、複雑な炭水化物の合成を阻害することでこれを行うと考えられる。ルピナス植物を対象とした研究では、亜致死レベルのグリホサートに曝露すると、デンプン含量が減少し、ショ糖が増加することが示された[110]。このメカニズムは、炭素と窒素の有機物への取り込みに重要な酵素であるホスホエノールピルビン酸カルボキシラーゼ(PEPC)の阻害に起因している。
PEPは、PEPCと5-エノールピルビン酸合成酵素(EPSPS)の両方の基質であり、グリホサートによってシキメート経路で障害され、植物や微生物における芳香族アミノ酸の合成障害につながる酵素である。これは、雑草に対するグリホサートの主な毒性効果であると考えられている。PEPC[111]とEPSPS[112,113]の両方とも活性部位に必須のグリシンを持っており、これがグリホサートで置換されると、酵素活性が著しく阻害されることになる。これはおそらく、これらの酵素のその抑制のための主な説明である。
大腸菌の EPSPS の DNA コードをグリシンからアラニンに変更すると、グリホサートの阻害作用は完全に廃止される[113]。他の微生物はこの同じメカニズムで抵抗性を獲得しており、これがGMOラウンドアップ・レーディクロップスの細菌遺伝子改変の基礎となっている[114]。
大腸菌PEPCの末端グリシンをアスパラギン酸などの負に帯電したアミノ酸で置換すると、酵素活性の完全な停止をもたらした[111]。これらの著者は次のように書いている:”PEPCは、その極端なC末端で主鎖の遊離CO2基を超える追加の負電荷を許容しないように見える” 末端のグリシンに対するグリホサート置換は負の電荷を付加する:それはC末端にCH2PO3H-アニオンを付加する。
我々は、これらの2つの重要な経路における基質としてのその遮断によるPEPの蓄積は、フルクトースをPEPに変換する経路の阻害につながるフルクトースの蓄積につながり、より多くのフルクトースがフルクトース1,6ビスフォスフェートにリン酸化され、解糖経路に供給されることを余儀なくされるだろうと仮定している。これはまた、おそらく食事中のフルクトースの多くが腸内微生物によって代謝されずに残っていることを意味し、代わりにフルクトースを代謝するために肝臓に大きな負担をかけ、我々はフルクトース誘発性脂肪肝疾患で見ているパンデミックを説明している[115-117]。
硫黄ディスビオーシス
大腸の健康に対する酪酸の利点に対する認識が高まっているため、タンパク質および炭水化物の異なる供給源の酪酸産生に対する効果を調査するために、ラットを用いた摂食研究が実施された[118]。
ラットには、炭水化物としてフラクトオリゴ糖または馬鈴薯澱粉のいずれかを、タンパク質源としてカゼインまたは米または大豆のいずれかと一緒に与えた。酪酸の産生亢進は、カゼインまたは大豆と比較して米タンパク質と関連していた。我々は、この観察された違いの要因として、米と比較してカゼインと大豆ではグリホサート汚染が増加していたのではないかと仮説を立てた。
大豆のほとんどがグリホサート耐性になるように設計されており、カゼインは高用量のグリホサートを飼料に与えられた牛に由来することを考えると、これはもっともらしいことである。さらに、カゼインと大豆の両方において、ポリメチオニンの補給が低酪酸収量を補正することを発見した。
もっともらしい仮説は、グリホサートがメチオニン合成を阻害したということであるが、補給はこれを補った。硫黄含有アミノ酸のメチオニンは、肝臓の重要な抗酸化物質グルタチオンの適切なレベルを維持するために不可欠であるだけでなく、胆汁酸の重要な成分であるスルホン化アミノ酸、タウリンと同様に。
ニンジン細胞株を用いた研究では、グリホサートはメチオニンレベルを 50~65%低下させることが示された[119]。グリホサートが腸内微生物によるメチオニンの合成に影響を与えるメカニズムは、3′-ホスホアデノシン5′-ホスホ硫酸(PAPS)還元酵素のグリホサートの抑制を介したものである。大腸菌を対象とした研究では、グリホサートの存在下で3.75倍の活性低下を示した[120]。
この酵素はメチオニン合成の律速酵素である。酵母 PAPS 還元酵素の活性部位の構造解析により、この酵素は、3′-P ループ(51GLTG54)にある 2 つのグリシン、いわゆる Arg ループの中間にある 1 つのグリシン、および C 末端触媒モチーフである ECGIH にある 1 つのグリシンという 4 つのグリシン残基を総称して含む 3 つの高度に保存されたモチーフを持っていることが明らかになった[121]。
低硫黄食と高硫黄食を比較した牛の研究では、牛が牧草を食べている間は硫黄は有益であったが、仕上げ期に入ると過剰な硫黄が問題となり、体重増加の減少を引き起こすことが示された[122]。おそらく、牛たちは遺伝子組み換えされたトウモロコシと大豆飼料で構成された飼料に切り替えられたのであろうが、その飼料はグリホセ ートで大きく汚染されていた可能性が高いものであった。
高硫黄食を与えられた牛は、仕上飼料の採用後、ルーメン内の硫黄還元細菌、特にデスルフォビブリオ・デスルフリカンスの有意な増加(p = 0.03)が見られた。高硫黄食はプロピオン酸に比べて酪酸と酢酸の両方が減少し、硫化水素ガスの産生が増加した。モリブデンが亜硫酸酸化酵素を触媒することから、グリホサートによるモリブデンのキレート化がこのアンバランスに寄与していると考えられる。
さらに、銅はデスルホビブリオ種の増殖を抑制する[123]ので、グリホサートによる銅キレート化はデスルホビブリオ種の増殖を促進する可能性がある。デスルホビブリオの種は、エネルギー源としてフルクトースを利用して硫酸を硫化水素ガスに還元することができる[124]。
二酸化硫黄、亜硫酸塩、カラギーナン(海藻から抽出された硫酸化多糖類)は、現代の西洋の食生活によく見られる[125]。これらの酸化硫黄化合物の高用量投与は、慢性的なグリホサート中毒と組み合わせて、メタン生成種を犠牲にして大腸内の硫黄還元細菌の過剰増殖をもたらすことが予想され、これは潰瘍性大腸炎に直接リンクしている[125]。
単離されたヒト大腸細胞を用いた試験管内試験研究では、硫化水素は酪酸代謝を特異的に阻害するが、これらの細胞によるグルコース酸化は阻害しないことが実証された[126]。H2Sは運動ニューロンに対するALSの損傷に関与している[127]。ALS患者の髄液やSOD1G93A変異マウスの組織で高レベルのH2Sが検出されている[127]。
亜硫酸塩の毒性効果の一つは、チアミンを破壊することであり、チアミン欠乏症の問題につながる[128,129]。チアミン欠乏症がALSの原因因子として疑われるようになったのは最近のことである。チアミン欠乏の徴候を示した2人の患者の死亡を受けて 2015年の論文で報告された研究者たちは、ALS患者122人のチアミン欠乏の潜在的な役割を調査した[130]。
その結果、18%の患者で重度のチアミン欠乏が認められ、さらに10%の患者で軽度の欠乏が認められた。微生物のチアミン合成酵素がユビキチンと共有していることは、C末端にユニークな高度に保存された二重グリシンリピートを持っていることに大きな意味があると考えられる[46,131]。驚くべきことに、1980年代初頭にグリホサートが導入されて以来、北欧の複数の鳥類を苦しめてきた麻痺性疾患は、明らかにチアミン欠乏と関連している[132]。
肝臓病と果糖代謝
ALS患者は血清中の尿酸値が例外的に低い傾向があり、尿酸値は疾患の重症度と逆相関している[133]。尿酸塩は肝臓でトリグリセリド合成と並行してプリン体の分解を経て合成される。律速酵素であるキサンチンオキシダーゼは、補酵素としてモリブデンに依存している。
我々は以前にモリブデンの欠乏は、障害された亜硫酸オキシダーゼ活性のために腸内の亜硫酸塩の毒性を説明することができることを見ていた。不良キサンチンオキシダーゼは、尿酸塩の減少した合成と一緒に、脂肪にフルクトースを代謝する肝臓の無力につながる可能性がある。
驚くべきことに、いくつかの研究でアルコール摂取がALSを予防することが示されている。431,943人の参加者を対象としたメタアナリシスでは、アルコール摂取とALSの関連性についてオッズ比0.57が得られている[134]。そのメカニズムは依然として不明である。しかし、アルコールは肝臓に深刻なストレスを与え、脂肪肝炎、肝線維症、肝がんを引き起こすため、このような保護は代償がないわけではない。
興味深いことに、果糖とアルコールは両方とも肝臓で尿酸合成とトリグリセリド産生を増加させ、それぞれ非アルコール性脂肪性肝疾患とアルコール性脂肪性肝疾患を引き起こす[135]。しかし、NAD+とNADHの比率には逆の効果があり、果糖はNAD+を増加させ、アルコールはNADHを減少させる。トリプトファン(シキメート経路の産物)は肝臓でNADHの重要な前駆体であるが、トリプトファンもキヌレニンに変換され、炎症を起こした腸に侵入したマクロファージに貯蔵される[136]。
したがって、Samsel と Seneff[30]で議論されているように、シキメート経路の抑制とグリホサートによる炎症性腸の誘導の両方が、肝臓への NADH の供給障害につながるはずである。肝臓のNADHが不足すると肝臓が果糖の代謝を妨げるというのはもっともらしいが、アルコールはNAD+からNADHへの還元を通じて供給を更新することでこの問題を部分的に修正し、同時に肝臓病を促進する。
また、NADの前駆体としてビールに多く含まれるナイアシンを介して、より直接的な利益があるかもしれない。アルコールはまた、肝臓でのフルクトース代謝への正の効果を介して直接的にも間接的にも、尿酸塩の合成を促進する。尿酸は、アストロサイトにおける核内因子(赤血球由来2)-like2(Nrf2)シグナル伝達経路の誘導を介して、その一部で神経保護的である[137]。
モリブデン欠乏症、NADH欠乏症、NADHに対するNAD+の比率の不均衡に加えて、大腸内のデスルホビブリオ種の過剰活性もまた、フルクトースや他の糖の肝臓代謝障害に寄与する可能性がある。H2Sは非常に拡散性が高く、発生源の場所から近隣の組織に容易に移行することができる。消化管で産生された高レベルの内因性H2S[138]は、肝臓に容易に拡散するであろう。
肝臓は大量のH2Sを消費し、NAD+/NADH比を低下させるが、利用可能な酸素の多くを消費することが実験的に示されている[139]。したがって、肝臓がH2Sに大量に曝露されると、酸化的リン酸化を利用して肝門脈を介して腸から届いた糖を処理する能力が低下することが予測できる。高果糖食で腸内の果糖代謝が悪い場合、果糖処理の負担を主に骨格筋細胞に転嫁することになる。
ALSの前症状期に肝臓が果糖の多くを除去できたとしても、その間に肝臓は過剰な脂肪沈着物を蓄積し、慢性的な果糖曝露による累積的な損傷に悩まされることになる。ラットモデルでは、銅欠乏は肝臓での高ショ糖食の悪影響を増強し、肝臓の炎症や線維化とともに脂肪肝疾患を引き起こした[140]。
最終的には、肝臓病により、フルクトースの低血清レベルをさらに維持することができなくなる。変異型SOD1G93AマウスのALSモデルにおける肝機能の調査では、臓器の萎縮と貯蔵脂肪の蓄積とともに、肝臓におけるナチュラルキラーT(NKT)細胞のレベルが劇的に増加することが明らかになった。これらのマウスの症状を呈する前の肝リンパ球でさえも、NKTリガンドによってex vivoで刺激を受けると、有意に高いレベルのサイトカインが分泌された[141]。
ミネラルの不均衡
SOD1とCcOの両方が銅に依存していることから、銅の欠乏や過剰がALSに関与している可能性があると考えられる。SOD1は銅(Cu)と亜鉛(Zn)の両方に結合して機能することに依存している。したがって、これらの必須ミネラルのバイオアベイラビリティーの不均衡が障害を引き起こす可能性がある。驚くべきことに、銅の欠乏[16,142-144]と亜鉛の欠乏[145]の両方がALSの病理学的な役割を果たしていることが、研究文献で強く証明されている。
CcOとCu,Zn SOD(SOD1)は、ヒトにおける2つの主要な銅結合酵素である。ヒトSOD1とヒトCCSの両方を含む二重変異マウス(G93AxCCS変異マウス)では、CCSの過剰発現により、より多くの銅がSODに流用され、ヒトSOD1酵素の過剰発現による銅欠乏の問題を悪化させている[142]。
銅ATPase ATP7Aは、小腸とゴルジコンパートメントと個々の細胞の形質膜の間にCuの輸送の両方からCu(I)吸収を調節する重要なCu調節タンパク質である[146-148]。ATP7Aの原因メンケス病、致命的な乳児期発症の神経変性銅疾患、Cuと多くの関連する全身の病理学の障害ビリヤリー輸送によって特徴付けられる変異[149,150]。
ATP7Aは、反対側にチャネルを横切ってポンピングされるように、Cu結合ドメインからCuを受け入れるためのプラットフォームを形成する第2の膜貫通ドメイン内のグリシン-グリシンキンクが含まれている[149]。これらのグリシンのいずれかに対するグリホサートの置換は、この機能を混乱させることが予想される。
ALSモデルのマウスにおける銅のホメオスタシスを研究した注目すべき論文では、これらのマウスにおけるヒトSOD1のコピー数が多いために銅に対する需要が高く、結果として一般的な銅欠乏症を引き起こすことが提案されている[16]。研究チームは、G93AxCCSマウスが単一変異型SOD1マウスよりもはるかに早くALSを発症することを示した。
G93AxCCSマウスは、ヒトのCCSを持たないマウスに比べて約8倍の速さで死亡することが明らかになった。銅の分布は親和性の勾配によって決定され、SODは銅に対して最も強い親和性を持っている[151]。CCSの過剰発現はミトコンドリアへの銅の輸入を阻害し、CcOから銅を奪い、それによって複合体IVを破壊するという仮説が立てられている[143,144]。
実際、SODG93AxCCSマウスではCcO活性が大幅に低下した[143]。驚くべきことに、これらの二重変異マウスは、Cu複合体であるCuATSMを補給することで、より長く生存することができた。このことはALS患者の治療法に希望を与えているが、Cuの過剰摂取は毒性を引き起こす可能性があるので注意が必要である。
我々 は多くの研究文献から抗酸化トリペプチド、グルタチオンについて聞くが、同じように重要な可能性のあるトリペプチド、グリシル ヒスチジニル-リジン (GHK) についてはあまり書かれていない、最も頻繁に GHK-Cu と呼ばれるその能力のために Cu[152]。このCuをキレートするトリペプチドは、細胞にCuを提供する上で重要な役割を果たしている。
これらのトリペプチドの両方がグリシンを含んでおり、1つは、それらのグリシンのためのグリホサート置換の結果について疑問に思う必要がある。それは、GHK-Cuのグリホサートベースのバージョンは、このようにSOD1とCcOには利用できないように、はるかにタイトにCuを結合するだろうと予測することができる。
TrumbullとBeckmanによる魅力的な論文は、病気のプロセスにおけるZn欠乏のための役割について議論している[145]。彼らの説得力のある引数は、Zn欠損野生型SOD1は、NOとO2からペルオキシナイトライト合成を誘導する上でより破壊的であることを示している。彼らはさらに、Znサイトを占めるCuはCuサイトのCuよりもはるかに多くの酸化還元活性であることを主張している。
彼らは、そのようなd-ペニシラミン[153]とCuATSM[16]などのCuキレート剤で観察された利点は、主にZnサイトからCuを抽出する能力に起因する可能性があることを示唆している。家族性ALS患者におけるSOD1の変異型のいくつかは、Znに対する親和性が大幅に低下している(30倍にもなる)[154]。Crowらは、これがチロシンのニトロシル化の亢進につながると提案しているが、別の可能性として、CuによるZn部位の汚染が増加している可能性がある。
マンガン(Mn)はALSに関連して不適切に分布しているようである[155-157]。マンガン症は、マンガン製錬業者や鉱山労働者、溶接工の間での過剰なマンガン曝露によって引き起こされることが知られている神経学的状態であり[158]、ALSとパーキンソン病の両方の症状として現れる。
Kapakiらの研究[155]では、ALS患者では対照群に比べて脳脊髄液(脳脊髄液)と血清中のCu濃度が低下しているのに対し、血清中のMn濃度は上昇していることが明らかになった。Kapakiら[156]では、死後の脊髄のMn濃度を調べたところ、特に前角で有意に高いMn濃度を示した。Kihiraら[157]では、ALS患者と対照群の間で脊柱全体のMn濃度は同程度であったが、ALSに関連して前角と後角の間にアンバランスが見られ、前角では過剰な濃度となっていた。Roosら[159]は、脳脊髄液と血液中のMn濃度を調べたところ、ALSに関連して脳脊髄液のMn濃度が実質的に高いことを発見した(5.67μg/L vs. 2.08μg/L)。
これらの奇妙な分布から、MnはALS患者では循環経路ではなく神経線維に優先的に沿った経路で脊柱や脳脊髄液に分布している可能性が示唆された。SamselおよびSeneff[22]では、異常なMn分布経路とパーキンソン病との関連は、肝臓チトクロームP450(CYP)酵素の障害を介したグリホサートによる胆汁の流れの乱れに起因していた。
通常、肝臓は胆汁酸と結合してMnを体内に再分配する。このルートがブロックされている場合、Mnは最初に脳幹の核に到達し、そこから、神経線維を介して、脳と脊髄の他の部分に広がるように迷走神経を介してエクスポートされる。
このような経路は、脳脊髄液の高濃度と前角で観察される過剰なMnの両方を説明するであろう、脊柱の中央管に隣接している。ALSとコレスタシスを直接関連づけた論文は見つからなかったが、血清ビリルビン
ウシ伝染性海綿状脳症の基礎となる病理学の理論には実質的な類似性がある[162]。CuとFeのバイオアベイラビリティーが低いMnが豊富な環境では、過剰なMn吸収と酸化的環境が組み合わさって、Cu金属タンパク質であるプリオンタンパク質(PrP)の発現を上昇させるMn3+誘発性連鎖反応を誘導する。
Mn3+はPrP上の空孔Cuドメインを置換し、PrPのフォールディングのミスフォールディングを引き起こし、結果としてプリオン病を引き起こす。同様のシナリオで、ミスフォールディングしたSOD1の蓄積を説明することが可能であると思われる。プリオンタンパク質内の特異的に毒性のあるペプチドは、「疎水性コア」としても知られているグリシンに富んだ領域内の短いパリンドロームとして同定されている[163]。
このパリンドロームの配列はAGAAAAGAであり、複数の種にわたって高度に保存されている。この配列のタンパク質からの切除により、その毒性効果とプリオンの伝播に影響を与える能力が除去されることが示されている[164]。
グリシンは、この配列内の有毒種として特異的に標的とされている[165]。グリホサートが回文中のグリシンのいずれかに置換されていると、ミスフォールディングと毒性を説明できる可能性がある。
グリホサートは強力な金属キレート剤[166]であり、Mn[23]、Zn[24]、Cu[25]と強力に結合することができ、グリホサートに曝露された植物ではこれらの金属を利用できないようにする[24]。
グリホサートは、pHが十分に低い場合、これらの金属を放出する。しかし、Cuの場合、グリホサートはpHが極酸性レベル(pH1.0)まで下がるまで金属イオンを放出しない[167]。したがって、グリホサートは他の金属キレート剤と同様に、CuとZnの両方をSOD1とCcOに利用できないように振る舞うことが予測できる。
これに神経線維に沿ってMnが分布しているために脊柱内のMn濃度が上昇していることが組み合わされると、ALSに関連した深刻なミネラルのアンバランスが描かれることになる。より一般的には、ペプチド配列内に埋め込まれた強力な金属キレート剤の存在は、生物がそれらの金属に依存する酵素への金属分布を適切に管理することをはるかに困難にするだろう。
SOD1の複数のグリシン残基のいずれかにグリホサートが置換されると、SOD1の金属に対する結合能が強く変化することが予想される。グリホサートで置換されたSOD1は、結合した金属を放出することをはるかに嫌うだろうと予測される。このような障害の可能性については検証が必要である。
進行性神経筋系障害
筋障害
フルクトースはグルコースに比べて糖化剤としての反応性が8倍も高く、フルクトース代謝の産物であるメチルグリオキサールはフルクトースよりもさらに一桁以上のダメージを与える[168]。
ALS患者の多くは筋肉のグルコース取り込み率と乳酸出力が増加しており、筋肉の代謝が異常になっている[169]。フルクトースはアルドース還元酵素(ポリオール)経路を介して内因性に産生される。
ALS患者の骨格筋細胞は、ポリオール経路を介してグルコースからフルクトースへの代謝が亢進していることを示している[170]。また、骨格筋では、アルドラーゼ[169]が関与する果糖分解経路を介して、さらにフルクトースの代謝が促進されていることが示されている。
Dharらによる2013年の研究では、フルクトースを多く含む食事(総カロリーの60%)を与えられたラットでは、高血圧や抗酸化物質であるグルタチオンの減少とともに、大動脈と腎臓のメチルグリオキサールのレベルが上昇したことが示された[172]。
早引き筋は遅引き筋とは対照的に、早引き筋は糖の代謝に酸化的リン酸化ではなく解糖をはるかに多く利用するII型線維を多く持っている。これはまた、より多くのフルクトースを処理して乳酸に代謝し、他の細胞に分配するために輸出することができることを意味する。
しかし、これはまた、彼らがフルクトースやメチルグリオキサールのようなその誘導体による糖化損傷を受けやすいことを示唆している。マウスモデルでは、特に速筋は、ALS末期のALSの間、カルシウムの活性化に応答して力のレベルの低下を産生した[173]。
筋力に関する別の研究では、グリシンアナログとして作用するグリホサートが、ALSのマウスモデルにおける150 Bioinfo Proteom Img Anal |Volume 2: Issue 2で、症状前に、早転筋の内側腓腹筋(MG)の収縮力が有意に低下することが明らかにされた[174]。
ALSに関連した症状は90日目にしか現れないが、変異型SOD1マウスは40日目にはすでにMGの収縮力が35%低下していた。
運動器の数の減少も同時に見られたが、その程度は著しく低かった。この頃には、遅筋のヒラメ筋の衰えは最小限にとどまっていた。この効果は、グリホサートのミオシンへの取り込みが着実に増加していることによって説明することができる。
ミオシンはATP依存性の運動タンパク質で、筋肉のアクチンベースの運動に関与している。
G699のアラニンをグリシンに置換することを含むミオシンの突然変異に関する画期的な研究では、このわずかな変化が、この分子運動タンパク質の運動性を99%低下させることが明らかになった[175]。
この研究で著者らは、このG699A置換によって影響を受けたミオシンモーターの割合を操作することができた。彼らは、わずか2%のモーターが改変されただけで、全体的な運動力に50%の減少があったことを発見した。
ミオシンは、本質的にグリシンに依存する唯一の分子モーターではない。キネシンとダイニンは、脊柱の運動ニューロンを骨格筋の筋細胞のシナプスに接続する長い軸索において極めて重要である。運動ニューロンのキネシンは長い軸索に沿ってソーマからシナプスにミトコンドリアを輸送し、ダイニンは使用済みのミトコンドリアをソーマに輸送する。
キネシン[176]とダイニン[177]の両方とも、モーターとしての適切な機能に不可欠な高度に保存されたグリシンを持っている。ダイニンは、損傷したミトコンドリアをリソソーム処理を介して廃棄するためにソーマに戻すことを制御している。
天然農薬ロテノンの神経細胞への毒性効果に関する研究では、ダイニンの発現を阻害することにより、ミトコンドリアの軸索輸送を阻害し、この効果が神経伝達の障害に先行することが実証された[178]。ダイナクチンはダイニンモーターの効率を高める多タンパク質複合体である。
ダイネクチンサブユニットの残基59のグリシンの突然変異は、進行が遅いALSの一般的な形態を作り出す[179,180]。このサブユニットのG59Sを発現するトランスジェニックマウスでは、運動ニューロンの異常と変性が発現する[181-183]。
スタチン薬の使用とALSまたは「ALS様症候群」との間に関連性が見出されている[184]。スタチン薬の一般的な副作用が筋肉の損傷であることはよく知られており、速筋が優先的に影響を受ける[185]。
スタチン使用者は、たとえ自覚症状がなくても、血清乳酸とピルビン酸の比率が異常に高いことがわかっている[186]。血清乳酸値の上昇もまた、特に非安静下でのALSと関連している[187]。スタチンは、フルクトースに由来する脂肪酸を緩衝するのに十分なコレステロールを合成する能力が低下するため、フルクトースを代謝する肝臓の能力を損なうことが予想される。研究では、変異型SOD1G93Aマウスは、ALSの顕在症状が現れる前であっても高コレステロール血症を示すことが示されている[188]。
シナプスにおけるグルタミン酸の興奮毒性
運動ニューロンのインパルスの柔軟な制御を理解するために、研究者は特に、受容体イオンチャネルを介した化学的シグナル伝達が軸索による高速神経インパルスの伝搬に取って代わるシナプスに注目してきた。軸索の末端では、次の中継ステップのためにシナプス後のニューロンで生成された新たなインパルスを刺激したり、抑制したりするために、神経伝達物質が短時間に生成される。アミノ酸のL-グルタミン酸は、中枢神経系で最も一般的な興奮性神経伝達物質である。
各シナプスの物理的寸法と化学的完全性は、アストロサイト、各シナプスの周りをしっかりと包み込むグリア細胞によって維持されており、グルタミン酸を素早く取り込んでシナプスから除去することができる。取り込まれた後、アストロサイト内のグルタミン酸はグルタミンに変換され、後にシナプス前の神経終末にリサイクルされてグルタミン酸に変換されて再利用されるか、細胞エネルギー源としてクレブスサイクルに導入される[189]。
最近のALS研究では、特にN-メチル-D-アスパラギン酸(NMDA)受容体に作用するグルタミン酸が多すぎると、カルシウム過負荷によるストレスが生じ、過剰に活性化した運動ニューロンを破壊してしまうと考えられているため、これらのプロセスを正確に解明することに集中してきた[190]。過度のCa2+流入はミトコンドリアにストレスを与えることが知られており、ミトコンドリアは酸化的代謝を介して過剰な毒性のあるスーパーオキシドや他の活性酸素を生成し、SODや他の保護システムに挑戦し、ミトコンドリア膜の障害や細胞のアポトーシスを引き起こす可能性がある[189-191]。
この過剰反応は、ALSの神経細胞変性を開始する役割を果たしていると考えられている。グリシンはNMDA受容体のアゴニストでもあり、グリシン濃度の上昇は海馬ニューロンにおけるNMDA受容体を介した興奮性シナプス後電流を顕著に増加させることができる[192]。低親和性で非競合的なNMDA受容体拮抗薬であるメマンチンの治療は、ALSマウスモデルにおいて有望な結果を示している[193,194]。
ラット脳の海馬ニューロンに対するグリホサート曝露の影響に関するCattaniらによる重要な研究では、生体内でのグリホサート曝露はNMDA誘発性グルタミン酸毒性に直接関連するいくつかの影響を示した[191]。
第一に、グリホサートは受容体部位でグリシンアナログとして作用することで、シナプスに放出されるグルタミン酸の量を増加させた。第二に、グリホサートはシナプスからのグルタミン酸のアストロサイト再取り込みと、グルタミン合成酵素を介したグリア細胞内での代謝分解を減少させた。最後に、シナプス後のニューロンへのCa2+からのイオン電流が増加し、酸化的負荷とミトコンドリアへのダメージ、すなわちニューロン死につながる興奮毒性が追加された。
NMDA受容体とL型電圧依存性Ca2+チャネル(L-VDCC)の両方がグリホサートに反応してカルシウム過負荷を誘発した。Cattani研究[191]で観察されたアストロサイトにおけるグルタミン合成酵素に対する阻害効果は、マンガンがグルタミン合成酵素を触媒するため、グリホサートによるマンガンキレート[22]に直接起因するものである。
グリホサートはまた、重要な抗酸化物質であるグルタチオンのレベルを低下させ、酸化的損傷を特徴づけるチオバルビツール酸反応種(TBARS)を増加させた。脳や脊髄の運動ニューロンにおけるこのような累積的な損傷は、他の原因によるALSと区別がつかない可能性が高い。
LPS、sALS、タンパク質の破壊
2012年に実施された家禽の微生物相を対象とした研究では、ラウンドアップの最小阻害濃度(MIC)のグリホサートは、多くのメカニズムを介してマイクロバイオームのバランスを破壊するのに十分であることが明らかになった[103]。この研究では、病原性の高い細菌はグリホサートに対して高い抵抗性を示し、有益な細菌は特にその影響を受けやすいことが明確に示された。さらに、グリホサートがカルシウム、マグネシウム、マンガン、鉄などのミネラルに対するキレート効果を介して間接的に細菌のバランスに影響を与えていることが議論された。
細菌は、グリシンアナログとして作用するグリホサートの細胞間ホメオスタティックバランスを必要としている。 www.ommegaonline.org 151 Bioinfo Proteom Img Anal |Volume 2: Issue 2 金属イオンを生存のために必要としている。グリホサートが引き起こす微生物バランスの変化が重要である。リポ多糖類(LPS)やエンドトキシンは、グラム陰性菌の外膜の主要成分である。
毒性効果を引き起こす LPS の成分である脂質 A の最も病原性の高い形態は、大腸菌やサルモネラ菌などの病原性細菌に見られるが、これらの細菌はいずれもグリホサートに対する耐性があり、MIC 値が 5 mg/ml であるのに対し、様々な有益な細菌の場合は 0.15,0.30,0.075 µg/ml となっている。
グリホサートに対するこの高いレベルの抵抗性は、病原性グラム陰性株の増殖を可能にするだろう。侵入してきた病原体は、もともとパターン認識受容体(PRR)によって検出され、自然免疫応答を開始する。Toll様受容体(TLR)は、多数の病原体を認識するPRRのグループであり、TLR4はLPSに反応する受容体である[195]。TLR4のLPS活性化には、コモレキュラーMD2とCD14も必要である[196]。TLR4は歴史的に病原体認識と関連してきたが、Ahmedらは最近、脳損傷や組織損傷イベントに応答して、その活性化が神経炎症と根本的に関与していることを示した[197]。
神経疾患に関連して、LiuとBingはパーキンソン病におけるLPSの役割を研究し、LPSは進行性のドーパミン作動性ニューロン喪失を誘導するだけでなく、行動障害を引き起こすことを動物実験で報告している[198]。2009,Zhangらは、sALS患者の循環LPSレベルが対照群に比べて統計的に有意に高いことを発見した。中等度の障害しかない患者でも統計学的に有意に高い値が認められ、ALSの病態におけるLPSの役割を示唆している[199]。
ALSと前頭側頭葉変性(FTLD)の特徴は、ユビキチン化したタンパク質の凝集であり、TDP-43はその重要な構成要素である。ALS患者では統計的に高いレベルの循環LPSが観察されているが、グラム陰性菌の増加による不衛生な状態から生じるエンドトキシンであるLPSもまた、主要なALSタンパク質、特にTDP-43とアミノ酸グルタミン酸の破壊に関与していることが示されている。
Correia[200]の研究によると、LPSによる炎症はTDP-43の転位と凝集の両方を促進する。具体的には、LPS処理はミクログリアとアストロサイトの両方の培養物においてTDP-43タンパク質の量を増加させたが、mRNAレベルではそれに対応する増加は見られなかったことが報告されている。さらに、ミクログリアとアストロサイトのLPS処理は、TDP-43の細胞質的な誤局在化を促進した。ミクログリアでは、LPS曝露により、細胞質のTDP-43パンクテート凝集体が形成された[200]。
グルタミン酸とALSとの関連性については、以前にも指摘されている。LPSはグルタミン酸と相乗的に作用し、グルタミン酸の毒性を有意に増加させる[201]ので、sALSのようにLPSの循環量が多い患者はグルタミン酸の毒性が増加する危険性がある。
その他の要因 ALSにおけるコラーゲン
複数の研究により、ALS患者のコラーゲンに異常があることが明らかにされている[202-205]。Fieldらは、ALS患者の皮膚から採取されたコラーゲンが欠損していることを指摘し、少なくともBMAA曝露に起因する症例については、コラーゲンのタンパク質合成中にBMAAがL-セリンに取って代わられることが説明になるのではないかと提案している。
これらの著者は次のように書いている:「我々は、sALSコラーゲンに見られる異常は、BMAAの誤挿入とそれに続くコラーゲン蛋白質の誤った折り畳みに起因するのではないかという仮説を立てた」[204] 体内の総蛋白質量の25%はコラーゲンであり、コラーゲンのアミノ酸残基の25%はグリシン残基である。したがって、コラーゲン合成中にグリホサートをグリシンに置換すると、コラーゲンの三重らせん構造が大きく破壊されることが予想される。
小野らの研究では、ALS患者の脊柱のコラーゲンの質を、他の神経疾患を持つ患者および神経疾患を持たない対照群と比較して調べた[202]。その結果、ALS患者ではコラーゲンの束がより断片化されて広く分離し、毛細血管の周囲の空間でフィブリルがランダムに配向していることがわかったが、どちらの対照群でもこれらの病態は観察されなかった。また、ALS患者では脊髄全体のコラーゲンが有意に少なかった(p<0.001)。
OnoらによるALS患者の皮膚のグリコサミノグリカンに関する別の研究では、非硫酸化グリコサミノグリカンであるヒアルロン酸のレベルが異常に高いことが明らかになった[203]。このグリコサミノグリカンはゴルジ体ではなく細胞膜で合成されるというユニークな性質を持っており、GDP-フコースのような核化糖のゴルジ体への輸送障害があっても障害されることはなかった。
GGGGCCリピート拡大
C9またはf72遺伝子におけるGGGCCリピート拡大は、家族性ALSに関連する最も一般的な遺伝的欠陥であり、この現象を取り上げずしてALSのレビュー論文は完成しないであろう[206]。この特徴は家族性ALS患者の約40%、散発性ALS患者の8~10%に見られる。この遺伝子の野生型の発現には、通常、この配列の30回程度の繰り返しが含まれているが、多くのALS患者では数百のGGGGGCCのコピーが発見されている。
これは、DNA複製の際のコピーエラーによるものではなく、一本鎖DNAのエラーが修復されている間に遺伝子が拡大していくことによって起こるものである。したがって、酸化ストレスは、遺伝子が活発に発現している時に、DNAの一本鎖が露出している時に拡大を誘導するのではないかという仮説が立てられている。
この繰り返しの拡大は、ハンチントン病に関連する遺伝的欠陥を彷彿とさせる[207]。ハンチントン病では、トリヌクレオチド配列CAG[208]の暴走リピートが存在する。この配列はグルタミンをコードしており、したがって、この遺伝子の翻訳はポリグルタミンの長いペプチドの合成につながり、これが毒性効果に結びついていると考えられている。
他のタンパク質のポリCAG配列に関連する神経変性疾患は、少なくとも8つある。8-オキソグアニン・グリコシラーゼ(OGG1)というタンパク質の欠陥が、CAG配列の病的拡大の原因である可能性が高い[209]。
OGG1がヘキサヌクレオチドGGGCCの拡張にも関与しているかどうかはまだ明らかになっていない。OGG1は、グアニンの8-オキソグアニンへの酸化という、酸化損傷によってDNAに生じる最も一般的な欠陥を修復する役割を担っている。OGG1は一本鎖DNA配列から欠陥のあるヌクレオチドを除去し、その後、DNAポリメラーゼが欠損したグアニンを充填する。しかし、ヘアピンターンの形成を含む複雑なメカニズムのため、修復機構は、不用意に追加のヌクレオチドを局所的なCAG配列の複製として再挿入することができる。
したがって、グリシンアナログとして作用する過剰なグリホサートは、Seneff、 S.、 er al)。152 Bioinfo Proteom Img Anal |Volume 2: Issue 2 酸化と欠陥OGG1は、時間の経過とともにCAGリピート配列の継続的な長さにつながる可能性がある。
過度の酸化は、銅不足やグリホサートが保存されているグリシンを置換することでミトコンドリアのCcO活性が低下し、スーパーオキサイドが漏出することによると考えられる[86]。OGG1 の A ドメインのαAβB ループの 42 位には保存されたグリシンがあり、グアニンと 8-オキソグアニンの識別に必須である[210]。
このグリシンをグリホサートで置換することで酵素の機能が阻害されるかどうかは疑う余地がないが、それがどのようにしてリピート拡大を誘導する能力を説明しているのかは明らかではない。しかし、8-オキソグアニン部位も修復するOGG2は保存されているグリシンを欠いており、病理学的なリピート現象を引き起こすことはない[211]。
開始コドンが欠落しているにもかかわらず、GGGGGCCのポリヘキサヌクレオチド配列は、表2に示すように、3つの異なるジペプチド配列をもたらす3つの異なるフレームシフトで、実際に複数のポリジペプチドに翻訳することに成功している。
これらのそれぞれは、アラニン、プロリン、アルギニンという3つの他のアミノ酸のうちの1つとグリシンを交互に持っている。これらのポリペプチドは、実際にTDP-43およびFUSを含むプラーク領域で凝集し、蓄積する。
表[212-214]に記載された関連文献で説明されているように、これら3つのアミノ酸のすべてが、疾患を引き起こす天然に存在する類似体を有することは、重要なことかもしれない。循環から除去されるペプチドに高濃度でこれらのアミノ酸を組み込むプロセスは、細胞質から問題のある類似体を追い出そうとする戦略であるかもしれないというのは、もっともらしいと思われる。
確かに、これらのポリペプチドへのグリシンの高濃度取り込み率は、細胞からグリホサートを除去するのに役立つはずである。ハンチントン病のCAG配列はポリグルタミンを産生し、非コード化アミノ酸6-ジアゾ-5-オキソ-L-ノルロイシン(DON)は天然に存在するグルタミンの類似体である[215]。
自己免疫疾患としてのALS
ALSは、自己免疫疾患としての認識が高まっている[216]。運動神経末端に対する体液性免疫応答は、運動ニューロンにおけるカルシウムの恒常性を乱す生理的変化を引き起こす。最終的には、これがアポトーシスによる細胞死を引き起こす。
神経細胞の機能に関連する複数のタンパク質に対する自己抗体がALSと関連して発見されており、その中にはFas、ニューロフィラメント、電圧依存性Ca2+チャネル、ガングリオシド、アセチルコリン受容体などが含まれる[148,217-222]。
さらに、喘息、セリアック病、早期発症糖尿病、多発性硬化症、重症筋無力症、シェーグレン症候群、全身性エリテマトーデス、潰瘍性大腸炎などの他の自己免疫疾患は、将来ALSと診断されるリスクを高める[223]。ALS患者のIgGと筋繊維をインキュベートすると、Ca2+電流応答のピークが減衰する[224]。これは、L型電圧ゲーテッドカルシウムチャネルとの抗体反応によるものと考えられる。
確認として、ウサギ骨格筋から得られたL型電圧ゲーテッドカルシウムチャネルの抗体反応を試験した研究では、ALS患者48人のうち75%がこのタンパク質と反応する抗体を産生していたのに対し、正常者では25人中1人だけが陽性で、他の疾患を持つ対照患者35人中1人だけがこの反応を陽性としていた[222]。
抗体である免疫グロブリンG(IgG)は、体内で最も支配的な免疫グロブリンである。IgG/抗原複合体の細胞受容体への結合は、細胞免疫応答を誘発する。IgGはグリコシル化されたタンパク質であり、付着したグリカンは、ガラクトース、シアル酸、フコース、硫酸、およびN-アセチルグルコサミン(GlcNAc)の二分鎖残基の付加を含む修飾に基づく複合シグナルを伝達する。
細胞が抗原シグナルに応答する程度は、これらの修飾の特定の構成に決定的に依存する。フコシル化されたグリカンがALS IgGにおいて過少に発現していることは以前に述べた[83]。ALS患者由来のユニークな過少フコシル化糖鎖は、エフェクター細胞上のCD16に対するIgGの親和性を高め、抗体依存性細胞毒性(ADCC)を増強する。G93A-SOD1マウスの脳ミクログリアと神経細胞の間のシナプスに局在するALS IgGは、神経細胞の損傷に関与している可能性が高い。
腹部脊髄運動ニューロン-神経芽細胞腫ハイブリッド細胞を用いた試験管内試験(試験管内試験)研究では、ALS患者のIgGがアポトーシスを誘導してこれらの細胞を死滅させることが示されている[225]。オリゴ糖のガラクトシル化レベルの低下は、エリテマトーデスや関節リウマチと関連している[226,227]。これは、抗炎症作用を有する末端シアル化を阻害する[228]。
ALSに関連して観察されるコアフコシル化の欠如は、フコースがもはや存在しなくなると可能になる抗体と受容体の間の炭水化物-炭水化物相互作用により、受容体への結合が大幅に強化される(最大100倍)[229]。
アルツハイマー病、ハンチントン病、パーキンソン病、多発性硬化症、ALSなど、いくつかの異なる神経変性疾患の患者は、がんを発症する全体的なリスクが大幅に低い[230]。少なくともALSの場合は、アフコシル化IgGがこの効果を媒介している可能性がある。
不思議なことに、最近、IgGから化学的にフコースを除去したIgGを癌患者に投与して、癌に対する攻撃的な免疫攻撃を誘導するという新しい癌治療法が話題になっている[231]。これらの抗腫瘍療法は、将来的にALSのリスクを高めることにつながるのではないかという懸念がある。重症筋無力症は、筋力低下と疲労を引き起こす自己免疫性神経筋疾患である。
通常、シナプス後の神経筋接合部にあるニコチン性アセチルコリン受容体に対する抗体が循環することによって引き起こされる。これらの受容体に対する抗体は、ALSとの関連でも発見されている[221]。
興味深いことに、Mohanらはアセチルコリン受容体に対する抗体もまた、分子模倣によりミオシン重鎖と交差反応することを発見している[232]。このことは、筋力に壊滅的な影響を与えると予測されていたミオシン重鎖の699位のグリシンへのグリホサート置換[175]の可能性を示唆しているが、このことは、最初に欠損したミオシンタンパク質に対する免疫反応を誘導し、それが分子模倣を介してアセチルコリン受容体に対して活性化する可能性を示唆している。
これが真実であれば、欠損ミオシンタンパク質の蓄積による筋活動の遮断を可能にするエレガントな方法である。コリン作動性抗体によるシナプスの喪失は、慢性疲労症候群の強い特徴である代謝低下をもたらす[233]。
これとは対照的に、脳内の代謝低下はALSと関連している[235]。我々は、自己免疫疾患には抗体と受容体の両方の糖化パターンの障害が関与しており、これらはグリホサートによるグリコシル化ペプチド合成時のタンパク質機能の破壊に起因していると仮説を立てている。
エピジェネティクスの役割
エピジェネティクスは、遺伝子の転写後修飾とグリシンアナログとして作用するグリホサートを介して、転写活性が多くの遺伝子だけでなく多数のシグナル伝達経路にわたってどのように変化するかを説明している
www.ommegaonline.org 153 Bioinfo Proteom Img Anal |Volume 2: Issue 2 proteins.
食事、毒性暴露、汚染物質、薬、ライフイベント、軽度外傷性脳損傷(mTBI)などの環境ストレス因子はすべて、遺伝暗号を永久に変更することなく、DNAとそのタンパク質を化学的に修飾することで遺伝子発現を刺激する能力を持っている[236]。これらのストレス因子や環境曝露はエピジェネティックな変化を誘発し、その結果、遺伝子がオンになったり(遺伝子転写)オフになったり(サイレンシング)することをシグナルすることができる。
エピジェネティックな変化の動態は、静的な遺伝子突然変異とは異なり、酵素およびそのシグナル伝達経路を標的とすることによって逆転させることができる。エピジェネティクスには、ゲノムが遺伝子発現の変化を介して、シグナル伝達機構を介して環境と相互作用し、環境に適応するプロセスを含むことができる。
これらのメカニズムの多くは発生期に発見されているが、同様の現象が成人でも発見されつつある[237-241]。さらに、エピジェネティクスの調節における変化は、環境に誘導されたパーキンソン病と関連しており、エピジェネティクスはその病態形成の重要な因子となっている[242]。Lam et al 2016)は、一卵性双生児において、ALSへの遺伝的素因がALSの発症を保証するものではないことを示した。
この研究では、環境因子またはエピジェネティック因子が炎症性サイトカイン遺伝子発現の変化に主な役割を果たしていると結論づけた[243]。ChoiとKimは双子の研究で、彼らの結論に遺伝的要因が寄与していないことを発見し、エピジェネティクスとそれが表現型に果たす役割の重要性を強調した[244]。
さらに、遺伝子発現の変動は、環境の多様な誘因によって影響を受けうることが認識された。さらに、これらの著者らは、環境要素が遺伝子エピジェネティック・マシーンに及ぼす影響は、実際には隣接する遺伝子の転写活性を変化させることができ、”エピジェネティック・メカニズムは、遺伝子発現の変化を通じて、生物が環境に応答することを可能にする “と述べている。Jiangらは、エピジェネティクスを「神経科学の新しいフロンティア」と呼んでいる[245]。
グリホサートが体内の多数のエピジェネティック経路に及ぼす役割については、さらに解明する必要がある。これらの結果や疫学研究は、ALSに関与する特定の病因因子に関する貴重な情報を提供してくれることであろう。グリホサートなどの環境因子が遺伝子活性にどのように影響を与えるかを完全に理解することが、エピジェネティックな経路を操作してALSの謎を解き明かすための薬や治療法を開発するための鍵となるであろう。
議論
ALSは死亡率の高い衰弱性疾患である。複数の原因因子が複雑に絡み合っているため、症状が出る前に最も重要な初期因子を特定することは困難である。
しかし、一度ALSと診断されてしまうと、病気の進行を止めることは不可能に近いため、予防のためには、これらの初期の代謝異常を理解する必要がある。図2に示すように、腸内の代謝異常、特に腸内微生物によるフルクトースの代謝異常が、その後の骨格筋の損傷に続き、骨格筋に供給する運動ニューロンの損傷を引き起こすことを提案する。
フルクトースは、PEPカルボキシラーゼを介してCO2から炭素を固定化するシキメート経路と別の微生物経路の両方の基質であるPEPへの前駆体である。これらの経路はいずれもグリホサートによって障害されることが示されており、そのメカニズムは標的酵素の高度に保存されたグリシンへの置換が関与していると考えられている。
ALSは人生の中で比較的遅い時期に発症する。私たちの見解では、ALSの発症過程は数十年かけてゆっくりと4つの異なる段階を経て進行していくと考えている。
初期段階
最も初期の段階では、主に腸内で作用する。大腸粘膜へのフコースとキシロースの供給障害と大腸細胞への酪酸のバイオアベイラビリティーの低下は、大腸ムチンの菲薄化と腸管バリア機能の低下をもたらす。
第二段階
第二段階では、肝臓がNADHの重大な欠乏とグリホサート、LPS、H2 Sへの毒性曝露に苦しんでいる間に、肝臓が過剰なフルクトースを除去しようとするため、肝疾患が関与している。
第三段階
肝線維化とステアトーシスが臨界期に達すると、肝臓はもはや確実に循環から果糖をクリアすることができなくなり、病気の第三段階につながる。特に、痩せ型で身体的に健康な人の場合、骨格筋が果糖クリアランスのタスクを引き受けることになり、主に嫌気的に果糖を乳酸塩に変換することによって、速く収縮する筋肉は、酸化的リン酸化よりも解糖を好むため、優先的に害を受ける。筋肉の過労は、アミノ酸トランスポーターを介してグリホサートの取り込みを奨励し、ミオシンへのグリホサートの組み込みは、深刻な収縮能力を阻害する。これは、制御する運動ニューロンとのシナプスで興奮性刺激の要求を増加させ、疾患の第4期および最終期を前倒しする。
最終段階
最終段階では、複数の脱線が脊髄運動ニューロンを障害する。障害を受けた筋肉からの過剰な要求は、エネルギー要求量の増加につながり、その結果、抗酸化物質の要求量も増加する。
SOD1とCcOの両方には、グリホサートによって破壊される可能性のある重要なグリシンが含まれており、グリホサートによるグリシンへの置換によっても破壊される可能性のあるRNA結合タンパク質によって編成された、酸化的損傷の増加とストレス顆粒への不可逆的なタンパク質の凝集を引き起こす。
また、ミトコンドリアの長い軸索を下って戻ってくる輸送に関与する分子モーターも深刻な障害を受けるだろうオリゴデンドロサイトにおけるグリホサートの汚染は、軸索へのミエリン供給の障害をもたらす。タンパク質分解に抵抗するグリホサート汚染タンパク質に対する自己抗体が、この病気のプロセスに寄与している。
過剰なグルタミン酸の発現によるシナプスから軸索に沿って逆流するダイバック効果も考えられる。酸化的に損傷を受けたミトコンドリアは、それらを細胞のソーマに輸送して消去することができないため、軸索内にロックダウンられた状態になっている。さらに、グリホサートのキレート作用によるCuとZnの供給障害も一役買っている。
驚くべきことに、ALSの最も一般的な遺伝的マーカーは、病理学的に繰り返されるDNA配列GGGGGCCからの多重フレームシフトタンパク質合成から、様々なグリシン含有ポリジペプチド配列の合成を含む。挑発的には、これはグリホサートに汚染されたジペプチド配列の構築と隔離を介してグリホサートをトラップし、クリアするための戦略である可能性がある。
グリシンアナログとして作用するグリホサート( Seneff、 S.、 er al)。154 Bioinfo Proteom Img Anal |Volume 2: Issue 2
図3:グリホサートへの曝露がALSの発症に寄与する複数の方法の概要を図示したもの。
原文参照
これで、ALS発症のリスクが高い人のプロファイルが描けるようになったようだ。確かに、既知の遺伝的マーカーのいずれかがリスクを増加させることは間違いない。しかし、これとは無関係に、食事の選択が重要な役割を果たしているかもしれない。
果糖、特に遺伝子組み換えトウモロコシ由来の高果糖コーンシロップを多く含む食生活、マンガンと硫黄と銅の比率が高い食生活、亜硫酸塩とカラギーナンを多く含む加工食品の食生活、痩せ型の体型、禁酒、体力を過度に重視した食生活が、高リスクのプロファイルを形成していることが予想される。
農業従事者、特にグリホサート耐性作物を扱う人は特に脆弱である。環境にさらされていない人にとっては、リスクを減らすための簡単な方法は、食事のフルクトースとショ糖を大幅に減らした100%認証の有機全食の食事に切り替えることである。
結論
世界保健機関(WHO)世界銀行、ハーバード大学公衆衛生大学院によると、神経変性疾患(NDG)は、開発途上地域における疾病負担の第8位の原因になると予想されている。また、NDG疾患は、今世紀半ばまでにがんを抜き、死因の第2位になると予想されている[246]。
NDG疾患は2030年には7,000万人を超え、2050年には1億人を超えると予想されている[247]。NDG疾患は明らかに私たちの社会における健康上の重荷であり、ある程度の健康危機である。
この論文では、グリホサートの金属キレート作用、腸内微生物の破壊、果糖代謝の障害、重要な栄養素、特に芳香族アミノ酸とその誘導体の供給の妨害、肝臓への毒性効果、そして最も重要なことに、タンパク質合成時にグリシンの代わりになる可能性があるという特性から、グリホサートの慢性曝露がALSを引き起こす可能性のあるメカニズムを提示した。
グリホサートと ALS の間の複数の関連性は、図 3 に示したグラフィカルな概要で説明されている。私たちは、グリホサートに数十年にわたって慢性的に曝露された後に、腸の機能異常から始まり、肝疾患、筋不全、そして最終的には脊柱の運動ニューロンへの広範な損傷に至るカスケードがどのようにしてALSと診断されるかを示していた。
他のNDG疾患は、誤って折り畳まれたタンパク質が神経組織内の包接体に蓄積されるという特徴を持つ点で、ALSとかなり重複している。私たちは、ALSを超えて多発性NDGの憂慮すべき増加には、グリホサートが強い要因になっていると考えている。
特にグリホサートがグリシンアナログとして作用するグリホサートが、タンパク質合成中にグリシンに置換されることで、グリホサートが誘発すると予想される陰湿で破壊的な効果を考えると、www.ommegaonline.org 155 Bioinfo Proteom Img Anal |Volume 2: Issue 2 規制機関は、雑草の防除やその他の目的でのグリホサートの使用を禁止することを真剣に検討すべきである。
謝辞
この研究は、Qmulusプロジェクトの支援の下、Quanta Computers、 Inc.によって一部資金提供された。著者らは、プロテオームの枯渇とALSとの関連性に関する重要な論文を私たちに知らせてくれたDeborah Wotherspoonに感謝したいと思う。