
TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities
www.ncbi.nlm.nih.gov/pmc/articles/PMC6291370/
要旨
20年前、神経変性疾患の定義的特徴としてタンパク質のミスフォールディングと凝集体の蓄積が認識されたことを契機に、中枢神経系(中枢神経系)の神経細胞やその他の非神経細胞においてタンパク質の品質管理がどのように維持されているのかを徹底的に検討することになった。オートファジーは細胞の自己消化経路であり、中枢神経系のプロテオスタシスに特に重要な役割を果たしており、オートファジー異常はアルツハイマー病(アルツハイマー病)パーキンソン病(PD)ハンチントン病(HD)の神経変性の特徴として報告されている。
転写因子EB(TFEB)は、オートファゴソーム形成、リソソーム形成、リソソーム機能に必要な遺伝子の発現を促進するオートファジーの主要な転写調節因子の一つであり、中枢神経系で高発現している。この7年間でTFEBが注目されており、TFEBの機能不全が多くの神経変性疾患の発症に関与していることが明らかになってきた。
本総説では、アルツハイマー病、PD、HD、X-linked spinal and bulbar muscle atrophy、筋萎縮症、筋萎縮性側索硬化症などの研究を中心に、TFEBの機能障害が神経変性疾患にどのように関与しているのかについて、現在の理解を明らかにした。
TFEBはオートファジー活性化の中心的な役割を果たしていることから、TFEBの機能障害の基盤を理解することで、TFEBがどのようにして治療の対象となるのかが明らかになってきており、神経変性疾患への幅広い応用が期待される治療法の開発に向けての刺激的な機会となることが期待される。
キーワード
神経変性、オートファジー、転写因子EB、アルツハイマー病、パーキンソン病、ハンチントン病、ポリグルタミン、脊髄・大腿筋萎縮症、筋萎縮性側索硬化症、リソソーム、シヌクレイノパシー
ニューロンの生存は、効率的なタンパク質の品質管理を維持することに依存している。オートファジーは、進化的に保存されたリソソーム分解経路であり、神経細胞で非常に活発に活動しており、アルツハイマー病、パーキンソン病、ハンチントン病を含む多くの神経変性疾患の特徴である、凝集しやすいタンパク質と機能不全のミトコンドリアを除去する機能を持っている(図1)。実際、最近の研究では、神経細胞のオートファジーが神経変性疾患の標的であること、また、オートファジー経路の構成タンパク質や調節因子をコードする遺伝子内で神経変性疾患に関連した突然変異が起こりやすいことから、神経変性疾患のエフェクターであることが強く示唆されている。
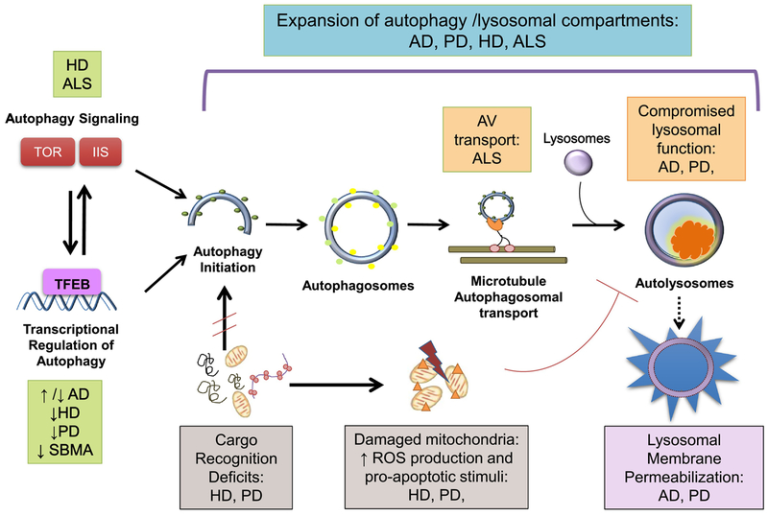
図1 神経変性疾患におけるオートファジー機能不全
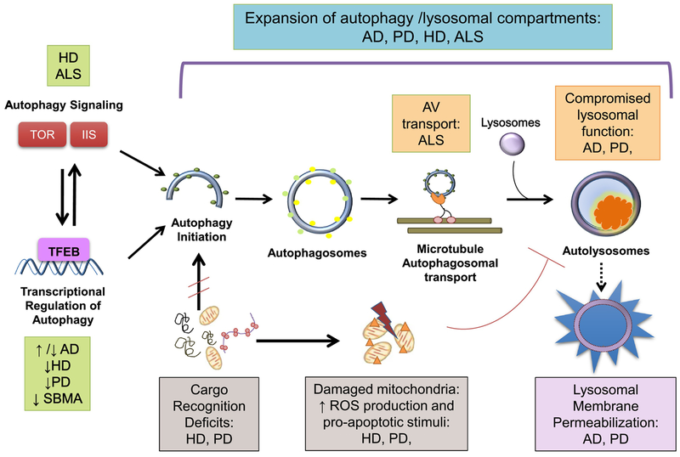
オートファジー小胞(AV)の増加は、アルツハイマー病(AD)、パーキンソン病(PD)、ハンチントン病(HD) 球脊髄性筋萎縮症(SBMA)および筋萎縮性側索硬化症(ALS)を含む多くの神経変性疾患に共通の所見である。しかし、これらの神経変性疾患で観察されるオートファジー経路の機能不全は、異なる分子機構によって説明されている。
1) オートファジーシグナル伝達経路は、HD、ALS、および加齢脳で障害されており、ラパマイシン(TOR)およびインスリン/IGF1シグナル伝達(IIS)の機序的標的が有意に影響を受けていることが明らかになった。
2) オートファジー転写ネットワーク活性はHD、PD、加齢脳で低下する。これらの所見とは対照的に、アルツハイマー病ではオートファジー遺伝子のアップレギュレーションが示唆されている。マスターオートファジーレギュレーター転写因子E-B(TFEB)シグナリングの変化は、PD、HD、およびSBMAのために報告されている。
3) カーゴ認識の欠損、特に欠損ミトコンドリアの欠損は、HDとパーキンソン病で発生する。損傷を受けたミトコンドリアが蓄積すると、反応性酸素種(ROS)の負荷が増加し、リソソソーム機能が低下し、リソソソームの透過性が低下し、リソソソームの内容物が細胞質に漏出する。HDにおける「空の」オートファゴソームの蓄積もまた、神経細胞の恒常性を損なう。
4) 正常なオートファジー誘導とオートファゴソーム形成にもかかわらず、オートファジー液胞(AV)のリソソームへの成熟と微小管輸送の障害はALSの特徴である。
5)リソソソーム機能不全は、特に加齢に伴いプロテオスタシスのクリアランス機構が低下することで、アルツハイマー病やHDにおけるオートファジーの機能障害に寄与している。また、リソソソーム内腔に未消化物(小器官、リポスフスチンなど)が蓄積すると、リソソソームの分解能力がさらに低下し、リソソソーム膜の不安定化と漏出が促進される。
転写因子EB(TFEB)は、リソソソーム機能とオートファジーのマスターレギュレーターとして登場した。細胞のホメオスタシス経路を制御する重要な転写因子として、複雑な制御ネットワークが TFEB の活性と機能を支配しているが、まだ不完全に定義されている。
かなりの研究が、細胞代謝におけるTFEBの役割を理解しようとしてきたが(MartinaおよびPuertollano 2013;Medina et al 2015;Settembre et al 2013;Settembre et al 2012)中枢神経系(中枢神経系)ホメオスタシスにおけるTFEBの関与を探求し始めたのは、最近になってからである。
TFEBは、ニューロンおよびアストロサイトの両方において活性であるため、中枢神経系において広く発現している(Decressac et al 2013; Reddy et al 2016; Wang et al 2016b)。2014)神経変性疾患ではTFEBの局在、活性、機能が変化している(Cortes et al 2014b; Decressac et al 2013; Tsunemi et al 2012; Wang et al 2016b)。
このレビューでは、神経変性疾患におけるTFEBの役割に関する現在の理解を明らかにし、TFEBがこれらの悲惨な疾患の治療のための潜在的な治療標的となり得るかどうかを検討する。
TFEBの活性はどのように制御されているのか?
TFEBは、転写因子の基本的ならせん-ループ-らせん-ロイシンジッパーファミリーの一員であり、通常は細胞質に局在するが、飢餓状態やリソソソーム機能が低下した場合には核に転座する(Sardiello et al 2009; Settembre et al 2011)。TFEBの過剰発現は、オートファゴソームの数を増加させ、新しいリソソソームの生成を促進し、オートファジーのフラックスを増加させる。
CLEAR(Coordinated Lysososomal Enhancement And Regulation)シグナル伝達ネットワークを介して、TFEBはオートファジーリソソソーム遺伝子の発現を調節し、細胞のクリアランスおよび代謝を促進する(Sardiello et al 2009; Settembre et al 2011)。実際、オートファジーおよびリソソソーム関連遺伝子に加えて、CLEAR遺伝子ネットワークのより深い検討は、細胞の異化、ミトコンドリア機能、および細胞ストレス応答に関与する経路を浮き彫りにしている(Palmieri et al 2011)。
これと一致して、TFEBは、最近、肝臓における脂質異化および分解を直接調節することが示され(Settembre et al 2013)および骨格筋におけるグルコースホメオスタシスおよびミトコンドリア生合成を促進することが示された(Mansueto et al 2017)。したがって、細胞のコンテキストは、TFEB媒介転写の特定の標的を決定する上で重要な役割を果たす可能性があり、神経変性におけるTFEBの役割を解読するために、中枢神経系特異的TFEB標的の詳細な解析の必要性を強調している。
TFEBは基本的な細胞恒常性経路を調節するマスター転写因子として重要な役割を果たしているにもかかわらず、TFEBの活性を支配する制御ネットワークは複雑であり、まだ完全には定義されていない。TFEBは翻訳後修飾を受けており、タンパク質間の相互作用や細胞内局在を直接制御している。現在のTFEBの生物学的理解では、リン酸化がTFEB活性の主要なドライバーであると考えられている。特に、セリン211のリン酸化は、TFEBの活性化状態を決定する上で重要な役割を果たしており(Martina et al 2012; Roczniak-Ferguson et al 2012)細胞質のシャペロン14-3-3との相互作用を促進し、TFEBを細胞質に保持している。セリン142のリン酸化もまた、TFEBの細胞質保持に必要とされる可能性がある(Settembre et al 2011; Settembre et al 2012)。エレガントな試験管内試験研究は、ラパマイシン複合体1(mTORC1)のすべての重要な機械的標的、およびおそらく細胞外シグナル調節キナーゼ2(MAPK1としても知られている)を含む、これらの残基でTFEBを直接リン酸化する栄養感知細胞キナーゼを同定した(Martina et al 2012; Roczniak-Ferguson et al 2012; Settembre et al 2013; Settembre et al 2011)。非常に最近、MAP4K3は、アミノ酸供給をTFEB活性化状態にリンクさせ、TFEBの重要な上流調節因子であることが発見された(Hsu et al 2018)。MAP4K3は、セリン4上のTFEBをリン酸化し、このリン酸化イベントは、14-3-3での細胞質隔離を介してその完全な不活性化を保証するために、mTORC1がそのセリン211残基上のTFEBをリン酸化するために必要とされる(Hsu et al 2018)。
飢餓またはリソソームストレス時には、mTORC1は不活性化され、リソソームカルシウム放出は、TFEBを標的とするホスファターゼカルシニューリンを活性化する。このシグナル伝達カスケードは、TFEBの脱リン酸化で絶頂を迎え、その核内転座を促進し、CLEARネットワークにおけるTFEB標的遺伝子のその後の遺伝子転写を促進する(Medina et al 2015)。TFEBは、細胞の代謝需要を管理するためにCLEARネットワークの転写を調整するバイオエネルギーシグナリングの中心的なハブとして生じている(Palmieri et al 2011; Settembre et al 2012)。さらに最近では、リジン116でのTFEBのアセチル化が、ミクログリアにおけるTFEB機能の新規な翻訳後修飾因子であることが発見された(Bao et al 2016)。様々な予測データベースによる解析により、TFEBのアミノ酸配列中の他の潜在的なリン酸化許容残基が同定されたが、そのほとんどは現在研究されていないままである。これらの新規残基の翻訳後修飾のTFEBホメオスタシスへの寄与(もしあれば)およびニューロンおよび他の中枢神経系細胞型におけるミスフォールドタンパククリアランスおよび代謝におけるそれらの役割の検討は、最も重要であろう。
中枢神経系におけるTFEBの機能とは?
細胞代謝におけるTFEBの役割を理解するためにかなりの研究が行われてきたが(Martina and Puertollano, 2013; Medina et al 2015; Settembre et al 2013; Settembre et al 2012)神経細胞の恒常性維持におけるTFEBの潜在的な関与を探求し始めたのは最近になってからである。
TFEBは、中枢神経系において高度に発現し(Decressac et al 2013; Reddy et al 2016; Wang et al 2016b)ニューロンおよびアストロサイトの両方において活性であることが知られている(Decressac et al 2013; Xiao et al 2014)。重要なことに、TFEB活性および局在は、神経変性疾患において変化している(Cortes et al 2014b;Decressac et al 2013;Tsunemi et al 2012;Wang et al 2016b)
TFEB機能の障害は、疾患の病因にリンクされた欠陥細胞クリアランス表現型に関与していることを示唆している。TFEBの適度な過剰発現は、有意な神経毒性を伴わずに達成することができ(Pi et al 2017)神経変性疾患モデルにおけるマウス中枢神経系におけるTFEB活性変調の様々な研究を可能にする(Decressac et al 2013; Pi et al 2017; Polito et al 2014; Xiao et al 2014)。これまでのところ、TFEBの変異は、ヒト患者における腎細胞癌とのみ関連しているが(Kauffman et al 2014)最近のGWASは、TFEBのイントロニック領域変異をアフリカ系アメリカ人集団における後期発症アルツハイマー病リスクとリンクしたが(Mez et al 2017)TFEBの変異は、ヒト患者における腎細胞癌と関連していない。これまでのところ、この特定のバリアントがTFEBの機能に及ぼす影響は誰も調べていないが、この潜在的な修飾因子が検出されたことは、疾患に関連したTFEBの遺伝的変化の可能性を広げるものである。
アルツハイマー病
アルツハイマー病は認知症の非常に一般的な原因であり、加齢に伴う脳の変性によって特徴づけられる。アルツハイマー病の脳病理学的特徴は、高リン酸化微小管関連タンパク質タウ(リン酸化タウ)とアミロイドβ-42(アミロイドβ42)ペプチドの細胞外沈着物である神経斑で構成される脳内細線維状のもつれである。
アミロイド前駆体タンパク質(APP)プレセニリン1およびプレセニリン2をコードする遺伝子の遺伝的変異がアルツハイマー病の家族性症例を説明し、影響を受けた脳領域では、オートファジーネットワークの深遠な転写異常とオートファジー液胞の広範な蓄積が表示され、しばしば未消化の貨物を運んでいる(Lipinski et al 2010; Wolfe et al 2013)。
アルツハイマー病におけるオートファジー機能不全のための現在の作業モデルは、リソソーム機能が損なわれた(遺伝的アルツハイマー病関連変異および/または年齢に関連したまたは環境誘発性のプロテオスタシスの変化によって引き起こされる)オートファジー流束のボトルネックを作り出し、未熟なオートファジー小胞およびエンドリソソーム中間体の病理学的蓄積に直接つながるという仮説を立てている(Bordi et al 2016;Cataldo et al 1996;Cataldo et al 2000;Wolfe et al 2013)。
アルツハイマー病発症におけるTFEBの役割は、現在、激しい研究の対象となっている。TFEBの機能と局在の重要な変化がアルツハイマー病のモデルで報告されているが、アルツハイマー病におけるTFEBの直接的な関与についての利用可能な証拠は、非常に相関性が高く、時には矛盾したままである。
アルツハイマー病患者のリンパ球および単球(損傷を受けた中枢神経系領域に移動し、アルツハイマー病の進行を制御する可能性が高い免疫細胞)の分析は、明らかに低下したTFEBの発現を明らかにし、TFEBの機能不全がアルツハイマー病で観察されたリソソソーム欠損の根底にある可能性があることを示唆している(Tiribuzi et al 2014)。
この結論と一致して、アルツハイマー病患者の脳サンプルのサブセル分画は、病理の程度と海馬核TFEBのレベルの間の強い逆相関を持つ核TFEBの選択的な損失を文書化した(Wang et al 2016b)。TFEBの同様の核内排除は、ダブルプレセニリンノックアウト細胞の試験管内試験モデルにおいて最近報告された(Reddy et al 2016)。さらに、CLEARネットワーク活性は、アルツハイマー病患者線維芽細胞およびiPSC由来のヒトADニューロンにおいて有意に減少しており(図3)TFEBの細胞質保持がアルツハイマー病発症に寄与する可能性があることを示唆している(Reddy et al 2016)。
計算モデルおよびDNAプルダウン結合アッセイは、遅発型アルツハイマー病の最もよく知られた主要な危険因子対立遺伝子であるAPOE ɛ4が、TFEBのトランザクティベーションのすべての確立された標的およびオートファジー機能のすべてのキープレイヤーであるSQSTM、MAP1LC3B、およびLAMP2遺伝子のプロモーター内のCLEAR要素への結合のためにTFEBと競合する可能性があることを示唆している(Parcon et al 2017)(図2)。APOE ε4/ε4キャリアは、他のAPOE対立遺伝子を有するアルツハイマー病患者と比較して、より早い疾患発症および著しく増加したタンパク質凝集を有することから、この知見はかなり重要であり、これはAPOE ε4/ε4 アルツハイマー病患者におけるCLEARネットワークの阻害を強く示唆している。
この仮説と一致して、マウスにおける神経細胞特異的TFEB切除は、ヒトAD患者における神経病理学的所見を強く想起させる脳内のアミロイドβおよびペアヘリカルフィラメント(PHF)p-Tau蓄積をもたらす(Reddy et al 2016)。しかし、現在までのところ、TFEB機能障害のアルツハイマー病への寄与を直接取り上げた研究はない。中枢神経系における細胞型特異的ノックアウトまたは過剰発現による、アルツハイマー病神経変性および神経病理に対するTFEBのモジュレーションの効果は、アルツハイマー病病態へのTFEBの機能不全の寄与についての重要な洞察をもたらす可能性がある。
図2 神経変性疾患におけるTFEBの制御異常
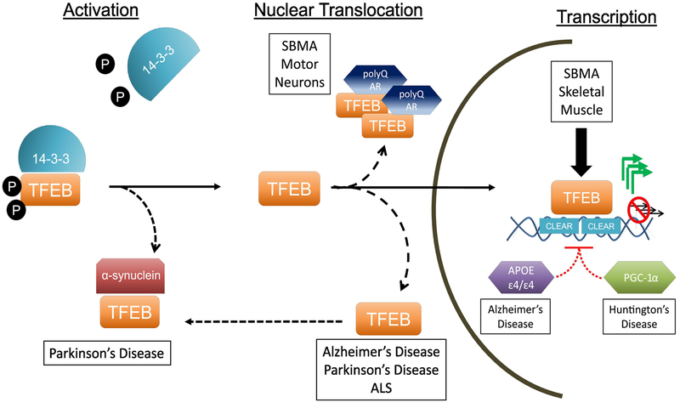
TFEBの機能は、それ自身の活性化、核内への移動、標的遺伝子の転写活性化の3つの段階で厳密に制御されている。リン酸化されたTFEBは、活性化されていない状態では、14-3-3-3タンパク質と相互作用し、細胞質に隔離されたままである。活性化されると(リソソソームストレスやmTOR阻害下で)TFEBは脱リン酸化されて14-3-3複合体から解離し、核内局在化シグナルを明らかにする。
TFEBは核内に移動し、標的遺伝子のCLEARネットワークの転写を促進する。これらのステップのうちのいくつかは神経変性疾患において機能不全となることが報告されており、TFEBの隔離(パーキンソン病では)核の排除(SBMA、アルツハイマー病、PD、ALSでは)転写不全(SBMA、アルツハイマー病、HDでは)が含まれている。これらの観察結果は、TFEBの制御異常が神経変性蛋白質障害の特徴であることを示している。
図3 神経変性におけるTFEBの非細胞自律的な制御異常
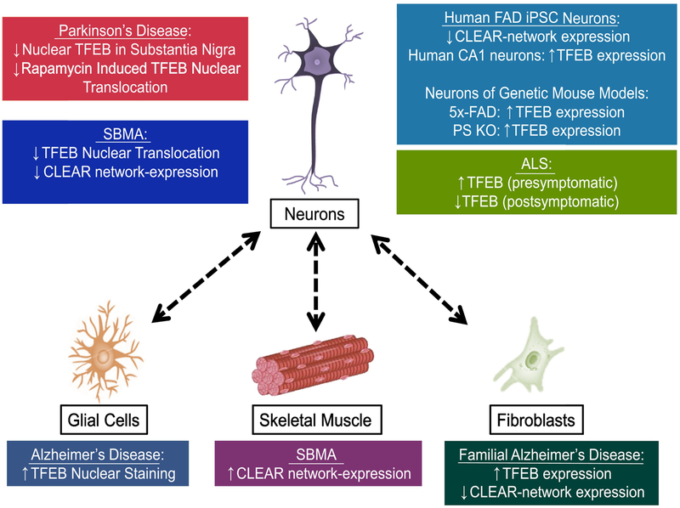
非神経細胞が神経変性におけるニューロン機能不全に寄与する非細胞自律神経毒性は、ほとんどの疾患における疾患発症の主要な特徴として浮上してきている。多くの研究では、神経変性疾患におけるTFEBの細胞型特異的な調節障害が報告されており、TFEBの生物学的性質には細胞の文脈が重要な役割を果たしていることが示唆されている。
このことは、特にアルツハイマー病や神経変性疾患では、異なる疾患関連組織が表裏一体のパターンでTFEBの制御異常を示すことが明らかになっている。TFEB機能の細胞型特異的な制御や、異なる組織間でTFEBの状態を伝達するシグナル伝達経路を理解することは、神経変性疾患の治療法開発のための重要なターゲットとなる可能性がある。家族性アルツハイマー病:家族性アルツハイマー病、iPSC:誘導多能性幹細胞
TFEB活性の減少の観察とは対照的に、家族性プレセニリン-1 A246E変異を有するアルツハイマー病患者由来の線維芽細胞は、増加したTFEB発現を示した(Coffey et al 2014)。プレセニリン-1条件ノックアウトマウスの別の研究はさらにTFEBレベルの変化が検出されなかったが、CLEARネットワーク遺伝子のサブセットの有意なアップレギュレーションを明らかにした(Zhang et al 2012)。
TFEB標的の同様のトランスクリプトームアップレギュレーションは、5つの家族性アルツハイマー病変異を有する5x-家族性ADマウスで報告された(Landel et al 2014)。レーザーキャプチャーマイクロダイセクションによるアルツハイマー病被験者の海馬のヒトCA1錐体ニューロンの最近のトランスクリプトーム解析は、TFEBとMitF転写因子ファミリーの密接な関連メンバーであるTFE3の発現の増加、およびオートファジー遺伝子標的の発現の増加を示した(Bordi et al 2016)。これらの知見は、特に影響を受けた脳領域において、アルツハイマー病におけるTFEBの機能獲得を強く支持している(図3)。
アルツハイマー病におけるTFEB機能障害 相関か因果か?
矛盾するが、家族性アルツハイマー病プレセニリン変異は、慢性的なリソソソームアルカリ化を産生することによってリソソーム機能を損なう可能性があり(Coffey et al 2014;Lee et al 2010;Zhang et al 2012年)それが代償メカニズムとしてTFEBの関与と活性化を誘発する可能性があるので、これら2つの相反する結果のセットは、相互に排他的ではないかもしれない(Sardiello et al 2009;Settembre et al 2011)。二次的な(または並行して)加齢に伴う TFEB 核排除とそれに伴う機能低下が散発性 アルツハイマー病 でも起こるかどうかは、現在のところ不明であり、研究されるべきである。さらに、TFEBの活性化状態は、神経変性において高度に細胞型特異的である(Cortes et al 2014b)ことから、TFEBの機能が隣接する細胞型で異なる可能性があり、アルツハイマー病におけるTFEBの機能不全の徹底的な解剖を可能にするために、細胞中枢神経系集団の分離を必要とすることが示されている。実際、これは運動ニューロンと骨格筋の間のXリンク脊椎&大腿筋萎縮で文書化されているように、TFEBの機能が通信非ニューロン細胞とニューロンで全く反対であることは、前例がないわけではない(Cortes et al 2014b)。最後に、死後のアルツハイマー病患者の脳組織の分析は、破局的なニューロンの崩壊に関連付けられている疾患の非常に後期段階を表している。TFEBの過剰活性化は、したがって、病気の病因を駆動しない最終的な細胞応答を反映している可能性がある。
セリンスレオニンキナーゼであるグリコーゲン合成酵素キナーゼ3(GSK3)は、APPとタウを直接リン酸化してアミロイドβ42産生を促進することにより、アルツハイマー病発症において役割を果たすことが提案されている(Blallock et al 2004;Parr et al 2012)。GSKは2011年にTFEB活性の仮説的な調節因子として同定され(Settembre et al 2011年)GSK阻害剤VIIIは、試験管内試験で核内TFEB蓄積を増加させ、APPレベルを低下させることができる(Parr et al 2012)。これらの知見は、アルツハイマー病におけるGSK活性の変化を媒介としたTFEBの機能不全を示唆しているが、TFEBがGSKを媒介としたリン酸化の直接的な標的であるかどうか、GSKによるAPPの減少がTFEBに依存しているかどうか、そして最も重要なことは、この経路が生体内試験でのアルツハイマー病発症に本当に関連しているかどうかを決定するためには、さらなる研究が必要である。
アルツハイマー病におけるTFEBの治療標的
アミロイドβ42ペプチドとp-Tauの両方がオートファジー媒介分解の基質である(Berger et al 2006; Caccamo et al 2010)ことから、オートファジーの調節がアルツハイマー病において有益である可能性が示唆される。例えば、APPマウスの脳へのBeclin-1(ATG6としても知られるオートファジー開始因子)のレンチウイルス送達は、細胞内アミロイドβ42およびアミロイドプラーク蓄積を有意に減少させたが、Beclin-1の減少はAPP/Beclin-1ヘテロ接合ヌルマウスのアルツハイマー病およびオートファジー表現型を悪化させた(Pickford et al 2008)。よく知られたmTOR阻害剤であり、オートファジー誘導剤であるラパマイシンの治療は、海馬のアミロイドβとタウの病理を減少させ、中枢神経系のオートファジーを活性化させ、3xTg-ADマウスの学習と記憶障害を改善した(Caccamo et al 2010)。また、ラパマイシンはショウジョウバエのタウ症モデルにおいてもタウ毒性を減少させ、ハエの生存率と退行性眼の表現型を改善し、オートファジーを介した変異タウタンパク質のクリアランスを促進した(Berger et al 2006)。これらの研究におけるTFEBの特異的な役割は検討されなかったが、mTORの阻害は、TFEBの核内転座および活性化の重要な決定因子である(Settembre et al 2011)。さらに、これらの研究は、マウスのオートファジー誘導に対するアルツハイマー病脳の応答性を強調し、オートファジー調節がアルツハイマー病治療開発のための更なる検討に値することを示唆している。
ベクリン-1遺伝子の投与量を変更し、mTORを阻害することによるオートファジーの調節の結果にもかかわらず、オートファジー転写ネットワークがアルツハイマー病において深刻な障害を受けていることを認識することが重要である(Lipinski et al 2010)主要なオートファジー阻害剤であるmTORの過剰活性化(Caccamo et al 2010;Lafay-Chebassier et al 2005)オートファジー経路自体がアルツハイマー病の発症において影響を受けていることを示唆している。
広範な生化学的および超微細構造解析は、リソソームにおけるオートファジーカーゴの分解障害に基づくオートファジーの機能喪失を明らかにしてきた(Boland et al 2008;Lee et al 2010;Nixon et al 2005;Yang et al 2014)。
したがって、アルツハイマー病脳におけるオートファジーの神経保護的誘導は、最初の小胞形成からリソソーム融合とリソソーム分解機能に至るまでの全体のオートファジー経路が促進される場合にのみ、これらの基礎となるカーゴクリアランスの欠損の修正が必要となるかもしれない。
現在利用可能なオートファジー治療薬の多くはオートファジー活性化のみを標的としているため、リソソソーム全体のクリアランスを向上させることができない治療効果となる可能性が高い。
TFEBはオートファジー誘導をアップレギュレートし、リソソーム数、機能、クリアランスの増加を促進するので、TFEBは理想的な治療標的となるはずである(Sardiello et al 2009)。
これと一致して、TFEBは、アルツハイマー病および関連するタオパチーのモデルで有望であることが示されている。TFEBの中枢神経系送達は、神経原線維のもつれの病理を効果的に減少させ、タオパチーのrTg4510マウスモデルにおいて、行動表現型、シナプス欠損、および神経変性を救済した(Polito et al 2014)。TFEB過剰発現は、毒性のあるリン酸化タウ種を選択的に標的とするように思われたが、健康なニューロンには劇症的な影響を及ぼさなかった(Polito et al 2014)。
生体内試験での毒性タウ種のTFEB媒介分解に対する同様の特異性は、遺伝的ニューロン特異的TFEB過剰発現モデルにおいて報告された(Wang et al 2016a)。この系では、P301Sタウオパシーマウスモデルにおけるニューロン標的化TFEB過剰発現は、大脳皮質および海馬における毒性リン酸化タウおよびリポフスチンレベルの有意な減少をもたらし、記憶および学習テストにおけるパフォーマンスの有意な改善とともにもたらされた(Wang et al 2016a)。
上述したように、TFEBの状態は、非神経細胞とニューロンの間で異なることがあるので、非神経細胞におけるTFEBの変調の研究は何を示しているのであろうか?興味深いことに、アルツハイマー病の非神経細胞はまた、TFEBの誘導から利益を得るかもしれない。アストロサイトは、シナプスクリアランスと神経機能を調節し、ニューロンの生存に重要な役割を果たしており、その機能はアルツハイマー病中に変化する。
APP/PS1トランスジェニックマウスの海馬へのアストロサイト特異的なTFEBの送達は、TFEB過剰発現アストロサイトにおけるアミロイドβの取り込みとクリアランスを促進することにより、間質性アミロイドβレベルと海馬プラーク負荷を減少させた(Xiao et al 2014)TFEBが生体内でアストロサイトのアミロイドβクリアランスを促進することができることを示唆している(図3)。
ミクログリア、中枢神経系の常駐マクロファージは、脳を調査する上で重要な役割を果たし、アルツハイマー病におけるアミロイドプラーク沈着のサイトに募集されている(Meyer-Luehmann et al 2008)。SIRT1によるTFEBの脱アセチル化は、リソソソームバイオジェネシスおよびフィブリルアミロイドβのミクログリア分解を増強し、APP/PS1マウスのex vivo脳スライスにおいてアミロイドプラークの減少を誘発する(Bao et al 2016)。
しかし、最近の研究は、変更された代謝調節とmTORC1機能の障害に起因するミクログリアにおける過剰なオートファジー活性化は、アルツハイマー病において劇症的であり得ることを示している(Ulland et al 2017)神経変性における異なる細胞型におけるオートファジー障害の複雑さを強調している。アルツハイマー病海馬におけるTFEBの核染色は、異常なmTORC1シグナリングに起因するTFEBの過剰活性化も考慮に値するが、おそらく、神経細胞の破片やタンパク質の凝集の清掃をサポートするために、グリアでより顕著であるように見える(ボルディ et al 2016)。
いずれにせよ、アルツハイマー病の病理学へのアストログリア細胞タイプの貢献は激しい調査中であるが、それは非ニューロン細胞がニューロンよりもアミロイドβクリアランスを制御する上でより強力なエンティティである可能性があることを推測したくなる。アストロサイトは、アルツハイマー病の初期段階ではシナプス裂け目から可溶性アミロイドβをクリアーする役割を果たしているようにさえ見えるが、ミクログリアはアルツハイマー病病理の後期段階ではフィブリルアミロイドβ42をクリアーすることができる。これらの知見は、異なる疾患ステージで異なる細胞タイプのTFEBを誘導することで、調整されたTFEBアップレギュレーションの治療アプローチの可能性をサポートしている。
TFEB の新規な低分子誘導剤の発見は、アルツハイマー病 の分野で大きな関心を生成している。GSK3の化学的阻害を介したTFEBの活性化は、APPおよび有毒なC末端APPフラグメントのリソソームクリアランスを促進する(Parr et al 2012年)TFEBによるリソソームクリアランスの誘導は、アルツハイマー病に関連した蛋白質障害を効果的にクリアすることができることを示唆している。これと一致して、最近同定されたTFEBの化学的活性化剤であるヒドロキシプロピル-β-シクロデキストリン(Song et al 2014)は、2つのAPP疾患関連変異を発現するADマウスにおいて、毒性タンパク質のクリアランスおよび臨床症状を改善した(Yao et al 2012)。APP/PS1トランスジェニックマウスモデルにおけるTFEB活性化によって媒介される同様の神経保護効果は、高麗人参に見出される化合物であるジペノサイドXVIIについて最近報告された(Meng et al 2016)。
さらに、TFEB誘導に起因するリソソームクリアランスの利益に加えて、他のTFEBの作用がアルツハイマー病治療の利益に大きく関連している可能性がある。TFEB媒介転写の標的として知られるリソソソームプロトンポンプの発現増加(Sardiello et al 2009)は、家族性アルツハイマー病で観察されるリソソソーム酸性化の障害を潜在的に是正する可能性がある(Lee et al 2010)。TFEBはまた、アルツハイマー病で障害されることが知られているプロセスであるリソソソームカルシウム放出を誘導する(Medina et al 2015)。興味深いことに、TFEBは、PTENキナーゼのアップレギュレーションとそれに伴うAktとmTORの阻害を介して自動制御ループを生成する可能性があり、さらにTFEBの活性化で最高潮に達する(Polito et al 2014)。PTENは神経の分化とシナプス可塑性に必須であるため、TFEBを介したPTENのアップレギュレーションは、アルツハイマー病ニューロンにさらに利益をもたらす可能性がある。
パーキンソン病および関連する神経症
パーキンソン病(PD)は、主に黒質部のドーパミン作動性ニューロンに影響を及ぼす神経変性疾患である。パーキンソン病の主要な特徴は、主に誤って折り畳まれて凝集したα-シヌクレインからなる、レビー小体として知られるタンパク質性の細胞質内包物が黒質ドーパミン作動性ニューロンに蓄積することである(Kalia and Lang, 2016)。オートファジー-リソソソーム欠損はパーキンソン病の特徴であり(図1)オートファジー小胞の蓄積およびリソソソーム機能の障害の報告がある(Dehay et al 2010;Winslow et al 2010)。また、パーキンソン病病理における多くの分子プレーヤーは、α-シヌクレイン、パーキン、DJ-1,およびLRRK2を含むオートファジー経路そのものを調節することが報告されている(Kalia and Lang, 2016)。
アルツハイマー病と同様に、TFEBは、パーキンソン病患者の死後実質性黒質において選択的に核から排除されているようであり、リソソーム機能のマーカーの漸進的な低下と密接に関連しており、TFEBの誤局在化がパーキンソン病で起こることを示唆している。α-シヌクレインは、14-3-3-3タンパク質と構造的にも機能的にも相同性があり(Ostrerova et al 1999)TFEBと関連し、細胞質へのTFEBの保持を促進することが知られているシャペロンファミリーである(Martina et al 2012)。興味深いことに、ラットのPD様神経変性モデルにおいて、α-シヌクレインとTFEBは生体内で共沈した(Decressac er al)。 この仮説と一致して、α-シヌクレイン病理を示すParkin Q311X変異マウスのラパマイシン処理は、黒質におけるTFEB核転座および機能の障害を明らかにした(Siddiqui et al 2015)異常なα-シヌクレイン-TFEB相互作用は、様々な形態のα-シヌクレイン毒性における共通の特徴であるかもしれないことを示唆している。同様に、減少したTFEB核転座反応は、X-linked spinal & bulbar muscular atrophyの細胞モデル(Cortes et al 2014b)においても報告されており(以下で詳細に議論)異なる神経変性疾患に寄与する潜在的な共有メカニズムとしてTFEBの核からの隔離を強調している。このモデルを支持するために、ある研究では、コントロールとパーキンソン病の腹側分割領域(PD神経変性に一般的に抵抗性のある脳領域)の間で、TFEBの機能および細胞内局在が変化していないことが明らかになった(Decressac et al 2013)。さらなる実験が必要であり、α-シヌクレイン毒性に対して交互に感受性または抵抗性を有するパーキンソン病脳領域におけるTFEBインタラクチュードームを評価しうる。
α-シヌクレインはオートファジーを介した分解の基質として知られており、リソソソーム機能は様々なPDモデルにおいて障害されている。例えば、MPTP誘発性ドーパミン作動性神経毒性マウスでは、オートファジーによる液胞の蓄積とドーパミン作動性細胞死が、リソソソーム数の著しい減少に先行し、最終的にはリソソソームの透過性化と細胞クリアランスの障害をもたらした(Dehay et al 2010)。ウイルス性TFEBの過剰発現による神経細胞のリソソソームバイオジェネシスの誘導は、リソソソーム破壊とオートファージ性空胞蓄積を抑制し、神経保護とドーパミン神経細胞のα-シヌクレイン凝集体の減少を達成した(Dehay et al 2010)。ラパマイシン治療はまた、このモデルにおけるPD関連ドーパミン神経変性を減衰させ、リソソームのレベルと機能を回復させた(Dehay et al 2010)。TFEBの活性化剤である2-ヒドロキシプロピル-β-シクロデキストリン(Song et al 2014)は、最近、試験管内試験でα-シヌクレインのオートファジークリアランスを促進することが示された(Kilpatrick et al 2014)。2015)FDA承認のmTOR阻害剤である制御皮質衝撃-79による治療は、ドーパミン作動性α-シヌクレイン毒性のラットモデルにおいて、TFEB核局在の回復と同時に、発症後の病勢進行をブロックした(Decressac et al 2013)。これらの知見は、発症後に効果的な介入治療を行うことができれば貴重な進歩となるため、治療への応用の可能性という点で心強いものである。
ミトコンドリアキナーゼPINK1および細胞質E3リガーゼParkinの機能喪失変異は、早期発症家族性パーキンソン病の原因となり、損傷したミトコンドリアの蓄積と関連している(Kalia and Lang, 2016)。PINK1/Parkin経路は、ミトコンドリアのオートファジー(マイトファジー)を介して損傷したミトコンドリアを選択的に標的化し、除去するように機能するが、PINK1/Parkin活性化とマイトファジーの間の下流のステップは、まだ十分に理解されていない。ミトコンドリア脱分極の古典的な方法であるオリゴマイシン/アンチマイシン処理によるマイトファジーの誘導は、PINK1/Parkin依存性の方法でTFEB核転座を誘導する(Nezich et al 2015)。興味深いことに、この経路はまた、作動するために無傷のAtg5機能を必要とし、mTORC1シグナリングとは独立しているようである(Nezich et al 2015)。この新規なマイトファジー-TFEBシグナル伝達軸がPD病因にどのように関与しているかは、現在のところ不明である。
障害されたマイトファジーは、Parkin Q311Xマウスにおいて観察され、そして減少したTFEB発現と関連している(Siddiqui et al 2015)。興味深いことに、CLEARネットワークの解析により、TFAMおよびNrf1などのミトコンドリアタンパク質が、TFEB媒介のトランザクティベーションの標的であることが明らかになった(Palmieri et al 2011)。これらのミトコンドリア因子の発現は、Parkin Q311Xマウスにおいて有意に減少し、ラパマイシン処置によって救済され得る(Siddiqui et al 2015)。実際、Parkin Q311X substantia nigraの分析はまた、PGC-1α/TFEB経路の活性の低下を示した(Siddiqui et al 2015)が、これは転写抑制因子PARISの調節障害に起因していた。これらの結果は、し、TFEBがミトコンドリアタンパク質をコードする遺伝子をトランスポークすることができるので、TFEBはミトコンドリア生物学的には調節の標的および調節のエフェクターの両方であることを示している(Settembre et al 2013; Tsunemi et al 2012)。PGC-1α/TFEB軸の同様の欠損は、ハンチントン病で報告されており(Tsunemi et al 2012)神経変性におけるTFEBシグナル伝達の中心的役割をさらに強調している。
パーキンソン病におけるTFEB機能障害の正確な役割および治療標的としての可能性については、まだ調査中である。しかしながら、X-linked parkinsonism with spasticity(XPDS)およびKufor-Rakeb症候群として知られている2つの早期発症型のパーキンソン病の遺伝的バージョンは、液胞ATPaseのサブユニットの変異によって引き起こされ、リソソソーム酸性化とPD様神経病理学の発達との間の直接的な関連性を示している(Kalia and Lang, 2016)。このことは、オートファジーの制御による細胞クリアランスの転写制御と、リソソーム膜のイオンポンプをコードする遺伝子の発現を制御することによるリソソーム酸性化の制御を有するTFEBが、パーキンソン病における治療開発のための特に魅力的なターゲットであることを示唆している。
ポリグルタミンリピート拡大疾患
ポリグルタミンリピート疾患は、関与する遺伝子のコード領域に存在するCAGリピートトラクトの拡張によって引き起こされる成人発症の進行性神経変性疾患である。このようにして得られるそれぞれのタンパク質は、拡張したポリグルタミン(polyQ)トラクトを含み、突然変異疾患タンパク質のミスフォールディングおよび蓄積により機能増加毒性効果を誘発する。疾患の繰り返しの長さが長くなると、発症年齢は変異型CAGの拡張トラクトの大きさと反比例する。ハンチントン病(HD)X-linked spinal and bulbar muscular atrophy(SBMA)dentatorubralpalludoluysian atrophy(DRPLA)および6つのspinocerebellar ataxias(SCA1,2,3,6,7,17)を含む9つの認知されたCAG-polyQ障害がある。原因となる突然変異タンパク質は幅広い発現パターンを示すため、中枢神経系内外の様々な細胞型で容易に検出することができる。しかしながら、広範な発現にもかかわらず、すべてのpolyQ障害は、各障害における特定のニューロン集団を標的とした選択的な神経毒性のパターンを示す。オートファジー機能不全はポリグルタミン疾患で観察され、神経細胞の死に至る共通の発症機序が示唆されている(図1)。
ハンチントン病
最も一般的なpolyQ病であるハンチントン病(HD)は、不随意運動、認知機能の低下、および精神疾患を特徴とする常染色体優性神経変性疾患である。HDは、ハンチンチン(Htt)タンパク質のアミノ末端領域でのCAGトリヌクレオチドリピートの拡大(36回以上のCAGリピート)によって引き起こされる。様々な証拠のラインは、HD病理の一部としてオートファジー機能不全を強く示唆し、オートファジー機能不全がポリQ-Htt神経毒性に寄与する可能性を示唆している(Cortes and La Spada, 2014)。ある研究では、オートファジーカーゴ認識の欠陥が、オートファジーカーゴ受容体とポリQ-Httタンパク質との間の異常な相互作用によって媒介されている可能性が高いと提案されている(Martinez-Vicente et al 2010)。興味深いことに、Httタンパク質は、オートファジー経路タンパク質そのものであるように見えるが、mas Httは、ULK1(Ochaba et al 2014)およびAtg1オートファジー開始複合体(Rui et al 2015)のための足場として機能するオートファジーアダプタータンパク質として機能することが提案されている。
HDにおけるTFEBの調節障害の最初の証拠は2011年に報告された。HD N171-82Qトランスジェニックマウスの線条体において、PGC1αを介した転写の阻害により、TFEBの発現と機能が障害されることが示された(Tsunemi et al 2012)。TFEBの誘導は試験管内試験ではpolyQ-Httの凝集を劇的に減少させるのに十分であり、PGC-1αの過剰発現は、TFEBの転写・活性化を促進することで、HDマウスの線条体においてpolyQ-Httのターンオーバーを促進し、タンパク質の凝集を消失させた(Tsunemi and La Spada, 2012)(図2)。TFEBとPGC-1αの関係は複雑であり、TFEBはPGC-1αの転写を直接活性化することで脂質代謝の転写制御を行い(Settembre et al 2013年)TFEBとPGC-1αの双方向の活性化という二重のフィードバックループが生じていることがわかってきた。興味深いことに、HD患者の重要な臨床的特徴の一つは、著しい体重減少や筋萎縮を含む生体エネルギーの調節障害であり、polyQ-Htt – PGC-1α転写によるTFEB活性の阻害が、HDのこれらの観察された代謝症状に寄与している可能性を高めているが、この現象はさらなる研究が必要である。
全長およびアミノ末端の切断されたポリQ-Httは、オートファジー経路によって効率的に分解される。実際、オートファジーの調節はHD治療薬開発の長年のターゲットとなっており、様々なHD動物モデルにおいて、行動運動異常や神経病理の改善などの重要な効果が達成されている。有望な化合物には、既知のmTOR阻害剤である制御皮質衝撃-779(Ravikumar et al 2004年)およびmTOR非依存性オートファジー活性化剤であるトレハロース(田中 et al 2004)およびリルメニジン(Rose et al 2010)がある。制御皮質衝撃-779とトレハロースはいずれもTFEBの弱い活性化因子であるが、観察された神経保護効果に対するTFEBの役割については言及されていない。TFEBの過剰発現は、試験管内試験においてポリQ-Httの効率的なクリアランスを促進することができ(Sardiello et al 2009; Tsunemi et al 2012)さらに最近ではzQ175 HDマウスの線条体において(Vodicka et al 2016)。
球脊髄性筋萎縮症
X-linked Spinal and Bulbar Muscular Atrophy 球脊髄性筋萎縮症(SBMA)は、ケネディ病としても知られている神経筋疾患で、アンドロゲン受容体(AR)遺伝子の最初のエクソンのCAGトリプレットの拡大によって引き起こされる。SBMAは、脊髄と脳幹の下部運動ニューロン変性による成人期の近位筋力低下を特徴とする。
最近、安定細胞株、トランスジェニックマウス、および患者由来の神経前駆細胞(NPC)の網羅的な解析により、SBMAにおけるTFEBシグナル伝達の深遠な転写阻害が明らかになった(Cortes et al 2014b)。興味深いことに、SBMA細胞および運動ニューロンは、オートファジー開始およびオートファゴソーム形成に有能であるように見えるが、オートファジー分解をうまく完了することができず、リソソソーム機能が損なわれていることを示している。我々は、TFEBとARの間に新たな相互作用があることを明らかにし、polyQ-ARによるTFEBの制御障害がSBMAに見られるオートファジーフラックスの障害を説明する可能性を示唆した(図2)。重要なことに、我々は患者由来のNPCにおいてTFEBを過剰発現させることでオートファジーフラックスを回復させた(Cortes et al 2014b)ことから、SBMAおよびオートファジーフラックスの阻害を特徴とする他の疾患における治療開発の重要なターゲットとしてTFEB調節の可能性を強調している。実際、AR97Qを発現するSBMAトランスジェニックマウスを植物抽出物であるパネオフローリンで独立に処理すると、TFEB発現を強くアップレギュレートすることにより、行動および病理学的神経筋表現型に対して部分的に治療効果を発揮した(Tohnai et al 2014)。重要なことに、我々はまた、正常なQ長ARとTFEBとの間の相互作用の証拠を発見し、正常なARが過剰発現された場合に、増強されたTFEBシグナル伝達およびオートファジー経路活性の増加を検出した。Cortes et al 2014b)。我々のデータは、ARが通常、テストステロンに応答してTFEBを機能的および空間的に調節し、その活性を促進するためにTFEBと相互作用し得ることを示唆している。ARは通常、多くの転写調節因子と相互作用し、SBMAハエモデルの研究では、ポリQ-ARが相互作用する調節因子の機能を低下させることで神経毒性を促進する可能性があることが示されている(Nedelsky et al 2010)ので、ARとTFEBが共有する共活性化蛋白質の利用可能性の低下は、SBMAにおけるTFEBのトランザクティベーション機能の低下をもたらす可能性がある。ARが前立腺癌におけるTFEB転写も促進することができる相互フィードバックループが最近記載され、オートファジー/リソソーム遺伝子の複雑なアンドロゲン依存性転写調節を示唆している(Blessing et al 2017)。しかしながら、SBMA疾患の病因に対するこの新規なシグナル伝達経路の役割は、まだ探索されていない。
骨格筋は、SBMA病因において主要な役割を果たし、polyQ-AR毒性の重要な部位として運動ニューロンを優先している(Cortes et al 2014a; Lieberman et al 2014)。興味深いことに、SBMA運動ニューロンおよび患者由来のNPCにおけるTFEB活性が有意に減少した一方で、症状のあるSBMA YAC AR100トランスジェニックマウスからの大腿四頭筋サンプルの分析は、反対の劇的なTFEB標的遺伝子のアップレギュレーションをもたらした(Cortes et al 2014b)SBMAノックインAR113Qマウスにおける研究(Chua et al 2014)と一致していた。これは、疾患SBMA筋細胞におけるTFEBの超骨格的誘導の筋特異的プロセスを示唆している(図3)。制御されていないオートファジーは、筋ジストロフィーのモデル(Sandri et al 2013)において筋肉の消耗の根底にあると考えられているので、オートファジーの過剰な活性化は、SBMA骨格筋の表現型に寄与している可能性がある。この仮説と一致して、SBMAノックインAR113QマウスにおけるBeclin-1ハプロイン不全によるオートファジー活性のグローバルな低下は、このモデルにおいて骨格筋繊維のサイズを増加させ、寿命を有意に延長した(Yu et al 2011)。SBMA AR113Qノックインマウスに高脂肪食を与えると、過剰な筋TFEB活性が減少し、SBMA骨格筋の代謝変化のいくつかを救済した(Rocchi et al 2016)。これは、TFEB機能を変更するための治療的アプローチとしての食事操作の最初の証拠であり、SBMAおよびTFEB機能障害に関連する他の疾患における治療開発のためのプルーフオブコンセプトを提供する。
異なる組織タイプにおけるポリQARによるTFEBの調節障害の様々な側面の原因となるメカニズムは不明のままであるが、オートファジー療法の全身投与がSBMAにおいて劇症的な影響を及ぼす可能性があることが懸念される。SBMAの骨格筋と運動ニューロンの間のクロストークを理解し、組織特異的な方法でTFEB活性を調節するキープレイヤーを特定することは、オートファジー療法の開発を進めるための合理的な根拠を得るために不可欠である。しかし、重要なことは、SBMAにおける運動ニューロン毒性の非細胞自律性と骨格筋へのアクセス性により、退化した運動ニューロンを治療するための薬剤の送達が容易になるということである。
筋萎縮性晩発性頭蓋炎
筋萎縮性側索硬化症(ALS)は、ルー・ゲーリック病としても知られており、成人の最も一般的な運動ニューロン障害であり、運動野、脳幹、脊髄の運動ニューロンの選択的な喪失を特徴としている。ALSは手足や呼吸筋の衰弱や萎縮を引き起こし、最終的には死に至る。ほとんどのALS症例は原因不明の「散発性」であるが、最大10%の症例は家族性である。これらの家族性症例は、特にスーパーオキシドジスムターゼ1(SOD1)TAR DNA結合タンパク質(TDP-43)融合肉腫/翻訳肉腫(FUS/TLS)および第9染色体オープンリーディングフレーム72(C9orf72)を含む多くの遺伝子の突然変異に起因する(RameshおよびPandey 2017)。興味深いことに、いくつかのオートファジー遺伝子の変異は、オートファジーアダプターp62(Sequesterome 1,SQSTM1)およびオプティニューリン(OPTN)、プロテオスタシスおよびストレス顆粒調節因子バロシン含有タンパク質(VCP)、オートファジー受容体ユビキリン-2(UBQLN2)、およびオートファジー調節因子TANK-結合キナーゼ1(TBK1)を含む、ALSと関連している(Peters et al 2015年)。
ALSでは、死後患者組織の複数の研究により、運動ニューロンにおけるオートファジー異常の証拠が明らかにされている(Ramesh and Pandey, 2017)。オートファゴソームの蓄積は、散発性および家族性ALS患者の退化した運動ニューロンで頻繁に見られ、p62陽性の介在物に隣接して発見されることが多い(Li et al 2008; Morimoto et al 2007; Sasaki, 2011)。損傷を受けたミトコンドリアの蓄積もALSで観察されるが、HDやアルツハイマー病とは異なり、損傷を受けたミトコンドリアは通常オートファジーの液胞内に存在する(Liu et al 2004; Vande Velde et al 2008)。このことは、ALSにおけるオートファジー機能不全には、貨物認識プロセスが無傷であるため、異なるメカニズムが関与していることを示唆している。その代わりに、軸索輸送やオートファゴソームとリソソームの融合の欠陥が、オートファゴ液胞の蓄積の原因となっている可能性がある(Ligon et al 2005; Williamson and Cleveland, 1999) (図1)。しかしながら、mTORシグナル伝達の減少の証拠は、ALSマウスにおいて記録されている(Hetz et al 2009; Li et al 2008; Morimoto et al 2007)が、オートファジーの超生理学的活性化と一致する。ALS疾患の病態形成におけるオートファジー機能不全の根底にある複雑なメカニズムを反映して、オートファジーを標的とした治療法は、動物モデルにおいて、改善を示す報告もあれば、利益を示さないか、または神経筋疾患の有意な悪化を示す報告もあり、混合した結果を示している(Ramesh and Pandey, 2017)。
運動ニューロンの進行性喪失に寄与する家族性ALS1の重要な神経病理学的特徴は、変異型SOD1タンパク質凝集体の異常蓄積である。最近、ALS SOD1-G39Aトランスジェニックマウスからの症候前および症候後の脊髄溶解物の分析により、TFEB発現のステージ依存的な変化が明らかになった(Chen et al 2015)。TFEBは、疾患の初期段階ではアップレギュレーションされたが、その後、ニューロン特異的と思われる方法で疾患の中期および末期ではダウンレギュレーションされた(Chen et al 2015)。試験管内試験でのTFEBの過剰発現は、この系でのSOD1-凝集クリアランスに対するオートファジーの効果は検討されなかったが、Beclin-1発現を増加させることにより、細胞の生存および増殖を増加させた。それにもかかわらず、TFEB局在の制御異常は、ALS患者の脳サンプルの細胞内分画分析において、核内TFEBレベルが60%も減少したことが報告されている(Wang et al 2016b)。これらの所見は、2つの異なる病原体を代表するかもしれない散発性と家族性のALS患者のより大きなシリーズで再現する必要がある;しかしながら、TFEBの核内排除は、ALS疾患の病原体の顕著な特徴であるかもしれない。
C9orf72は 2011年にALSの原因となるヘキサヌクレオチドリピートが発見されて以来、精力的な研究が行われていたが、C9orf72タンパク質の細胞機能の完全な理解は未だに得られていない。最近の証拠は、C9orf72がオートファジーを負に制御する可能性があることを示唆している(Amick et al 2016;Sellier et al 2016;Sullivan et al 2016;Ugolino et al 2016)。2つの独立したC9orf72ノックアウトマウスモデルは、肝臓、脾臓、および脳においてオートファジーの増加を示し(Sullivan et al 2016; Ugolino et al 2016)オートファジー調節におけるC9orf72タンパク質の役割を支持した。1つのノックアウトモデルにおいて、このオートファジーの増加は、脳におけるTFEBタンパク質発現の上昇と関連していたが、これは、TFEBに対するC9orf72タンパク質の直接的な効果ではなく、mTOR活性の阻害によるものであった(Amick et al 2016; Ugolino et al 2016)。C9orf72は、アミノ酸枯渇に応答してリソソームに局在することが報告された(Amick et al 2016)エネルギー感知および細胞代謝におけるC9orf72の潜在的な新規な役割を示唆している。これは、別のALS関連遺伝子であるTDP-43がオートファジーおよびTFEB活性の負の調節因子として作用することを示唆する最近のデータと一致する(Xia et al 2016)。しかしながら、この発見の妥当性およびALS疾患の病態形成との関連性は、まだ不明であるが、依然として関心の高いトピックである。一方、C9orf72タンパク質は、Ulk1オートファジー開始複合体と物理的に相互作用することにより、オートファジーの正の調節因子として作用する可能性があり(Webster et al 2016)C9orf72タンパク質とオートファジーとの間に複雑な、おそらく二重の関係があることを示唆している。実際、これらの研究は、観察されたオートファジー効果に必要なものとして、SMCR8として知られる新規なC9orf72インタラクタをまとめて同定した(Amick et al 2016; Sullivan et al 2016; Yang et al 2016)が、このインタラクタの関連性および運動ニューロン疾患の病因におけるオートファジーへの影響は不明のままであるが、このインタラクタは、C9orf72タンパク質とオートファジーとの間の複雑な関係を示唆している。C9orf72 FALSにおけるオートファジー機能不全の性質およびC9orf72遺伝子産物の正常な機能を明らかにし、その後、TFEBがこの形態のFALSのALS病原性カスケードに適合するかどうかおよびどのように適合するかを決定するための更なる研究が必要であることは明らかである。
C9orf72型ALSにおけるオートファジー経路の状態は不明であるが、最近の他の遺伝性ALSの研究では、オートファジーが介在する疾患関連タンパク質の集合体のクリアランスが有益であることがわかっているが、これらの効果はTFEBの機能とは無関係に得られることがわかっている。例えば、ヒートショックプロテインB8の誘導は、試験管内試験でのTFEBの発現または局在を変化させることなく、TDP-43のクリアランスを媒介する(Cripppa et al 2016)。新規オートファジー調節因子であるMASKは、TFEBに依存しない方法でv-ATPaseサブユニットの発現をブーストして、FUS神経変性のショウジョウバエモデルにおける眼の変性を緩和することができる(Zhu et al 2017)。さらに、食事制限はオートファジーの欠陥を修正し、ダイナクチンG59S ALSマウスモデルにおいて、おそらくTFEBの活性化を介して、全神経機能を部分的に回復させたが、神経保護におけるTFEBの役割は検討されなかった(Wiesner et al 2015)。TBK1は、オートファジーアダプターの効率的なリクルートとオートファジー(マイトファジー)によるミトコンドリアの適切な分解に必要である。TBK1のALS関連変異は、オプティヌアリンとの相互作用を破壊し、マイトファジーの障害がTBK1-ALS疾患の病因の根底にある可能性を示唆している(Oakes et al 2017)。しかしながら、これまでのところ、TBK1-ALSの文脈でTFEB活性が変化したという証拠はない。したがって、運動ニューロン疾患におけるTFEBの調節異常の研究は大きな関心事であるが、ALS疾患の病態形成におけるオートファジーの正確な役割は、ALS動物モデルにおける前臨床研究の矛盾した結果に例示されるように複雑である。ALSにおけるオートファジー機能障害の具体的なメカニズムを明らかにするためには、これらの矛盾した結果を明らかにし、TFEBやオートファジー自体が治療の対象となるかどうかを判断するための今後の研究が必要であると考えられる。
最後に
オートファジー機能不全は、有害なタンパク質凝集体の蓄積を特徴とするほぼすべての神経変性疾患の特徴的な特徴である。また、TFEB機能障害は、特にアルツハイマー病、PD、HDを含む神経変性プロテインオパチーにおいても共通のメカニズムであると考えられているが、ほとんどの神経変性疾患においてTFEB機能障害の疾患発症への寄与をより明確にする必要がある。
TFEB活性は、ウイルスまたは遺伝子の過剰発現を介して脳内で調節され得、神経変性疾患の多数の多様な動物モデルにおいて、病理学的および行動表現型の有意な改善を達成する(Bao et al 2016;Decressac et al 2013;Wang et al 2016a;Xiao et al 2014)。
しかしながら、TFEBの基本的な生物学については、不明な点が多く残されている。例えば、リン酸化がTFEBの局在および機能の主要な調節因子であることは長い間知られてきたが(Martina et al 2012; Sardiello et al 2009; Settembre et al 2012)アセチル化もまた、TFEB活性の強力な調節因子として作用する可能性がある。
TFEBは、核内でサーチュイン1によって脱アセチル化され、フィブリラーアミロイドβに曝露されたミクログリアにおいて、リソソソームバイオジェネシスを増強する(Bao et al 2016)。さらに、健康および疾患におけるTFEBの翻訳後修飾の研究は、TFEBの機能に光を当てる可能性があり、治療開発のための魅力的な戦略をもたらす可能性がある。
最後に、多くの報告では、それぞれの細胞環境において異なる相互作用体が存在することにより、細胞タイプに特異的なTFEB制御ネットワークが存在することが示唆されている。これらのニューロンとアストログリアのTFEBの相互作用ネットワークは現在調査中であり、中枢神経系におけるTFEBの生理機能の理解に役立つはずである。
TFEBの機能を標的とする新規な化学的活性化剤の同定は、TFEBが「薬物化可能な」標的であることを示唆しており(Meng et al 2016;Song et al 2014)血液脳関門浸透性および生理活性を有するTFEB活性化化合物の最近の発見(Song et al 2016)により、この分野では、牽引可能な神経治療的介入としてのTFEB調節を試験するように設定されている。
実際、クルクミン由来の化合物であるC1は、mTORに依存しない哺乳類の中枢神経系においてロバストなTFEB活性化を達成し、このように長期的なmTOR阻害の望ましくない副作用の多くを回避する。
重要なことに、C1は経口投与により活性化されることが明らかになり、ヒト患者での使用がさらに容易になり、治療法開発のための魅力的な候補となっている。もちろん、TFEBの制御生物学の多くは、リソソームで起こる相互作用や修飾を含んでおり、この重要性を増している
オルガネラがどのようにTFEB制御カスケードに組み込まれているかをよりよく理解することが重要であることを強調している。そのために、リソソームからのカルシウムイオンの流出を調節し、TFEBの強力な活性化因子であることが判明した過渡受容体ポテンシャルムコリピン1(TRPML1)チャネルに関する最近の研究(Di Paola et al 2017)は、TFEB調節生物学へのより決定的な洞察が、生理学的に関連するTFEBおよびオートファジー調節のためのより魅力的な標的をもたらす可能性があることを強調している。
したがって、我々は、得られた知識を成功した神経治療薬に活用する機会を視野に入れて、神経変性で起こるオートファジーの調節障害におけるTFEBの役割を明らかにする取り組みが、今後10年間でさらに加速すると予測する。