Targeting Autophagy to Overcome Human Diseases
www.ncbi.nlm.nih.gov/pmc/articles/PMC6387456/
要旨
オートファジーは進化的に保存されてきた細胞のプロセスであり、ストレスの影響を受けた時に細胞のバランスを正しく維持するために、損傷した小器官や余分なタンパク質を分解する。
オートファジーは、オートファゴソームと呼ばれる二重膜小胞の形成を伴い、細胞質の貨物を捕捉してリソソームに送り、そこで分解された生成物は細胞質にリサイクルされる。分解された細胞成分に基づいて、オートファジーのいくつかの選択的なタイプ(マイトファジー、リボファジー、レティキュロファジー、リゾファジー、ペキソファジー、リポファジー、グリコファジー)を同定することができる。
オートファジーの調節障害は、炎症、老化、代謝性疾患、神経変性疾患、癌などの様々な疾患を引き起こす可能性がある。オートファジー過程の様々な段階を制御する分子機構や、疾患の発症に果たす役割についての理解は、まだ初期の段階にある。
また、オートファジー関連タンパク質の機能については、まだ解明されていない点が多い。本総説では、オートファジーの主要な細胞・分子機能、オートファジーの選択的なタイプ、細胞生理におけるオートファジーの役割を検出するための主な試験管内試験法について述べる。
また、いくつかの疾患におけるオートファジーの挙動の重要性をまとめ、標的治療のための新たな知見を提供する。
キーワード
オートファジー、細胞生存、細胞死、標的治療、炎症、代謝性疾患、神経変性疾患、自己免疫疾患、老化、癌
1. はじめに
オートファジーの調節機能の低下は、ヒトの様々な疾患の発症に関与している。ここでは、細胞のオートファジーを制御するメカニズムの概要を明らかにし、細胞が有害な部分や不要な部分を排除しながら、継続的な再生サイクルの中で自食することを可能にする生物学的プロセスを明らかにしてきた。オートファジーは保守的なプロセスであり、細胞のバランスや生理機能を維持・調節する上で重要な役割を果たしている。オートファジーの制御は、変化した小器官や異常なタンパク質の分解のために全身的に配置されている。オートファジープロセスの変化による機能不全は、腫瘍、代謝異常、神経変性疾患、炎症性疾患などの疾患の原因となる可能性がある[1]。最近の研究では、乳がんや卵巣がんはBeclin1遺伝子の変異と関連していることが示されている[2]。クローン病はATG16L1遺伝子の変異と関連している[3]。p62タンパク質の変異は筋萎縮性側索硬化症と関連している[7];PINK1変異によるマイトファジー欠損はパーキンソン病と関連している[8];糖尿病はまた、膵β細胞の正常な機能の変化を誘導するオートファジーの調節障害と関連しており、この疾患ではインスリン抵抗性の合併症を引き起こす[9]。オートファジーの早期不活性化は、正常細胞の腫瘍細胞への形質転換を誘導する可能性がある。微小環境が腫瘍細胞の増殖に代謝的に不利な場合や、腫瘍細胞が毒性のある薬剤で治療されている場合には、オートファジーの活性化が細胞の生存に有利になる[10]。オートファジーの病態における役割を理解することは、治療戦略の評価や疾患克服のための個々の治療法を改善するために非常に重要である[11]。
2. オートファジー、アポトーシス、壊死経路のクロストーク
オートファジーは、不要なタンパク質や損傷した小器官を排除することで、細胞の恒常性維持を可能にする異化プロセスである。このプロセスは、生理的条件(栄養素の不足、成長因子の欠乏)や様々なストレス刺激(酸素欠乏、酸化ストレスの誘発、毒性物質への暴露)に反応して活性化される。この現象は、エネルギーバランスの維持を確保することによって、細胞にとって重要であり、それはまた、小さなエネルギーの変動が細胞自体に有害である可能性があることを避けることができる。基底オートファジー細胞レベル、すなわち、刺激がなくても存在するものは、余分な小器官と非有用な凝集タンパク質の除去を介して、タンパク質や小器官の正常なターンオーバーを可能にする。このプロセスはまた、栄養素が不足しているときに細胞がエネルギーを生成することを可能にし、発生時(生存反応)に生体エネルギーのサポートを提供する。オートファジーが阻害されると、細胞内に損傷を受けた小器官の蓄積が観察され、タンパク質の凝集と適切なエネルギー供給の不足が細胞死をもたらす。オートファジーが制御されていない細胞プロセスでは、重要な細胞小器官や有用な生体高分子が分解され、抗アポトーシス因子が消化され、結果的に生理的生存メカニズムに干渉し、細胞自体の死、すなわちオートファジー細胞死を引き起こす可能性がある。いくつかの研究では、ショウジョウバエの発生期には、アポトーシスや壊死が活性化されていないのに対し、細胞死の生理学的なモデルとしてオートファジーが関与していることが報告されている[12]。しかし、「オートファジーによる細胞死」という用語は、慎重かつ節度を持って使用されるべきである。オートファジーは典型的には生存を促進するプロセスであるため、オートファジーが細胞死に因果的な役割を持っていることを証明するには、十分な証拠が必要である[13]。細胞死の他のメカニズムが原因ではないことを証明し、遺伝的または化学的手段によるオートファジーの阻害が細胞死を防ぐことを検証しなければならない。さらに、形態学的な基準だけでは細胞死を評価するには十分ではなく、複数のアッセイを用いて細胞死を測定し、特に細胞生存率アッセイを行うことが重要である[14]。
アポトーシス、オートファジー、壊死など、いくつかの種類の細胞死を制御するメカニズムは、しばしば別々のものであるが、時としてそれらは重なり合っており、細胞から受けた過剰な刺激に反応して、同時にまたはその後に活性化されることがある。多くの研究者が、オートファジーとアポトーシスの間の関連性を強調している [15]。同じ細胞内で、アポトーシスまたはオートファジー経路は、オートファジーが保護的または細胞死を誘発する場合、同じ刺激に応答して同時にまたは連続して発生する可能性がある。ストレスシグナルの後、細胞は、アポトーシス細胞死を抑制する生存オートファジーを活性化する(図1A)。他のケースでは、オートファジーがストレスによって誘発された損傷から細胞を防御できず、アポトーシスによって死滅する(図1B)。あるいは、オートファジーとアポトーシスが共に働き、両方のメカニズムで細胞が死ぬ場合もある(図1C)。オートファジーとアポトーシスが相互に関連している可能性のある細胞下区画は、小胞体[16]、ミトコンドリア[17]、リソソソーム[18]である。
図1 オートファジーとアポトーシスのクロストーク
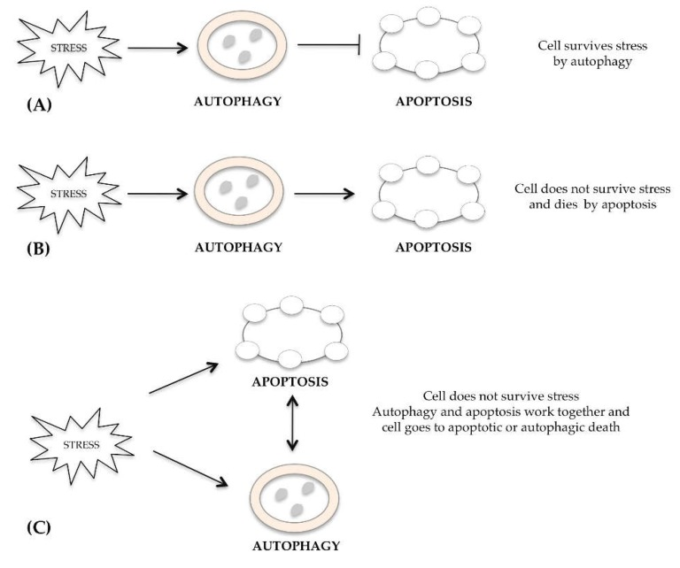
(A)ストレスシグナルの後、細胞はオートファジー経路を活性化して生存し、アポトーシスによる死を阻止する
(B)オートファジー機構が失敗してアポトーシスによって細胞が死ぬ
(C)ダメージを受けるとオートファジーとアポトーシスの両方が活性化し、共に働く;細胞は生存せずに死ぬ。
オートファジーとアポトーシスのクロストークは、損傷の程度と細胞の感受性によるものである。細胞死の種類には複数のシグナル伝達経路があり、それぞれが独立して制御している。細胞がストレスを受けると、アポトーシスに先立ってオートファジーが誘導されるが、オートファジーが阻害されたり、効果がない場合にはアポトーシスや壊死が誘導される。アポトーシス、壊死、オートファジーの主な特徴を表1にまとめた。
表1 アポトーシス、壊死、オートファジーの主な形態的特徴、生化学的特徴、分子的特徴
| タイプ | 形態学的特徴 | 生化学的特徴 | コアレギュレーター |
|---|---|---|---|
| アポトーシス |
|
|
ポジティブ:
負:
|
| 壊死 |
|
|
ポジティブ:
|
| オートファジー |
|
|
ポジティブ:
|
3. オートファジーの分子制御
細胞がオートファジーを受けているとき、小さな二重層膜(ファゴフォア)が形成され(図2A)これは、小器官および/または排除されるべき高分子を包み始める(図2B)。この膜は、オートファゴソーム、すなわち、二重膜の空胞を形成し、閉じる(図2C)。オートファゴソームは、加水分解リソソーム酵素が液胞の内膜とオートファゴソームの内容(図2F)を消化するオートファゴリゾーム(図2E)を形成するためにリソソーム(図2D)とマージする。放出された高分子は、生合成や細胞エネルギーの供給に利用することができる[19]。
図2 細胞のオートファジー段階の説明
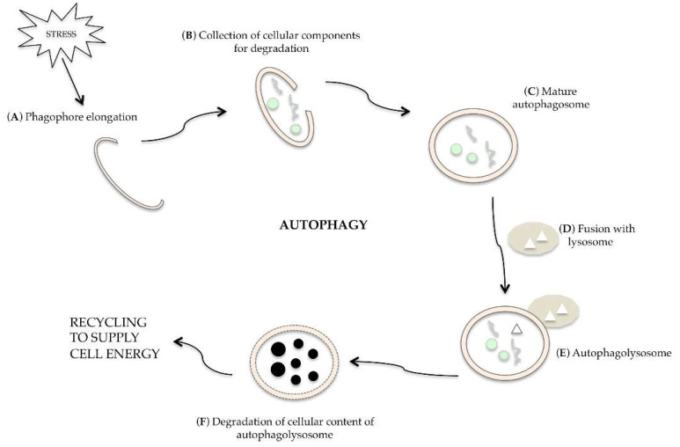
様々なストレスがかかると、ファゴフォアと呼ばれる小さな二重層膜が細胞内に伸び、細胞内の物質を集めて分解し(B)オートファゴソームを形成する(C)。リソソソームと融合した後(D)オートファゴソームが形成され(E)加水分解酵素の作用により細胞成分が分解され(F)細胞エネルギーの供給にリサイクルされる。
酵母Saccharomyces cerevisiaeの遺伝子スクリーニング試験により、オートファジーを制御するタンパク質産物を持つ約30の遺伝子(正確にはATG遺伝子、AuTophaGy関連遺伝子と呼ばれる)が同定され、オートファジーの分子制御を理解することが可能になった[20,21]。これらの遺伝子のほとんどは、ミバエから哺乳類までの高等真核生物において相同配列を有しており、オートファジーの分子機構が進化において高度に保存されていることが示唆されている[22]。
栄養素が利用可能な場合、哺乳類のラパマイシン標的(mTOR)とプロテインキナーゼA(PKA)は、unc-51様キナーゼ1(ULK1)複合体と呼ばれる誘導複合体のリン酸化と阻害を介してオートファジーをネガティブに制御する [23]。栄養素が不足すると、AMP-ATP比が上昇し、活性酸素種(ROS)の産生が増加し、ニコチンアミド・アデニン・ジヌクレオチド(NAD+)が増加し、mTOR活性が阻害され、AMP活性化プロテインキナーゼ(AMPK)がスイッチオンしてオートファジーが開始されるように、様々な化学メディエーターがストレスによって放出される(図3)。ファゴフォア形成の誘導段階は、ULK1,ATG13,200 kDaの焦点接着キナーゼファミリー相互作用タンパク質(FIP200)およびATG101タンパク質を含むULK1複合体によって媒介される[24]。その結果、ULK1複合体は、もはやmTORによってリン酸化されるのではなく、AMPKによってリン酸化され、オートファゴソームの核形成を調節する複合体に作用するようになる[25]。それは、Beclin1,ATG14,液胞タンパク質選別(VPS)15,VPS34,BECN1制御オートファジータンパク質1(AMBRA1)の活性化分子、および紫外線照射抵抗性関連遺伝子(UVRAG)によって形成されるホスファチジルイノシトール3キナーゼ複合体(PtdIns3K)を活性化することができる(図3A)[26]。このキナーゼ複合体の誘導は、脂質ホスファチジルイノシトール-3-リン酸(PI3P)を生成し、それは次に、空胞の形成に不可欠な他のタンパク質をリクルートする(リン脂質と相互作用することができるWD-repeatタンパク質がホスホイノシチド(WIPI)タンパク質と相互作用するような)[27]。
図3 オートファジーの分子経路の模式図
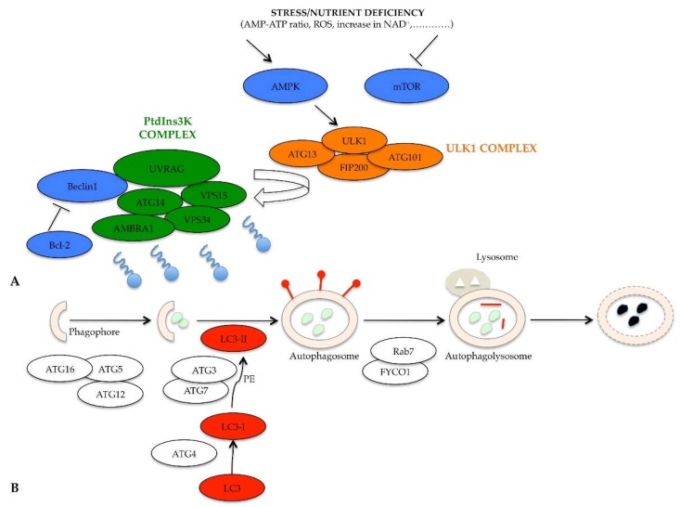
(A) mTOR阻害、AMPK活性化、ULK1複合体、PtdIns3K活性化を特徴とする上流の活性化、(B) ATG12-ATG5-ATG16複合体によるファゴフォア伸長の制御、ATG8/LC3複合体によるオートファゴソームの成熟化、Rab7,FYCO1輸送タンパク質を介したプロセスの終着点。
mTORの阻害とAMPKの活性化に加えて、PtdIns3K複合体の活性は、Beclin1-Bcl-2複合体によってさらに制御されている[28]。栄養素が豊富な場合、Beclin1タンパク質はBcl-2タンパク質と複合体を形成し、これがBeclin1のリクルートとPtdIns3K複合体の機能を阻害する;代わりに、栄養不足がある場合、Beclin1-Bcl-2複合体は解離し、Beclin1は自由に活動する(図3A)[29]。ファゴフォアとオートファゴソームの形成段階は、基本的にATG12-ATG5-ATG16複合体によって制御されており、ファゴフォアを形成するために膜が長くなることが必要である(図3B)[30]。ATG9,ATG2およびWIPI 1/2を含む膜貫通タンパク質系もまた、ファゴフォア構造の伸長に必要である[31]。プロセスの進行に不可欠な第二の共役複合体は、ATG8タンパク質であり、哺乳類では微小管関連タンパク質1軽鎖3(MAP1-LC3または短鎖LC3)として同定されている[32]。LC3タンパク質のC末端はATG4プロテアーゼによって切断され、LC3-Iと呼ばれる新しい形になる[33]。後者は、ATG3/ATG7システムにより、ホスファチジルエタノールアミン(PE)と結合する。脂質の共役はLC3-IをLC3-IIに変換し、成熟したオートファゴソームの外側に露出する[34]。オートファゴソームが完成すると、オートファゴソームは微小管に沿ってリソソームに向かって移動を開始し、融合が行われる[35]。微小管に沿った輸送は、LC3,Rab7,およびFYCO1によって形成されたアダプタータンパク質複合体によって媒介される [36]。オートファゴリソソーム形成後、LC3-IIタンパク質は内包され、リソソソーム酵素の作用によりPE残基が剥離され、細胞質に放出され、その結果、発現が低下する(図3B)[37]。
4. 選択的オートファジー
栄養素の不在によって引き起こされる不利な条件を生き抜くために必要な細胞エネルギーは、損傷を受けた細胞成分の非選択的な分解から得られる。しかし、小胞体を損傷するストレスや細胞代謝の変化のような他の状況では、排除されるべき細胞質オルガネラがオートファジーマシンによって特異的に同定され、「選択的オートファジー」と呼ばれるメカニズムによって排除される[38]。選択的オートファジーには、除去される小器官によって異なる種類がある。ミトコンドリアの場合はマイトファジー、リボソームの場合はリボファジー、小胞体の場合はレティキュロファジー、リゾソームの場合はリゾファジー、ペルオキシソームの場合はペキソファジー、脂質の場合はリポファジー、グリコーゲンの場合はグリコファジー、誤って折り畳まれたタンパク質の場合はアグレファジー、感染した病原体の場合はゼノファジーなどである[39]。
小器官は、ユビキチン依存性または非依存性のメカニズムによって選択的に分解され得る[40]。最初のケースでは、標的オルガネラは、ポリユビキチン化鎖を有しており、この鎖は、BRCA1遺伝子1(NBR1)の近傍であるp62,核ドメイン10タンパク質52(NDP52)およびオプティニューリンなどの基質特異的な受容体に向かってそれを誘導する[41,42,43,44]。これらのタンパク質は、一方ではUB結合ドメイン(UBD)を介して損傷した小器官上のポリユビキチン化鎖(UB)を認識して結合し、他方ではLC3相互作用領域(LIR)を介してオートファゴソーム内のLC3-IIタンパク質を認識して結合することができるため、分子アダプターとして機能している(図4A)。このように、オートファゴソームの拡張に関わる役割とは別に、LC3-IIは貨物選択にも重要な役割を果たしている。
図4
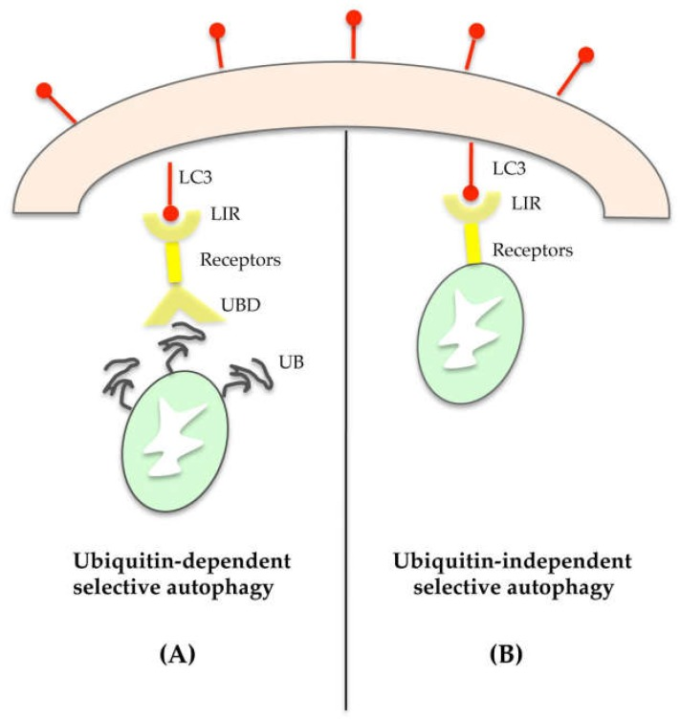
選択的オートファジーは、ユビキチン依存性(A)とユビキチン非依存性(B)がある。ユビキチン依存性メカニズム(A)では、標的オルガネラは、ポリユビキチン化鎖(UB)を介して受容体(p62,NBR1,NDP52,オプティニューリンなど)と相互作用する。受容体タンパク質は、損傷したオルガネラの側ではUB結合ドメイン(UBD)を介してUB鎖に結合し、反対側ではLC3-相互作用領域(LIR)を介してオートファゴソームのLC3-IIタンパク質に結合する。ユビキチン非依存機構(B)では、損傷したオルガネラ上に局在する受容体(BNIP3, NIX)がオートファゴソーム上のLC3と貨物を直接結びつける。
p62は、シークエストソーム1(SQSTM1)としても知られる62 kDaのタンパク質であり、一般的にユビキチン化されたタンパク質に局在し、オートファゴソームの空胞に隔離されている[45]。p62は、神経系、内分泌系、生殖系、免疫系などの組織全体に広く発現しており、疾患の発症に重要な役割を果たしている[46]。実際、追加の研究では、p62はLC3や他のATGタンパク質との相互作用なしに小胞体上のオートファゴソーム形成部位に局在することが示されている[47]。オートファジーカーゴの送達におけるその役割のため、p62の発現はオートファジー分解と逆相関しており、オートファジーフラックスの指標として使用することができる[14]。
もう一つのカーゴ受容体はNBR1であり、ポリユビキチン化鎖を結合するC末端ドメインとして、またATG8と相互作用する配列として、p62といくつかのドメインと特徴を共有している[48]。最近では、NBR1とp62が協働して、ポリユビキチン化されたタンパク質凝集体をオートファゴソームに選択的に標的化し、ペキソファジーを介してペルオキシソームを分解することが実証されている[49,50]。
ユビキチンに依存しないメカニズムに関しては、小器官上の多くのオートファジー受容体は、そのカーゴをオートファゴソームと直接リンクさせている(図4B)。例として、マイトファジーは、オートファゴソームのLC3-IIと直接相互作用するBcl-2/アデノウイルスE1B 19-kDa interacting protein 3(BNIP3)またはBNIP3-like(NIXとして知られている)を関与させることができる[51]。
非選択的オートファジーは、細胞の生存、身体的健康および人間の健康に不可欠であるが、選択的オートファジーは、多種多様な疾患において直接的な役割を果たしている[52]。
5. 哺乳類細胞におけるオートファジーの検出方法
オートファジーは非常に複雑で動的なプロセスであり、試験管内試験または生体内試験モデルでの解析が困難な場合がある。近年、オートファジーの複雑な研究を提唱してきた多くの研究者は、オートファジーアッセイのモニタリングの解釈に使用するための長い出版物を作成するための技術的および科学的貢献を提供していた[14]。体外でのオートファジー研究に適した技術、混乱を招くアーテファクトの形成を避けるために従うべき技術的手段、および正しいデータ解釈のためのガイドラインの概要を以下に説明する。オートファジーをモニターするための主な体外での方法を表2にまとめた。
表2 オートファジーをモニタリングするための主な試験管内試験の方法
| 方法 | 説明 |
|---|---|
| 光学および電子顕微鏡 | 細胞質内の液胞、それらの内容、成熟の段階、およびオートファジーコンパートメントの代謝回転を表示します |
| GFP-LC3蛍光顕微鏡 | 液胞/リソソームの局在を監視する |
| LC3ウエスタンブロッティング | オートファジーモジュレーターの有無にかかわらず、オートファジーフラックスを監視します |
| フローサイトメトリー | 蛍光プローブによるオートファゴソームの定量化 |
| ウエスタンブロット | p62および関連するLC3結合タンパク質の代謝回転 |
| キナーゼアッセイまたはウエスタンブロッティング | mTOR、AMPKおよびULK1キナーゼ活性 |
| ATG遺伝子サイレンシング | 現象に関与するATG標的タンパク質を特定することができます |
図5 天然化合物処理後のオートファジー腫瘍細胞の光学顕微鏡観察
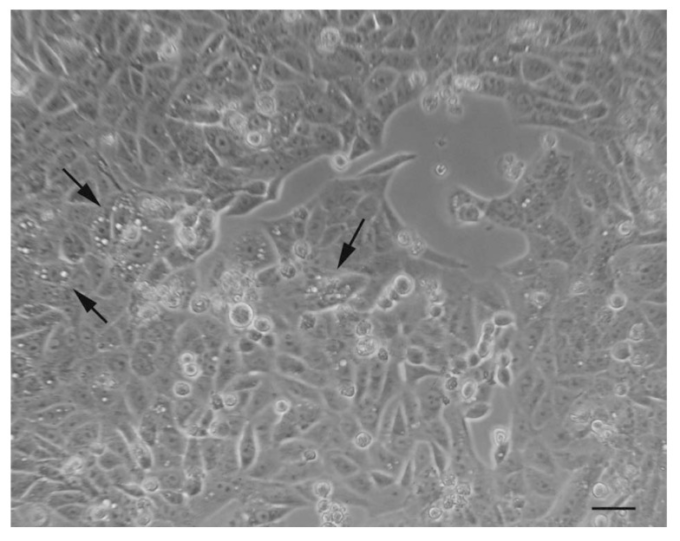
矢印で示すように、細胞質の内部には目に見える穴が開いている。スケールバー:20μm。
透過型電子顕微鏡による同じサンプルの詳細な分析は、細胞質小器官および消化およびリサイクルされるべき物質を含む単一および二重膜を有する細胞質の空胞を示している(図6A)。標準的な包接手順に従って調製されたサンプルの観察は、オートファゴ液胞の形成と成熟の異なる段階(開始、核生成、ファゴフォア形成、オートファゴソーム、オートファゴソームとリソソームおよびオートファゴリゾソームとの融合)を定義することができる[53]。急速凍結法(凍結破壊法として知られている)を用いて試料を調製すると、液胞分子膜組織の深い理解が得られる(図6B)。
図6 天然化合物処理後のオートファジー腫瘍細胞の透過電子顕微鏡観察
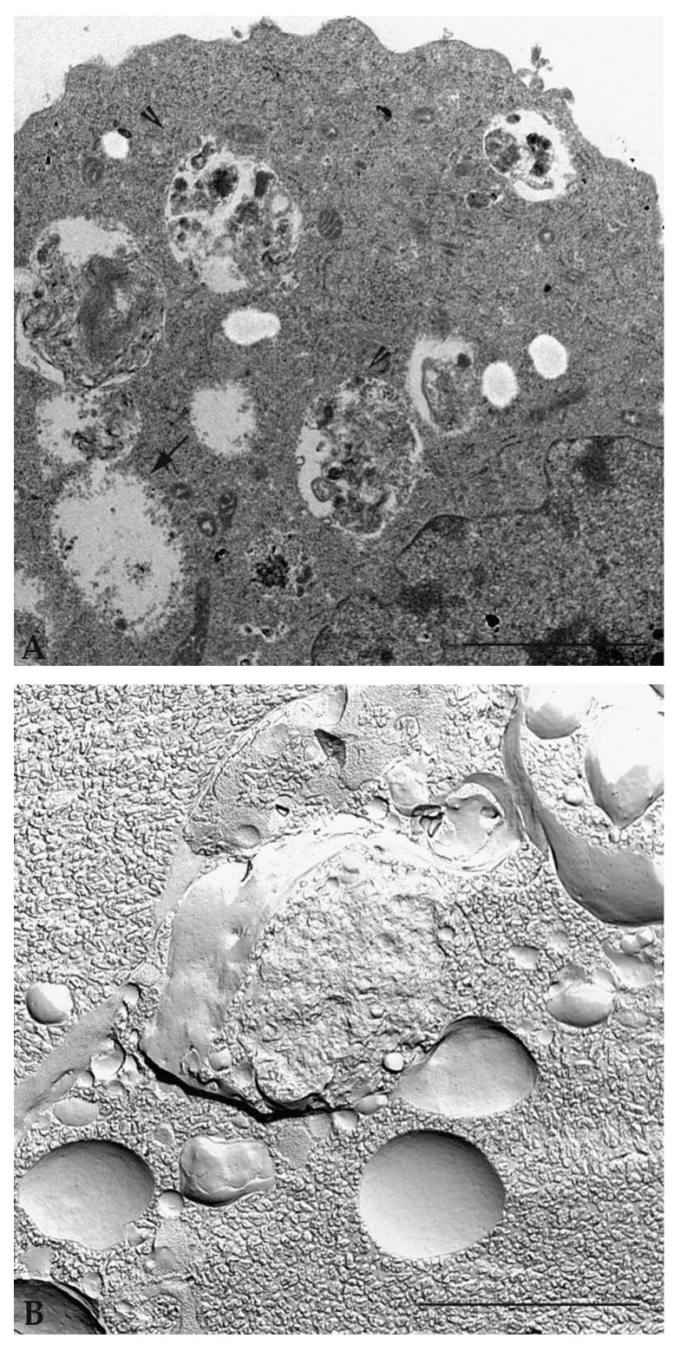
(A)極薄切片の観察では、空の液胞(矢印)消化されて強く電子化した液胞(矢印)(B)凍結法で作製した試料の観察では、多層膜を持つ液胞が観察されている。スケールバー:2μm。
オートファゴソームは、それを識別する特定のマーカーを用いて認識することもできる。オートファゴソームの特徴的なタンパク質は、タンパク質に特異的な蛍光抗体を用いて、蛍光顕微鏡で観察することで同定することができる。また、LC3 タンパク質の局在性や、LC3-I から LC3-II への変換などの分子ターンオーバーの変化を定量化し、選択的な薬剤によるオートファジー機構の活性化や阻害をウエスタンブロッティング法により行うことができる。オートファジーの二重の役割を細胞生存または細胞死のメカニズムとして明らかにするために、ATG遺伝子をサイレンシングすることで興味深い結果が得られている。これらの遺伝子はオートファジーを制御する遺伝子であるため、一度興味深い遺伝子をサイレンシングすると、プロオートファジーまたはプロアポトーシスタンパク質の発現がどのように変化するかを評価することが可能となる。これらの結果から、オートファジーが細胞の生存または細胞死のいずれかの目的のために、調べた生物学的プロセスに関与しているかどうかを理解することができるようになるだろう[54]。
このような技術の迅速な説明から、オートファジーのメカニズムは非常に動的で複雑であり、特に研究がオートファジーによる細胞の生存か細胞死か、またはオートファジーとアポトーシスを区別する必要がある場合には、容易に理解することができる。評価の誤りを避けるために、複数の検査を同時に行うことが推奨されている。
6. オートファジー、炎症と老化
オートファジーは、病原性微生物などの外因性刺激、または活性酸素、ミトコンドリア損傷、環境刺激物質などの内因性メディエーターによって活性化される主要な自然免疫経路であるインフラマソームの主要な調節因子として同定されている[55]。インフラマソームの活性化は、ヌクレオチドオリゴマー化ドメイン(NOD)様受容体(NLR)アダプタータンパク質、およびプロカスパーゼ-1を含むタンパク質複合体の形成およびオリゴマー化を伴う。この活性化により、カスパーゼ-1が切断され活性化され、その後、インターロイキン(IL)-1βやIL-18などの炎症性サイトカインが自然免疫細胞から放出されるようになる [56]。特に、内因性メディエーターが大規模な炎症反応を誘導すると、組織損傷を引き起こし、炎症性疾患の発症を促進する。したがって、インフラマソームのネガティブまたはポジティブな調節は、健康状態を良好に保つために不可欠である。
複数の研究で実証されているように、オートファジーは様々なメカニズムを介してインフラマソームの活性化をネガティブに制御することができる。
- 例えば、ミトコンドリアのような損傷を受けた小器官を除去することで、活性酸素の放出が減少し、その後のインフラマソーム活性化が抑制される。
- p62に依存したインフラマソーム複合体とミトコンドリアの分解。
- プロ-IL-1βをオートファゴソームに隔離して分解することにより。この経路は、オートファジーが介在するIL-1β分泌の減少に寄与している[57,58,59]。
オートファジーの欠乏は炎症性疾患の原因となる;遺伝子研究により、ATG16L1遺伝子の多型が腸の慢性炎症性疾患であるクローン病と関連している可能性が示されている[60,61,62]。残念ながら、クローン病の病態生理はまだ調査中であり、実際、他の遺伝子や素因となる環境因子が患者の発症に影響を与えることがいくつかの著者によって示されている[63,64]。したがって、炎症性腸疾患におけるオートファジーの本当の役割を区別するためには、ある程度の注意が必要である。
全体的に、これらのデータは、炎症性腸疾患の発症には、炎症性腸疾患とオートファジーが相互に調節し、宿主から自己防衛するための炎症反応と、組織障害や炎症性疾患を誘発しうる過剰な炎症反応の防止とのバランスを有利にすることを示唆している[67]。
最近の研究では、老化を特徴づけるオートファジー活性の低下は、マクロファージにおける機能不全ミトコンドリアの蓄積、活性酸素、およびNLRP3インフラマソームの活性化によるものであることが示されている[68]。これらの因子は、動脈硬化や2型糖尿病などの老化疾患のリスクを高める素因となる。特に、老化に関連する3つのシグナル伝達経路、すなわち、インスリン/インスリン様成長因子1(IGF-1)シグナル伝達、mTOR、およびSirtuin-1(Sirt-1)ネットワークの活性低下が同定されている。加齢に伴う疾患には、レスベラトロール、カテキン、エピガロカテキン-3-ガレート、ルテオロイシド、またはプロポリスなどの抗老化天然化合物を含む食品を豊富に含む食事に従うことが推奨される[69]。最近の研究では、赤ワインの天然ポリフェノールであるレスベラトロールが、加齢に伴う疾患の有望な治療標的であるSirt-1などのNAD+依存性脱アセチル化酵素を活性化することが実証されている[70]。さらに、この天然物はNLRP3の活性化を抑制することでオートファジー活性化を刺激することができた[71]。
7. メタボリックシンドローム疾患におけるオートファジーの役割
メタボリックシンドロームにおけるオートファジーの役割は特に興味深い。メタボリックシンドロームは、NHS(英国保健医療局)で定義されているように、特に先進国では多因子性疾患であり、これらの危険因子のうち少なくとも3つを特徴としている。
- 高血圧(85/130mmHg以上)。
- 高トリグリセリド(150mg/dL以上)。
- 高密度リポタンパク質(HDL)コレステロールが低い(40/50mg/dL未満)。
- 高血糖(100mg/dL以上)。
- 体脂肪の内臓分布 [72]。
これらの因子が同時に存在すると、患者は2型糖尿病、肥満、およびその結果としての心臓病を発症する危険性にさらされる。この病理学の発症は、家族の素因と悪いライフスタイルによって非常に頻繁に好まれており、不正確でバランスの悪い食事と運動不足が特徴である。メタボリックシンドロームにおけるオートファジーの役割を理解するためには、2型糖尿病を定義する生理学的な欠陥の上に住む必要がある。2型糖尿病は、一方ではインスリン分泌の不足によって特徴づけられ、他方ではインスリン抵抗性(生産されたインスリンが満足に標的臓器に作用しない)によって特徴づけられる。これら2つのプロセスの複合作用が、2型糖尿病(糖尿病とも呼ばれる)の典型的な高血糖の原因となっている。血糖値の上昇は、全身レベルで、標的臓器の細胞において、以下の機能障害を決定する:活性酸素種の増加と抗酸化システムの減少(酸化ストレス活性化)ミトコンドリアの機能障害、炎症プロセスの活性化、腎症、足潰瘍、脳卒中、神経障害、網膜症、心臓病を引き起こす(図7)。
図7 メタボリックシンドロームにおけるオートファジーの関与を模式的に示す
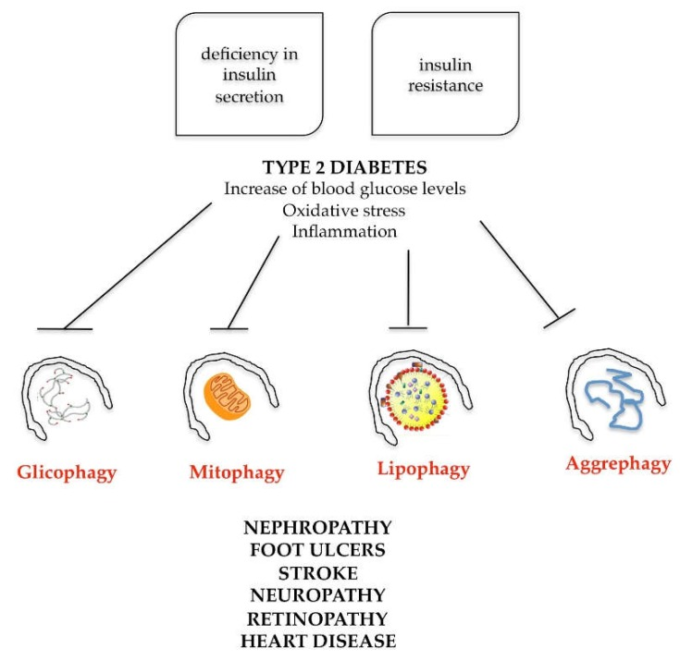
2型糖尿病は、インスリン分泌の欠乏やインスリン抵抗性により、血糖値、酸化ストレス、炎症が増加する。これらの細胞イベントは、結果として全身的な影響を伴うグリコファジー、マイトファジー、リポファジーまたはアグレファジーなどの異なるタイプの選択的オートファジーを阻害することができる。
2型糖尿病の進行は、インスリン分泌不全およびインスリン抵抗性の発症を通じてオートファジーと関連している[73]。オートファジーは、膵β細胞の構造と機能を維持し、アポトーシス細胞死から守るという、膵β細胞に対する保護的な役割を持っている。さらに、リポファジー、アグレファジー、マイトファジー、グリコファジーなどの選択的オートファジー機構は、インスリン標的臓器(肝臓、脂肪組織、骨格筋、腎臓)を高血糖に由来する酸化ストレス損傷から保護する。したがって、複数のオートファジー機構の変化は、代謝と細胞バランスを変更し、糖尿病、肥満、心血管合併症などの病理学的状況の確立を支持している[74]。
新陳代謝が活発な臓器に対するオートファジーの保護効果は、動物実験を通して科学的に証明されている。例えば肝臓では、オートファジーは肝細胞の適切な機能とグリコーゲンとトリグリセリドの代謝の規則性を保証する。肝細胞がATG7遺伝子の欠失を受けた動物モデル研究では、変化して変形したミトコンドリア、脂質滴の蓄積、およびユビキチン化されたタンパク質凝集体の数の増加が示されている[75]。膵臓のレベルでは、オートファジーはβ細胞の構造、質量、機能の維持を確実にし、小胞体の変化によって生じるストレスレベルを減少させる [76]。脂肪組織では、オートファジーは脂肪細胞の分化を確実にする:脂肪細胞におけるATG5およびATG7遺伝子の除去は、脂質の異常な蓄積を引き起こし、白色脂肪組織から褐色脂肪組織への異常な変態を引き起こす[77]。最後に、骨格筋では、オートファジーにより、グルコースに対するより大きな耐性と、質量自体の良好な維持が保証される。骨格筋におけるATG5およびATG7遺伝子の欠失は、マウスの筋肉量および脂肪量を減少させ、また、脂肪組織のレベルでの損傷(白色脂肪組織の褐色への変換およびより大きなβ酸化)を引き起こす[78]。最近のレビューでは、代謝性疾患における多機能性p62の役割に関連する研究がまとめられており、p62が潜在的な標的である可能性を示唆している。p62タンパク質は、膵β細胞をアポトーシスから保護し、インスリン抵抗性を低下させ、オートファジー活性化により心筋細胞のタンパク質の質を制御し、上皮成長因子受容体(EGFR)活性化を阻害することで糖尿病性腎症の発症を抑制する保護的な役割を果たしている[79]。
メタボリックシンドロームにおけるオートファジーの中心的役割を考えると、この疾患と闘うための最良の治療戦略は、カロリー制限と断続的な絶食、身体運動、そして必要に応じて薬理学的治療(図8)を通じたオートファジーの標的としての利用である(80)[80]。
図8 絶食とカロリー制限、運動または薬理学的治療によるオートファジー調節の模式図
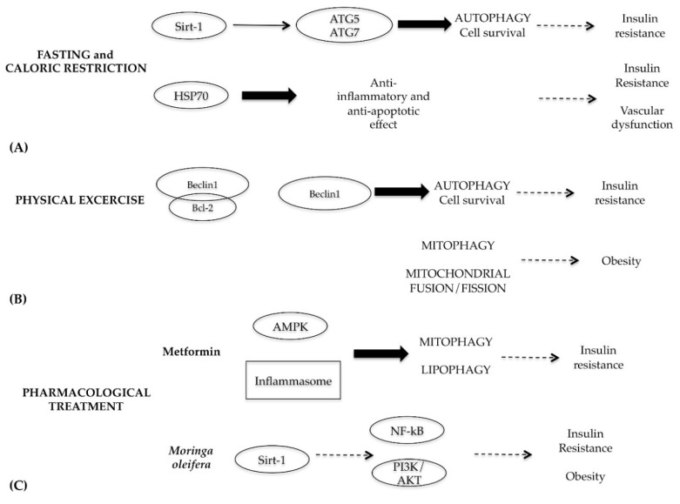
(A) 絶食とカロリー制限は、それぞれオートファジー効果と抗アポトーシス効果を持つSirt-1とHSP70タンパク質に作用する;(B) 運動はオートファジーを誘導し、Beclin1とBcl-2複合体を破壊し、ミトコンドリアの融合/分裂のバランスをとる;(C) メトホルミンまたはモリンガオレイフェラの天然抽出物による治療は、ミトファジーとリポファジーを回復させる。
断食およびカロリー制限は、Sirt-1およびAMPK(図8A)の活性化を通じてオートファジーの生理学的レベルを回復させることができるので、人間の健康に有益な効果がある[81] [82,83]。Sirt-1は、グルコース代謝とインスリン分泌に関与するNAD+依存性脱アセチラーゼである;このタンパク質は、インスリンに対する抵抗性が高い細胞では阻害される[84]。このタンパク質は、NADH酸化の亢進に起因するNAD+濃度の上昇を検出する代謝センサーとして機能する。カロリー制限によって活性化されると、Sirt-1はATG5やATG7などの必須オートファジー調節因子を脱アセチル化し、そのオートファジー効果を刺激することができる[85]。さらに、空腹時やカロリー制限は、炎症性サイトカインや酸化ストレスの調節を通じて、代謝性疾患に伴う血管機能障害や心血管リスクを低下させることができる[86]。特に、絶食は、抗炎症性および抗アポトーシス特性を有するヒートショックプロテイン70(HSP70)などのストレス誘発性タンパク質の転写を増加させる。糖尿病患者の骨格筋では、HSP70のレベルが低下し、これはインスリン抵抗性と関連している;そのため、絶食によるHSP70レベルの上昇はインスリン抵抗性とその結果として生じる血管機能障害を緩和する[87]。
2型糖尿病および心血管系合併症を有する人々における身体運動の有益な効果は十分に文書化されている。運動は、酸化ストレス、エネルギーの不均衡、細胞内カルシウムレベル、タンパク質のリモデリングをわずかに増加させるイベントのカスケードを活性化する。これらのイベントはすべて、ミトコンドリアとタンパク質のターンオーバーと代謝の改変を促進するオートファジー生存機構を活性化することができる。これらの適応応答は、一般的に脂質およびグルコースのホメオスタシスの最適化および抵抗性パフォーマンスの改善につながる[88]。特に骨格筋細胞では、身体活動はオートファジーのスタートアップ段階に不可欠なBeclin1-Bcl-2複合体を破壊することで機能する(図8B)[89]。
身体運動は肥満者において有益な役割を果たしている。肥満の被験者の骨格細胞では、マイトファジーの欠陥は、酸化ストレス、ミトコンドリアの蓄積と誤作動、および分裂と融合のミトコンドリアプロセスの間の平衡の変化(ミトコンドリアの健康を良好な状態に維持するための基本的なバランス)につながる可能性がある。分子および細胞メカニズムは明確には解明されていないが、肥満の骨格筋におけるアポトーシスイベントを減少させるミトファジーおよび融合・分裂プロセスの生理学的レベルを回復させることによって、身体運動が介入する[90]。
薬理学的介入に関しては、糖尿病患者は多くの副作用を有する血糖降下薬であるメトホルミンで治療される。AMPKタンパク質に作用するメトホルミンは、マイトファジーとリポファジーの両方を活性化し、インフラマソームの活性化をブロックすることができる[91]。メトホルミンの副作用や薬剤耐性の発現を考慮すると、オートファジーを制御する分子経路に関連する新たな知見に照らして、血糖降下作用を有する天然物質の探索は常に進化している[92]。最近の研究では、フラボノイド、イソチオシアネート、ポリフェノールを豊富に含むモリンガオレイフェラ植物の抽出物が、抗炎症作用、血糖降下作用、血中脂質減少作用を含む複数の機能を示すことが明らかになった[93]。これらの多機能性の原因となる分子機構は、NF-kBおよびPI3K/AKT経路の不活性化と、Sirt-1によって媒介されるミトコンドリア呼吸鎖に対する保護作用の調節である(図8C)[94]。
8. オートファジーと神経変性疾患
アルツハイマー病、パーキンソン病、ハンチントン病、筋萎縮性側索硬化症などの病的な神経変性疾患の典型的な症状として、誤ったタンパク質の凝集や一部の神経細胞集団の喪失が挙げられる。オートファジーは、損傷した小器官や凝集したタンパク質を分解する細胞内の主要なシステムであり、神経変性疾患の発生に関与していることが報告されている。神経変性疾患では、オートファゴリゾームにおけるオートファゴソームの成熟機構の変化が見出されている。
ATG5やATG7遺伝子の欠乏がマウス中枢神経系の神経変性を引き起こすことが示されている[95]。
さらに、オートファジーは、パーキンソン病の変異αシヌクレイン、ハンチントン病の変異ハンチン、筋萎縮性側索硬化症の変異TAR DNA結合タンパク質43(TPD-43)など、変性疾患に関連する様々なタンパク質の分解に重要な役割を果たしている。
アルツハイマー病では、細胞外アミロイドβプラークや、高リン酸化タウタンパク質の凝集体からなる細胞内神経原線維のもつれの存在が明らかにされている[96]。健康な脳ではオートファゴソーム小胞はあまり目立たないが、アルツハイマー病では多数のオートファゴソームが目立っている。オートファジー液胞の蓄積は、オートファジー誘導ではなく、クリアランスの障害によるものであり、アルツハイマー病の治療法として、オートファジー調節の後期段階にあることが示唆されている。オートファゴソームとリソソームの融合に関与する膜貫通タンパク質はプレセニリン1(PS1)である。この変異タンパク質はアルツハイマー病の発症に重要な役割を果たしていると考えられており、実際、PS1のSer367リン酸化喪失は2つの空胞の融合を阻害している[97]。また、PS1はリソソームの酸性化を制御することでCa2+の恒常性を維持していることも明らかにしている[98]。
リソソームの酸性化の低下は、リソソームの機能を変化させ、オートファゴソームとの融合を妨げる。この変化はアルツハイマー病と同様の神経細胞ジストロフィーを引き起こすが、これらの観察から、このオルガネラとその正しい機能が、凝集したタンパク質の除去にどのように重要であるかを理解している。
他のタンパク質はアルツハイマー病と相関しており、例えば、小胞輸送に関与するホスファチジルイノシトールタンパク質に結合するクラスリン基がエンドサイトーシスを調節し、タウタンパク質の凝集を防ぐ、Beclin1のようなものである。ベクリン1はオートファゴソームの形成に重要な役割を果たしており、重要なアポトーシス活性化成分であるカスパーゼ3は、ベクリン1タンパク質を分離し、オートファジープロセスの進行を停止させることができる。
アルツハイマー病は、遺伝子や生化学的な欠陥が酸化ストレスや慢性炎症を引き起こして発症することが知られている。これについては、抗酸化系の増加とそれに対応する酵素レベルの増加が、動物モデルにおいて神経変性疾患に有益な効果をもたらすと考えられている。アルツハイマー病の病態に関与し、酸化ストレスに反応して活性化する核内因子として、赤血球由来2様核内因子(Nrf2)がある[99]。この因子は、オートファジー受容体NDP52を介してオートファジーを活性化し、凝集したタウタンパク質を除去すると考えられている[100,101]。
パーキンソン病は広範囲に及ぶ神経変性疾患であり、結節性黒質部のドーパミン作動性ニューロンの重度の消失と、レビー小体と呼ばれるポリユビキネートおよびα-シヌクレインタンパク質の内包物が続くことを特徴としている。これらの小体はパーキンソン病だけでなく、他の神経疾患にも特徴的であるようである [102]。
パーキンソン病におけるオートファジーの役割は、神経細胞におけるリソソームおよびオートファゴソームの変化の存在によって実証されている;この証拠を裏付けるために、リソソソームが機能的に変化すると、α-シヌクレインの量が上昇し、オートファジー経路の変化を示している。リソソソームヒドロラーゼをコードする遺伝子GBA常染色体劣性突然変異は、オートファゴソーム-リソソーム経路の変化とα-シヌクレインの凝集を誘導することが示されている[103]。EB転写因子(TFEB)は、オートファゴソームの形成やリソソソーム融合に関連する遺伝子を正に制御し、リソソームエキソサイトーシスのクリアランスを増加させる因子として同定されている[104]。最近では、その過剰発現がリソソソーム障害を軽減し、α-シヌクレインに関連した神経障害を改善することが示されている。
常染色体優性型パーキンソン病の共通の原因は、ロイシンリッチリピートキナーゼ2(LRRK2)やVPS35 D620Nの変異によるものである。LRRK2のアップレギュレーションはオートファジーの流れを変化させ、VPS35 D620Nの変異はWASH複合体の変化を通じてATG9タンパク質の輸送を変化させることが示されている。パーキンソン病の常染色体劣性型は、パーキンRBR E3ユビキチンタンパク質リガーゼ(PARK2)とPTEN誘導性putative kinase 1(PINK1)の変異によって引き起こされ、損傷したミトコンドリアの分解(マイトファジー)を調節している。PINK1は上流因子として作用し、脱分極したミトコンドリアに特異的に蓄積する一方で、PARK2はユビキチンのミトコンドリア基質への移行を触媒する [105,106]。
ハンチントン病は、ハンチンスチン蛋白質(HTT)をコードする遺伝子のGAGトリヌクレオチドリピートによって引き起こされる神経変性疾患である。ハンチンチンはオートファジー蛋白質の輸送に重要な役割を持っている。HTTが欠乏すると、ミトコンドリアを巻き込んだオートファゴソームの異常な蓄積を引き起こす。ハンチントン病では、ATG7遺伝子多型、すなわちBeclin1とオートファジーの選択基質p62/SQSTM1の両方に変化が見られる。これらの系の調節障害は、ハンチントン病の早期発症においても非常に重要である。逆に、非変異型HTTはp62と結合し、ULK1と相互作用し、オートファジーを誘導することができる[107]。
筋萎縮性側索硬化症は、脊髄や脳の運動ニューロンの変性が進行し、萎縮や筋麻痺に至る神経筋疾患である。現在までに、オートファジーやマイトファジーに関わる遺伝子を含め、約30の遺伝子が関与しているとされている。特に、タンパク質の凝集体や損傷したミトコンドリアのクリアランスの変化が観察されている。筋萎縮性側索硬化症に関連する遺伝子は、TDP43やFUSなどのRNAと結合するタンパク質をコードしており、リボスタシスの変化が本疾患に有利であることが示唆されている。筋萎縮性側索硬化症の発症時には、プロテアーゼ、ミトコンドリア機能、細胞骨格の完全性、細胞内交通の変化も関与している。オートファジーに関与する筋萎縮性側索硬化症遺伝子は、SQSTM1,TBK1,OPTNである。これに代えて、C9ORF72,VCP、CHMP2B、VAPB、ALS2,DCTN1などの小胞輸送を調節する遺伝子も、直接的にも間接的にもオートファジー機構に関与している可能性がある。筋萎縮性側索硬化症では、患者の運動ニューロンでは、タンパク質の凝集体の蓄積や、腫脹またはジストロフィー性のミトコンドリアがしばしば見られる。現在、この疾患の治療法はない[108]。
9. 癌におけるプロサバイバルオートファジー
がんにおけるオートファジーの役割は議論の余地のある複雑なものであり、がんの種類、病期、および遺伝的背景に依存する [10]。癌発生の初期段階では、オートファジーは、活性酸素によるDNAやタンパク質への損傷から細胞を保護し、正常細胞の腫瘍細胞への変態を遅らせる[109]。ベクリン1のモノアレル欠失マウスは、自然発生的な腫瘍発生の感受性が高いことが確認されている[110]。
オートファジーは、促進、進行、転移などの後期段階では、活性酸素による代謝ストレス生成物を排除し、がん細胞の生存に必要な栄養素を供給するため、腫瘍細胞の増殖を促進する効果があると考えられている[111]。
オートファジーの役割は、特にがん幹細胞(CSC)において興味深いものであり、泌尿器系膀胱がんや乳がんなどの複数のがん種で観察されるように、より分化したがん細胞集団と比較してオートファジーのレベルが高いことが特徴である[112,113]。これらの高レベルのオートファジーは、休眠状態などのCSCの特性を維持するために不可欠である。
オートファジーは、固形腫瘍の酸素不足領域や急性骨髄性白血病浸潤後の骨髄領域において、低酸素ストレスへの癌細胞の適応の際に制御的役割を果たしている[114]。
さらに、オートファジーの発現により、がん細胞は化学療法による毒素作用から身を守り、薬物治療によって誘導されるアポトーシス効果(オートファジー媒介性抵抗性)をブロックすることができるようになる[115]。例えば、シス白金抵抗性の卵巣がん細胞は、オートファジーフラックスの増強されたレベルで特徴づけられる[116]。
最後に、アポトーシス抵抗性腫瘍細胞に対してより大きな治療結果を得るために使用されるオートファジー細胞死を誘導することができる薬剤がある[117,118]。
腫瘍塊内に様々な細胞および分子プロセスが共存していることから、オートファジー/アポトーシススイッチの主要分子(Beclin1,カスパーゼ、p53,PI3K/AKT/mTOR、およびp62)の同定に向けた研究が進められており、これは抗がん剤標的治療の計画に有用である[119,120]。
前臨床研究では、治療戦略におけるオートファジー抑制の重要性が強調されている。KRASは細胞増殖を制御する遺伝子であり、変異すると細胞は増殖を続け、がんに発展する。BRAFは、セリン/スレオニンプロテインキナーゼB-Rafとして知られるタンパク質をコードする遺伝子である。このタンパク質は、細胞増殖に向けた細胞シグナルの活性化に関与している;多くのヒト腫瘍では、この遺伝子は欠損している[121,122]。このような変異を示すヒト細胞株では、高レベルのオートファジーが観察された。KRASとTp53の状態も非常に重要である;実際、ATG5またはATG7遺伝子を欠損したKRAS変異マウスでは、膵臓の前悪性病変が明らかになり、一方、KRASとTp53欠損マウスでは、ATG5またはATG7遺伝子の欠損により、悪性腫瘍の獲得と腺癌の発生が誘導された[123]。これらの結果は、腫瘍のBRAFまたはKRAS変異におけるオートファジー阻害が、潜在的な抗がん剤標的となり得ることを示唆している。
近年、オートファジーを誘導または阻害することができる多くの化合物が薬理学的介入のために同定されているが、それらの使用は前臨床データによって広く支持されなければならない[124]。mTORの強力かつ選択的な阻害剤であるTorin1,およびmTORC1阻害剤であるRapamycinは、オートファジーを刺激することができる[125]。オートファジーは、3-メチルアデニン、LY294002,Wortmannin(PI3K特異的阻害剤)SBI-0206965(ULK1阻害剤)Spautin1(UB特異的ペプチダーゼ阻害剤)Vinblastine(リソソソームとオートファゴソームの融合を阻害する)によって複数のレベルで阻害される。E64dとペスタチンA(リソソソームプロテアーゼ阻害剤)バフィロマイシンA1(ATPアーゼポンプ阻害剤)クロロチン(オートファゴソーム分解を阻害するリソ運動性化合物)(図9)[124,126]。
図9 オートファジー単相のモジュレーション
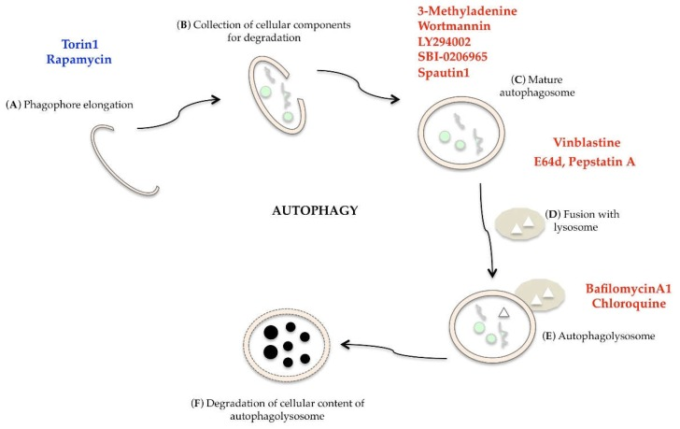
オートファジー誘導剤は青字(Torin1,Rapamycin)オートファジー阻害剤は赤字(3-メチルアデニン、Wortmannin、LY294002,SBI-0206965,Spautin1,Vinblastine、E64d、PestatinA、BafilomycinA1,Chloroquine)である。
クロロキン抗マラリア薬の派生薬であるヒドロキシクロロキンは、臨床試験が試みられている(表3);オートファゴリソソーム内のリソソーム酵素分解作用を阻害することができる。クロロキンの代わりにヒドロキシクロロキンが使用されているのは、オートファジー阻害作用の方が毒性が低いためである。多くの臨床試験で、単独または他の化学療法薬と併用して投与した場合の有効性が示されており、他の試験では、膠芽腫、星細胞腫、肺癌、膵臓癌などの様々な固形癌に対して有効であることが証明されている[10]。
表3 ヒドロキシクロロキンの一部の臨床試験の一覧
| 治療の組み合わせ | 腫瘍の種類 | ClinicalTrials.govでのトライアル |
|---|---|---|
| シロリムスまたはボリノスタット+ HCQ | 進行した固形腫瘍 | NCT01266057 |
| シングルHCQ | 膠芽腫および星状細胞腫 | NCT02432417 |
| HCQ +ボリノスタット | 悪性固形腫瘍 | NCT01023737 |
| シスプラチン、エトポシド+ HCQ | ステージ4の小細胞肺がん | NCT00969306 |
| HCQ +アブラキサン+ゲムシタビン | 膵臓腺癌 | NCT01978184 |
| ソラフェニブ+ HCQ | 難治性または再発性固形腫瘍 | NCT01634893 |
さらに、生物由来の天然物の多くは、オートファジーを調節する生物学的および薬理学的特性を有している;これらの潜在的な応用は、試験管内試験モデルで研究され、臨床応用に結びつく可能性がある[127]。
10. オートファジーと自己免疫疾患
全身性エリテマトーデス(SLE)などの自己免疫疾患は、予後や治療成績に大きな変化をもたらすことが特徴である。これらの疾患には、適応側のB細胞やT細胞、自然側の樹状細胞、マクロファージ、好中球などの免疫系全体が関与しており、その結果として抗核体が産生される。代謝経路は免疫系の分化と活性化の重要な調節因子であるため、それらが変化すると自己免疫疾患の発症につながる可能性がある[128]。免疫系分化の代謝制御は、mTOR依存性オートファジーを活性化する造血幹細胞から始まる[129]。mTORは栄養感知キナーゼであり、代謝シグナルを遺伝的パターンにリンクさせることで、細胞の成長と分化を制御する。特に、mTORは2つの複合体を形成している:Th1とTh17の発生を促進するmTORC1とTh2の発生を媒介するmTORC2;両方の複合体は調節性T細胞(Treg)の発生を制限する[130]。代謝ストレス時には、Rab4Aによるエンドソームトラフィックの活性化が、CD4およびCD3表面受容体とダイナミン関連タンパク質1(Drp1)ミトコンドリア分裂開始因子を介してオートファジーを促進する。Drp1のリソソーム分解はマイトファジーを減少させ、その結果として大きな伸長ミトコンドリアが蓄積され、全身性エリテマトーデスT細胞では活性酸素が発生する[131]。活性酸素はmTORC1を活性化し、DN Tcellsによる炎症性壊死、IL-4とIL-17の分泌、CD8メモリーT細胞とTreg細胞の枯渇を引き起こす [132,133]。肝臓では、この活性化は抗核抗体の産生と病気の発症に先行している。これらのデータは、Rab4を介したDrp1の枯渇には病原性の役割があることを示し、SLE治療の潜在的なターゲットとなることを示している[131]。SLEのT細胞におけるオートファジーは十分に調べられているが、B細胞におけるオートファジーはあまり研究されていない。Clarkeと共著者は、マウスおよびヒトの全身性エリテマトーデスB細胞でオートファジーが強化されていることを示し、それがプラスマブラストの発達に必要であることを示している[135]。彼らは、ATG7欠損マウスで分離されたB細胞が試験管内試験刺激を受けても形質細胞への効果的な分化に失敗したことから、形質細胞分化におけるオートファジーの重要な役割を実証した。同様に、オートファジー阻害後に刺激したヒトB細胞は形質細胞に分化しなかった。このことから、SLEマウスモデルのB細胞の初期段階では、発症前にオートファジーの活性化が加齢とともに増加していることを発見した[135]。
末梢のTh17細胞とTreg細胞の間の不均衡は、SLEの発症や他の自己免疫疾患に重要である[136]。IL-17A、IL-17F、IL-22を産生するTh17細胞は、自己免疫応答を促進し、B細胞による自己抗体産生を促進する [137]。Treg細胞は、IL-10およびTGF-βを放出して免疫恒常性を制御する。最も重要なTreg特異的転写因子はフォークヘッドボックスプロテイン3(Foxp3)であり、Treg細胞におけるFoxp3の発現低下はSLEにおけるTreg機能不全の原因となっている[138]。代謝チェックポイントおよび自己免疫制御におけるオートファジーの極めて重要な役割を考えると、特にSLEの発症においては、最近の研究ではオートファジーを標的とした治療法が取り上げられている。
いくつかの研究では、 ヒドロキシクロロキンがSLEや関節リウマチ治療などの他の免疫疾患の主役であることが報告されている[139]。SLEにおけるTh17細胞とTreg細胞はともにオートファジー活性化を示しており、クロロキンでオートファジー経路を遮断することで、Th17/Treg応答のバランスを取り戻すことができる。このように、オートファジーの阻害は、SLEで観察されたTh17細胞の過剰活性化を逆転させてIL-17の分泌を減少させ、Treg細胞のFoxp3発現を増加させ、Treg細胞の機能を維持する。このようにして免疫バランスが回復し、炎症性サイトカインや自己抗体の血清レベルが低下する[140]。
SLE患者はmTOR活性化に起因するTreg機能障害を有する。mTORに作用するラパマイシンはT細胞の増殖を抑制し、シロリムスの一般名で医薬品として開発されている[141]。シロリムスは、全身性エリテマトーデスを発症しやすいマウスの自己免疫を抑制することが生体内試験試験で明らかになった[142]。最近の前向き研究では、重症で持続性のあるSLE患者におけるシロリムスの有効性と安全性が示された[143]。
SLEは悪性腫瘍(主にリンパ腫)の発生率の増加と関連している可能性があることを考慮すると、免疫抑制剤による治療は二次がんの発生にも寄与している可能性がある[144]。新たなデータは、ラパマイシンが白血病やリンパ腫に対して有効な抗腫瘍剤であることを示唆しており、したがって、SLE患者の治療に免疫抑制剤として使用できる可能性があることを示唆している[145]。しかし、オートファジーの薬理学的調節が正の効果をもたらすか負の効果をもたらすかは必ずしも明らかではない;したがって、特定の標的治療法を開発するためには、より詳細な研究が必要である。
11. 結論
オートファジーは、進化を通して高度に保存された分解プロセスである。1974年には、Christian de Duve博士がオートファジーを「自食」のプロセスとして記述し 2016年には大隅良典博士がオートファジーの必須遺伝子の同定に向けた顕著な貢献が認められた[146,147]。
オートファジーは、細胞のエネルギーや生理的バランスを維持する上で基本的な役割を果たしていることが多くの研究で明らかにされており、この現象の変化が病気の頻度を高める原因の一つとなっている。多くの薬理学的治療や適切な生活習慣は、オートファジーの生理学的レベルを回復させることを目的としており、このプロセスが公衆および健康上の関心の高い潜在的かつ有望なターゲットであることを示唆している。
