Contents
Blood–brain barrier breakdown in Alzheimer’s disease and other neurodegenerative disorders
www.ncbi.nlm.nih.gov/pmc/articles/PMC5829048/
要旨
血液脳関門(BBB)は、脳微小血管内の連続的な内皮膜であり、細胞間の接触を封鎖しており、硬膜血管細胞と血管周囲のアストロサイトエンドフィートによって被覆されている。BBBは、全身循環に存在する因子からニューロンを保護し、シナプスやニューロンの適切な機能に必要な高度に制御された中枢神経系の内部環境を維持している。
BBBの障害は、神経毒性のある血液由来の破片、細胞、微生物病原体の脳内への流入を可能にし、炎症反応や免疫反応と関連しており、神経変性の複数の経路を開始させる可能性がある。本レビューでは、生きているヒトの脳における神経画像研究、死後組織およびバイオマーカー研究で、アルツハイマー病、パーキンソン病、ハンチントン病、筋萎縮性側索硬化症、多発性硬化症、HIV-1関連認知症および慢性外傷性脳症におけるBBB破壊を実証した研究について論じている。
BBB破壊が神経細胞の損傷、シナプス機能不全、神経細胞の接続性の喪失、神経変性を引き起こす原因となるメカニズムについて解説している。また、治療薬送達における健康なBBBの重要性と、神経医薬品の脳への送達に関連して、疾患に起因する病的なBBB破壊の悪影響についても簡単に論じている。
最後に、将来の方向性、この分野のギャップ、BBBを標的とすることで神経疾患の経過を制御する機会が提示されている。
はじめに
ヒトの脳には、酸素、エネルギー代謝物、栄養素を脳細胞に供給し、脳から全身循環に二酸化炭素やその他の代謝性廃棄物を除去する血管が〜644キロ含まれている1,2。全身のわずか2%を占めるにもかかわらず、脳は体内のブドウ糖と酸素の〜20%を消費し、その活性化された領域への血流と酸素の供給を急速に増加させることができ、神経血管のカップリング2,3として知られているプロセス。毛細血管は、最小の脳血管(図1)であり、脳血管の長さの約85%を占め、血液脳関門(BBB)(図1)1の主要な部位である。ヒトの脳では、毛細血管は約12m2の内皮細胞表面積を有しており、これは血液から脳への溶質の輸送に利用可能であり、その逆もまた然りである。ヒトの脳の毛細血管間の平均距離は~40μm4であり、したがって、溶質の平衡化は、分子がBBBを横切ると、脳の間質空間全体でほぼ瞬時に行われる。
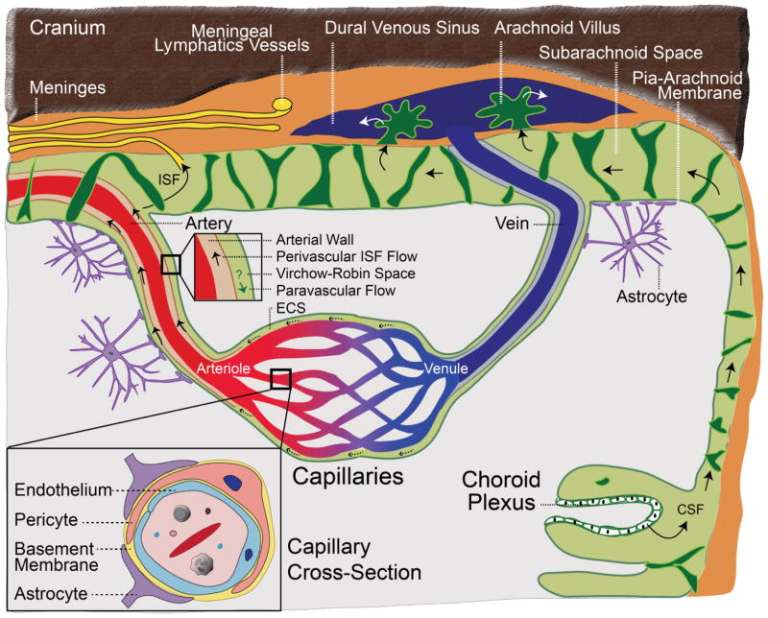
図1 血液脳関門
脳の毛細血管は、血液脳関門(BBB)の重要な部位である。毛細血管断面図(大)は、毛細血管壁に巻き付いているアストロサイトのエンドフィーを示している。動脈断面図(小図)は、血流とは反対方向の動脈壁を通る間質液(ISF)の血管周囲流を示している;血管周囲流は、血流と同じ方向にも発生する可能性がある。脳脊髄液は脈絡叢によって産生され、脳室からくも膜下腔に流れ込み、髄膜リンパ系および/またはクモ膜絨毛を通って静脈血に排出される。ISFは脳室(図示せず)やくも膜下腔で脳脊髄液と交換することができる。ECS、細胞外空間。
内皮BBBは、高い経内皮電気抵抗と低い傍細胞および細胞間透過性5(図2)の結果となる細胞間の接触を緊密に封じられている。内皮単分子膜は、硬膜細胞(毛細血管の周皮細胞、動脈や動脈の血管平滑筋細胞)とアストロサイトエンドフィート6,7によって被覆されている。高度に透過性の全身性毛細血管8とは対照的に、脳毛細血管は、それらがBBB(図2)を越えてそれらの輸送を容易にする脳内皮に特殊なキャリアおよび/または受容体を持っていない限り、緊密に密封された内皮と一緒に、脳内へのほとんどの血液由来の分子のエントリを制限する経サイトーシスによる内皮のバルクフローの低率を示している。
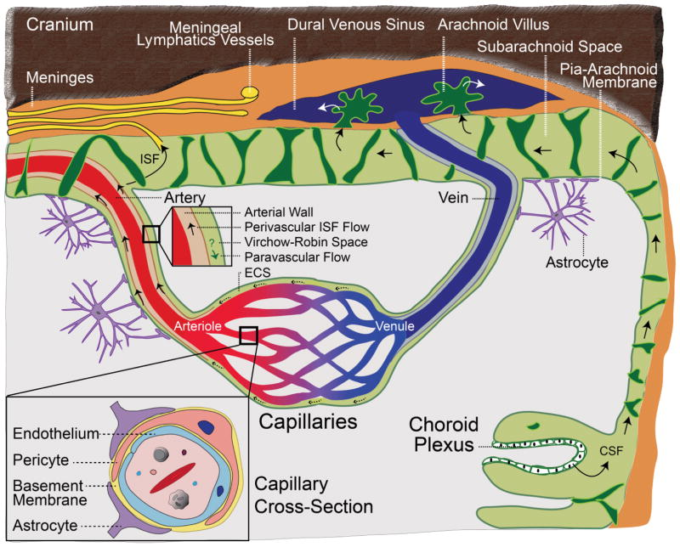
図2 毛細血管内皮の重要な輸送特性
a|タイトジャンクション(TJアドヒデンスジャンクション(AJジャンクション接着分子(JAM)は、溶質の自由な傍細胞間の交換を妨げている。c|代謝物、栄養素、ビタミン、ヌクレオチドなどの基質を基質特異性と濃度勾配に応じて輸送する溶質キャリア媒介輸送(CMT)。e|NLS1(ナトリウム依存性リゾホスファチジルコリンシンポーター1)はω3必須脂肪酸を脳内に輸送している。水はアクアポリン(AQP)受容体を介して輸送される。水はアクアポリン(AQP)受容体を介して輸送される:内皮細胞ではAQP1,アストロサイトエンドフィートではAQP4。h|神経毒性物質は、ホスファチジルイノシトール結合型クラスリン集合タンパク質(PICALM)を介したトランスサイトーシスとLDL受容体関連タンパク質-1(LRP1)によってクリアされ、アルツハイマー病と関連した有毒なアミロイドβ(アミロイドβ)種が除去される。興奮性酸性アミノ酸CMTトランスポーターEAAT1とEAAT2は神経毒性のあるグルタミン酸とアスパラギン酸をクリアする。しかし、先進的な糖化最終産物のための受容体(RAGE)は、アルツハイマー病ではアップレギュレートされ、循環アミロイドβの再突入を媒介し、脳のアミロイドβレベルを増加させる。i|脳細胞外空間(ECS)(点線矢印)を横切って拡散する溶質は、経血管輸送(c-e、g-i)を介して、動脈壁内の血管周囲ISFの流れ(実線矢印)によって、血流の逆方向に、クリアされ、最終的には、脳脊髄液で満たされたくも膜下空間に到達し、髄膜リンパ管と頸部リンパ節に排出される。
BBBの完全性を維持することは、適切なシナプス機能、情報処理、およびニューロン結合にとって重要な脳間質液(ISF)の化学組成を厳密に制御するために非常に重要である。BBBの完全性の損失は、増加した血管透過性の結果と減少した脳血流と障害された血行動態応答2,3,5,7,9に関連付けられている。BBBの破壊は、有毒な血液由来分子、細胞、微生物の脳内への侵入を可能にし、炎症反応や免疫反応と関連しており、神経変性の複数の経路を開始する可能性がある。
本総説では、まず、BBBの分子構造と輸送生理について簡単に説明し、次に、血管病理学、神経画像、死後およびバイオマーカー研究により、いくつかの神経変性疾患におけるBBBの破壊を明らかにした。アルツハイマー病(アルツハイマー病パーキンソン病(PDハンチントン病(HD筋萎縮性側索硬化症(ALS自己免疫性神経変性疾患とされる多発性硬化症(MS)などの神経変性疾患や、HIV-1関連認知症、慢性外傷性脳症(CTE)などの神経変性疾患でのBBBの破壊を明らかにしている10。我々は、BBB破壊が神経変性につながる病態メカニズムに焦点を当て、簡単に治療薬送達のためのBBB機能障害の意味合いに注意してほしい。最後に、将来の方向性、この分野におけるギャップ、およびBBBを標的とすることで神経疾患を制御する機会について議論する。本レビューでは、アルツハイマー病や神経変性疾患における脳血流の低下と変化した血行動態反応の役割を検討しておらず、また、他の場所で広範囲にレビューされているアルツハイマー病や神経変性の実験モデルにおけるBBB障害を取り上げていない2,3,9,11。
BBB分子構造
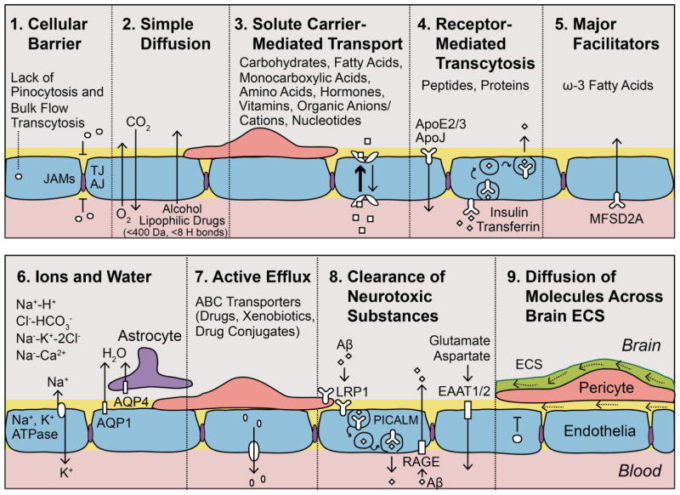
脳内皮細胞はタイトジャンクションとアドヒーレンスジャンクションでつながっている。タイトジャンクションには、オクルーディン、クラウディン-1,クラウディン-3,クラウディン-5,クラウディン-12,および膜関連グアニル酸キナーゼタイトジャンクションタンパク質ZO1,ZO2,ZO3が関与しており、アドヒデンスジャンクションには、カドヘリン、血小板内皮細胞接着分子(PECAM-1および接合部接着分子(JAM)JAMA、JAMB、およびJAMC6が関与している。ピノサイトーシスとバルクフロー流体トランスサイトーシスの乏しさは、酸素と二酸化炭素が急速にそれを横切って拡散するものの、脳内皮(図2)を横切る溶質の限られた交換に寄与している。小動脈12と毛細血管13は、脳の酸素供給の主要な供給源である。さらに、分子量が400 Da未満、または8個未満の水素結合を含む化合物(エタノールなど)は、単純な膜貫通拡散によってBBBを横断することができる4。
溶質担体媒介輸送(CMT)は、炭水化物、アミノ酸、脂肪酸、モノカルボン酸、ヌクレオチド、ホルモン、ビタミン、有機陰イオンおよび陽イオンのBBB横断輸送を促進する。受容体介在性トランスサイトーシス(RMT)は、血液から脳へ(トランスフェリンとインスリン)4,脳から血液へ(アポリポ蛋白質)6という両方向への蛋白質とペプチドの経内皮輸送を可能にする。ナトリウム依存性リゾホスファチジルコリンシンポーター1(NLS1,また、主要なファシリテータースーパーファミリードメイン含有タンパク質2aとして知られている重要なトランスポーターは、また、BBB形成のために重要である脳14に必須のω3脂肪酸を輸送する15(図2)。
BBBの隔膜上のナトリウムポンプ(Na+, K+-ATPase)は、カリウムと引き換えに、脳ISFへのナトリウムの流入を調節している16。他のイオントランスポーターは、ナトリウム、カリウム、塩化物、カルシウムイオンの輸送を調節し、BBBで重炭酸イオンの水素イオンと塩化物のためのナトリウムの交換を容易にする。BBBの内腔側で発現したATP結合カセット(ABC)トランスポーターは、内皮から血液中への積極的な排出を介して、薬物、異生物活性剤や薬物複合体の脳蓄積を防ぐ6,17。CMTは、BBB全体でアミロイドβ(アミロイドβ、アルツハイマー病に関連付けられているいくつかの形態)6,19-25のRMTクリアランスがこれらの潜在的に有毒物質の脳レベルを低く保つのに対し、興奮性アミノ酸(そのようなグルタミン酸やアスパラギン酸など)18の中枢神経系から血液へのクリアランスを容易にする(図2)。多くのことがBBBとその輸送生理の分子アーキテクチャについて言うことができるが、これらのトピックは、他の場所で詳細に検討されているとして、唯一の簡単な概要は、ここで与えられている6,7。
脳によって生成された分子は、脳の細胞外空間を横切って拡散し、2つのメカニズムによって脳からクリアされる:図2(b-d、f-h)5,6,26に示されているメカニズムを介して、BBBを横切って経血管輸送、、および動脈血管壁の基底膜内の血液の流れに逆方向に移動するISFの血管周囲輸送(図1)26-28。1980年代と1990年代に行われた研究では、血管周囲のISFの流れによって運ばれた溶質は、脳脊髄液(脳脊髄液)で満たされたくも膜下腔に到達し、深部頸部リンパに排出されることが示された29,30。過去3年間のさらなる研究では、頸部リンパ節32-34に排出される髄膜リンパ管31によるISFと高分子のクリアランスにおける硬膜リンパ系の役割が確認されている(図1)。生理的条件下では、血管周囲ISF経路は、80-85%が経血管BBB輸送によって除去されるのに対し、マウスの脳19,35からアミロイドβのアルツハイマー病関連形態のクリアランスの15-20%を担当している。1985年には、溶質は急速に血液36の流れに同じ方向に、Virchow-Robin空間を介してくも膜下空間からの血管傍輸送によって脳全体に配布することが示された。その後の研究では、このparavascular循環を記述するために用語 “グリンパティック “システムを導入し、中枢神経系の溶質輸送は、アストロサイト37,38上のアクアポリン-4(AQP4)水チャネルによって制御されたパラ動脈からパラ静脈方向に、脳の細胞外空間を介して脳脊髄液の対流を介して発生することを示唆した。提案されているグリンパ系のメカニズムは、しかしながら、最新の研究39-42によってサポートされていない、と大脳実質全体のパラ動脈からパラ静脈の細胞外空間への脳脊髄液の対流、圧力駆動の流体の流れは、証明されていない43-45,39,40のままである。さらに、マウスとラットにおけるAqp4の欠失は、くも膜下腔から脳への蛍光性溶質の輸送に障害を与えないことから、アストロサイトエンドフィートによる水分産生は、実質細胞外腔内での溶質輸送の調節には役割を持っていないことが示唆される39。これらの論争を解決するためには、さらなる実験的研究が必要である。
神経変性における血管病理
脳血管機能障害と血管病理は、アルツハイマー病関連のアミロイドβとタウの病理学5,6,46-53に加えて、アルツハイマー病における認知機能の低下と神経細胞の喪失に寄与している。多くの証拠は、アルツハイマー病における脳血管機能障害が単に併存する血管性痴呆に起因するものではないことを示している。例えば、脳血管と神経変性疾患との関連性の1つの研究では、米国国立アルツハイマー病調整センターのデータベースを使用して、単一の神経変性疾患(アルツハイマー病、前頭側頭葉変性症、α-synucleinopathy、海馬硬化症、プリオン病、脳血管疾患)52の剖検に基づいた診断を受けた5,715人の患者を識別するために使用された。混合性認知症を認めないアルツハイマー病と診断された4,629人のサブグループでは、80%が脳血管疾患、小血管疾患、出血、アテローム性動脈硬化症、動脈硬化症、脳アミロイド血管症(脳アミロイド血管症)52を示す裂孔や多発性微小梗塞を含む血管病理を有していた。アルツハイマー病または脳血管疾患のいずれかの剖検に基づく診断を受けた患者の2つのサブグループは、冠動脈疾患、高コレステロール血症、糖尿病などの血管危険因子の有意に類似した有病率を示した52。
脳血管障害はアルツハイマー病認知症の主要な危険因子であり、ほとんどの認知領域で低スコアと関連している51。BBB破壊の重要な原因であり、アルツハイマー病の3つの病理学的特徴の1つである脳アミロイド血管症54は、認知機能の低下に寄与する様々な血管病理を誘導する26。さらに、前臨床アルツハイマー病では、血管バイオマーカーの変化は、認知障害の発生前に、アミロイド沈着、アミロイドβ42の脳脊髄液レベルの低下、およびタウおよびリン酸化タウの脳脊髄液レベルの増加48を含む標準的なアルツハイマー病バイオマーカーの検出可能な増加の前に起こる。脳の小血管疾患は、後述するように、アルツハイマー病患者において顕著であり、世界の全認知症の約50%に寄与している3,55-58。
アルツハイマー病のツーヒット血管仮説によると、血管の損傷は最初の障害であり、BBB機能障害と脳灌流の低下を引き起こし、その結果、神経細胞の損傷と脳内のアミロイドβ蓄積につながる5,6,47,50。脳血管障害はライフスタイルに影響され、アポリポ蛋白E(APOE*ε4)のε4対立遺伝子の保有などの遺伝的危険因子、血管危険因子(高血圧、糖尿病、脂質異常症など)および環境危険因子(公害など)によって加速されるアルツハイマー病病理を促進するために独立しておよび/または相乗的にアミロイドβと作用する可能性がある47,50。
血管病理は他の神経変性疾患にも寄与している52。例えば、脳血管疾患は、α-シヌクレインの蓄積と黒質のドーパミン作動性ニューロンの変性を特徴とする第二の一般的な神経変性疾患であるPD52の病態形成に関与している。血管疾患や血管リスク因子がPD59の運動機能障害や認知機能障害を悪化させる。脳血管疾患、BBB障害、神経血管異常は、変異型ハンチンチン蛋白質の凝集による運動異常、認知異常、精神異常、代謝異常を伴う常染色体優性の神経変性疾患であるHD60,61にも認められる。機能不全BBBを横切るT細胞、B細胞、末梢マクロファージのBBB破壊とトラフィッキングは、MS33の病理学的特徴である。BBBの破壊はALS62で記載されており、CTE64の特徴であり、HIV-1関連認知症では、HIV-1に感染した単球やマクロファージの脳内への侵入を可能にしている63。
BBB破壊の神経画像学的証拠
本節では、アルツハイマー病や他の神経変性疾患におけるBBBの完全性と機能に関する最近のPETとMRIの研究を検討する(表1)。
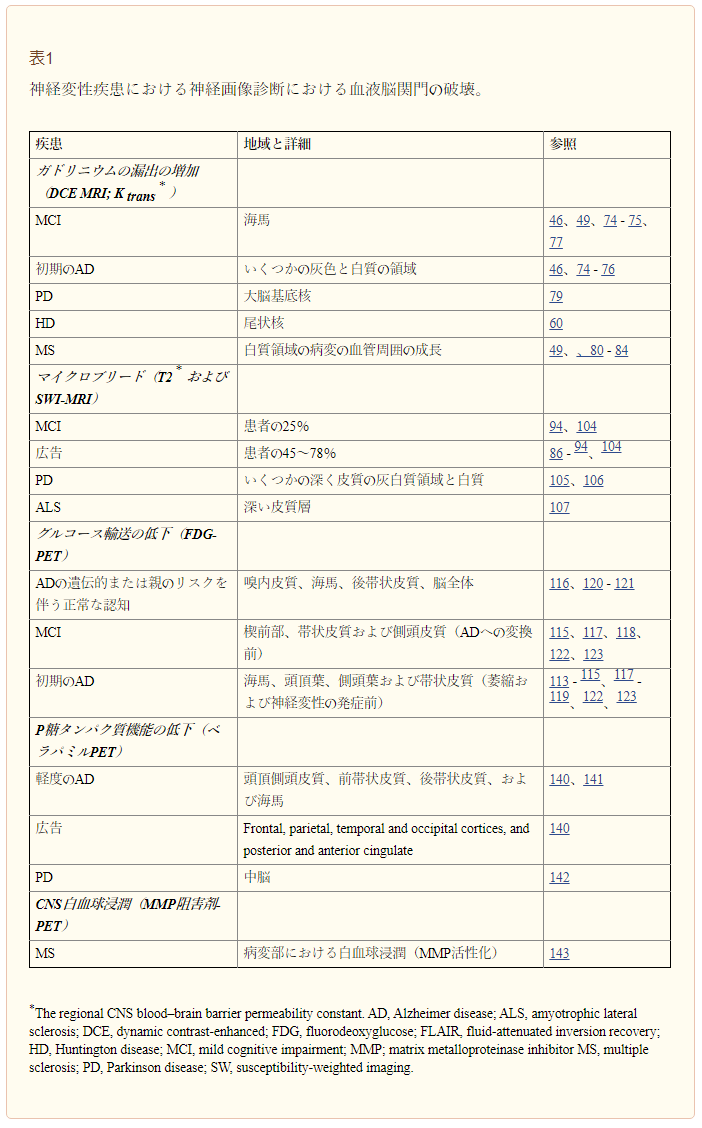
表1 神経変性疾患における神経画像診断における血液脳関門の障害
ガドリニウムに対するBBB透過性の増加
記憶と学習の中心である海馬のBBB破壊は、動的造影強調(DCE)MRIを使用して軽度認知障害(MCI)を持つ個人で観察されている。この手法では、ガドリニウム造影剤の脳内への漏出により、Patlak分析法49,65,66を用いて、局所的な中枢神経系のBBB透過性定数Ktransを定量化することができる。MCIを持つ個人の海馬におけるBBB破壊を年齢をマッチさせた対照群と比較した研究では、BBB破壊の程度は血管リスク因子49の影響を受けていないことがわかったが、可溶性血小板由来成長因子受容体β(PDGFRβ)の脳脊髄液レベルの上昇と相関していた49,67,Pericyte損傷のマーカー49,67。海馬でのBBB破壊は海馬萎縮49に先立って発生したが、これは一般的にアルツハイマー病初期に見られる68,69であり、BBB破壊が神経変性に先行する可能性があることを示唆している。この概念は、時間の経過とともに神経変性の変化を引き起こすBBB破壊の実験モデルからのデータによって支持されている70-73。初期アルツハイマー病患者のフォローアップDCE-MRI研究では、いくつかの灰白質および白質領域でBBB破壊が確認された46,74-76(表1)。これらの知見と一致するように、ヒトにおける初期の造影MRI研究では、健康なコントロールと比較してMCIを持つ個人の海馬におけるBBB透過性の増加を示した77,と造影剤は、血液から脳への脳脊髄液経路を介して可能性の高いアルツハイマー病を持つ個人の脳内に蓄積することを示唆した78。
DCE-MRI研究は、健常対照者と比較してパーキンソン病患者の大脳基底核におけるガドリニウムのBBBリーク(Patlak定量法49,65,66を使用して)の増加を検出している79。HD患者では、DCE-MRI解析により、尾状核におけるBBB透過性の増加と疾患負担スコアの増加、および灰白質動脈脳血量の増加との間に正の相関があることが明らかになっている60。DCE-MRI研究は、同様にMS49,80-82の白質におけるBBB透過性の増加の存在を確立している、特に活動的なMS病変で83,84(表1)。脳脊髄液におけるマトリックスメタロプロテアーゼ-9(MMP-9)活性の増加は、MSにおけるBBB破壊に寄与することが示唆されている82,85。生きているヒトの脳におけるBBB破壊の病的役割を理解するためには、血管の変化、アルツハイマー病、PD、HD、MSにおける神経学的欠損の進行、脳の構造的・機能的連結性の変化との関係を調べるために、将来の縦断的なDCE-MRI研究が必要である。DCE-MRI研究をALS、HIV-1関連認知症、CTE患者にまで拡大することで、神経変性疾患における局所BBB破壊が病因的な役割を果たしているかどうかを明らかにすることができるだろう。
微小脳出血
血管の損傷は、アルツハイマー病86-93,MCI94,およびアルツハイマー病94の遺伝的リスクが増加しているAPOE*ε4個体で頻繁に見られる脳微小出血(微小出血)として顕在化した顕著なBBB破壊につながる可能性がある。脳アミロイド血管症は、アルツハイマー病における血管変性と小葉微小出血の主な原因の一つであり、BBB破壊、梗塞、白質変化、認知障害26に寄与している。微小脳出血の位置はその病因に関係している。脳アミロイド血管症は大脳基底核、視床、小脳と脳幹(他の場所でレビュー95)で小葉微小球症と高血圧性血管障害の原因となる微小球症を引き起こす原因となる。アルツハイマー病における微小球状出血は主に小葉88,92,96-99(脳アミロイド血管症関連微小球状出血に類似)であり、主に後頭葉92,98,99に認められる。18F-florbetapir PETによって検出される脳内アミロイド沈着は、MCIおよびアルツハイマー病99の患者における微小血球症の数と正の相関がある。しかし、アルツハイマー病86-93またはMCI94の患者における微小血球症の有病率が高いことを報告したいくつかの研究では、アミロイドPET画像化86-93を行っていないため、微小血球症と脳アミロイド血管症の重症度を直接比較することはできなかった。
皮質表在性サイデローシス(すなわち、ヘモシデリンの髄膜下層沈着物の検出)は、脳アミロイド血管症の代替イメージングバイオマーカーとして示唆されている100,89,101,102。皮質表在性サイデローシス、小葉微小球症、アミロイドプラーク負荷の程度は、認知的に正常な対照者よりもアルツハイマー病患者の方が高く(MRIやアミロイドPET研究94で示されているMRIによる表在性サイデローシスの証拠は、病理学的に脳アミロイド血管症が確認された3人の患者でも観察されている。微小球症と表在性サイデローシスの地形と有病率をアルツハイマー病の脳アミロイド血管症に明確に関連付けるためには、以下で議論するように、より多くのアミロイドPETと高磁場強度MRIの研究が必要である。
微小出血は脳の小血管疾患を定義する基準としてよく用いられる103。T2*加重および感受性加重イメージング(SWI)MRI上の小さな低緊張領域は、微小出血の後、血管周囲空間でマクロファージによって貪食された血液由来のヘモシデリン沈着物を表していると考えられている96。MRIの磁場の強さは脳微小出血を検出する能力を決定する104。例えば、3T MRIの研究では、アルツハイマー病患者の約45%88,90,91,94とMCI94を持つ個人の25%が微小出血を発症することを示しているが、7T MRIの研究では、アルツハイマー病患者の78%が微小出血87を持っていることが判明した。現在の研究のほとんどは1.5Tおよび3TのMRIで行われているため、MCIおよびアルツハイマー病における微小出血の発生率は過小評価されている可能性が高い。脳組織の高分解能共焦点顕微鏡は、1.5Tまたは3T MRI62で容易に見逃されている直径20〜30μmの小さな毛細血管出血を検出することができる。
脳微小出血は、パーキンソン病患者の深部灰白質領域(尾状部、視床、頭頂部、淡蒼白球を含む皮質領域および白質全体でT2*重み付けMRIおよびSWI-MRIによって検出されている。微小出血の発生率は、認知症のないパーキンソン病患者や対照群に比べてPD認知症患者の方が高く、白質病変の程度と関連している105,106。
また、T2強調MRIでは大脳皮質に微小出血の可能性があると考えられているが、これはALS患者でも認められている62。また、高分解能T2強調7T MRIを用いた研究では、ALS患者の脳と脊髄に微小出血が認められている107。
ブドウ糖輸送障害
ブドウ糖は脳にとって重要なエネルギー基質である。グルコースの脳への取り込みは、PETトレーサーとして放射性標識されたグルコースアナログ、18F-フルオロ-2-デオキシグルコース(FDG)を用いて測定される46。FDGは、ニューロンではなくBBBの内皮でのみ発現し、ニューロンではない溶質キャリアファミリー2,促進型グルコーストランスポーターメンバー1(グルコーストランスポーター1(GLUT1)としても知られている)を介して脳に入る6,108。GLUT1に加えて、FDGの脳への取り込みは、脳萎縮性変化2に先立って、MCIと早期アルツハイマー病で減少している脳血流2,5に依存している。
グルコースとFDGの両方が急速にGLUT-1を介して脳に輸送されているが、脳細胞によって貪欲に取り込まれ、その後、細胞内ヘキソキナーゼ127,128によってリン酸化され、脳内の彼らのその後の代謝運命は完全に異なっている129。グルコース-6-リン酸は、解糖経路で急速に代謝されるのに対し、FDG-6-リン酸は、グルコース-6-リン酸異性化酵素の基質ではないため、そのさらなる代謝130,127,131,128を妨げるフルクトース-6-リン酸に変換することはできない。したがって、FDG の全身投与後約 45-90 分後には、この化合物の 90-97%が FDG-6-リン酸またはそのエピマーの形でマウス128 またはラット131,132 脳内に残存し、残りは FDG である。脳細胞はグルコース-6-ホスファターゼの活性が非常に低く、細胞膜を横切るFDG-6-リン酸の輸送が悪いため133,134,FDG-6-リン酸は脳細胞内にロックダウンられたまま132,135,脳からゆっくりとのみ排除される。BBBを介してFDGの脳への取り込みは、GLUT1ではなく、直接神経細胞の取り込みに依存しているので、アルツハイマー病脳におけるFDGの取り込みの減少は、血管障害(つまり、障害されたBBB機能)を指している。重要なのは、GLUT1レベルは実質的にアルツハイマー病109-112の脳微小血管で減少している。FDGのBBB輸送および脳への取り込みの減少は、後にアルツハイマー病の診断に変換するMCI患者、および早期アルツハイマー病患者における神経変性および脳萎縮に先行する。この血管欠損は、前臨床アルツハイマー病136の病期分類において考慮されるべきである。
FDG-PET研究はまた、MCI患者は、検出可能な神経変性変化、脳萎縮および/またはアルツハイマー病113への転換に先立って、いくつかの脳領域(楔前部、帯状後部、右角回および両側の側頭連合皮質を含む)でグルコース取り込みが減少していることを示している。アルツハイマー病患者の後帯状回と副頭頂葉におけるFDG取り込みの減少は、部分的なボリューム効果の補正の有無にかかわらず観察され、これらの減少は脳萎縮に起因するものではないことを確認している114。縦断的なFDG-PETの所見はさらに、正常な老化の間に海馬のグルコース取り込みの減少は、臨床的なアルツハイマー病診断の前に認知機能の低下を予測することができることを示唆している115。同様に、早期発症の常染色体優性アルツハイマー病に関連するプレセニリン-1(PSEN1)変異の無症候性キャリアは、脳萎縮がない場合にFDG取り込みにおけるアルツハイマー病様の減少を示している116。海馬、頭頂側頭葉皮質および/または後帯状皮質で減少したブドウ糖の取り込みが繰り返し初期のアルツハイマー病117,118でFDG-PETによって示されている、アルツハイマー病119,120の遺伝的リスクを持つ個人で、アルツハイマー病121および/またはMCIの陽性の家族歴を持つだけでなく、アルツハイマー病122,123を開発するために進んだ認知機能障害を持たない個人で、。FDG脳内取り込みのパターンはまた、正常な認知を有する個人をMCIおよびアルツハイマー病117を有する個人から識別することができる。神経変性に先立つFDG-PETの変化は、BBB125,126を介してグルコース輸送の減少を反映して、ヒト113-116だけでなく、アルツハイマー病124のトランスジェニックマウスモデルで発見されていない。
さらに、Src2a1+/-マウス(その野生型と比較して脳血管内のGLUT1レベルの50%を発現している)における実験的研究は、アミロイドβ108によって加速される二次的な神経変性に続く急速なBBBの破壊を示している。BBBにおけるGLUT1がヒトADにおける治療標的であるかどうか、またはヒトにおけるこのトランスポーターの薬理学的アップレギュレーションが、動物モデルでできるようにBBBの破壊、神経変性、認知障害を防ぐことができるかどうかを探る試みはこれまで行われていない。さらに、他の神経変性疾患におけるグルコース輸送の役割については、これまで検討されておらず、今後の研究で追求されるべきである。
P糖タンパク質の機能障害
P-糖タンパク質(ABCB1遺伝子にコードされる)は、薬物や外来物質の内皮から血液中への積極的な排出を仲介し、脳内への蓄積を防ぐ6,17。P糖タンパク質はBBBを越えてアミロイドβをクリアするが、これにはLDL受容体関連タンパク質-1(LRP1)137-139が必要である。P-糖タンパク質の機能は11C-ベラパミル-PETで臨床的に評価される。アルツハイマー病におけるベラパミル-PETの研究は、前頭葉、頭頂部、側頭葉、後頭皮質、および後・前帯状突起140でベラパミルの取り込みが増加したことを実証している。同様に、軽度のアルツハイマー病患者を対象としたベラパミル-PET研究では、頭頂側頭葉、前頭葉、後帯状皮質と海馬でP-糖タンパク質の活性が大幅に低下していることが明らかになった141。さらに、ベラパミル-PET研究では、パーキンソン病患者の中脳におけるP-糖タンパク質活性の低下が示されており、これはBBB機能障害を示している142。これらの研究をまとめると、P-糖タンパク質機能の低下がアルツハイマー病の病因に関与していることが示唆されている ・脳内に蓄積される外来物質(高レベルの物質はニューロンを傷つけ、炎症を促進する)を可能にすること、および/またはBBBを介したアミロイドβクリアランスを減少させることのいずれかによって-。このように、P-糖タンパク質とLRP1は、アルツハイマー病における重要な治療標的である可能性があり、おそらくパーキンソン病においても重要である。
中枢神経系白血球浸潤
PET トレーサーとして放射性標識 MMP を使用した研究では、白血球浸潤143 と関連する初期の MS 病変で MMP 活性の増加を示した。さらに、MS患者では、脳静脈排水障害144 と灰白質の脳血管反応性が低下しており、灰白質萎縮145 と相関している。中枢神経系への白血球浸潤は他の神経変性疾患、特にアルツハイマー病146-149,HIV-1関連痴呆150,CTE151でも見られることから、これらの疾患の経過の中でいつこのような細胞浸潤が起こるかを明らかにするためには、同様のMMP-PETニューロイメージング研究が有用であると考えられる。しかし、そのような研究はまだ不足している。
死後のBBB障害の証拠
本節では、アルツハイマー病や他の神経変性疾患患者の死後組織の解析から得られたBBB障害の証拠を検討する。これらの研究では、BBB障害は脳毛細血管の漏出、BBB関連細胞(周皮細胞や内皮細胞を含む)の変性、循環白血球や赤血球の脳内浸潤、異常な血管新生や分子の変化によって示されている(表2)。
表2 神経変性疾患の死後組織分析における血液脳関門の障害
原文参照
毛細血管の漏出
アルツハイマー病患者の死後脳組織のいくつかの研究では、(様々な分析方法を用いて:免疫組織化学、免疫ブロッティング、プルシアンブルー染色)フィブリノーゲン、トロンビン、アルブミン、IgG、およびヘモシデリン146,147,152-157などの鉄含有タンパク質の蓄積を含む、前頭前野および内耳野皮質と海馬における血液由来タンパク質の毛細管漏出が発見されている。これらの血液由来のタンパク質は、しばしばアルツハイマー病関連アミロイドβ147,153,155の堆積物と共局在化して発見される。BBB破壊の証拠は、アルツハイマー病の主要な遺伝的危険因子であるAPOE*ε4対立遺伝子を持つ個体で最も顕著である。対照的に、最も一般的な対立遺伝子、APOE*ε3のためのホモ接合個体は、アルツハイマー病のリスクを低減し、BBB内訳147,152,156,158の減少の程度を示している。他の場所でレビューされているように11,複数の実験的研究は、BBBの破壊がβアミロイドーシス159-162とAPOE*ε4トランスジェニックマウス71,163,164のADモデルで毛細血管の漏れを引き起こすことを確認している。
パーキンソン病患者の脳組織の死後分析では、線条体にフィブリノゲンまたはフィブリン165,IgG166,ヘモシデリン165,167が血管周囲に沈着しており、これはBBB破壊を示唆している。毛細血管周囲のフィブリン沈着もHD60患者の脳組織で認められた。フィブリノーゲン、トロンビン、IgGおよびヘモシデリンの沈着は、散発性または家族性のALS患者の脳および脊髄組織で発見されている62,107,168と、運動ニューロン変性の発症前のALSのトランスジェニックマウスモデルで発見されている11,169。
最後に、MS患者では、特にタイトジャンクションに異常がある血管に沿って、活動性病変と非活動性病変の両方で毛細血管内のフィブリノーゲンの漏出がみられる170。CTE患者の死後最初の研究では、Virchow-Robin空間に脳浮腫とヘモシデリンを含む血管周囲マクロファージが発見されている171。
周皮細胞変性
アルツハイマー病患者からの脳組織の電子顕微鏡研究は、浸透親水性物質の大規模な蓄積に関連する大脳皮質の周皮細胞の変性を明らかにした。これらの変化は、血液由来タンパク質のファゴサイトーシスの増加、ミトコンドリアの変化、およびピノサイトーシス小胞の数の増加を示唆している172,173。周皮細胞マーカーPDGFRβの免疫染色は、APOE*ε4対立遺伝子の数(APOE*ε3のホモ接合性と比較して)156の数にリンクされた遺伝子投与効果の証拠を示したアルツハイマー病155患者からの脳サンプルにおける脳毛細血管上の周皮細胞の被覆率と数の減少を明らかにした。アルツハイマー病の皮質組織の免疫測定では、前膜157,アルツハイマー病のコースの初期に影響を受ける領域での周皮細胞マーカーPDGFRβの損失が確認された。周皮細胞はBBBの完全性7,176を維持し、その変性はBBBの破壊70,174,175につながる。さらに、ペリサイトは脳からアミロイドβをクリアし、その損失は、アルツハイマー病159のマウスモデルにおけるアミロイドβとタウ病理の発症と進行を加速させる。
ALS患者の脊髄や脳組織の免疫組織学的分析でも、顕著なペリサイト変性が認められている62,168。HIV-1関連認知症やHIV脳炎患者の脳組織の死後の研究では、微小血管の変性とBBB破壊の証拠が発見されており、その中にはペリサイトの被覆率の低下も含まれている177。血管周囲空間の拡大64,151,178および深部血管の壁細胞の鉱化171を含む血管障害もまた、CTE患者の脳組織で発見されている64,151,171,178。
内皮変性
毛細血管長の減少(内皮変性を示唆するタイトジャンクションタンパク質の発現低下、および毛細血管基底膜の変化は、アルツハイマー病5,155,156,158,173,179,180を持つ患者からの脳組織で報告されている。これらの変化は、アルツハイマー病180の血管分化の調節因子であるホメオボックスタンパク質MEOX2をコードするMEOX2の脳内皮細胞発現の低下によって引き起こされる、異常な脳血管新生を反映しているかもしれない。広範囲の前駆細胞形成因子もまた、(減少したMEOX2発現の存在下で)アポトーシス180を調節するAFX1転写因子の発現の増加を介して、減少した脳毛細血管密度および細胞死につながるアルツハイマー病181患者の脳から単離された微小血管において発現している。健康な内皮を維持するPericyte由来の可溶性因子もまた、動物モデルで示されているように、潜在的に内皮変性に寄与する可能性があり、Pericyte変性のためにアルツハイマー病脳で欠落している可能性がある70。
また、微小血管の変化(内皮細胞の厚さ、長さ、密度の減少タイトジャンクションタンパク質の喪失や異常、基底膜の変化を伴う内皮変性もPD166患者の脳組織で報告されている。ALS患者の脊髄または脳組織を免疫組織学的に解析すると、タイトジャンクションの減少、毛細血管基底膜の変化、血管周囲空間の拡大168,182,183,およびアストロサイトエンドフィートの毛細血管からの解離を伴う内皮変性が明らかになった107。内皮タイトジャンクションタンパク質claudin-5とオクルーディンの発現低下がHD60で示されている。また、内皮タイトジャンクション蛋白質ZO1の発現が低下したり不連続になったりする内皮変性は、正常な白質と比較して、活動的なMS病変と非活動的なMS病変で示されている170。HIV-1関連認知症またはHIV脳炎患者の脳組織の死後研究では、周皮細胞の被覆率の低下177,タイトジャンクションの減少および破壊150,184,毛細血管基底膜の変化150など、微小血管の変性およびBBBの破壊の証拠が発見されている。
細胞浸潤
赤血球の滲出は、アルツハイマー病146,PD165およびALS62で認められている。末梢マクロファージによる浸潤もアルツハイマー病149,172およびHIV脳炎150で示されている。さらに、好中球はアルツハイマー病148でBBBを越えることができる。これらの知見をまとめると、アルツハイマー病や他の神経変性疾患におけるBBBの破壊は、微小出血やヘモシデリン(赤血球によって運ばれるヘモグロビンに由来する)の沈着を引き起こす赤血球の押し出しを可能にするだけでなく、脳内の自然免疫応答を活性化することが示唆されている。非アルツハイマー病神経変性疾患におけるこれらの免疫系の応答は、そうでなければ破壊されたBBBを越えて脳に入るであろう病原体を識別し、排除することに向けられているかどうか(そして、アルツハイマー病のモデルでは感染プロセスを取り囲むことと引き換えにアミロイド沈着を加速することが示されている185,186)今後の研究で決定されるであろう。
異常な血管新生
プロ血管新生因子の増加レベルは、アルツハイマー病脳181で報告されている。しかし、失われた毛細血管ネットワークの成功した更新は、おそらく進行中の周皮細胞変性7とMEOX2180の低内皮発現に起因するアルツハイマー病脳では、上記で議論したように、損なわれている。血管新生のマーカーの変化によって示されるように、異常な血管新生は、また、PD187,188で黒質実質部、脊髄局、および被蓋骨で発見されている。パーキンソン病における運動症状を緩和するための視床下核の脳深部刺激の有効性は、毛細血管の長さと密度の増加、内皮細胞の厚さの増加などの微小血管構造189,166の改善にさえ起因しているかもしれない。また、深部脳刺激を受けたパーキンソン病患者の死後脳サンプルでは、未治療のパーキンソン病患者と比較して、血管周囲IgGの漏出が減少していることが示された166。
HD60では、毛細血管(直径5~10μmの血管)の密度が増加し、より大きな微小血管(直径10~20μm)の数が減少しており、異常な血管新生を示唆していた。HDの異常血管化は大脳皮質と黒質60,61にも認められた。
分子変化
いくつかの研究では、アルツハイマー病脳内皮がGLUT1,BBB108を介して減少したグルコース輸送につながるBBB特異的なグルコーストランスポーター109-112,の低レベルを発現していることが示されている。アルツハイマー病脳微小血管はまた、LRP1,BBB19,20,156,190,191で主要なアミロイドβクリアランス受容体の減少発現を示している(その変化はまた、アミロイドーシス、オランダ型(HCHWA-D)20を持つ遺伝性脳出血患者に存在している)。LRP1発現の低下は、脳からのアミロイドβクリアランスの低下をもたらし、脳内への蓄積を促進する20,22,24。したがって、LRP1は血管を介したアミロイドβクリアランスを促進するための重要なターゲットである192。このメカニズムは、抗アミロイドβ抗体に基づく現在のアミロイドβクリアランス治療の有効性、特に、脳から血液へのアミロイドβクリアランスがBBB193,194,195を介して必要とされる末梢アミロイドβシンク作用メカニズムを有する治療のために重要である可能性がある(FIG.2)。
アルツハイマー病患者では、脳内皮と硬膜細胞の両方で、脳微小血管内の高度な糖化最終生成物受容体(RAGE)のレベルが上昇している190,191,196。RAGEはアミロイドβをLRP1とは逆方向に輸送し、循環するアミロイドβの脳内への再侵入を仲介し、炎症を促進する。実験研究はまた、アルツハイマー病200を持つ患者におけるRAGEブロッカーの進行中の第III相試験の開始につながったアルツハイマー病196-199の主要な治療標的としてRAGEを同定した。
対照群と比較して、アルツハイマー病患者では、脳内皮と末梢血においてシクロフィリンA(炎症性サイトカイン)とマトリックスメタロプロテアーゼ9(MMP9)の両方のレベルが上昇している。これらの増加はAPOE*ε4キャリアで特に顕著であり156,トランスジェニックAPOE*ε4マウスのそれに匹敵する所見71であり、これらの増加はシクロフィリンAとMMP9が関与するBBB分解経路の活性化を示唆している。このシクロフィリンA-MMP9経路の活性化は、非無症候性APOE*ε4キャリアの脳脊髄液分析により確認されており、このキャリアはBBB破壊と関連していることが201,脳組織におけるシクロフィリンA mRNAレベルの分析により確認されている202。シクロフィリンA阻害剤であるアリスポリビルは、C型肝炎の追加治療薬として第III相臨床試験で有望性が示されていることから 203,これらの研究は、APOE*ε4 アルツハイマー病キャリアにおけるBBBの安定化にも有用である可能性を示唆している。BBBのシクロフィリンA-MMP9経路の阻害が、ヒトAPOE*ε4 アルツハイマー病キャリアにおける神経変性過程に影響を与えるかどうか(ヒト化APOE*ε4トランスジェニックマウス71)は、今後の研究の興味深い課題である。
HIV-1関連認知症やHIV脳炎患者の死後調査では、P-糖タンパク質204の脳内皮発現が低下していることが報告されている。さらに、HD患者では、変異型ハンチン凝集体が脳内皮細胞、血管周囲マクロファージ、血管平滑筋細胞、血管基底ラミナ60に蓄積し、HD205が進行した患者の脳内では遺伝的に無関係な胎児の神経移植片に蓄積している。これらのデータは、脳血管系と免疫系が変異型ハンチンの拡散に寄与していることを示唆するとともに、血管細胞を含む非神経細胞が変異型ハンチンの拡散に寄与していることを示唆している。
BBB障害の脳脊髄液エビデンス
ここでは、アルツハイマー病や他の神経変性疾患におけるBBB破壊の脳脊髄液バイオマーカーを検討する(表3)。アルツハイマー病および他の神経変性疾患における異常な血管新生、内皮機能不全、壁細胞傷害、炎症性サイトカインおよびケモカインの他の脳脊髄液バイオマーカーは、他の場所で詳細にレビューされている50,206,207が、ここでは検討されていない。
表3 神経変性疾患における脳脊髄液 ELISAにおける血液脳関門障害について
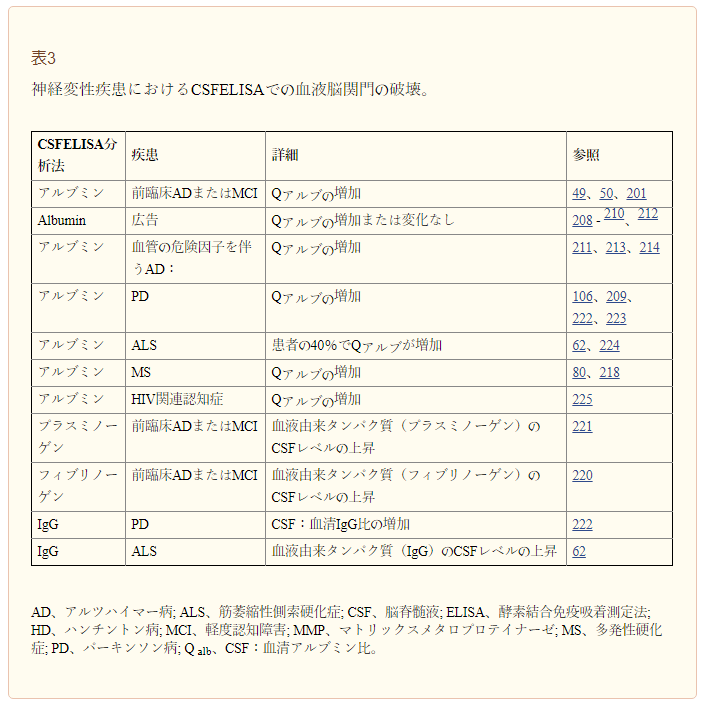
アルブミンは血液由来のタンパク質であるため、アルブミン商(Qalb)として知られる血清アルブミンレベルに対する脳脊髄液アルブミンの比率の増加は、BBB破壊の指標として頻繁に使用されている50。いくつかの研究では、前臨床アルツハイマー病201,MCI49,およびアルツハイマー病208-210を有する患者でQalbが上昇していることが報告されている。しかし、他の研究では、軽度の動脈性高血圧、糖尿病、虚血性心疾患211,213または脂質異常症214を含む血管危険因子211-214が存在しない限り、アルツハイマー病211患者におけるQalbの上昇は認められなかった。しかし、アルツハイマー病患者の大多数は血管危険因子を有している。65歳の患者の65%、85歳の患者の80%となっている3,56,57。血管危険因子とQalb201,208との関係を具体的に検討していない研究もあるが、血管危険因子は、DCE-MRIのKtrans透過性解析49で血液から脳の血管外細胞空間へのガドリニウムの流出によって測定されるように、BBB破壊の程度を悪化させることはなかった。これらの観察は、BBB破壊が血管リスク因子とは無関係にアルツハイマー病と関連しているという見解を支持するものである。今後の研究では、併存疾患や血管危険因子3,47,215による虚血性血管障害がアルツハイマー病におけるBBB破壊を増強するかどうかを慎重に検討すべきである。
しかし、脳脊髄液アルブミンレベルは、脳マクロファージ、ミクログリア、アストロサイト、ニューロン、およびオリゴデンドロサイト(コンドロイチン硫酸プロテオグリカン4(NG2としても知られている)を発現する細胞)216-218によるアルブミンの取り込みと同様に、タンパク質分解による切断によって影響を受ける可能性がある。したがって、QalbはBBB破壊の程度を過小評価している可能性がある。一方、脳脊髄液の再吸収および/または産生量の低下はQalbを上昇させ、BBB破壊を反映していない可能性のある偽陽性結果をもたらす可能性がある206。この考え方を支持するために、7人のアルツハイマー病患者を対象とした1つの研究では、これらの患者では脳脊髄液産生率がかなり低下していることが判明した219。脳脊髄液ターンオーバーの減少が一部のアルツハイマー病患者におけるQalbの増加の根底にあるかどうかを決定的に決定するためには、脳脊髄液動態の詳細なヒト研究が必要である。DCE-MRI49,マイクロブリードT2*加重MRI87,および/または代替的な脳脊髄液血液由来バイオマーカー(フィブリノーゲン220やプラスミノーゲン221など、以前にMCI患者および初期アルツハイマー病患者のBBB破壊を検出するためにそれぞれ使用されてきたもの)の測定を含むBBB完全性の感度の高い検査は、さらに検討されるべきである。
いくつかの重要な脳脊髄液バイオマーカー研究では、早期パーキンソン病を有する非認知症患者におけるQalb106,209,222,223の増加、および脳脊髄液:血清IgG比222の増加が対照群と比較して報告されている。4つの独立した研究では、ALS患者138人中55人(~40%)でQalbが増加したことが報告されている。ALS患者ではアルブミン、IgG、その他の血液由来タンパク質の脳脊髄液レベルの上昇も報告されている62,224。QalbはMS218患者でも上昇しており、この変化はDCE-MRI80で検出される白質BBB透過性の増加と相関している。HIV-1関連認知症患者では、Qalbの上昇は神経フィラメント軽鎖の脳脊髄液レベルで測定される軸索損傷と関連していた225。最後に、認知機能低下201に先立つMS82,85およびAPOE*ε4キャリアの患者における脳脊髄液中のMMP9活性の増加は、BBB破壊と関連している。
BBB破壊と神経変性
上述の神経変性疾患は、BBB破壊をもたらす血管壁の病理学的変化を共有している。内皮の変性は、タイトジャンクションタンパク質の損失および/または経細胞分裂を介した経内皮バルクフローの増加をもたらす5,6。これに伴う周皮細胞の変性はBBBの破壊を引き起こし7,70,174-176,プラスミノーゲン、トロンビン、フィブリノーゲンなどの神経毒性を持つ血液由来のタンパク質が中枢神経系の様々な領域に侵入することにより、神経変性の複数の経路(図3)を開始する(表1-3)。
図3 血液脳関門(BBB)の破壊は神経変性を促進する
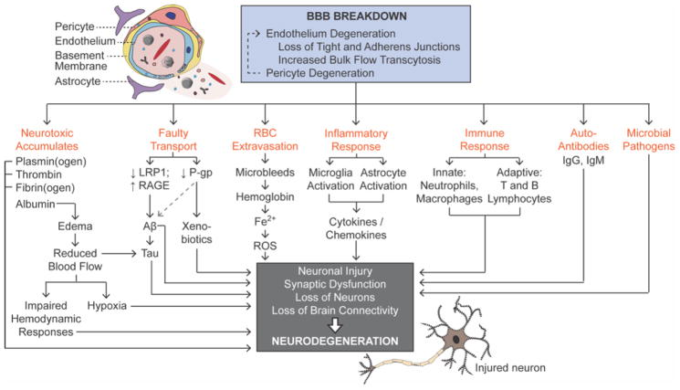
BBB破壊は、タイトとアドヒアレンスジャンクションの損失とバルクフロートランスサイトーシスの増加を伴う、周皮細胞と内皮の変性によって特徴づけられる。BBB破壊は、微生物病原体の侵入、神経毒性物質の蓄積、不良なBBB輸送、赤血球の extravasation、神経毒性遊離鉄(Fe2+)の放出につながり、活性酸素種と酸化ストレスを発生させる。炎症性および免疫応答は、自己抗体の生成につながる。CMT、溶質担体媒介輸送;ECS、細胞外空間、L-DOPA、L-3,4-ジヒドロキシフェニルアラニン;RMT、受容体媒介トランスサイトーシス。
循環中のプラスミノーゲンから生成されるプラスミンは、神経細胞のマトリックスタンパク質であるラミニンを分解し、それによって神経細胞の損傷を促進する226。高濃度のトロンビンは神経毒性と記憶障害を媒介し227,二重結合の破壊を促進する228。フィブリノーゲンは軸索の引っ込み229とBBB損傷を引き起こし、神経炎症を促進する230。さらに、フィブリン枯渇はMSのトランスジェニックマウスモデルにおいて神経炎症と脱髄の発症を遅らせる231,フィブリンによる治療は一次ミクログリアと骨髄由来マクロファージの両方においてM1型活性化と抗原提示遺伝子の誘導を誘導する232。MS における脳病理の発症における凝固・線溶タンパク質の役割については、別の場所で検討されている233。
アルブミンの流入は、脳の微小循環と血流を閉塞する血管周囲水腫につながる。ターンでは、これらの低酸素状態は、神経細胞の損傷と神経変性に寄与する血行動態応答の障害につながる2,13。赤血球の滲出(微小血球)は、ほぼすべての神経変性疾患92,104で見られ、有害な鉄含有タンパク質(ヘモグロビンなど)の血管周囲への蓄積を引き起こし、それらが分解される際に遊離鉄(Fe2+)が放出され107,62,73,活性酸素種(ROS)を発生させ、神経細胞を酸化ストレス234にさらす。
アルツハイマー病、PD、およびHIV-1関連認知症140-142,204などの神経変性疾患では、BBBでのP-糖タンパク質媒介の活性エフラックス輸送の機能不全は、有害な外来物質(環境汚染物質、食品添加物、農薬、薬物など)の脳内蓄積につながる。BBB20,25,137でP-糖タンパク質とLRP1のレベルを低下させ、脳微小血管6,190,191,196,199でRAGEのレベルを増加させ、アルツハイマー病とその蓄積にリンクされている有毒なアミロイドβ種の誤ったクリアランスにつながる脳内。血流の減少とアミロイドβレベルの増加の両方は、アルツハイマー病2のもう一つの重要な病理学的特徴であるタウ病理を促進することができる。他のタンパク質(アルツハイマー病やCTEではタウ、パーキンソン病ではα-シヌクレイン、HDではハンチンチン)のクリアランス不良が、中枢神経系でのそれぞれの蓄積に寄与するかどうかは、現時点では明らかになっていない。
興味深いことに、実験的研究では、α-シヌクレインは遊離ペプチドとしてBBBを介して脳内外に輸送されることが示唆されている235。さらに、全身投与されたα-シヌクレインオリゴマー、リボンおよびフィブリルは、異なるシヌクレイン病を引き起こし、それらはすべてBBBを横断することができることを示唆している236。ヒトでは、α-シヌクレインを含む細胞外小胞が脳脊髄液と血液中で発見されており、血液と脳脊髄液の間でこのタンパク質が双方向に輸送されていることが示唆されている237,238。赤血球はα-シヌクレイン含有細胞外小胞237 の供給源であり、またPD165,166の患者では線条体への赤血球の滲出が検出されていることから、滲出した赤血球はヒトにおけるα-シヌクレイン症の発症に寄与している可能性がある。α-シヌクレインのレベルは中枢神経系よりも循環系の方が2桁高いことから238,BBBを介したα-シヌクレインの輸送はパーキンソン病の病態に関与している可能性があり、新たな治療標的となる可能性がある。
神経毒性物質の蓄積と血流の低下はミクログリアやアストロサイトを活性化し、神経毒性サイトカインやケモカインの分泌を伴う炎症反応を引き起こす47。さらに、いくつかの疾患(アルツハイマー病など)では、末梢マクロファージ147,149と好中球148の脳浸潤は、自然免疫応答の活性化を示唆している。末梢マクロファージの浸潤に加えて、TおよびBリンパ球のBBBへの流入がMS患者で認められ、適応免疫応答を示唆している33。全体として、これらの研究は、BBBの破壊は、脳に循環白血球のエントリを可能にすることを示唆している。
BBBの破壊は、ヒトではいくつかの抗-脳内自己抗体239の生成につながるが、神経変性疾患の発症におけるそれらの役割は完全には解明されていない。さらに、BBBの破壊は、循環する病原体が脳に侵入してニューロンを傷つけることを可能にし、および/または、アルツハイマー病185,186の動物モデルで示されているように、βアミロイド症を悪化させるアミロイド反応を誘発することができる。
BBB破壊がどのように薬物送達に影響を与えるか
BBBを越えて治療薬をうまく送達するためには、機能的にも構造的にも健康な血管、正常な血管化、適切な血流、および中枢神経系への薬物送達を容易にするための溶質キャリア媒介輸送(CMT)または受容体媒介トランスサイトーシス(RMT)システムの採用が必要である(図4)。既存のCMTおよびRMT BBBシステムを使用する戦略は、神経治療薬の脳への浸透性および効力を高めるために探索されてきた(図2)。例えば、大規模な中性アミノ酸CMTトランスポーターは、PD240で脳にL-3,4-ジヒドロキシフェニルアラニン(L-DOPA)を送達し、トランスフェリンRMTは、様々な神経学的状態で脳に治療用抗体を送達することができる4,241-243。他にも、ナノ粒子を用いた治療薬の中枢神経系への送達を改善するために、ナノ粒子244および/または集束超音波245,246によるBBBの開通などのアプローチが試みられている。
図4 血液脳関門(BBB)機能不全-ドラッグデリバリーへの影響
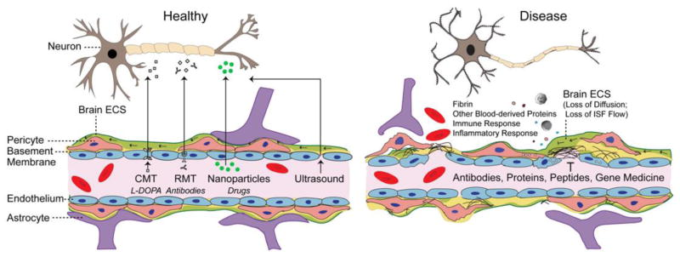
健康なBBB(左)では、バリアを突破して神経医薬品を脳に届けるための戦略は、キャリア媒介型トランスポーター(CMT受容体媒介型トランスポーター(RMTナノ粒子、および/または例えば焦点を合わせた超音波によるBBBの一過性の開口に依存している。病理的条件(右)の下では、破壊されたBBBは、拡大した血管周囲空間への血液由来の破片や細胞の蓄積を導く。これにより、脳の細胞外空間(ECS)を横断する濃度勾配駆動拡散による中枢神経系全体の分子の正常な分布が阻害され、治療用の抗体、タンパク質、ペプチド、遺伝子医薬などの薬剤が効率的に神経細胞の標的に到達するのを妨げる間質液(ISF)やISFの流れの領域的な形成が阻害される。詳細は本文を参照のこと。
神経内科医は一般的に、疾患に起因するBBB破壊は、治療用抗体、タンパク質、ペプチド、低分子、および/または遺伝子医薬品を、BBBを操作することなく、患部の神経細胞に送達する機会を提供する可能性があると想定している。しかしながら、神経変性の影響を受けた脳領域では、しばしば神経変性の前に発症し、疾患の進行に伴って持続する血管の機能的および構造的変化を特徴とする病的なBBB破壊が生じる。これらの血管変化には、
- 内皮の変性
- BBBにおけるタイトジャンクションおよびアドヒデンスジャンクションの発現低下
- 内皮バルクフローのトランスサイトーシスの増加
- BBBトランスポーターの発現障害
- 周皮細胞の変性
- 有害物質の血管周囲蓄積
- 炎症
- 免疫応答(図3)
が含まれ、これらはすべて、脳への治療薬の送達を妨げる。病的状況下では、血液由来の生成物、水分および電解質が肥大した血管周囲空間に蓄積し、脳の細胞外空間を横切る溶質の正常な拡散、ISFの形成およびISFの流れを阻害し、結果として中枢神経系全体への溶質の分布が障害される(図4)。
疾患によって引き起こされたBBBの障害、細胞外空間における溶質輸送の障害、およびISFの局所的な流れの低下の結果として、治療薬(抗体、タンパク質、ペプチドおよび低分子を含む)は、拡大した血管周囲空間内の病的に変化した脳組織に、他の血液由来の残骸とともにロックダウンられ、神経細胞の標的に到達するのを妨げる可能性がある。また、神経変性疾患におけるCMTおよびRMTシステムの機能低下は、薬物送達のための治療薬としての使用をさらに複雑にしている。したがって、脳血管の健全性を向上させ、細胞外空間やISF循環を介した拡散性を回復させるためには、健康な血管を持つ脳領域や、疾患を患った脳領域における損傷した血管系の安定化が必要となる。
遺伝学的研究からの洞察
このレビューでは、BBB破壊は神経変性疾患の重要な病原性の特徴として位置づけている(TABLES 1-3)。重要なことは、BBB破壊は、(まれに)脳内皮細胞および/または血管壁の壁細胞の一次的な遺伝的欠損を伴う遺伝性単発性神経疾患にも見られるということである6。これらの疾患の既知の遺伝的病因は、BBB遺伝的欠損および神経変性を支える共通の因果関係メカニズムについての貴重な洞察を提供する。例えば、SLC2A1遺伝子(溶質キャリアファミリー2,促進されたグルコーストランスポーターメンバー1(GLUT1)をコードする)の変異は、GLUT1欠乏症候群につながるが、これは、乳児期の発作の発症、小頭症、軽度の運動障害、およびBBB破壊の早期発症に関連した発達遅延247によって特徴付けられる108。MFSD2A遺伝子(必須脂肪酸ω3の脳内皮トランスポーターであるナトリウム依存性リゾホスファチジルコリンシンポーター1(NLS1)をコードする)の変異は、致死的または非致死的な小頭症、認知障害、痙縮と不在発話248,249および早期のBBB破壊15を導く。甲状腺ホルモンを中枢神経系に輸送する内皮モノカルボン酸トランスポーター-8(MCT8;SLC16A2によってコードされる)の遺伝的欠損は、重度の精神運動遅滞を伴うアラン・ヘルドン・ダドリー症候群につながる250。OCLN(密結合BBBタンパク質オクルディンをコードする)の変異は、早期発症の発作、小頭症、灰白質石灰化をもたらし、JAM3遺伝子(別の密結合BBBタンパク質JAMCをコードする)の変異は、BBBからの漏出による脳出血と独立膜下石灰化をもたらす251,252。ぺリサイトにおけるPDGFRβ遺伝子の変異はBBBの破壊につながり、発作や運動・認知障害を誘発する基底核における早期発症の微小血管石灰化を特徴とする原発性家族性脳石灰化を引き起こす253,254。
結論
対照的に、我々は、非単発性のヒト神経変性疾患におけるBBB破壊の基礎となる分子機構については、限られた知見しか持っていない(TABLES 1-3)。ほとんどのメカニズムに関する知見は、これらの疾患の動物モデルから得られている6,11。しかし、海馬サブフィールド46,49,65,66,および地域の脳血流と血行動態応答2,拡大血管周囲空間55,およびこれらの血管変化の検出性を高めるために高強度7T磁石を使用して、微小出血の発生率と分布を決定するための地域の脳血流と血行動態応答2,および改善されたイメージング技術の開発は、87,104,これらの血管変化の検出性を高めるために高強度7T磁石を使用して、ヒトにおける将来の神経血管研究のためのかなりの約束を保持している。他の神経変性疾患で使用するための新しい分子リガンド(アルツハイマー病255およびFDG-PET113-123およびベラパミルPET140-142のために現在使用されているアミロイドおよびタウPETリガンドに加えて)の開発(例えば、生体内試験143でBBBにおけるMMP活性を可視化するリガンド、および/または疾患過程で影響を受ける他のBBBトランスポーター、受容体および/または接合タンパク質の活性)は、神経変性における脳血管系の役割についての重要な機械論的洞察を提供すると期待されている。また、脳脊髄液や血液中の血管損傷や修復の新しいバイオマーカーの開発や、神経血管ユニットの他の全身的および細胞特異的バイオマーカー(アストロサイト、神経細胞、オリゴデンドロサイト、ミクログリア、炎症性バイオマーカー、および/またはアルツハイマー病50におけるアミロイドβやタウなどの標準的な疾患バイオマーカーを含む)との関連性を明らかにするための研究も、神経変性や認知機能低下に対する血管の寄与についての理解を深めることになるだろう。
神経変性疾患、認知症、運動性中枢神経系の変化の発症における血管系の役割とは何かという重要な問題に加えて、神経変性過程や認知機能低下の予測における神経血管イメージングや分子バイオマーカーの予後や診断の価値についても新たな疑問が生じてきている。中枢神経系の血管変化が、ヒトの単発性神経BBB疾患6のように、複雑な神経変性疾患における神経変性、脳接続性の喪失、神経細胞の損傷と喪失の発症と進行に寄与する初期の病原性イベントを駆動するとすれば、いくつかの動物モデル71,73,192,196,199で示されているように、神経BBBの停止を治療的に標的とすることで、ヒトにおける神経疾患の経過を逆転させることができるのかという疑問が残る。遺伝学、血管リスク因子、環境、生活習慣が、正常な老化と疾患の間のBBB機能にどのように影響を与え、これが神経障害とどのように関係しているかは、今後の研究のもう一つの重要な焦点である。
本レビューから得られた知見は、健康な脳が正常に機能するためには、健康な血管系が必要であることを示唆している。動物モデルの研究では、BBBとその関連細胞の高度なRNA-seq分子アトラスの開発が始まっているように6,256,ヒトにおける同様の研究は、ヒトBBBの分子レベルでの機能を理解するために追求されるべきである。幹細胞技術を用いて、遺伝的リスクを有する様々な神経変性疾患患者や散発性疾患患者の人工多能性幹細胞(iPSCs)を用いた試験管内試験ヒトBBBモデルを開発することは、神経変性疾患の血管機能を安定化させるための創薬やBBBを標的とした新たなドラッグデリバリーアプローチの開発を進めることになるだろう。今後、BBBは、他のアプローチと組み合わせて、神経変性疾患の予防、停止、最終的には神経変性過程や臨床障害を逆転させるための重要な治療機会と考えている。
キーポイント
- 血液脳関門は、全身循環に存在する因子からニューロンを保護し、シナプスやニューロンの適切な機能に必要な高度に制御された脳内環境を維持している。
- 血液脳関門の破壊は、神経毒性のある血液由来の産物、細胞、病原体の脳内への侵入を可能にし、炎症反応や免疫反応と関連しており、神経変性の複数の経路を開始させる可能性がある。
- 神経イメージング研究では、アルツハイマー病やその他の神経変性疾患における初期の血液脳関門機能障害が実証されており、バイオマーカー・バイオ流体データにも裏付けられており、死後の組織解析でも一貫して観察されている。
- 神経変性疾患における血液脳関門機能障害には、血液脳関門透過性の亢進、微小出血、ブドウ糖輸送障害、P-糖タンパク質機能の障害、血液由来の血管周囲堆積物、細胞浸潤、周皮細胞および内皮細胞の変性などがある。
用語集の項目
血液脳関門 (BBB)
血管壁細胞と血管周囲アストロサイトのエンドフィートによって被覆された、細胞間の接触が密封された脳血管系の連続的な内皮膜で、循環血液と脳の区画を分離する機能を持ち、血液から脳へ、脳から血液への溶質の輸送を厳密に制御する。
ペリサイト
脳の毛細血管内皮を包み込み、血液脳関門の形成と維持に重要な壁細胞である。
神経変性疾患
中枢神経系の様々な領域で神経細胞の変性変化や喪失を引き起こす進行性の神経機能障害。
タイトジャンクション(TJ)
脳内の内皮細胞を緊密に接続し、低い傍細胞透過性と高い経内皮電気抵抗を持つ解剖学的血液脳関門を形成する内皮タンパク質である。
細胞膜拡散 (Transmembrane diffusion)
細胞膜を横切る受動的な輸送の一種で、分子の正味の移動がそれぞれの濃度勾配の下で起こる。
キャリア媒介輸送(CMT)
特定の膜キャリアタンパク質を介して血液脳関門を通過して濃度勾配を下降する分子の輸送
受容体介在性経細胞交代(RMT)
経内皮経細胞交代の際にリガンドと一緒に内在化する膜受容体を介して、血液脳関門を通過する分子の高度に特異的な輸送を行う。
脳脊髄液(脳脊髄液)
脈絡叢で継続的に生成され、脳室系全体を流れる流体である。
脳アミロイド血管症
アルツハイマー型認知症において、小脳動脈や毛細血管の血管壁にアミロイドが沈着して血管変性や小葉微小出血を引き起こし、血液脳関門破壊、梗塞、白質変化、認知機能障害などを引き起こす。
アルツハイマー病血管障害のツーヒット血管仮説
血管損傷は、二次的な神経細胞障害を直接引き起こす初期の障害であり、一方ではアルツハイマー病のアミロイドβ(Aβ)に依存しない方法で、二次的な神経細胞障害を引き起こす(ヒット1)、他方ではAβクリアランスの障害とAb産生の増加による脳内Aβ蓄積を引き起こす(ヒット2)。
アポリポ蛋白Eε4(APOE*ε4)
散発性晩期アルツハイマー病の主要な遺伝的危険因子
動的造影磁気共鳴イメージング(DCE-MRI)
ガドリニウム造影剤に対する局所的なBBB透過性を定量化するために用いた動的MRIシーケンス
T2*重み付け感受性強調画像(SWI)MRI
ヘモシデリン沈着物が低濃度の信号を発生させるMRIシーケンス;生きているヒトの脳の脳微小血流の生体内試験での局所的な測定を可能にする。
18F-フルオロ-2-デオキシグルコース(FDG)
グルコースの18F(F)放射性標識アナログである2-デオキシグルコース(2DG)は、グルコースとは対照的に脳内で代謝されない;FDGは、GLUT1グルコーストランスポーターを介して血液脳関門を越えて脳に取り込まれるグルコースの推定値を提供するために、グルコースのサロゲートPETリガンドとして臨床で使用されている。
低密度リポ蛋白質受容体関連蛋白質-1(LRP1)
血液脳関門におけるアルツハイマー型アミロイドβの主要な排出トランスポーターであり、脳から血液へのアミロイドβのクリアランスに関与している。
放射性14C標識PETリガンドであるベラパミルポジトロン断層撮影法(PET)
生きたヒトの脳の血液脳関門におけるP-糖タンパク質の機能を生体内試験で検出することができる。
高度糖化最終生成物の受容体(RAGE)
アルツハイマー型認知症アミロイドβ(Aβ)の血液脳関門での主要な流入トランスポーターであり、Aβの脳内蓄積、炎症反応、血流抑制、血液脳関門破壊に寄与する。
RNA配列決定
トランスクリプトーム法による生物学的試料中のRNA転写産物の存在と量を明らかにするためのアプローチ
誘導多能性幹細胞(iPSCs)
成体細胞を胚性多能性の状態に再プログラムしたもので、研究研究や治療のために関心のある細胞型に分化させることを目的としている。
