
コンテンツ
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7369141/
オンライン公開 2020年6月16日.
要旨
背景
前臨床試験、臨床試験、レビューにより、3′,5′-環状アデノシン一リン酸(cAMP)と3′,5′-環状グアノシン一リン酸(cGMP)をホスホジエステラーゼ阻害剤で増加させることがアルツハイマー病の疾患修飾につながることが示唆されている。アルツハイマー病では、cAMP/タンパク質キナーゼA(PKA)とcGMP/タンパク質キナーゼG(PKG)のシグナル伝達が障害され、cAMP/PKAとcGMP/PKGはcAMP応答エレメント結合タンパク質(CREB)を活性化する。CREBはミトコンドリアや核内DNAと結合し、シナプス形成、記憶、神経細胞生存遺伝子(脳由来神経栄養因子など)やペルオキシソーム増殖因子活性化受容体-γ coactivator-1α(PGC1α)を誘導する。 cAMP/PKAとcGMP/PKGはSirtuin-1を活性化し、PGC1αを活性化する。PGC1αはミトコンドリアの生合成や抗酸化遺伝子(Nrf2など)を誘導し、BACE1を抑制する。
目的と方法
ホスホジエステラーゼ阻害薬がアルツハイマー病を予防または治療するかどうかについて、有効性を検証した臨床試験、疫学、メタアナリシスをレビューし、批判的に検討する。
結果
カフェインとシロスタゾールはアルツハイマー病リスクを低下させる可能性がある。とシルデナフィルの臨床試験は有望だが、予備的で結論は出ていない。PF-04447943とBI409,306は効果なし。フィンポセチン、シロスタゾール、ニセルゴリンの臨床試験は混合している。デプレニル/セレギリンの試験では短期的な効果しか示されていない。
広範囲ホスホジエステラーゼ阻害剤プロペントフィリンは、5つの第III相臨床試験で、軽度から中等度のアルツハイマー病患者の認知、認知症の重症度、日常生活動作、グローバル評価を、ADAS-CogやCIBIC-Plusを含む複数の尺度で改善することが18ヶ月間の第III相臨床試験で示されている。
しかし、2冊の本は、MedScapeの記事に基づいて主張した18ヶ月の第III相臨床試験が失敗したので、プロペントフィリンは中止された。現在、プロペントフィリンは、アルツハイマー病のように、年齢に関連した野生型アミロイドβ沈着を伴うイヌの認知機能障害の治療に使用されている。
結論
ホスホジエステラーゼ阻害薬は、アルツハイマー病を予防し、治療する可能性がある。
キーワード
アルツハイマー病、アミロイド前駆体タンパク質分泌酵素、臨床試験第III相、3′、5′-環状AMPホスホジエステラーゼ、3′、5′-環状-GMPホスホジエステラーゼ、環状AMP応答エレメント結合タンパク質、環状ヌクレオチド、グリコーゲン合成酵素キナーゼ3,NF-E2関連因子2,NF-κB、ペルオキシソーム増殖因子活性化受容体γ-コアクチベーター1-α、サーチュイン1
背景
散発性アルツハイマー病は、高齢者の認知症の最も一般的な原因である。脳アミロイドβ(アミロイドβ)の生成、オリゴマーへの凝集および神経斑への沈着、タウの高リン酸化による神経原線維のもつれ[1, 2]、鉄およびアルミニウムの蓄積[3, 4]、感染症[5-11]、酸化ストレスおよび脂質、RNA、DNAおよびタンパク質の損傷[12-16]、異常なカルシウムおよび亜鉛シグナル伝達[17-21]などが関与する。ミトコンドリア機能不全[22-24]、小胞体ストレス[25-27]、リソソーム機能不全[17, 20, 28-30]、オートファジー機能不全[31]、神経炎症[32-38]、神経細胞の細胞周期停止再突入[39-48]、インスリンおよびインスリン様成長因子1抵抗性[49-51]、シナプス機能不全[52]、およびニューロン死。
環状ヌクレオチドは、中枢神経系における様々な生理学的プロセスを調節するためのセカンドメッセンジャーとして作用する内因性の細胞シグナル伝達分子である。Ca2+を固定化し、一過性受容体電位メラスタチン2(TRPM2)チャネル開放型環状アデニン二リン酸リボース(cADPR)[53-57]と細胞質DNA誘導性環状グアノシン一リン酸アデノシン一リン酸(環状GMP-AMP、またはcGAMP)[14-16,58-62]を含む、アルツハイマー病の病因に関与している可能性がある複数の環状ヌクレオチドがある。これらのうち、環状プリンヌクレオチド3′,5′-環状アデノシン一リン酸(cAMP)と3′,5′-環状グアノシン一リン酸(cGMP)については、最も多くのエビデンスがある。
生理的なcAMPおよびcGMPシグナル伝達
生理学的には、Gαsを含む複合体がアデニル酸シクラーゼ(AC)を活性化するとcAMPが生成される。アデニル酸シクラーゼは、アデノシン三リン酸(ATP)からcAMPを生成し、cAMPによって直接活性化された交換タンパク質(EPAC)とプロテインキナーゼA(PKA)を活性化する。PKAホロ酵素は、2つの触媒サブユニットと2つの調節サブユニットから構成されている。cAMPに結合すると、調節サブユニットは触媒的なPKAサブユニットを放出し、多くの標的をリン酸化する [63]。
cGMPシグナル伝達に関しては、一酸化窒素合成酵素(NOS)が一酸化窒素(NO)を産生し、NOは細胞質内の可溶性グアニルシクラーゼ(sGC)を活性化し、sGCは細胞質内のcGMPを産生する[64, 65]。また、心房性ナトリウム利尿ペプチド(ANP)およびB型ナトリウム利尿ペプチド(BNP)は、粒子状の血漿膜局在型受容体グアニルシクラーゼA(GC-A)を活性化し[66]、C型ナトリウム利尿ペプチド(CNP)は粒子状のグアニルシクラーゼB(GC-B)を活性化する[67]。粒子状グアニルシクラーゼGC-AおよびGC-Bは、細胞膜に近接してcGMPを産生する [67, 68]。
ホスホジエステラーゼ
ホスホジエステラーゼ(PDE)酵素は、環状プリンヌクレオチドを分解する。PDE1,2,3,10,および11は、cAMPおよびcGMPの両方を分解する [69,70]。PDE4,7,および8はcAMPを特異的に分解する。PDE5,6,9はcGMPを特異的に分解する [69,70]。PDE5 は NOS/NO/sGC シグナルによって生成された細胞質の cGMP を分解し、PDE9 は Natriuretic peptide/pGC シグナルによって生成された細胞質周囲の cGMP を分解する [71]。cGMPの効果に関する研究の解釈を複雑にしているのは、低濃度のcGMPはPDE3を阻害してcAMPレベルを上昇させ、一方、中程度の高用量のcGMPはPDE2を活性化してcAMPレベルを低下させるということである[72]。
様々な薬物や天然に存在する化合物は、1つ以上のPDE酵素を阻害する。アルツハイマー病のためのPDE阻害に関する様々な前臨床研究に基づいて、Heckmanらによるレビューでは、PDE2,PDE4,およびPDE5阻害剤がアルツハイマー病の治療に最も有望であると結論づけられている[73]。様々なPDE阻害剤でcAMPおよび/またはcGMPシグナル伝達を増加させることの複数の利点は、cAMP応答エレメント結合タンパク質(CREB)であるサーチュイン1(SIRT1)に対するこれらの経路の効果に起因している。ペルオキシソーム増殖因子活性化受容体-γコアクチベーター-1α(PGC1α)核内因子エリスロイド2関連因子2(Nrf2)活性化B細胞の核内因子κ-光鎖エンハンサー(NFκB)グリコーゲン合成酵素キナーゼ3β(GSK3β)シグナル伝達(図1)。1). 以下では、これらの分子経路に対するcAMPおよびcGMPレベルの影響について考察する。
図1 アルツハイマー病に関連するcAMPおよびcGMPシグナル伝達の下流標的
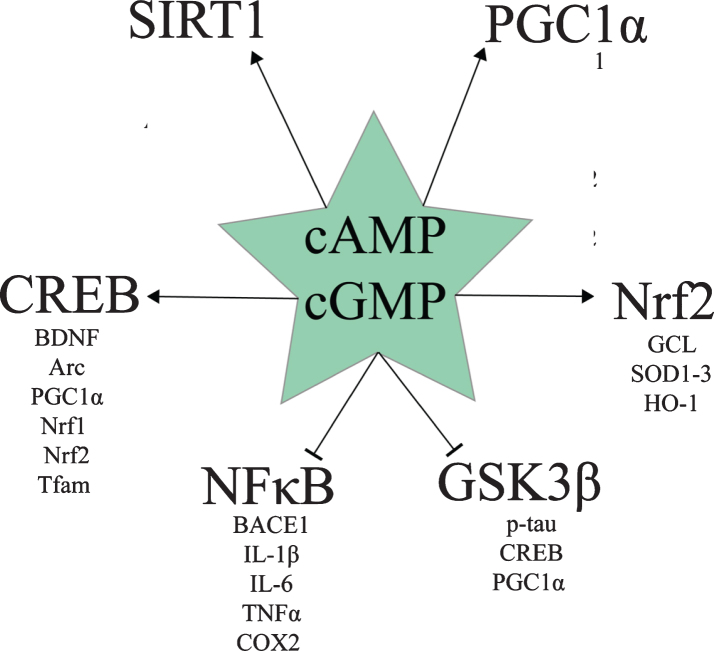
cAMP、cGMP、CREB
cAMP/PKAシグナルとcGMP/PKGシグナルの両方がCREBをSer133でリン酸化することで活性化する[74-79]。pSer133-CREBタンパク質レベルは、アルツハイマー病海馬[80]、アルツハイマー病前頭前野、アルツハイマー病末梢血単核細胞[81]で低下する。活性化されたCREBは、シナプス形成や記憶形成に関与する遺伝子、例えば脳由来神経栄養因子(BDNF)や活性制御細胞骨格関連タンパク質(Arc)などを誘導し、神経細胞の生存を誘導する[84]。BDNFやBcl2などの遺伝子[84-90]や、PGC1α、核呼吸因子1(Nrf1)、Nrf2,ミトコンドリア転写因子A(Tfam)などのミトコンドリア生合成や抗酸化防御に関与する遺伝子[83,91]。
cAMP、cGMP、SIRT1,PGC1α
cAMP/PKAおよびcGMP/PKGシグナリングの両方は、CREBおよびSIRT1を介したPGC1α活性化を介して、ミトコンドリア生合成および抗酸化遺伝子発現を促進する[92-105]。PGC1α転写を誘導するCREB [74-79]の活性化[83,91]に加えて、cAMP/PKAおよびcGMP/PKGシグナリングの両方がSIRT1を活性化する[94-96,98,104-110]。SIRT1は、プロアミロイド原性βセクレターゼ(BACE1)の調節を低下させ、抗アミロイド原性A崩壊酵素およびメタロプロテアーゼドメイン含有タンパク質10(ADAM10)の調節を上昇させる[106]だけでなく、PGC1αを脱アセチル化し、それによって翻訳後に活性化させる[97,111-118]など、アルツハイマー病において複数の有益な効果を発揮する。
PGC1αは、Nrf1,Nrf2,およびTfamを含むミトコンドリア生合成の中核的な調節因子を誘導する[93]。PGC1αは、フォークヘッドボックスO3(Foxo3a)に結合し、SIRT1によって脱アセチル化された抗酸化遺伝子、すなわちマンガンスーパーオキシドジスムターゼ(MnSOD)カタラーゼ、ペルオキシレドキシン3および5,チオレドキシン2,チオレドキシン還元酵素2,アンカップリングプロテイン2(UCP2)を誘導する[112]。 脳内PGC1α過剰発現は、グルタチオンペルオキシダーゼ1(GPx1)UCP4,およびUCP5をアップレギュレートすることも示されている[119]。PGC1αはまた、酸化的呼吸[92,103]および脂肪酸β酸化に関与する遺伝子を誘導する[120]。逆に、ペルオキシソーム増殖因子活性化受容体γ(PPARγ)依存性のメカニズムでは、PGC1αはBACE1の発現を抑制する[121,122]。
BACE1はアミロイドβ生成の律速酵素であり[123]、神経ミトコンドリア生合成および抗酸化酵素発現はアルツハイマー病では著しく阻害されるため、PGC1αシグナル伝達のこれらの下流効果はアルツハイマー病治療にとって重要である。アルツハイマー病患者の海馬では、神経細胞のミトコンドリア数の減少とPGC1αの発現低下、および下流のNrf1,Nrf2,Tfam [93]、およびMnSOD [23,112]の発現低下によって示されるように、ミトコンドリアの生合成が障害されている。アルツハイマー病におけるPGC1αシグナル伝達のこの欠損は、非常に早い時期に始まるようである。PGC1α、Nrf1,Nrf2,およびTfamは、アミロイドβオリゴマーの有意な濃度が形成される前であっても、トランスジェニックADマウスモデルでは、生後1ヶ月の早さでmRNAレベルでダウンレギュレートされた[91]。アルツハイマー病や血管性痴呆の脳のようなげっ歯類の脳の低灌流状態では[119, 124-127]、PGC1α神経過剰発現は、認知障害や低酸素ニューロンの代謝活性を改善した[119]、薬理学的にPGC1αシグナル伝達を調節することの治療の可能性を示している。
治療的には、PDE阻害剤は、pSer133-CREBおよびPGC1αシグナル伝達におけるこのアルツハイマー病欠損を調節する方法を提供する。APPswe M17細胞において、cAMPはPKA依存的な方法でCREBリン酸化およびPGC1α発現を救済した[93]。さらに、cAMPシグナル伝達は、シナプス形成、記憶形成、ニューロン生存、ミトコンドリア生合成だけでなく、ミトコンドリア生合成とマイトファジーの間の生理的バランスの回復を促進する可能性がある。例えば、6ヶ月齢のAPP/PS1マウスに15mg/kg/日のcAMP上昇型PDE7阻害剤S14を経口投与したところ、ミトコンドリアの生合成を増加させ、アミロイドβ誘発性の過剰なマイトファジーを減少させることで、記憶障害、神経新生の増加、およびミトコンドリア量の回復がみられた[128]。ミトコンドリアの生合成とマイトファジーの間のこのバランスは、PGC1αダウンレギュレーション[93,129]によって示されるように、アルツハイマー病患者の海馬錐体ニューロンで混乱しているように見えるだけでなく、いくつかの生き残ったミトコンドリアの多くは、オートファゴソーム[22,130]に蓄積することを発見した。最終的に、低灌流化したげっ歯類の脳では、非特異的PDE阻害剤プロペントフィリンがミトコンドリアのATP産生を回復させる[131, 132]ことから、PDE阻害剤でCREBとPGC1αシグナル伝達を調節することの一つの結果として、ミトコンドリアのATP産生が増強されることが示唆される。
cAMP、cGMP、およびNrf2
CREBやPGC1αの下流にあるNrf2は、グルタミン酸-システインリガーゼ(グルタチオン合成の律速酵素)SOD1-3,ヘムオキシゲナーゼ1(HO-1)などの抗酸化防御、解毒、オートファジー、細胞保護に関わる様々な遺伝子を誘導する転写因子である[133-136]。Nrf2は核に局在しているときに活性である;しかしながら、核内のNrf2レベルはアルツハイマー病患者の海馬細胞では低下しており、アルツハイマー病におけるNrf2活性の低下を示唆している[137]。cAMPとcGMPの両方がNrf2シグナルを増加させるようである。
cAMPは、ほとんどの細胞型においてNrf2シグナル伝達を増加させるようである[138-140]。例えば、マウスおよびヒト肝細胞において、8-ブロモアデノシン-cAMPアナログは、PKA依存的な方法でNrf2標的遺伝子発現および抗酸化応答エレメント(ARE)転写活性をアップレギュレートした[138]。α-メラノサイト刺激ホルモンまたはフォルスコリン(いずれもcAMPを増加させる)でケラチノサイトおよびメラノサイトを処理すると、Nrf2およびNrf2標的遺伝子発現が増加した[139]。PDE4阻害剤ロフルミラストは、呼吸器同期ウイルスに感染した気管支上皮細胞においてNrf2レベルを補充することが示されている[141,142]。したがって、cAMPは、ニューロンおよび他の脳細胞におけるNrf2シグナル伝達を増加させる可能性がある[138-140]。
cGMPはまた、ヒト気管支上皮細胞において、ヒ素はcGMP/PKG依存的な方法でNrf2を活性化したように、Nrf2シグナル伝達を活性化する可能性がある[143]。C6グリオーマ細胞では、ニトロ化したcGMPがNrf2を活性化した[145]。大腸がん細胞では、一酸化窒素がNrf2を活性化した [146]。このことは、多様な細胞タイプにおいて、cGMP、ニトロ化cGMP、およびNOによるNrf2の文脈依存的な活性化を示唆している。したがって、cAMPおよびcGMP(したがってPDE阻害剤)の両方が、脳細胞におけるNrf2活性を促進する可能性がある。
cAMP、cGMP、NFκB
アルツハイマー病 のもう一つの重要な治療ターゲットは NFκB である。NFκB は、c-Rel、RelA/p65,RelB、p105/NFKB1,および p100/NFKB2 の 5 つの可能なタンパク質サブユニットのうちの 2 つからなる転写因子ダイマーのファミリーである。RelA/p65サブユニットを含むNFκBダイマーは、炎症性遺伝子発現(例えば、プロ炎症性サイトカインIL-1β、IL-6,およびTNF-α、ケモカインIL-8,RANTES、MIP1,酵素COX2およびiNOS、接着分子VCAM1およびICAM1 [147])ならびにBACE1の発現を誘導する[34,35]。炎症性NFκBは、酸化ストレス[148]、トール様受容体(TLR)を介した多様な病原体および損傷関連分子パターン[149,150]、TLR2を介したフィブリルアミロイドβ[32]、プロ炎症性サイトカイン(例えば。TNF受容体を介したTNF-αおよびIL-1受容体を介したIL-1β)[147]、およびタンパク質チロシンキナーゼ[151]と連携したB細胞抗原受容体(BCR)などの他の免疫受容体[37,148]。MYD88[32]のようなシグナル伝達メディエーターを介して、TLRは、例えば、NFκBキナーゼ阻害剤(IKK)を活性化し、この阻害剤は、NFκBタンパク質α(IκBα)をリン酸化し、そのユビキチン化および分解を引き起こし、c-Rel、RelA/p65,およびRelBを含む二量体を放出して核に移動し、遺伝子転写を誘導する[151]。
cAMP/PKA シグナルは、ほとんどの細胞タイプと文脈で NFκB を阻害するが、すべての細胞タイプと文脈ではない [147, 152]。アルツハイマー病 に関連して、cAMP を上昇させると、TNF-αまたはリポ多糖類(LPS)で刺激されたミクログリアで NFκB を阻害することが示されている [147, 153, 154]。cAMPによる炎症性NFκBシグナル伝達の破壊は、複数のメカニズムを介して起こることが示されている[147, 155]。具体的には、cAMP/PKAリン酸化CREBは、IκBα遺伝子をアップレギュレートする[147,156,157]。 cAMPは、IκBリン酸化、ユビキチン化、および分解を減少させ、IKKβを阻害する[147,157]。リン酸化された CREB は、共通の必須結合パートナーである CREB 結合タンパク質 (CBP) のために NFκB を競合させ、遺伝子転写を CREB に向けて、NFκB 媒介の発現から遠ざける [147]。cAMPは、TNF-αなどの特定の遺伝子のプロモーターにおける活性化CREB-c-Jun二量体から抑制的CREB-ICER二量体への交換を誘導する[147]。 cAMPは、p65ホモ二量体がプロモーターに結合するのを防ぐc-Fosの発現を活性化する[147]。
cGMPはcAMPを介して間接的にNFκBを阻害する[155, 158]。血管平滑筋細胞において、NOドナーまたはCナトリウレチンペプチドを用いて有効なcGMPレベルを上げると、cGMPによるPDE3阻害、cAMP蓄積、およびcAMP/PKA依存性NFκB阻害がもたらされた[158]。
したがって、PDE阻害剤でcAMPおよびcGMPレベルを上昇させると、脳ミクログリアのNFκBを阻害し、アルツハイマー病における神経炎症およびβアミロイドーシスを減少させる可能性が高いと考えられる[34, 35, 147, 153, 154, 156-158]。
cAMP、cGMP、GSK3β
GSK3βは、タウ上の85個のリン酸化可能なセリン残基とスレオニン残基のうち45個の一次キナーゼである[159-162]。PKAがGSK3βよりも先にタウをリン酸化するとペアらせん形成が抑制され、GSK3βがPKAよりも先にタウをリン酸化するとペアらせん形成が促進されることが示唆されている[163]。さらに、GSK3βはCREBおよびPGC1αをリン酸化し、それによって阻害する[101,164-169]。
cAMP/PKAは、皮質ニューロン[170-172]、血小板[173]、マウス精子[174]、Rat1,NIH 3T3およびHEK293細胞[175]を含むほとんどの細胞型において、Ser9でリン酸化することによりGSK3β活性を抑制するが、マウスメラノーマ細胞[176]では抑制しないことを示唆している。複数のアルツハイマー病のげっ歯類モデル研究では、cGMPを上昇させるPDE5阻害剤シルデナフィルがGSK3βを阻害することが示されており、cGMPが脳内のGSK3βを典型的に阻害することが示唆されている[177-181]。
アルツハイマー病におけるGαs/AC/cAMP/PKAシグナル伝達の変化
しかしながら、CREB、PGC1α、Nrf2,NFκBおよびGSK3βシグナル伝達に対するcAMPおよびcGMPの有益な効果は、前臨床研究および神経病理学的証拠がAC/cAMP/PKAおよびNOS/NO/sGC/cGMP経路がアルツハイマー病患者の脳において病理学的に障害されている可能性を示唆しているため、アルツハイマー病の発症中に失われているようである[79,182-205]。前臨床モデルでは、BACE1/アミロイドβおよびタウ病理の両方がcAMPシグナル伝達を混乱させる。アミロイドβ25-35曝露は一過性にcAMPレベルを増加させ、cAMP/PKA依存性の方法で神経細胞のグルコース取り込みを抑制する[206]が、長期のアミロイドβ42曝露はcAMPレベルを減少させる[182]。アミロイドβ42の亜致死量(アミロイドβ25-35ではない)は、KCl-およびN-メチル-D-アスパラギン酸誘導性CREBリン酸化を阻害する [193]。BACE1の過剰発現はアミロイドβとは無関係にcAMPレベル、PKA活性、CREBリン酸化を減少させる [199]。ヒトのタウ過剰発現は、PKAを阻害し、CREBのリン酸化を減少させ、グルタミン酸受容体1[200]のリン酸化を減少させ(GluA1,リン酸化はその膜局在性を高め、導電性、長期増強、記憶形成を促進する[207])トロポミオシン受容体キナーゼB[200]のリン酸化を減少させ(TrkB、BDNFの受容体[208])BDNFのmRNAとタンパク質レベルをダウンレギュレーションする[200]。したがって、アミロイドβとタウの両方の病理学的研究は、cAMP/PKAシグナル伝達を障害しているように見える。
神経病理学的研究では、AC/cAMP/PKAシグナル伝達がアルツハイマー病患者の脳で障害されているように見える。そもそも、ACタンパク質および活性レベル(特にGαs刺激)は、一般的にアルツハイマー病患者の脳において減少することが明らかにされている[183-187,201-205]。基底、Gαs刺激、およびフォルスコリン刺激AC活性は、アルツハイマー病海馬で減少することが発見されている[201-203]。しかし、Gαs刺激ではなく、基底またはフォルスコリン刺激AC活性は、1つの研究でアルツハイマー病海馬、側頭皮質、および角回で減少することが判明した[204]。AC-Iタンパク質レベルが有意にアルツハイマー病海馬で減少した[205]。頭頂皮質膜AC-IおよびAC-IIタンパク質レベルは、Ca2 + /CaM刺激AC活性[183]と同様に、アルツハイマー病患者で有意に減少した。基本的なAC活性は、アルツハイマー病前頭前野、側頭皮質、および角回で減少することが判明した[184]。Gαs刺激ではなく、基底またはフォルスコリン刺激AC活性は、アルツハイマー病上側頭皮質[185]で減少した。Gαsを刺激したが、基底またはフォルスコリンを刺激しないAC活性は、アルツハイマー病大脳新皮質[186]で減少した。3H]フォルスコリン結合は、[187]に結合するために少ないAC触媒サブユニットを示唆し、アルツハイマー病前頭皮質で減少した。これらの知見は全体的にAC活性の低下がアルツハイマー病脳で発生する可能性があることを示唆している。
cAMPレベルは2つの研究[197,198]でアルツハイマー病患者の脳脊髄液(脳脊髄液)では変化しなかったが、タウ病理学[209]と相関する方法で1つの研究で上昇した。cAMPレベルの上昇は、アルツハイマー病の微小血管[189]、および血管アミロイドβ[188]と関連して前頭皮質と側頭皮質および海馬の大脳皮質および髄膜血管で認められた。これは、cAMPのレベルと局在がアルツハイマー病脳で微妙に調節障害される可能性があることを示唆している。
cAMPの下流では、PKA活性はほとんどの研究でアルツハイマー病において抑制されているようである[185, 190-192, 194, 200, 210, 211]。3H]cAMPの細胞質性ではなく粒子状のPKAへの結合は、アルツハイマー病患者の内耳皮質と小胞体で減少した[190]。可溶性PKA活性も粒子状PKA活性も、ある研究ではアルツハイマー病患者の上側頭皮質では変化していなかった[185]が、アルツハイマー病患者の大脳微小血管ではPKA活性は変化していなかった[210]。しかし、別の研究では、減少したPKA活性はアルツハイマー病患者の側頭皮質で認められた[192]。PKA活性の低下は、アルツハイマー病患者の内側側頭皮質でも観察された[191]。調節性51 kDa PKA-RIIαおよびPKA-RIIβおよび触媒性PKA-Cβタンパク質のレベルは、アルツハイマー病患者の内側側頭皮質で減少した[191]。触媒的PKA-Cα蛋白質レベルはアルツハイマー病患者の前頭前野で減少した[194]。しかし、PKA-RIIαのカルパインが切断された47 kDaフラグメントは、アルツハイマー病患者の内側側頭皮質で増加した[191]。培養海馬ニューロンでは、アミロイドβ曝露はPKAの阻害、PKA-RIIαタンパク質レベルの上昇、およびグルタミン酸誘発性CREBリン酸化の減少をもたらした(後者はcAMP上昇型PDE4阻害剤ロリプラムによって逆転した)[211]。ヒトのタウの過剰発現は核内のPKA-RIIαタンパク質を増加させることが判明しており[200]、Ca2+で活性化されたカルパインプロテアーゼ、アミロイドβ、タウの協調的な作用が核内での47kDaのPKA-RIIαフラグメントの発現を促進する可能性を示唆している。PKA-RIIαの発現をサイレンシングすると、PKA活性およびCREBリン酸化のタウ誘発性欠損が改善された [200] ことから、アルツハイマー病におけるこの核内47kDa PKA-RIIαのアップレギュレーションが核内PKA活性およびCREBリン酸化を抑制する可能性が示唆された。したがって、全体的なPKA活性および核内PKA活性は、アルツハイマー病において抑制されているようである[185, 190-192, 194, 200, 210, 211]。
アルツハイマー病におけるGC/cGMP/PKGシグナル伝達の変化
前臨床および神経病理学的データは、NOS/NO/sGC/cGMPシグナル伝達がアルツハイマー病においても障害される可能性があることを示唆している。アミロイドβは海馬スライスにおける長期増強に必要なNOS/NO/sGC/cGMP/CREB経路を阻害する[79]。NOS活性は、アルツハイマー病上前頭回と海馬[196]で減少することが判明している。NO活性化sGCの活性は、基底sGCや微粒子GC(pGC)ではなく、アルツハイマー病上側頭皮質で減少することが判明した[195]。
維持されたpGCシグナル伝達[195]の上流では、BNPレベルの上昇は、75-78歳の高齢者患者の認知障害と関連しているが、それ以上ではない[212,213]、および複数の研究では、血中または血漿中のBNPレベルの上昇と軽度認知障害(MCI)の発生、重症度、またはリスク、MCIのアルツハイマー病への転換、または認知症[213-223]との間の関連を発見している。例えば、ある研究では、血漿中BNPレベルが脳脊髄液 アミロイドβ42レベルと同様にMCIまたはアルツハイマー病の診断と関連していることを発見した;さらに、血漿中BNPレベルはMCIおよびアルツハイマー病患者で増加していることが発見された[213, 214]。このことは、BNP/pGC/cGMPシグナル伝達がMCIおよびアルツハイマー病において維持または増加している可能性を示唆している[195, 212-214]。
全体的に、cGMPレベルは、アルツハイマー病患者の脳脊髄液 [197,198]において減少し、これは、アミロイドβ42の脳脊髄液レベル[197]、うつ病症状[198]、およびMini-Mental Status Exam(MMSE)[197,198]によって測定された認知機能障害と相関していることが判明した。このことは、アルツハイマー病はNOS/NO/sGC/cGMPシグナル伝達の深遠で疾患の重症度と相関する減少によって特徴づけられることを示唆している。
アルツハイマー病を治療するためのcAMPおよびcGMPシグナル伝達の増加
このように、PDE阻害剤は、破壊されたcAMP/PKA、cGMP/PKG、CREB、PGC1α、Nrf2シグナルの回復、SIRT1の活性化、NFκB誘導BACE1発現抑制、アミロイドβ生成抑制、炎症抑制、GSK3β媒介タウリン酸化抑制、ニューロン生存促進などの効果が期待される。シナプス形成、ミトコンドリアの生合成とマイトファジーの生存、ATP 生成、抗酸化・解毒酵素の発現、記憶の形成-これらはすべて、アルツハイマー病 の発症を予防、停止、逆転させるための戦いにおいて重要な治療上の利点となるであろう。このナラティブレビューでは、PDE阻害薬がアルツハイマー病、MCI、または認知症を予防または治療することができるかどうかを調べるために、有効性を検証した臨床試験、疫学研究、およびメタアナリシスを概説する。
PDE阻害薬の臨床試験と疫学
ビンポセチン
ビンポセチンはPDE1を阻害し、cAMPとcGMPを上昇させる[224]。C. SzántayとC. Lörinczらによって1970年代に独自に発見された、ビンポセチンは、30年以上にわたって認知症や脳卒中の治療のためのヨーロッパ諸国で承認されており、それは栄養補助食品として米国で利用可能である[225]。前臨床データは、ビンポセチンがげっ歯類のADモデルにおける認知障害を救済することを示している[226]、CREBリン酸化[227]およびBDNF発現を増加させること[228]、BACE1をダウンレギュレートすること[226]、Nrf2 mRNA発現をアップレギュレートすること[229]、酸化ストレスを減少させること[226,228-239]、ミトコンドリア機能障害[240,241]、およびアポトーシスを減少させること[230,239]、GSK3βを阻害すること[226]を示している。228]、NFκB [229,231,232,239,242-250]、およびNLPR3 inflammasome [243]は、プロ炎症性サイトカインIL-1β [226,229,231,232,236,237,243]のレベルを低下させる。IL-6 [236, 237, 239, 251]、およびTNF-α [226, 229, 231, 232, 242, 243, 246]と、サイクリンD1のダウンレギュレーションとp27(Kip1)のアップレギュレーションによってG1期の細胞周期を停止させる[252]。
これらの有望な前臨床所見にもかかわらず、特にアルツハイマー病の治療のためのビンポセチンについての証拠は非常に少なく、そこにあるものはほとんどが失望されている。1989年の1年間の非盲検パイロット試験では、15人のアルツハイマー病患者は、30,45,または60mgのビンポセチンを1日1回投与してもCGI上での改善や低下の遅延を示さなかった[253]。多施設、二重盲検、プラセボ対照試験では、サブグループ分析により、アルツハイマー病患者におけるCGI、Short Cognitive Performance Test/Syndrom-Kurztest(SKT)Brief Cognitive Rating Scale、またはSandoz Clinical Assessment Geriatric(SCAG)に有意な治療効果は観察されなかったことが示された [254]。これらは、この記事を書いている時点で見つけることができたアルツハイマー病におけるビンポセチンの唯一の臨床試験であり、両方ともビンポセチンはアルツハイマー病には効果がないことを示しているようである。対照的に、全原因性認知症の治療のためのビンポセチンの証拠は決定的ではないが、比較的有望である。
2001年に実施された、包含基準を満たした39件の研究のうち3件のメタアナリシスでは、他の障害による認知症または認知機能障害を有する327人の患者にビンポセチンまたはプラセボが投与された[255]。ビンポセチン投与群では、Mini Mental Status Questionnaire(MMSQ)SCAG、CGIのスコアが有意に高かった。しかし、著者らは、ビンポセチンはアルツハイマー病の治療には適応がないと指摘しているが、これはこの適応で利用できるデータが少なく、一貫性がないためである[255]。
2003年のコクラン系統的レビューと、583名の認知症患者を対象としたビンポセチンの3つの利用可能な不確定二重盲検無作為化プラセボ対照臨床試験のメタアナリシスでは、30mgと60mgのビンポセチンは、13ヵ月までのCGIとSKTの注意力・記憶力尺度を含めて、有意な効果を示すことが示された。しかし、データが決定的ではないため、著者らはビンポセチンは認知症治療には適応がないと結論づけている[256]。
2012年の臨床試験では、ビンポセチンを18ヵ月間服用した中等度の重症MCI患者で、MMSEおよびアルツハイマー病評価尺度-認知サブスケール(ADAS-Cog)での認知、日常生活活動、ハミルトンうつ病尺度での気分、患者の変化のグローバル印象および臨床的変化のグローバル印象での患者および治験責任医師による病状の全体的な変化に有意な改善が認められた[257]。
2014年のパイロット研究において、ビンポセチン5mgを1日2回3ヶ月間投与したところ、プラセボと比較して、様々な病因の認知症またはてんかんのいずれかによる認知機能障害を有する56人の患者において、記憶力と集中力が有意に改善された[258]。
したがって、アルツハイマー病に対するビンポセチンに関するエビデンスは非常に限られているが、利用可能な2つの試験では、ビンポセチンはこの適応症には有効ではないことが示されているようである。これらの適応症に対する有効性をさらに評価するためには、より大きな患者コホートとのさらなる臨床試験が必要であるが、しかし、ビンポセチンは、MCIと認知症患者のための有望性を保持するように見える。
ニセルゴリン
エルゴット誘導体ニセルゴリンは、Ca2+/カルモジュリン依存性PDE1およびcGMP刺激PDE2活性を阻害する[259]が、心臓からの高Km cAMP分解PDEを活性化する[260]。また、Ca2+/Mg2+依存性の脳アデノシン三リン酸酵素(ATPase)を非競合的に阻害する。また、低濃度ではNa+/K+ ATPaseを活性化するが、高濃度では阻害するという脳内Na+/K+ ATPaseに対する複合的な効果を発揮する[260, 261]。また、強力なα1Aアドレナリン受容体アンタゴニストとしても作用する[262]。ニセルゴリンは、1970年代から50カ国以上で利用可能であり、アルツハイマー病、血管性痴呆、急性および慢性末梢循環障害、および脳梗塞を含む様々な条件の治療に使用されてきた[263,264]。
前臨床研究では、ニセルゴリンは、ADマウス[265]、BDNFをアップレギュレート[266]、年齢に関連したコリン作動性欠損を逆転させ、K+誘導アセチルコリン放出[267]を強化し、認知障害を回復することが示されている。[268]、カタラーゼの活性化[269]、酸化ストレス[265,269-271]、炎症[265,266]、アポトーシス[265,266,272]の減少、およびIL-1β、IL-6,およびTNF-αのレベルの低下[266]。
2001年のコクラン系統的レビューでは、14の不確定、二重盲検、無作為化、プラセボ対照試験から構成されており、軽度から中等度の認知症患者に対して、ニセルゴリンは6ヵ月後と12ヵ月後のSCAG、MMSE、CGIに有意な改善をもたらすことが示されている[273]。このメタアナリシスによると、合計342人の患者において、ADAS-Cogの6ヵ月後と12ヵ月後における有意な改善は認められなかったが、この尺度では有意ではない傾向が認められた[263, 273]。ADAS-Cogはもっぱらアルツハイマー病患者に使用されているので、これは重要である。ADAS-Cog上の違いが6ヶ月で有意ではなかったという主張は、もっぱらCrook et al 1996年、PharmaciaとUpjohnからの未発表報告[273]に基づいている。ニセルゴリンによる ADAS-Cog の変化が 12 ヶ月で有意ではなかったという主張は 2001 年の多施設、無作為化、二重盲検、プラセボ対照試験に基づいているが、この試験では、ニセルゴリン 30 mg を 1 日 2 回投与すると、6 ヶ月で ADAS-Cog 上の認知が有意に改善され、12 ヶ月を含めて、治療を受けた患者と対照患者の間の ADAS-Cog スコアの平均差がますます大きくなった [274]。Amaducci et al 1999年による一次ソースポスター要旨によると:”12ヶ月間治療縮小患者コホートでは、3,6,9,12ヶ月後の治療の間のADAS-Cogスコアの差はそれぞれ0.89,1.07,1.45と1.64であった、ますますニセルゴリンを支持して[274] “。ニセルゴリンを投与されたアルツハイマー病患者は、最初の6ヶ月の間に改善し、その後12ヶ月目までに悪化したことが指摘された[274]。しかし、ニセルゴリン投与されたアルツハイマー病患者のADAS-Cogの悪化は、プラセボ投与された患者よりも重度ではなかったが、12ヶ月目まで、そして12ヶ月目までを含めて、ますます明らかになってきた利点である [274]。言い換えれば、それはニセルゴリンは、6ヶ月でADAS-Cog上のアルツハイマー病症状の中程度の、しかし有意な改善をもたらし、12ヶ月では有意な改善をもたらさなかったが、むしろ12ヶ月でますます急激な減少を遅らせたことが表示される[274]。なぜこの研究が12ヶ月分析のために含まれていたが、コクラン・レビューの6ヶ月分析のために含まれていなかったのかは不明であり、また、これが6ヶ月でのADAS-Cog上の改善の傾向が有意ではなかったというコクラン・メタアナリシスの結論を変更するかどうかは明らかではない。6ヵ月後のADAS-Cogの有意な改善の所見は、コクラン・レビューの論文が2001年の雑誌に掲載された後に正式に発表された [275]。この正式な論文では、12ヵ月後のADAS-Cogの低下が遅くなる傾向については言及されていなかった [274, 275]。
しかし、さらに最近の2つの試験では、より小規模な患者群を研究し、否定的な結果が得られた。2017年の非対照パイロット試験では、初期アルツハイマー病患者16人に30mgのニセルゴリンを1日2回、平均して1.5年間投与した [276]。ベースラインと追跡調査の間に、認知症の重症度、日常生活活動、認知、抑うつ症状に統計学的に有意な差は認められなかった。しかし、ニセルゴリンは前頭葉と頭頂部の脳血流を増加させた[276]。初期アルツハイマー病患者22人を対象とした2019年の研究では、アセチルコリンエステラーゼ阻害薬単独と比較して、アセチルコリンエステラーゼ阻害薬+ニセルゴリンは左側頭極と中帯状回への脳血流を温存したが、認知症の重症度に有意差は認められなかった[277]。
ニセルゴリンは 2014年のシステマティックレビューおよびメタアナリシスにより、特に他のエルゴット誘導体と比較して、プラセボよりも多くの有害事象が報告されておらず、良好な安全性プロファイルを有していることが明らかにされた[278]。
したがって、ニセルゴリンは認知症患者に適応があると思われる[273]。ニセルゴリンが提供した6ヵ月後のADAS-Cogの改善が有意であったかどうかについては、様々な報告があり、12ヵ月後にはこの尺度での認知機能の低下が緩やかになるという有意でない傾向が見られた[273-275]。しかし、さらに最近の2つのパイロット試験では、期待はずれの結果が得られた [276, 277]。このことは、ニセルゴリンがアルツハイマー病の治療に有用であるかどうかを決定するものではない。より多くの患者を巻き込んだアルツハイマー病におけるニセルゴリンの更なる臨床試験が必要とされているようである。
デプレニル/セレギリン
デプレニルは、セレギリンとしても知られ、カルモジュリン依存性PDE1A2およびモノアミン酸化酵素(MAO)を阻害する [224, 279]。1960年代にハンガリーの製薬会社Chinoin社のZoltan Ecseriによって発見されたデプレニルは、パーキンソン病、大うつ病性障害、注意欠陥/多動性障害の治療に臨床的に使用されている[280-282]。前臨床研究では、デプレニルはCREBリン酸化を増加させ[283]、Nrf2を活性化させ[284-286]、カタラーゼを活性化させ[283]、酸化ストレスを減少させ[284-287]、炎症を減少させ[286]、ミトコンドリア機能障害を減少させ[287]、アポトーシスを減少させ[287]、紫外線誘発性PARP1活性化を増強させるが、PARP1タンパク質発現をダウンレギュレートさせることが明らかにされている[288]。
2003年に行われた最新のコクラン系統的レビューとメタアナリシスでは、17の不確定二重盲検無作為化プラセボ対照臨床試験が含まれており、セレギリンはアルツハイマー病患者の記憶力と認知力を4-6週と8-17週で有意に改善したが、21-30,35-56,65-82週では改善せず、4-6週では日常生活活動を改善したが、それ以降の時点では改善しなかったことが明らかにされた[289]。これら、17件のランダム化比較試験(RCT)では、6件のRCTでMMSE、2件のRCTでRandt Memory IndexとWechsler Memory Scaleを、1件のRCTでBuschke Selective Reminding Test、散文想起、Rey-AVL、ADAS-Cogを含む複数の尺度で記憶力の有意な改善が認められた。副作用は報告されたが、患者が試験から離脱しなければならないほど重篤なものはほとんどなく、セレギリンとプラセボで報告された有害事象の数と試験離脱の間には有意差はなかった。しかし、グローバル評価尺度(すなわち、Blessed Dementia Scale、Gottfries-Bråne-Steen尺度(GBS)Global Deterioration Scale、CGI)のメタ解析では、セレギリンとプラセボの間に有意差は認められなかった。バークスとフリッカーは、結論は、”アルツハイマー病のためのselegilineは失望を証明している。.そこには[]アルツハイマー病の患者のための臨床的に意味のある利益の証拠はない。このことは、評価されたアウトカム指標、すなわち認知、感情状態、日常生活活動、グローバルな評価にかかわらず、短期的なものであれ、長期的なものであれ(69週まで)評価されたものであれ[289]、同様である。それにもかかわらず、著者自身の結果は、これらの改善の短命の性質は失望しているように見えるが、これらの改善の短命の性質が表示されるが、アルツハイマー病患者のセレギリン治療と様々なスケールでの記憶、認知、日常生活活動の中で控えめで短期的に統計的に有意な改善を指摘しているように見える。
シロスタゾール
PDE3はcAMPとcGMPを分解し、主に小胞体膜に局在している[290, 291]。CilostazolはPDE3阻害剤であり、抗血小板剤として使用されている。1980年代後半に大塚製薬株式会社によって発明され、末梢血管疾患の間欠性跛行の治療および脳卒中の予防に臨床的に使用されている[292, 293]。Cilostazolは、前臨床試験において、脳アミロイド血管症マウスモデルにおける認知機能障害の救済と可溶性アミロイドβ脳流出の促進、酸化ストレスとアポトーシスの減少、アミロイドβのオリゴマー化と沈着の抑制[294, 295]、GSK3βの阻害[168, 173]、CREBの活性化[168, 295-299]、cAMP/PKA/CREB/PGC1αシグナルを介したミトコンドリア生合成の誘導[298-301]が示されている。シロスタゾールは、Nrf2と標的遺伝子の発現を誘導し [298, 302-305]、酸化ストレスを減少させ [306]、SIRT1タンパク質レベルを上昇させ [307]、アデノシン一リン酸活性化プロテインキナーゼを活性化させ [308, 309]、NFκBを阻害し [306-308, 310-312]、BACE1を低下させ [307]、IL-1β [305]、IL-6 [168, 305]、およびTNF-α [298, 305]のレベルを低下させるとされている。
疫学的には、シロスタゾールの服用は、65歳以上の患者における全原因性認知症の発症リスクの減少と用量依存的に関連しており、全体的な修正ハザード比は0.75(シロスタゾールを服用している65歳以上の患者は、シロスタゾールを服用していない患者に比べて認知症になる可能性が25%低いことを意味する)であり、高用量シロスタゾール服用者の修正ハザード比は0.53(高用量シロスタゾール服用者は認知症になる可能性が47%低いことを意味する)[313]であった。サブグループ解析の結果、虚血性心疾患または脳血管疾患を有するシロスタゾール使用者は認知症から有意に保護されていた(それぞれ修正ハザード比0.44および0.34)[313]。
あるレトロスペクティブ研究では、対照群では年間のMMSEスコアが約2ポイント悪化していたのに対し、シロスタゾール投与群では年間のMMSEスコアに変化がなく、有意差はなかった[314]。サブグループ解析の結果、対照MCI患者では年間4ポイントのMMSEスコアの低下がみられたのに対し、シロスタゾール治療を受けたMCI患者では年間約0.2ポイントのMMSEスコアの上昇がみられたが、これは有意差であった[314]。健康な対照群と認知症患者のMMSEスコアの差は有意ではなかったが、対照群の認知症患者では年間のMMSEスコアが∼1.1ポイント減少したのに対し、シロスタゾール治療を受けた認知症患者では年間のMMSEスコアが∼0.4ポイント増加したことは特筆に値する[314]。言い換えれば、シロスタゾール治療を受けた認知症患者は、シロスタゾール治療を受けたMCI患者よりもMMSEでわずかに改善しているように見えた(この傾向は有意ではなかったが)が、認知症患者におけるシロスタゾールの効果は、対照の認知症患者の方が対照のMCI患者よりも急激な悪化が少なかったために、単に有意ではなかったと考えられる[314]。
別のレトロスペクティブ解析では、ドネペジル単独投与とドネペジル+シロスタゾール投与を受けた軽度認知症患者を比較したところ、シロスタゾール投与群ではMMSEで測定した認知機能低下の速度が有意に遅かった[315]。
アセチルコリンエステラーゼ阻害剤を服用している安定したアルツハイマー病患者60名を対象とした12ヶ月間の症例対照試験では、シロスタゾールの追加使用はMMSEで測定される認知機能低下率の低下と有意に関連していた[316]。
アルツハイマー病患者10人を対象とした非盲検非対照パイロット試験では、6ヵ月後にMMSEスコアの改善が認められた [69, 317]。アルツハイマー病と脳血管疾患を有する患者20人を対象とした無作為化比較試験では、アスピリンやクロピドグレルと比較して、シロスタゾール100mg/日を服用することで、6ヵ月後の日本語ADAS-Cog、トレイルメイキングテストA、および改訂ウェクスラー記憶尺度(論理的記憶I)のスコアの低下を防ぐことが明らかになった。対照群のみ左中側頭回の脳血流が減少していたのに対し、シロスタゾール投与群では右前帯状葉の血流が有意に増加していた[318]。
軽度または中等度のアルツハイマー病と白質病変を有する36名の患者を対象とした6ヶ月間の無作為化二重盲検対照試験では、シロスタゾール100mgとドネペジルを併用した場合、ドネペジル単独投与と比較して頭頂葉と前頭葉のグルコース代謝の低下が抑制され、左下前頭前野のグルコース代謝が維持された[319]。しかし、MMSE、ADAS-Cog、または他の転帰指標では有意差は認められなかった。しかし、シロスタゾールによるグルコース代謝の改善は、ADAS-Cogのスコアの改善と相関していた[319]。シロスタゾールのさらなる試験が進行中である[69, 320]。
したがって、シロスタゾールの使用は認知症のリスク低下と関連している可能性がある[313]。MCI患者[314]、軽度認知症患者[315]、アルツハイマー病患者[69, 316-318]のMMSEにおける認知機能低下の速度を遅らせる可能性がある。しかし、アルツハイマー病患者30人を対象とした2つのパイロット試験(1つは対照試験、もう1つは対照試験ではない)では有効性が認められたが、白質病変を有する36人のアルツハイマー病患者を対象とした厳格な臨床試験では有効性が認められなかったため、アルツハイマー病におけるシロスタゾールに関する試験データは非常に限られており、全体的に残念な結果となっている[69, 317-319]。これらの知見に基づき、シロスタゾールの将来の臨床試験は、MCIまたは早期アルツハイマー病患者、特に併存する脳血管疾患を有する患者に焦点を当てるべきである。
デンブフィリン (denbufylline)
はPDE4を阻害し、cAMPを上昇させる[321]。不明確な理由で、についての前臨床試験のエビデンスが不足しているが、以下の前臨床試験のエビデンスとアルツハイマー病治療のためのPDE阻害剤に関する前臨床試験の最近のレビューによると、PDE4阻害は、認知機能の強化とアルツハイマー病治療の目的のための最も有望なPDE治療ターゲットの1つであることは間違いない[73]。例えば、PDE4阻害剤ロリプラムによる治療は、APP/PS1トランスジェニックADマウスにおいて、基底シナプス伝達、長期増強、作業記憶、参照記憶、連想記憶の長期的な改善をもたらした[322]。CA1海馬のアミロイドβ40に曝露されたラットでは,0.5mg/kgのロリプラムが記憶障害とCREBのリン酸化を回復させた[323]。CA1海馬にアミロイドβ25-35をマイクロインフューズしたラットでは,0.1,0.25および0.5mg/kg/日の腹腔内投与で記憶障害とCREBリン酸化が回復した[323]。 5 mg/kg/日の腹腔内PDE4阻害剤ロリプラムの用量依存的に記憶障害を反転させ[324,325]、CREBリン酸化とBcl2発現を回復させ、p65 NFκBとBax発現を減少させ[324]、活性酸素とマロンジアルデヒドレベルを減少させ、グルタチオンレベルとSOD活性を回復させ、チオレドキシンをアップレギュレートし、海馬における誘導性のiNOS経路を阻害した[325]。凝集した アミロイドβ42 を歯状回に注射したマウスでは、長型 PDE4D のレンチウイルス RNA サイレンシングにより、アミロイドβ42 誘導性の cAMP の低下と、モリス水迷路と新規物体認識テストでの記憶障害が改善された。APPswe/PS1dE9マウスにおいて、PDE4Dアイソフォーム特異的阻害剤GEBR-7bを0.001mg/kgを生後5ヶ月で3週間投与したところ、CREBのリン酸化、BDNFの発現、シナプス密度、アミロイドβレベル、タウのリン酸化、またはGSK3β活性化に影響を与えることなく、生後7ヶ月での物体位置試験における空間記憶が改善された[326]。これらの前臨床試験済みのPDE4阻害薬はいずれも臨床試験を実施していないが、これはおそらく嘔吐の副作用が臨床開発の妨げになっているためであろう [69]。しかしながら、これらのPDE4阻害薬の前臨床効果は、の前臨床効果を合理的に代表するものであろう。
他の薬剤を服用していない軽度から中等度のアルツハイマー病患者45名を対象とした多施設共同無作為化、並行群、二重盲検、プラセボ対照臨床試験において、100mgを1日2回3ヶ月間投与したところ、プラセボと比較してSCAG、CGI、桁記号代用テスト、MMSEのスコアが有意に改善した[327]。それはまた、デルタ活動を減少させ、イメージング上の遅い活動を加速(アルツハイマー病患者で増加している)症状の改善と相関する警戒心の増加を示している[327]。嘔吐やその他の副作用は報告されていない[327]。
226人のアルツハイマー病患者と110人の血管性または混合性認知症患者を対象とした試験では、25,50,100mgのを4ヶ月間投与したところ、MMSEスコアが有意に改善したが、これらの患者を単一群とした場合にのみ改善した[328]。を投与された全原因性認知症患者では、プラセボを投与された患者よりもを投与された患者の方がMMSEスコアの改善が有意に多かった。重大な有害事象は報告されていない[328]。
したがって、アルツハイマー病におけるに関する試験データは非常に限られているが、やや有望である。このPDE4阻害薬との関連で嘔吐は報告されていない[327, 328]。アルツハイマー病におけるDenbufyllineのさらなる臨床試験が必要であろう。
シルデナフィル
PDE5Aは、NOの下流にあるsGCによって産生される細胞質cGMPを特異的に分解する[71]。シルデナフィル(バイアグラとしても知られる)はPDE5を阻害し、それによって細胞質のcGMPレベルを上昇させる。1980年代後半にファイザーによって発見されたシルデナフィルは、勃起不全および肺動脈性高血圧症の治療に臨床的に使用されている[329, 330]。アルツハイマー病の前臨床齧歯類モデルにおいて、シルデナフィルは記憶力を回復させ[177-180, 331-335]、アミロイドβレベルを低下させ[178, 179, 333-336]、タウ高リン酸化[177, 178, 180, 331]、GSK3β[177-180]およびJNK[331]を阻害し、IL-1βを低下させることが示されている。IL-6,およびTNF-α分泌[335]、pSer133-CREB[180,333,335]、BDNF[180,336]、Arc[180]、およびBcl2[336]をアップレギュレートし、BACE1[179]、アミロイドβPP、カスパーゼ-3,およびBax[336]をダウンレギュレートし、二本鎖DNA切断およびアポトーシス細胞を減少させる[336]。アルツハイマー病に対するシルデナフィルの最近のレビューについては、Sanders 2020 [337]を参照のこと。
シルデナフィル50mgの単回投与は、アルツハイマー病患者の小集団において、右海馬の自発神経活動を低下させ、脳血管反応性を低下させ、脳血流および脳内酸素代謝率を上昇させることが示されている[338,339]。この非常に限られた予備的証拠に基づいて、アルツハイマー病患者におけるシルデナフィルの臨床試験が必要である。
シルデナフィルの用量制限効果は、シルデナフィルの高用量投与により、cGMP感受性PDE2が活性化されてcAMPが分解されるほどcGMPレベルが上昇することである[72]。この作用は、代謝状態でのシルデナフィルの有効投与量を制限することが示されている[72]。したがって、将来の臨床試験では、シルデナフィルとプロペントフィリンなどのcGMP刺激型PDE2阻害薬との併用を試験すべきである[72,340]。
PF-04447943およびBI 409,306号
多施設共同、二重盲検、無作為化、プラセボ対照の第Ⅱ相試験において、ファイザー社のPDE9A阻害剤PF-04447943は、ADAS-Cog、Neuropsychiatric Inventory、またはCGI-Improvementスケールのスコアをプラセボよりも有意に改善することができなかった[341]。
2つの多施設、二重盲検、並行群、無作為化、プラセボ対照第II相試験において、ベーリンガーインゲルハイムが開発したPDE9阻害薬BI 409306を服用したMCIおよび軽度から中等度のアルツハイマー病患者は、12週間後の神経心理テスト電池の合計zスコア、ADAS-Cog11,Clinical Dementia Rating scale-Sum of Boxes、またはAlzheimer’s Disease Cooperative Study-Activities of Daily Livingスケールにおいて、有意な差を示さなかった[342]。
アルツハイマー病の前臨床モデルにおいて、PDE9阻害はシナプス可塑性および記憶を救済し、樹状突起棘密度変性および細胞毒性を減少させることが示されている[213, 341, 343, 344]。しかしながら、PDE9阻害薬PF-04447943およびBI 409306は、MCIおよびアルツハイマー病患者において効果がないことが示されている[341, 342]。これは、PDE9Aがナトリウム利尿ペプチドによって誘導されたpGCによって生成された細胞質周囲膜のcGMPを調節しているのであって、NO/sGCによって生成された細胞質のcGMPを調節しているのではないためであると考えられる[71]。議論されているように、NOS/NO/sGC経路および細胞質膜cGMPプールはアルツハイマー病において障害される[195,196]一方で、NP/pGC/cGMPシグナル伝達および細胞質膜周囲cGMPは維持または増加される[195,212-214]ので、PDE9阻害剤による細胞質膜周囲cGMPの増加は、PDE5阻害剤による細胞質膜cGMPプールの補充よりも実行可能性の低い治療アプローチになるかもしれない[213]。
もう一つの考慮点は、サイクリックヌクレオチドゲート(CNG)チャネルを活性化する細胞質周囲cGMPを産生するのは、NO活性化sGCではなく、NP活性化pGCであるということである[68]。したがって、PDE9の阻害は、PDE5の阻害とは異なり、CNG活性化を促進すると予想される(図2)[68,71]。CNGチャネルは、嗅覚ニューロンの細胞質へのCa2+イオンの流入を可能にする非特異的なカチオンチャネルである[345]。軸索Ca2+の過剰な流入はワレリア軸索変性を誘導し、細胞質Ca2+の過剰な流入は興奮毒性を誘導する[346-349]。嗅覚ニューロンは、燃焼由来のカーボンナノ粒子および微生物毒素を含む複数の散発性アルツハイマー病危険因子に曝される [350-353]。アルツハイマー病の病原因子は、エクソソームなどの細胞外小胞の中でニューロン間でトランスシナプス的に拡散し[354-358]、タウの病理はアルツハイマー病では脳内でステレオタイプな形で拡散する[2]。嗅覚ニューロンの軸索は嗅球[359]に投影されており、嗅球はアルツハイマー病の前臨床段階から重大な影響を受ける脳領域である[350, 351]。したがって、PDE9A阻害は、嗅覚ニューロンへのpGC/cGMP活性化CNG媒介のCa2+流入、軸索の変性、興奮毒性、およびおそらく嗅球へのアルツハイマー病病態の伝播を促進し、PDE9阻害薬を服用しているアルツハイマー病患者では、PDE9A阻害薬のプラスの効果が相殺され、正味の変化は得られない可能性がある。
図2 PDE5とPDE9のcGMPに対する効果
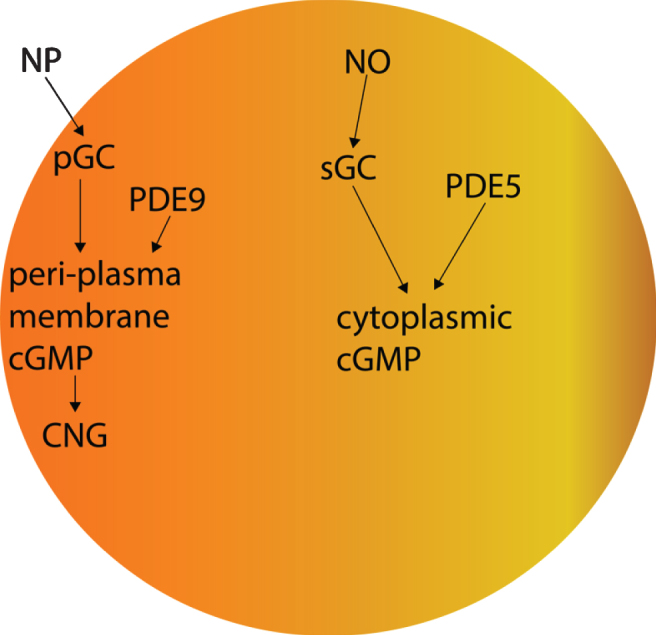
さらに、CNGサブユニットCNGA1は、アルツハイマー病患者の側頭皮質ではmRNAレベルで有意にアップレギュレートされており(図3)[360]、cGMP感受性CNGA3サブユニットはアルツハイマー病患者の海馬で有意にアップレギュレートされている(図4)[345,360]ので、CNGチャネルの発現がアルツハイマー病脳嗅覚ニューロンに限定されているかどうかは不明である。CNG mRNAは、非アルツハイマー病患者の側頭皮質ではほとんど発現していなかったが、発現していた場合には、典型的にはニューロンによって発現していた(図5-10)[360,361]。このことから、PDE9阻害剤は、散発性アルツハイマー病患者の側頭皮質と海馬において、pGC/cGMP/CNG/Ca2+を介した軸索変性と興奮毒性を促進する可能性を示唆している。
図3 アルツハイマー病内耳皮質でアップレギュレートされたCNGA1
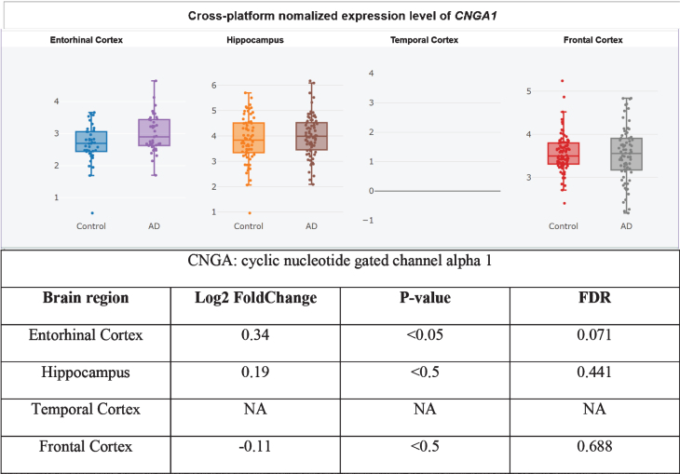
図4 アルツハイマー病海馬でアップレギュレートされたCNGA3
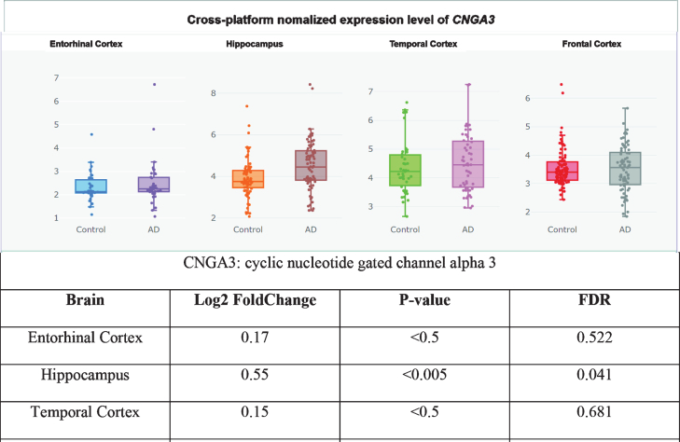
図5 非アルツハイマー病側頭皮質におけるCNGA1の細胞型特異的発現
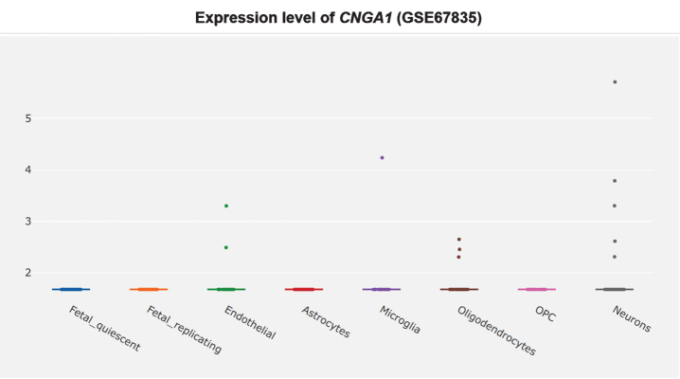
図10 非アルツハイマー病側頭皮質におけるCNGB3の細胞型特異的発現
図6 非アルツハイマー病側頭皮質におけるCNGA2の細胞型特異的発現
図7 非アルツハイマー病側頭皮質におけるCNGA3の細胞型特異的発現
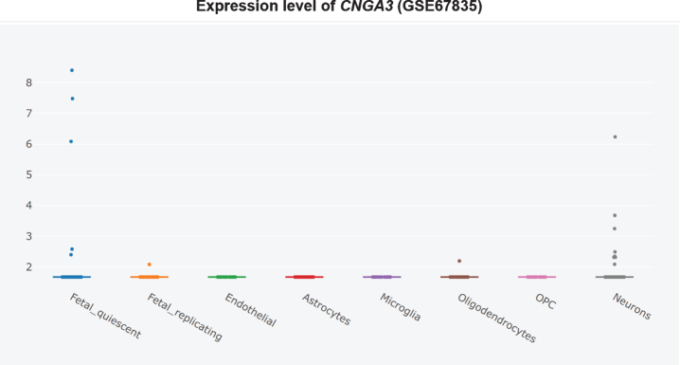
図8 非アルツハイマー病側頭皮質におけるCNGA4の細胞型特異的発現
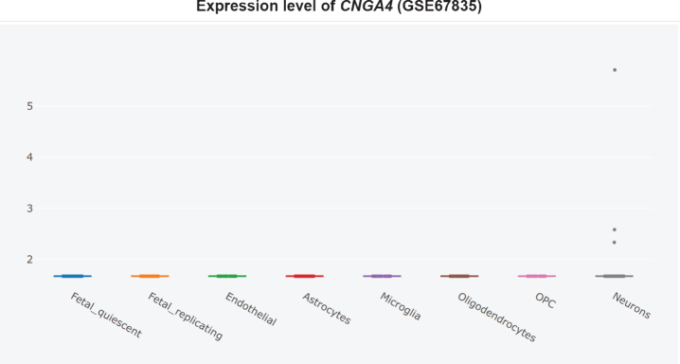
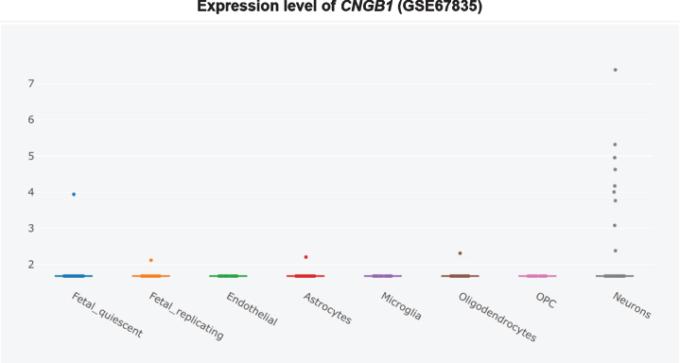
図9 非アルツハイマー病側頭皮質におけるCNGB1の細胞型特異的発現
したがって、PDE9阻害剤は、臨床試験では正味の効果がないに等しいほど多くの害をもたらす可能性がある。PDE9阻害剤は、アルツハイマー病の治療のためにさらに追求すべきではない。
カフェイン
カフェインは、非特異的/広スペクトルのPDE阻害剤である。プロペントフィリンと同様に、カフェインはメチルキサンチン誘導体である[362, 363]。アルツハイマー病トランスジェニックマウスでは、カフェインは部分的に線条体ではなく前頭皮質でのPKA活性とCREBリン酸化を救済した[364]、血漿、皮質、および海馬のアミロイドβレベルを減少させた[365,366]、BACE1とPS1発現を抑制した[366]、および作業記憶を改善した[366]。
カフェインがアルツハイマー病、認知機能低下、または認知症のリスクを低下させるかどうかについてのエビデンスは混在している。20の研究に登録された31,479人を対象とした2015年のメタアナリシスでは、コーヒーまたは紅茶からのカフェイン摂取は認知障害の発生率と有意な関連はなかった[367]。2016年の研究(n=6,467)では、カフェインの摂取は、65歳以上の女性における偶発的な認知症および認知機能低下のリスクの低下と関連していた[213,364,368]。11の前向き研究に登録された29,155人の参加者を対象とした2016年のメタアナリシスでは、カフェイン摂取と認知症または認知機能低下との関連は認められなかったが、カフェインの高摂取は偶発的なアルツハイマー病のリスク低下と有意に関連していた[369]。9つの前向きコホート研究に登録された34,282人を対象とした2017年のメタアナリシスでは、1日1~2杯のコーヒー摂取が認知障害のリスク低下と関連するJ字型の関連が認められたが、3杯以上のコーヒー摂取との関連は有意ではなかった[370]。しかし、8つの前向き研究の2018年の用量反応メタアナリシスでは、カフェインの摂取量とアルツハイマー病または認知症のリスクとの間に有意な関連は認められなかった[371]。それにもかかわらず 2019年の研究では、1日2杯未満のコーヒーを飲む場合と比較して、生涯を通じて1日2杯以上のコーヒーを飲むことは、認知的に接触している高齢者411人のアミロイドβプラーク負担の減少と有意に関連していることが明らかになった[372]。したがって、カフェイン摂取は認知症、認知機能低下、および/またはアルツハイマー病のリスク低下と関連している可能性がある。アルツハイマー病におけるカフェインの臨床試験は実施されていない。
プロペントフィリン(Propentofylline)
おそらく、アルツハイマー病の治療のための最も有望なPDE阻害剤はプロペントフィリンである。プロペントフィリンは、比較的強力で非特異的なcAMP/cGMP PDE阻害剤とアデノシン再取り込み阻害剤として作用するカフェインのようなメチルキサンチン誘導体である[340,362,363]。プロペントフィリンは、前臨床モデルにおいて、アミロイドβプラーク沈着、タウ高リン酸化、GSK3β活性化[373]、ミクログリア活性酸素生成[374-378]、グルタミン酸産生、LPS誘導ミクログリアIL-1βおよびTNF-α分泌[375-377]、アミロイドβ誘導IL-1β分泌[373]、およびミクログリア増殖[374-377]を抑制することが示されている。抗炎症性調節性T細胞増殖[379]、ATP産生の回復[131, 132]、記憶課題に対する脳代謝応答[380]を促進することが示されている。照射誘発G1/S遷移ブロックを増強することができる[381]。それはまた、アルミニウム誘発脳浮腫と低酸素様代謝変化[382]とアミロイドβ誘発記憶障害[383]だけでなく、神経成長因子の撤退またはアミロイドβ[384-386]のいずれかによって誘導されたpreferednerve細胞死を逆転させている。
プロペントフィリンは、おそらくこの原稿でレビューしたPDE阻害剤のいずれかの第III相臨床試験の最も印象的な歴史を持っている。6ヶ月から 14ヶ月間の4つの第III相、無作為化、二重盲検、プラセボ対照臨床試験の1998年のメタアナリシスでは、プロペントフィリン300 mgを1日3回、食事の1時間前に服用すると、アルツハイマー病患者と血管性痴呆患者の両方で一貫して有意な改善を提供した[375]。これらの効果は治療を中止した後も持続しており、これらの認知症患者では純粋に症状の改善というよりはむしろ疾患の改善が示唆されている[375]。サブグループ分析の結果、プロペントフィリン治療を受けたアルツハイマー病患者では、6ヵ月後の時点で、GBSで全人的機能、MMSEとSKTで認知、Nürnberger Altersbeobachtungs-skala(NAB質問票)で日常生活活動が有意に改善していたことが明らかになった。最終訪問時には、治療を受けたアルツハイマー病患者は、GBS、CGI項目II(全体的な改善)SKT、MMSE、およびNABを含むテストされたすべてのカテゴリーで有意な改善を示した[375]。
認知症患者を対象とした無作為化二重盲検プラセボ対照臨床試験の2003年コクラン系統的レビューでは、プロペントフィリン治療により、3ヵ月、6ヵ月、12ヵ月目の認知力(12ヵ月目のMMSEを含む)3ヵ月目のグローバル評価、3ヵ月、6ヵ月、12ヵ月目の認知症の重症度(12ヵ月目のCGIを含む)6ヵ月、12ヵ月目の日常生活動作が有意に改善したことが明らかになった[376]。しかし、ランダム化臨床試験の追加患者1200人のデータは、医薬品の製造元であるアベンティス社から著者に共有されていなかった[376]。2008年に行われたこの系統的文献検索の更新では、認知症患者を対象としたプロペントフィリンの新しい臨床試験は得られなかった[376]。
1999年に行われた、486人の軽度から中等度のアルツハイマー病患者を対象とした18ヵ月間の多国籍、無作為化、二重盲検、プラセボ対照臨床試験では、プロペントフィリン300mgを1日3回食前1時間に服用することで、ADAS-Cogの認知パフォーマンスとClinician’s Interview-Based Impression of Change Plus Caregiver Input(CIBIC-Plus)のグローバル機能が有意に改善された [376,387-391]。18ヵ月間の臨床試験でも治療中止後6ヵ月後も効果は持続しており、疾患修飾効果をさらに示唆している [387, 389, 391]。この試験の結果は、いくつかのポスター要旨[376, 387-391]で発表された。この試験はコクランレビューに掲載されているが、生データが不十分であったため、メタアナリシスには含まれていない[376]。
しかしながら、2007年の書籍の章では、2つの第III相臨床試験が成功した後(2つ以上あった)18ヵ月間の試験が “有益性を示すことができず、開発が中止されたと主張している[392]。著者はこの主張を引用しておらず、出典が不明である。しかし 2000年に発表されたMedScapeの記事は、18ヶ月間のプロペントフィリン長期使用試験(PLUS)は、アルツハイマー病患者における治療とプラセボの間に有意差を示さなかったことを主張し、アベンティスはこの薬の開発を中止するように導いた[393]。この記事によると、1990年代後半には、欧州医薬品評価機構(European Agency for the Evaluation of Medicinal Products)は、アルツハイマー病と血管性痴呆の治療薬としてのプロペントフィリンの販売申請をアベンティス社から却下され[393]、1999年には、メーカーは「規制上の懸念」に対処するためにアルツハイマー病患者を対象とした同薬の第III相試験を発表したが、これは後に取り消された[393]。この論文には参考文献は含まれていない。2008年の本は、その出典を引用せずに先行する主張を再現した[394]。これらの書籍もMedScapeの記事も、1999年のポスター要旨で発表された18ヵ月間の第III相臨床試験の成功については言及していない。2つの18ヶ月間の臨床試験があったのか、1つは成功したもの、もう1つは成功していないものであったのか、あるいはこのMedScapeの論文とその後の2冊の書籍が誤って報告した1つの臨床試験があったのかは、筆者には不明である。安全性に関しては、これらの試験では最大18ヵ月間、プロペンは良好な忍容性を示した [376, 387, 389, 391, 395]。
現在、英国では、プロペントフィリンがイヌの認知機能障害の治療に使用されている[396]が、これはイヌがアルツハイマー病様症状を伴う加齢に伴うアミロイドβプラークを自然に発症することから、齧歯類モデルと比較して優れたアルツハイマー病の前臨床モデルとして提案されている[397]。
経済分析では、スウェーデンの軽度から中等度のアルツハイマー病患者の標準治療にプロペントフィリンを追加することで、この集団のケアに関連する費用の3.8~7.6%を節約できることが明らかになった[398]。12ヶ月間の臨床試験に基づくカナダの経済分析では、認知症患者をプロペントフィリンで治療することで、健康状態が改善され、在宅ケアと介護者のコストが削減されることが明らかになった[399]。
プロペントフィリンは、測定されたPDEアイソフォーム(すなわち、PDE1,PDE2,PDE3,およびPDE4)のすべてのためにテストされた4つのPDE阻害キサンチン誘導体(すなわち、ペントキシフィリン、プロペントフィリン、トルバフィリン、およびアルビフィリン)のうち、最も効果的な阻害剤であり、それはcGMP刺激PDE2とPDE4を阻害する際に特に効果的であると[340]。これは、いくつかの理由のための顕著な選択性プロファイルである。まず、これはプロペントフィリンは、アルツハイマー病のPDE(すなわち、PDE2とPDE4)[69,73,340]で阻害するために最も有望なの1つ以上を阻害する唯一のPDE阻害剤をここでレビューした。第二に、複数のPDEを阻害し、cAMPとcGMPの両方をブーストすることは、CREB、PGC1α、Nrf2などのシグナル伝達の相乗的な改善を提供する可能性がある[72]。第三に、PDE5阻害剤シルデナフィルの高用量投与は、cGMP感受性PDE2を活性化する副作用があり[72]、cAMPの分解をもたらし、PKA媒介のCREBおよびPGC1αシグナル伝達を阻害する可能性がある[72, 82, 93]。このことは、プロペントフィリンとシルデナフィルがPDE2,PDE4およびPDE5を同時に阻害することで、アルツハイマー病および血管性認知症患者に相乗的な疾患修飾効果をもたらす可能性を示唆している。プロペントフィリン、シルデナフィル、ドネペジル、メマンチンは、アルツハイマー病治療のための将来の前臨床試験や臨床試験において、ドネペジルとメマンチン(標準治療薬)の単独投与と比較する必要がある。
結論
以上の議論から、特定のPDE阻害薬で環状プリンヌクレオチドレベルを調節することで、アルツハイマー病、MCI、認知症を予防・治療することができると結論づけることができる(議論したPDE阻害薬の臨床的有効性の要約は表1を参照のこと)。
カフェインおよびシロスタゾールは、偶発的な認知症、認知機能低下、およびアルツハイマー病のリスク低下と関連している可能性がある[213,313-316,364,368-370,372]ことから、PDE阻害薬がアルツハイマー病の予防に役立つ可能性が示唆されている。
とシルデナフィルの臨床試験は有望であるが、非常に予備的である[327, 328, 338, 339]ため、それらから結論を導き出すことはできない。PF-04447943およびBI 409,306の臨床試験は有効性の欠如を示している[341, 342]が、これはPDE9阻害が(細胞質のcGMPではなく)細胞質周囲膜のcGMPを増加させ、CNGを介したCa2+の流入を可能にするためかもしれない[68, 71, 213]。
ビンポセチン[253, 255-258]、シロスタゾール[69, 317-319]、およびニセルゴリンの臨床試験は予備的であり、混合している[273-277]が、シロスタゾールおよびニセルゴリンが最も有望であり、これら3つのさらなる研究の価値があると考えられる[273-277]。
デプレニル/セレギリンの臨床試験では、短期的な有益性は示されているが長期的な有益性は示されておらず、デプレニルはやや期待外れである[289]。対照的に、プロペントフィリンは5つの第III相臨床試験で、軽度から中等度のアルツハイマー病患者の認知、認知症の重症度、日常生活動作、およびグローバル評価を複数の尺度で改善することが示されている[375,376,387-391]。しかし 2007年と2008年にそれぞれ発表された2冊の本は、明らかに2000年に発表されたMedScapeの記事に基づいて、18ヵ月間の第IIIb相臨床試験で有効性が示されなかったため、プロペントフィリンは中止されたと主張している[392-394]。プロペントフィリンの18ヵ月間の第III相臨床試験が2件あったのか、それともMedscapeの記事が不正確だったのかは不明である。
最終的には、プロペントフィリンは軽度から中等度の散発性アルツハイマー病患者の治療に適応となるかもしれない。それにもかかわらず、プロペントフィリンは現在、イヌの認知機能障害の治療に使用されている[396]。ヒトのアルツハイマー病と同様に、イヌの認知機能障害は年齢に関連した野生型のアミロイドβ沈着を伴い、齧歯類モデルと比較してアルツハイマー病の優れた前臨床モデルとなっている[397]。皮肉なことに、プロペントフィリンがイヌの認知機能障害を治療するのであれば、ヒトの散発性アルツハイマー病を治療する可能性があるという仮説を示唆している。
表1 薬剤 ホスホジエステラーゼ阻害剤 臨床効果
| ドラッグ | ホスホジエステラーゼ阻害 | 臨床効果 |
| ビンポセチン | PDE1 [ 224 ] | ADの治療には効果がありません[ 253、254 ]。MCIと認知症の治療に有望である可能性があります[ 255–258 ]。 |
| ニセルゴリン | PDE1およびcGMPで刺激されたPDE2 [ 259 ] | 認知症の治療に効果的かもしれません[ 273 ]。ADの治療に有効かどうかは決定的ではありません[ 273–277 ]。 |
| デプレニル/セレギリン | PDE1A2 [ 224、279 ] | ADの短期的な改善のみ[ 289 ]。 |
| シロスタゾール | PDE3 | 偶発的な認知症のリスクが低いことに関連している可能性があります[ 313 ]。MCI、軽度の認知症、およびAD患者の認知機能低下を遅らせる可能性があります[ 69、314、315、316–318 ]。ADの治療に有効かどうかは決定的ではありません[ 69、317–319 ]。 |
| デンブフィリン | PDE4 [ 321 ] | [ADに対する有効かどうかを決定的328、327 ]。 |
| シルデナフィル | PDE5 [ 71 ] | ADに対するシルデナフィルの臨床試験はまだ実施されていません。 |
| PF-04447943およびBI409、306 | PDE9 | ADには効果がありません[ 341、342 ]。 |
| カフェイン | 広域スペクトルPDE阻害剤 | 臨床試験は実施されていません。認知症、認知機能低下、ADのリスクを低下させる可能性があります[ 213、364、367–372 ]。 |
| プロペントフィリン | 広域スペクトルPDE阻害剤[ 340 ] | 血管性認知症およびADの治療に効果的で適応となる可能性があります[ 375、376、387–391 ]。 |
プロペントフィリンの治療効果は複数の試験で治療中止後も数ヶ月間持続しており、アルツハイマー病患者における疾患修飾効果を示唆している[375, 387, 389, 391]。我々は、プロペントフィリンが強力で非特異的/広スペクトルのPDE阻害剤[340]として、細胞質のcAMPおよびcGMPを十分に上昇させ、神経のpSer133-CREB、SIRT1,PGC1α、およびNrf2シグナル伝達[74-81,92-105,138-140,143,144]を実質的に増強し、神経のシナプス形成を改善することから、この疾患修飾効果が観察された可能性を示唆している[84-90]。記憶[84]、ミトコンドリア生合成[83,91,93,103]、抗酸化[112]、解毒、生存遺伝子発現[84-90]を阻害する一方で、BACE1誘導性炎症性NFκB[34,35,147,153,154,156-158]およびタウリン酸化およびCREB阻害性GSK3β[159-162,164-168,170,171,173-175,177-181]を阻害することが知られている。
興味深いことに、前臨床研究のレビューによると 2020年時点で有効性試験を完了している唯一のPDE阻害薬で、アルツハイマー病治療のために阻害すべき最も有望なPDEの1つ以上を阻害することが知られているもの(すなわち、PDE2,PDE4,およびPDE5)[73]は、プロペントフィリン[340]のみである。プロペントフィリンは、3つの最も有望な治療標的のうちの2つであるPDE2およびPDE4[340]を阻害することが知られている[73]。プロペントフィリンもまた、これまでのところ最も有望な臨床試験が行われているPDE阻害薬である[375, 376, 387-391]ことは、おそらく驚くに値しない。しかし、プロペントフィリンがPDE5も阻害するかどうかは不明である[340]が、プロペントフィリンはcAMPとcGMPの両方を上昇させる非特異的/広スペクトルのPDE阻害薬であるため、阻害する可能性がある[340]。もしそうでない場合は、PDE5阻害剤シルデナフィルがプロペントフィリンとの併用が理想的な治療法かもしれない。さらに、cGMP刺激PDE2を強力に阻害することにより[340]、プロペントフィリンはシルデナフィルとの併用が理想的であり、シルデナフィルの用量制限的な副作用であるcGMP刺激PDE2の活性化を防ぐことができる[72]。将来の前臨床研究では、プロペントフィリンとシルデナフィルの併用と別個の併用と標準治療の効果を研究し、プロペントフィリンとシルデナフィルの併用治療が相乗効果をもたらすかどうかを判断するために、標準治療単独と比較して、アルツハイマー病におけるプロペントフィリンとシルデナフィルの併用と別個の併用と標準治療の効果を研究すべきである。今後の臨床試験では、軽度から中等度のアルツハイマー病患者を対象とした18ヶ月間の第III相臨床試験において、プロペントフィリンがADAS-Cog上のアルツハイマー病症状の重症度と全体的な印象を改善するかどうかを調査すべきである。