
Contents
pubmed.ncbi.nlm.nih.gov/35108575/
概要
アルツハイマー病(AD)は、認知症の中で最も一般的な疾患である。ADの脳病理は、臨床症状が現れる数十年前から始まっている。初期の病理学的特徴の1つは、バリアー漏出を特徴とする血液脳関門の機能障害であり、認知機能の低下と関連している。
本総説では、ADにおけるバリアー漏出の程度と臨床的関連性についての既存文献を要約する。まず、ADの動物モデルとその年齢や遺伝的背景に基づくバリアー漏出への感受性に焦点を当てる。
第二に、臨床および死後の研究におけるバリア機能不全を再検討し、患者におけるバリア漏出の原因となる変化をまとめ、ADにおけるバリア漏出の臨床的関連性を強調する。
第三に、ADにおける神経変性と認知機能低下にバリアー漏出を関連付けるシグナル伝達機構についてまとめる。最後に、臨床的関連性と潜在的な治療戦略について議論し、ADにおけるバリアー漏出の調査に関する将来の展望を示す。
バリアー漏出のメカニズム的ステップを明らかにすることは、ADおよびAD関連認知症におけるバリアー漏出を修復し、認知機能の低下を遅らせる新しい治療戦略を開発するために使用できる新しいターゲットを解明する可能性を持っている。
キーワード
アルツハイマー病、血液脳関門、バリア機能不全、バリアリーク、神経血管系、脳血管系
1.はじめに
1.1.アルツハイマー病:その背景、診断、病態生理、現在の課題
1.1.1.背景
100年以上前、ドイツの医師アロイス・アルツハイマーは「ある特異な病気」を記述し、後に彼の名を冠した(アルツハイマー病、AD)。アルツハイマーは、この病気を「プラークと神経原線維変化(NFT)を特徴的な病理学的徴候とする先天性認知症」と定義した(Alzheimer,1906,Alzheimer,1907;Fischer,1907,Fischer,1910)。
「認知症」とは、注意、記憶、コミュニケーション、問題解決、視覚認識、自己管理などの認知機能の低下を指す総称である(「認知症に関するファクトシート」)。アルツハイマー型認知症は、最も一般的な認知症であり、不可逆的で進行性の疾患であるため、患者は日常生活を送る上で他者に依存する無力な人となる(「Alzheimer’s Disease Fact Sheet(アルツハイマー病ファクトシート)」)。
初期段階では、AD患者は激越、不安、食欲不振、気分変動、睡眠障害、そして重要なことに認知能力の低下を経験する(「Alzheimer’s Disease Fact Sheet,」;Ehrenbergら,2018)。
病気の進行に伴い、患者の興奮が高まり、記憶喪失のエピソードがより頻繁に発生するようになる。これらのエピソードは、実行機能の低下、視空間能力の障害、さらに言語障害を伴う。不安、食欲不振、気分の落ち込み、睡眠障害などは、この進行したADの段階で落ち着く傾向がある。
ADの後期およびより重症の段階では、妄想や幻覚が出現し、患者は活動性の低い植物状態になる。ADは、診断時の年齢により、早期ADと後期ADに分類される。具体的には、早期発症型ADは30~60歳代で発症し、家族性の遺伝子変異が原因とされている。一方、遅発性ADは、様々な未知の環境要因や生活習慣により、60歳以降に発症する。
1.1.2.診断名
適切なADの診断には、一連の認知検査、画像検査、検査室検査が必要である(Sabbagh,Lue,Fayard,&Shi,2017)。
通常、患者または介護者が記憶に関連する懸念を報告した場合、臨床医は認知評価を推奨する。この評価では、ミニ認知機能評価、一般医認知機能評価、セントルイス大学精神状態検査(SLUMS)、臨床的認知症評価、ミニ精神状態検査(MMSE)など、一つまたは複数の標準的検査によって患者を評価する。
臨床医は、認知を損なう他の原因を除外するために、神経学的検査と代謝検査を実施することもある。さらに、脳脊髄液(CSF)バイオマーカー分析、構造的磁気共鳴画像法(MRI)、陽電子放射断層撮影法(PET)により、脳内のアミロイドやタウの沈着を調べることもある。
これらすべての最先端の診断方法にもかかわらず、ADの最終的な確定診断は、臨床症状、神経病理学的マーカー、プラークとタングルの分布に基づいており、神経病理学者、神経心理学者、神経学者のチームによって死後にのみ行うことができる(Braak&Braak,1991;Ehrenbergら,2018;Nelsonら,2012)。
1.1.3.病態生理
歴史的に、プラークと神経細胞間結合は、AD患者の脳の剖検標本において、変性した神経細胞と血管の周辺で観察されてきた(Divry,1927;Morel,1954;Scholz,1938)。
1960年代の電子顕微鏡と回折法による研究により、プラークとNFTはβプリーツと対のヘリカルフィラメントからなる大きな組織構造であることが示された(Eanes&Glenner,1968;Terry,1963;Terry,Gonatas,&Weiss,1964)。
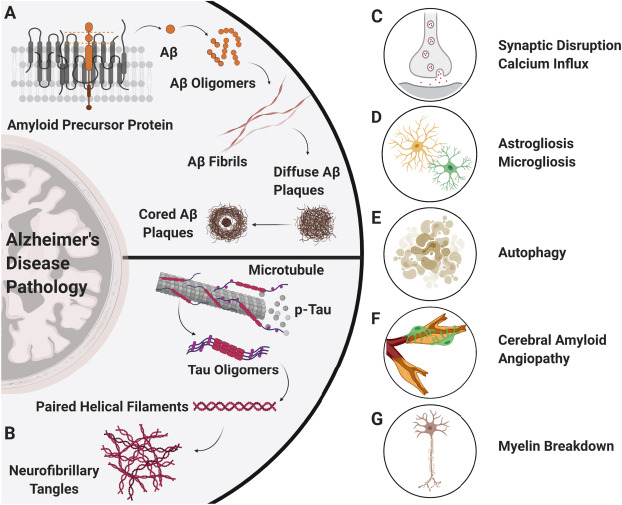
1980年代までに、複数の生化学的研究により、アミロイドベータ(Aβ、4 kDa)がオリゴマー、プロトフィブリル、フィブリルシートとしてこれらのプラークに蓄積することが判明した(図1A)(Glenner&Wong、1984;Masters、1984;Mastersら、1985;Roherら、1996)。
Aβは、アミロイド前駆体タンパク質(APP)が膜貫通領域内でαセクレターゼおよびγセクレターゼによって切断されるときに形成される36-43アミノ酸のペプチドである(Eschら、1990;Sisodia、Koo、Beyreuther、Unterbeck、&Price、1990)。
ADでは、APP処理の変化およびAβクリアランスの障害により、その年齢の個人に通常見られるAβ脳レベルと比較して4〜6倍高いAβ脳レベルが生じる(De Strooperら、1998;FuNATOら、1998;Paresce、Chung、&Maxfield、1997;Roherら 2009;Scheunerら、1996)。
いくつかの研究からのデータは、NFTが、異常に高リン酸化され、AD患者の脳に蓄積するタウオリゴマー、粒状タウ凝集体、およびペアヘリカルフィラメントを形成する微小管関連タンパク質タウ(〜50kDa)を含むことを示した(図1B;Grundke-Iqbal et al,1986;Grundke-Iqbal et al.,1986;Grundke-Iqbal,Johnson,Terry,Wisniewski,&Iqbal,1979;Kopkeet al.,1993;Maeda et al.,2007)である。
タウペプチドは31-32アミノ酸長で、ペプチドあたり2-3個のリン酸分子を持ち、脳内のタウ特異的キナーゼとホスファターゼによって維持されている(Goedert&Crowther,1989;Kopke et al.,1993)。
ADでは、総タウタンパク質および異常にリン酸化されたタウタンパク質のレベルは、健常対照者の脳に見られるタウタンパク質レベルよりも4〜8倍高くなることがある(Iqbal、Liu、&Gong. 2016)。
Aβとタウの両方の脳内蓄積は、AD患者の認知機能に影響を与えるいくつかの病態生理学的変化を引き起こす(図1C-G)。これらの病態生理学的変化の1つは、神経細胞およびアストロサイトにおける細胞内カルシウムの調節異常を伴い、興奮毒性およびアストログリア症を引き起こす(図1C)(Abdulら 2009;Balesら、1998;Mattsonら、1992)。
別の病態生理学的変化は、アストログリア症およびミクログリア症を含み、反応性アストロサイトおよびミクログリアがシナプス伝達に影響を与え、認知障害に寄与する(図1D;(DeKosky&Scheff,1990;Hong et al.,2016;Itagaki,McGeer,Akiyama,Zhu,&Selkoe,1989;Terry et al.,1991;Vogels,Murgoci,&Hromadka,2019)。第3の病態生理的変化は、神経細胞のリソソーム分解能力の低下により、オートファジーと神経細胞の喪失が起こることである(図1E)(Cataldo,Barnett,Mann,&Nixon,1996;Nixon et al,2005)。
第四の変化は、脳血管にAβが蓄積し、経時的に血管壁が破裂する脳アミロイド血管症(CAA)の発症である(図1F)(E. E.Smith&Greenberg,2009;Vinters,1987)。プラークやタングルが脳内の神経細胞に直接影響を与えるのに対し、CAAは脳内に微小出血、梗塞、白質病変を引き起こすことにより、シナプスや神経血管網を破壊する(E. E.Smith&Greenberg. 2009;Vinters、1987)。
第5に、CAAは神経血管機能障害を引き起こし、AD患者の認知機能を悪化させる(Brenowitz,Nelson,Besser,Heller,&Kukull,2015;K.Jellinger,2002)。
これらの変化以外にも、ミエリンの破壊はADにおけるシナプスの結合性に影響を与える(図1G)(Bartzokis et al. 2003)。これらの病態生理学的変化を総合すると、ADは多因子疾患であり、患者のための効果的な治療法を見つける上での課題を増やしている。

図1アルツハイマー病(AD)における中枢神経系の病態変化。アミロイドベータ(Aβ)とタウは、ADの病態進行を促進する2つのキープレイヤーであると提唱されている。A) Aβは、アミロイド前駆体タンパク質(APP)の異常な連続的切断によって形成され、他のAβサブユニットと結合してオリゴマー、プロトフィブリル、フィブリル、拡散型Aβ斑(グリア炎症に囲まれた状態)、神経突起を有するAβ斑を形成して神経変性を引き起こします。
B)一方、微小管に結合したタウタンパク質は異常な過リン酸化を受け、タウオリゴマー、ペアヘリカルフィラメント(PHF)、神経原線維変化を形成し、AD病態の原動力となる。Aβ、タウ、およびそれらのタンパク質凝集体は、脳の神経細胞とグリア細胞の両方に影響を与え、C)カルシウム流入の増加とシナプス伝達の障害、D)アストロサイトおよびミクログリアの炎症、E)オートファジー、F)脳アミロイド血管障害、G)神経細胞周囲のミエリン鞘の破壊を引き起こす。
1.1.4.現在の課題
AD治療薬の開発は5つの領域に集中している。
1)神経伝達系(38%)、2)Aβ病理(33%)、3)神経炎症(17%)、4)タウ病理(10%)、5)コレステロール代謝(2%)、(「Therapeutics」)である。
あらゆる努力にもかかわらず、現在FDAが承認した薬剤は5つしかなく、それらはすべて神経伝達系を標的としている(Di Santo,Prinelli,Adorni,Caltagirone,&Musicco,2013;Molino,Colucci,Fasanaro,Traini,&Amenta,2013)。
これらの薬剤のうち、ドネペジル、ガランタミン、リバスチグミンの3つはコリンエステラーゼ阻害剤であり、シナプス間隙のアセチルコリン濃度を高めることで、コリン性神経伝達を改善し、ADにおける認知機能低下に対抗することができる。
4つ目の薬剤であるメマンチンは、N-methyl-D-aspartate受容体拮抗薬で、過剰なグルタミン酸の作用を阻害し、グルタミン酸による脳のカルシウム興奮毒性を抑制する。5番目の薬剤であるaducanumabは、脳の実質的なAβ凝集体を標的とする(Sallowayら 2009)。
これらの薬剤はすべて、せいぜい認知に対する効果が緩やかで、AD患者の40~70%においてのみ認知症関連症状を低下させる(Di Santo et al.、2013;「アルツハイマー病治療薬の効果」)。さらに、これらの改善は、薬物療法の6~12カ月後に症状が悪化して収まる傾向があり、これらの薬剤はいずれも根本的な病態生理学的プロセスに影響を与えない(「アルツハイマー病治療薬の効果」)。
現在までのところ、疾患修飾効果を期待して開発された実験薬は、臨床試験において認知能力の有意な改善を示していない(Cummings,Lee,Ritter,Sabbagh,&Zhong,2019)。多大な研究努力にもかかわらず、これらの実験薬はどれも市場に出ていない。したがって、AD患者のAD病態の進行と認知機能の低下を遅らせる効果的な疾患修飾戦略が決定的に必要である。
1.2.血液脳関門の解剖学と生理学
1.2.1.解剖学
脳血管網は、脳表面で髄液と髄膜3層に囲まれた大脳動脈(直径200-1000μm)に端を発する(Bevan,Dodge,Walters,Wellman,&Bevan,1999)。
大骨頭動脈からの枝は、脳実質の深部に潜る貫通動脈(70-200μm)を形成する。これらの下行動脈は実質細動脈(20-70μm)を養い、細動脈はより小さい前毛細管(<35μm)に細分化され、最終的に毛細管(3-7μm)を形成する。
この毛細血管は、脳血管網のユニークな部分である。直径3-7μm、厚さ0.1μmの脳毛細血管は、脳血管系の最小の血管であり、血液脳関門の構造基盤を形成する(図2)(Rodriguez-Baeza,Reina-de la Torre,Poca,Marti,&Garnacho,2003)。
構造的には、脳毛細血管内皮細胞は、内腔側に厚さ0.2~2μmのグリコカリックス層、外腔側に50~100nmの血管基底膜に覆われている(図2A、2B)(Chappell,Westphal,&Jacob,2009;L.Xu,Nirwane,&Yao,2019)。
内腔側のグリコカリックス層は、短い膜結合型糖タンパク質だけでなく、長い硫酸化、非硫酸化グリコサミノグリカン鎖の足場であり、他のタンパク質、水分子、酵素、細胞を吸着して内皮細胞を栄養し、損傷、血流変化、毒性物質から保護する(Chappell et al,2009;Schaefer&Schaefer,2010)。
内皮側の基底膜は、コラーゲン、ラミニン、ニドーゲン、フィブロネクチンなどの繊維状プロテオグリカンから構成されている。ここでは、コラーゲンやラミニンのアイソフォームが自己集合し、ニドーゲン、フィブロネクチン、トロンボスポンジンなどのタンパク質や酵素によって保持された一次足場を形成する(図2B)(Thomsen,Routhe,&Moos,2017;L.Xu et al.,2019)。
ペルルカンやアグリンなどのプロテオグリカンは、さらにこのネットワークを安定化し、シグナル伝達機構を調節し、細胞-マトリックス相互作用を制御する(Barber&Lieth,1997;Liebner,Czupalla,&Wolburg,2011;Martin&Sanes,1997;Osada et al,2011;Thomsen et al.,2017)。
基底膜のプロテオグリカンは、毛細血管の血流と神経血管反応を調節するために、毛細血管ごとに周皮細胞をリクルートし、アンカーし、埋め込むこともしている。また、アストロサイトを接着・固定し、酸素やグルコースの供給を調整している(Attwell et al.2010;Hirschi&D’Amore,1996)。
プロテオグリカンがこれらの相互作用を媒介するのを助ける接着分子には、免疫グロブリン細胞接着分子(IgCAM)、接合部接着分子(JAM)、インテグリン、ならびにカドヘリンおよびセレクチンがある(Hynes、1986;Kramer&Marks、1989;Pytela、Pierschbacher、Ginsberg、Plow、&Ruoslahti、1986;Sarelius&Glading. 2015)。
コネキシンおよび他の接合タンパク質は、アストロサイトと周皮細胞の間の接合をさらに封鎖する(図2C-G)(Zhao&Gong,2015)。内皮細胞、周皮細胞、アストロサイト、および神経細胞は、共に「神経血管ユニット」(NVU)を形成し、バリア機能を調節して、脳に出入りするものを制御している(図2C-G)(Iadecola,2004;McConnell,Kersch,Woltjer,&Neuwelt,2017)。
*

図2 健康な状態の血液脳関門
毛細血管内皮の健康な神経血管単位が血液脳関門の構造基盤を形成している。A)脳毛細血管内皮細胞は、内腔側に厚さ200-2000 nmの糖タンパク質に富むグリコカリックス層、B)内腔側に厚さ50-100 nmのプロテオグリカンに富む血管基底膜に覆われている。
血管基底膜のプロテオグリカンは、C)アストロサイトとD)周皮細胞を毛細血管内皮細胞の周囲に固定する。E)粘着分子は、アストロサイトと周皮細胞の間、およびアストロサイトと周皮細胞の間の結合をさらに密閉する。
F-G)内皮界面では、クラウディン-5、オクルディン、ゾヌラオクルーデンス-1,2、3、血小板内皮細胞接着分子-1(PECAM-1)、血管細胞接着分子-1(VCAM-1)、血管内皮(VE)カドヘリンなどのタンパク質によって内皮間接合部がさらに密閉され、完全な血液脳関門を形成している。
1.2.2.生理機能
血液脳関門は、
- 1)内因性、外因性化合物の受動的な脳への取り込みを防ぐ物理的関門、
- 2)外因性物質の脳への取り込みを積極的に制限し、栄養供給を調節する生化学的関門、
- 3)血液脳関門で神経毒、薬剤、その他の化合物を分解する代謝的関門、
- 4)免疫細胞の脳への侵入を制限する免疫学的関門
を意味する。タイトジャンクションは、隣接する内皮細胞の間を密閉することで、物理的な障壁を形成している。この物理的バリアには細胞間隙がなく、ピノサイトーシス活性が低いため、化合物が通過するのは極めて困難である(図 2F)。
内皮細胞の内腔膜と外腔膜にある無数の流入・流出トランスポーターが生化学的バリアを構成している(Abbott,2013;Daneman&Prat,2015;Hartz&Bauer,2011)。
バリアのトランスポーターは、脳に出入りするものを厳密に制御している。内皮細胞内の第I相および第II相代謝酵素が代謝関門を構成している(Agundez,Jimenez-Jimenez,Alonso-Navarro,&Garcia-Martin,2014)。当初、脳には免疫細胞の侵入を防ぐ免疫関門があり、脳は「免疫優遇臓器」であると考えられていた。
この概念は正しくないことが証明され、代わりに、免疫学的バリアは、脳への免疫細胞の出入りを厳密に制御していることがわかった(Greenwood et al.)
*
健康な脳では、血液脳関門は、末梢と中枢神経系を隔てる活発でダイナミックなバリアであり、広範な化合物の脳への侵入を防ぎ、脳の恒常性を保ち、脳に酸素と栄養を供給し、老廃物を除去している。しかし、病気では、このバリアがしばしば損傷し、脳の恒常性の維持や適切な脳機能の維持に必要な機能を維持することができなくなる。
1.3.アルツハイマー病における血液脳関門の漏出
本総説では、ADの血液脳関門に生じる病態生理学的変化の総称として関門機能障害を定義する。これらの変化には、NVUの構造変化、Aβやグルコースの流入・流出輸送機構の変化、血中タンパク質の漏出などがあり、より詳細については、既存のレビュー(Chagnot,Barnes,&Montagne,2021;Jia et al,2020;Kadry,Noorani,&Cucullo,2020;Kirabali et al.,2020;Kyrtata,Emsley,Sparasci,Parkes,&Dickie,2021;Wang et al.,2021)。この記事の残りでは、特にバリアリーケージに焦点を当てる。
*
ADにおける血液脳関門の漏出は40年以上前に初めて観察されたが、漏出した関門がADの病理に寄与していることは最近まで確立されていなかった(Glenner,1979;Iadecola,2013)。
ADにおける一連の病態生理的変化は、NVUに影響を与え、血液脳関門の特性を変化させるため、関門機能障害、ひいてはリークを引き起こす(図3)(Kalaria&Harik,1992;Montagne et al,2020;Richard et al,2010)。
過去10年間、数多くの研究がADにおけるバリア漏出と認知機能低下の関連を示してきた(de la Torre,2017;Di Marco,Farkas,Martin,Venneri,&Frangi,2015;Love&Miners,2016;Montagne et al,2020;Nation et al,2019;Zhao&Gong,2015)。このセクションでは、ADの間にこれらのNVUの各構成要素に生じる最も顕著な変化について概説する。

図3 アルツハイマー病における血液脳関門
ADにおける神経血管系の病理学的変化には、A)凝固・血栓の増加、B)内皮細胞の構造変化と内皮間結合の破壊、C)基底膜のプロテオグリカンと糖タンパク質のレベルの変化、D)アストロサイト末端部の腫脹、内皮細胞周囲のアストロサイト被覆の消失などのアストロサイトの構造変化、E)クラスマトデンドロス、F)樹状突起結合の消失に加えて、G)内皮細胞周囲のペリサイト。
1.3.1.内皮細胞
ADでは、内皮細胞は、内皮間結合を破壊する構造変化を起こし、その結果、他のNVU構成要素との結合や相互作用を制限する(図3A、3B)(Oikari et al.、2020)。
例えば、いくつかの研究のデータは、CAAのような状態では、内皮細胞において血小板内皮細胞接着分子(PECAM-1/CD31)の脱落およびアポトーシスが起こることを示している(Magaki et al.,2018;Nielsen,Londos,Minthon,&Janciauskiene,2007;Xue et al.,2012)。
さらに、細胞間接着分子1(ICAM-1)や血管細胞接着分子1(VCAM-1)などの他の接着分子は、タイトジャンクションタンパクの破壊、アクチン応力繊維形成の増加、内皮細胞の変化(Clark、Mans、Pober、&Kluger. 2007;Haarmann et al. 2015)に寄与している。
これらの変化は、血液脳関門での副細胞輸送を混乱させる。さらに、内皮細胞は、マクロピノサイトーシス、クラスリン依存性エンドサイトーシス、カベオレ媒介トランスサイトーシス、または小胞輸送を介して血液由来物質の細胞外移動の増加も示す(De Bockら、2016;Gurnikら、2016;Panditら、2020;Zhouら、2021)。
傍細胞経路および細胞横断経路の増加は、ADにおけるリーキーなNVUに寄与する。糖鎖と基底膜の変化。糖衣と基底膜の構造的・生化学的変化は、ADのNVU全体における細胞-細胞間および細胞-マトリックス間の接着を変化させる(図3B、C)。
複数の研究からのデータは、これらの変化が脳内のプロテオグリカンレベルの上昇に起因していることを示している。Fillit,Kemeny,Luine,Weksler,and Zabriskie(1987)とJenkins and Bachelard(1988)は、AD患者の脳サンプルでは、非発症者のサンプルと比較してプロテオグリカンの総量が増加していることを示している。
例えば、グリピカン(60-70kDa)やシンデカン(20-30kDa)のような小さな細胞表面プロテオグリカンは、CAA病理を示す毛細血管の周りに多く存在する(Lashleyら,2006;Lorente-Geaら,2020;van Horssenら,2001;Watanabeら,2004)。
ヒアルロン酸(HA)は、AD患者のCSFサンプルで上昇するもう一つの糖鎖および毛細血管基底膜プロテオグリカンである(Nagga,Hansson,van Westen,Minthon,&Wennstrom,2014;Nielsen,Palmqvist,Minthon,Londos,&Wennstrom,2012)。
この点で、HAタンパク質レベルの上昇は、AD患者におけるBraakステージングの増加および認知低下の程度と関連していることは注目に値する(Reed et al.、2019)。コラーゲンとアグリンは、ADにおける基底膜の肥厚と断片化に寄与する他の2つのプロテオグリカンである(Christov,Ottman,Hamdheydari,&Grammas,2008;Keable et al.)
Agrinは、認知的に正常な被験者の脳微小血管に沿った均一な免疫反応と比較して、AD患者の脳微小血管内で「ぼろぼろで不規則な」免疫反応を示す(Donahueら、1999;van Horssenら 2001)。
アグリンの断片化もADの進行とともに増加し、脳サンプル中の可溶性/断片化アグリン濃度が高くなり、不溶性/全長アグリン濃度が減少する(Berzinら 2000;Sallowayら 2002)。まとめると、毛細血管基底膜のプロテオグリカン組成の変化は、ADにおけるバリアー保全に影響を及ぼすということである。
1.3.2.アストロサイト
ADでは、アストロサイトは、「クラスマトデンドロシス」、すなわち、細胞質空胞化、萎縮、およびアストロサイト端脚の膨張とともにアストロサイト突起のビーディングおよび崩壊につながる構造変化を受ける(図3D、3E)(Boespflug et al,2018;Cullen,1997;Higuchi,Miyakawa,Shimoji,&Katsuragi,1987;Mancardi,Perdelli,Rivano,Leonardi,&Bugiani,1980;Montagne,Zhao,&Zlokovic,2017;Sweeney,Sagare,&Zlokovic,2018;Yamashita,Miyakawa,&Katsuragi,1991)が挙げられる。
末端足部では、アクアポリン-4(AQP4)、内方整流カリウムチャネル(Kir4.1)、およびジストロフィン1などのアンカータンパク質のレベルが低下し、アストロサイト-内皮接続をさらに制限する(Boespflugら、2018;Wilcock,Vitek,&Colton,2009;Zeppenfeldら,2017)。
これらの構造変化は、NVUでのアストロサイトのアンカーリングを減少させ、脳血流および神経血管の相互作用を減少させる(Iadecola,2013)。
1.3.3.ニューロン
神経細胞は、シナプス付近の神経細胞-グリア界面でグルタミン酸、アセチルコリン、またはγ-アミノ酪酸などの神経伝達物質を分泌することにより、脳血管の組織と脳血流を調節する(Girouard&Iadecola,2006;Hendrikx et al,2019;Lacoste et al,2014;Muoio,Persson,&Sendeski,2014;Petzold&Murthy,2011)。
ADにおける広範なシナプス機能障害およびニューロン損失は、したがって、脳血管緊張および脳血流(図3F)に影響を与え得る(Babic,1999;Hamel,2004;Kirkwoodら,2016;Mesulamら,2019;Ohら,2019;Van Beek&Claassen,2011)。
初期の死後研究でも、AD患者の脳組織スライスの皮質ニューロンがNVUでアグリンやオクルディンなどのタンパク質に免疫反応することが示されている(Berzin et al.)一方、CSF分析では、ADにおける神経細胞傷害のマーカーと血液脳関門の機能障害との強い関連性は示されていない(Muszynski et al.、2017)。したがって、神経変性とバリア機能不全の関係については、さらなる研究が必要である。
1.3.4.周皮細胞
ADでは、周皮細胞は積極的な分解を受ける(Hallidayら、2016;Wilhelmusら 2007)。さらに、周皮細胞に沿って発現する神経/グリア抗原-2(NG2)などの細胞表面受容体は、広範な脱落を受け、これらの受容体は機能しなくなる(図3G)(Halliday et al.)
ヒト組織サンプルを用いた死後研究のデータは、変性した血管に沿った周皮細胞が、ADにおいてリポフスチン様物質およびAβ線維を蓄積することを示している(Baloyannis&Baloyannis,2012;Raha et al.,2017;Szpak et al.,2007)。
Aβ病理が進行すると、これらの周皮細胞は、Aβクラスターおよびコラーゲン線維を負荷した血管基底膜に十分に接着しなくなる(Szpakら 2007)。
In vitroでは、Aβ線維は、周皮細胞-基底膜相互作用をサポートするプロテオグリカンである可溶性NG2レベルを低下させる(Schultz、Nielsen、Minthon、&Wennstrom. 2014)。
AD患者(n=13)の海馬スライスは、非発明者(n=9)の海馬スライスと比較して、毛細血管あたりのNG2陽性周皮細胞が1.5倍低い(p<0.05)(Schultz et al.、2018)。
さらに、周皮細胞特異的可溶性タンパク質の低レベルは、AD患者のエピソード記憶、意味記憶、知覚速度、視空間能力、およびグローバル認知の低い認知スコアと直接関連している(Bourassa,Tremblay,Schneider,Bennett,&Calon,2020)。
1.3.5.血液脳関門の機能不全による関門漏出
私たちは、バリア機能障害を、NVUにおけるすべての病態生理学的変化の包括的用語として定義したが、バリアリークとは、特に、血液由来物質の脳への経細胞的および傍細胞的移動の増大を意味するものである。
血管周囲のAβクリアランスの障害、酸化ストレス、プロテアソーム分解の低下などの持続的なストレス因子は、NVUに持続的な傷害と変性を引き起こし、血液脳関門漏出を引き起こす可能性がある。
メタアナリシスでは、関門漏出は、ADにおける一般的な臨床および死後の観察であり、認知機能の低下を悪化させ、登録されたアルツハイマー病患者における実験的治療戦略の結果に影響を与えることが示されている(Farrall&Wardlaw,2009;Janelidze et al,2017)。
4.アルツハイマー病患者におけるバリアリーケージの死後証拠
本節では、ADの病理学的変化、バリア機能障害のマーカー、バリアリークとの関連について述べた研究を中心に紹介する。具体的には、バリアー漏出と相関する血液脳関門の構造的変化について述べる。次に、IgG、プロトロンビン、フィブリノゲンがバリアー漏出の代用マーカーであることを明らかにした研究に焦点を当てる。
4.1.タイトジャンクションタンパク質の変化
ZO-1、オクルディン、クローディン-5などのタイトジャンクション(TJ)タンパク質は、内皮細胞をしっかりと密封し、脳への溶質の細胞外輸送に対するバリアを形成している。ADでは、TJタンパク質は、分解、非局在化、破壊、または消失し、バリア漏出および認知機能低下に寄与する(Carranoら、2011;Debette、ら、2015;S.Leeら、2018)。
Fialaら(2002)は、AD患者(n=8)の組織スライスの脳毛細血管におけるZO-1の染色パターンが、非認知症者(n=5)の組織スライスの毛細血管におけるZO-1の線形かつ連続したパターンと比較して歪んでいることを示した。Romanitanら(2007)は、AD患者(n=4)の脳組織スライスの毛細血管におけるオクルディン発現が、非認知症者(n=5)と比較して増加することを報告した。
別の研究では、Viggarsら(2011)は、軽度(Braak I/II;n=28)、中等度(Braak III/IV;n=47)、および高度(Braak V/VI;n=17)のAD病変の患者の組織切片で検査した毛細管の76-80%がZO-1の染色パターンの乱れとclaudin-5の染色パターンの拡散を示していると報告している。
第4の研究において、Yamazakiら(2019)は、Aβ病理を持たない認知的に正常な人(n=10)と比較して、AD患者(n=19;p<0.05)の側頭、内腸、帯状皮質において、クローディン-5タンパク質レベル(p<0.05)が減少し、一方オクルディンタンパク質レベルが減少していることを見いだした。
*
CAAでは、TJタンパク質の発現パターンが破壊されている。例えば、Carranoら(2011)は、重度のAD(Braak III-VI)と軽度から重度の毛細血管CAA病変を有するアルツハイマー病患者の後頭葉皮質スライスにおいて、Aβで覆われた毛細血管周辺では、Aβを含まない毛細血管と比較してクローディン5、オクルディンおよびZO-1の免疫反応性が60-80%減少することを観察している。以上のことから、TJタンパク質の発現障害はバリア機能障害の根本的なメカニズムであり、AD患者の死後脳組織サンプルにおけるバリア漏出の指標となる可能性が示唆された。
4.2.基底膜の変化
毛細血管床に沿った変化したまたは「肥厚した」基底膜は、ADにおけるバリア漏出のもう一つの指標である(Mancardiら、1980;Scheibel&Duong、1988;Scheibel,Duong,&Tomiyasu,1987;Yamashitaら、1991)。Scheibelら(Scheibel et al.,1987;Scheibel&Duong,1988)は、AD患者の脳組織スライスの毛細血管に沿った複数の場所で基底膜の肥厚が「不規則」で「穿孔」しているが、これらの穿孔の下の内皮細胞ライニングは損傷していないことを見いだした。
さらに、KalariaとPax(1995)は、AD患者(n=18)の皮質組織スライスにおいて、コラーゲン-IVに対して免疫反応を示す肥厚した基底膜が崩壊した毛細血管の周りに観察されたが、非発達者(n=9)のスライスではこれらの包膜は存在しなかった。他の研究者は、AD患者(n=9)の脳毛細血管基底膜のコラーゲンI(8〜9倍)、コラーゲンIII(8〜9倍)、コラーゲンIV(5倍)のレベルが非認知症者(n=9)と比較して高いことを発見した(Christov et al. 2008)。
*
コラーゲンレベルの上昇は、基底膜の肥厚とCAAの病態を示唆するものである。Vinters(1987)の研究では、著者らはコラーゲンIVの沈着が重度のCAAの病理を持つ無傷の毛細血管の周囲に頻繁に存在することを示した。一方、van Horssenら(2001)は、AD患者から得た73個の後頭葉皮質中37個の前頭葉皮質中50個の検査で、コラーゲンIV沈着がCAA病理と関連していることを見いだした。
さらに、コラーゲンIVの沈着は、ADにおける微小出血の高い発生率と相関している。特に、Cullenら(Cullen,Kocsi and Stone,2005,Cullen,Kocsi and Stone,2006)は、AD患者(n=11)の皮質脳組織スライスにおいて、ヘムやフィブリノーゲンの周りにこれらのコラーゲンIV沈着物が「ハロー」として存在することを発見した。これらの研究を総合すると、コラーゲンの沈着や基底膜の肥厚は、ADにおけるバリアー漏出の指標となり得ることが示唆される。
4.3.アストロサイトの異常
ADでは、腫脹した反応性血管周囲アストロサイトは、しばしばフィブリノーゲンやIgGなどの障壁漏出マーカーと共局在し、TJタンパク質を破壊して障壁漏出を誘導するエンドセリン-1(ET-1)やVEGFなどの伝染性エンハンサーを放出する(Fouda,Fagan,&Ergul,2019;Michinaga&Koyama,2019;Tomimoto et al,1996)。
これらのエンハンサーのレベルは、非発明者のサンプルと比較して、AD患者の脳組織サンプルで上昇する。例えば、いくつかの研究からのデータは、非認知症患者からのものと比較して、AD患者からの皮質脳スライスにおけるET-1レベルの上昇を示す(Minami,Kimura,Iwamoto,&Arai,1995;Palmer,Barker,Kehoe,&Love,2012;W.W.Zhang et al.,1994)。
他の研究からのデータは、非発達者のものと比較して、AD患者の皮質組織スライスにおける細動脈、静脈、および毛細血管に沿ったVEGF免疫反応性の増強を示す(Provias and Jeynes,2008,Provias and Jeynes,2011,Provias and Jeynes,2014;Thirumangalakudi,Samany,Owoso,Wiskar,&Grammas,2006;Thomas,Miners,&Love,2015)。
また、ET-1およびVEGFのレベルが、脳血管障害およびバリアー漏出のマーカーと相関していることを強調する研究もある。この点に関して、Thomasら(2015)は、AD患者(n=20)の脳ホモジネートにおけるET-1タンパク質レベルが、脳低灌流の指標である低ミエリン関連糖タンパク質/タンパク質脂質比と相関していることを示した。
同様に、Zhangら(2016)は、CMBを発症したアルツハイマー病患者(n=47)では、CMBを発症していないアルツハイマー病患者(n=99)と比較してVEGF血清レベルが2倍高いことを報告している。Janelidzeら(2017)は、MCIおよびアルツハイマー病患者において、CSFのVEGFタンパク質レベルがQalb値と相関することを示した。
*
以上のことから、VEGFやET-1などの脳内伝染性調節因子レベルは、ADの脳血管機能障害やバリアー漏出の指標と相関することが示された。
4.4.周皮細胞の被覆の変化
ADでは、周皮細胞はしばしば構造的および機能的な異常を示す(Miners,Kehoe,Love,Zetterberg,&Blennow,2019)。1987年、Scheibelら(1987)は、死亡したアルツハイマー病患者(n=15)の脳組織スライスで見つかった不規則な形の毛細血管が、周皮細胞体に似た丸い円錐形の突出部を示したと報告した。
しかし、非痴呆者(n=10)の脳組織では、毛細血管床に沿ったこのような突出部は時々しか生じない(Scheibel,1987;Scheibel et al.)さらに、Stewart、Hayakawa、Akers、Vinters(1992)は、AD患者において毛細血管周囲の周皮細胞の増加、壁の厚さの減少、平均内径の減少、裂け目の増加、単位血管長あたりの内皮間結合数の増加、平均結合長の増加などを見出した。
一方、死後のAD脳組織のより最近の分析では、周皮細胞の減少がバリアリークを示すことが示されている(Hallidayら、2016;Miners,Schulz,&Love,2018;Sengilloら、2013)。
2013年、Sengilloら(2013)は、AD患者(n=6)の海馬および皮質脳組織スライスのペリサイト数が、非認知症者(n=6)のペリサイト数よりそれぞれ0.4倍および0.6倍低いことを報告している。
この研究では、AD患者の皮質および海馬スライスにおいて、周皮細胞数は、血管外IgGおよびフィブリンレベルと逆相関していた。別の研究では、同じグループが、周皮細胞数がIgGおよびフィブリン免疫反応性およびアルツハイマー病患者のAPOE e4状態と逆相関することを示した(Hallidayら、2016)。
別の研究では、Minersら(2018)は、AD患者(n=49)の頭頂葉前組織スライスにおいて、血小板由来成長因子受容体-β(PDGFRβ)免疫反応とフィブリノーゲン免疫反応および低脳酸素化レベルとは逆相関していることを示した。
一方、これらの相関は、非認知症者(n=37)では観察されなかった。Minersら(2019)はまた、可溶性PDGFRβのCSFレベルの上昇は、MCIおよびアルツハイマー病患者のサンプルにおけるQalbおよび海馬のKtrans値の上昇と相関することを実証した。これらの研究をまとめると、周皮細胞の構造変化および周皮細胞数の減少は、ADにおける脳血管機能障害およびバリアリークと相関することが示される。
4.5.IgG免疫反応
死後脳組織試料中のIgGの存在は、ADにおけるバリアー漏出の指標と考えられる最も早い時期に発表された所見の一つである。1970年代に石井ら(Ishii and Haga,1975,Ishii and Haga,1976)は、死亡したアルツハイマー病患者の脳組織スライスにIgGの滲出を認めたが、当時はこの所見の背景にある病態メカニズムは不明であった。
他の研究グループも同様の観察を行い、活性化した補体経路がアルツハイマー病患者の脳実質にIgGの蓄積を引き起こしているという仮説を立てた(Alafuzoff et al.,1987;Eikelenboom&Stam,1982;Mann,Francis,Hoffman,&Montes,1982;Powers,Sullivan,&Rosenthal,1982)。Goust,Mangum,and Powers(1984)は、溶出実験のデータに基づいて、脳内のIgGは、実質的な免疫反応からではなく、バリアーが低下したために、細胞外液のコンパートメントから来る可能性が高いことを示唆した。
1989年、Liu,Atack,and Rapoport(1989)は、IgGの脳への継続的蓄積は、逆行性神経細胞輸送または血液脳関門を持たない脳室周囲器官を介して起こることを示し、別の選択肢を提示した。このようなIgGの輸送形態は、脳の脳室周囲器官で血液-CSFバリアを形成する「タニーサイト」と呼ばれる特殊な上衣細胞の存在により、考えにくい(Langlet,Mullier,Bouret,Prevot,&Dehouck,2013)。
これらの研究を総合すると、脳実質におけるIgG免疫反応性は、ADで生じるいくつかの病的過程(例えば、実質内合成やミクログリア反応)と関連しており、したがって、関門漏出の特異的マーカーとはなりえない可能性があることが示された。
4.6.プロトロンビン免疫反応
プロトロンビンもまた、ADにおけるバリアー漏出の代替マーカーとしてしばしば用いられる血液中のタンパク質である。プロトロンビン(72-74kDa)は肝臓で合成され、凝固の開始時にトロンビン(37kDa)に変換される。Lewczuk,Reiber,and Ehrenreich(1998)は、健常者(n=18)において、プロトロンビンの95%が血液中に存在し(95-150 mg/L)、CSF中には1%以下(0.1-1 mg/L)しか存在しないことを示した(Smirnova et al,1997)。
一方、死後組織を用いた複数の研究により、プロトロンビンがアルツハイマー病患者の脳実質に蓄積していることが強調されている(Akiyama,Ikeda,Kondo,&McGeer,1992;Berzin et al,2000;Borroni et al,2002;Lewczuk et al,1999;Mari et al,1996;Zipser et al.2007)。これらの研究のいくつかは、脳実質におけるプロトロンビンの蓄積がバリアー漏出と関連していることを示した(Berzinら,2000;Lewczukら,1999;Zipserら,2007)。
具体的には、Berzinら(2000)は、異なるBraakステージ(Braak I/II,n=9;Braak III/IV,n=7;Braak V/VI,n=7)のアルツハイマー病患者から得た脳組織スライスにおけるプロトロンビン免疫反応を評価し、非発症者(n=12)と比較検討した。
その結果、AD患者の脳組織スライスでは、細く曲がりくねった毛細血管の周囲でプロトロンビン免疫反応性が上昇していたが、非認知症者の脳組織スライスでは上昇していないことが示された。Zipserら(2007)は、より大規模な死後脳組織のコホート(Braak I/II,n=27;Braak III/IV,n=29;Braak V/VI,n=27;非発明例,n=10)を調査し、非発明例と比較してBraakステージV/VIにおけるアルツハイマー病患者からのサンプルでは縮んだ内皮細胞により多くのプロトロンビン免疫反応が生じていることが確認されている。
一方、Braak病期I/IIのアルツハイマー病患者および非健常者の脳切片では、プロトロンビン免疫反応性は毛細血管内腔に限定されていた。これらの結果から、脳におけるプロトロンビンおよびトロンビンの免疫反応性は、ADにおけるバリアー漏出の潜在的な指標であることが示唆された。
プロトロンビンは脳内で合成される可能性があり、特に損傷した状態ではプロトロンビンを漏出指標として用いる場合には慎重な解釈が必要である(Arai,Miklossy,Klegeris,Guo,&McGeer,2006;Weinstein,Gold,Cunningham,&Gall,1995)。
4.7.フィブリノーゲン免疫反応
フィブリノーゲン(340kDa)は、死後脳組織のバリアー漏出を確認するために頻繁に使用される糖タンパク質である。フィブリノーゲンは肝臓で合成され、血液中を循環している。血管損傷時には、フィブリノゲンはフィブリン(0.15 kDa)に変換され、血栓を形成して血管損傷部位を塞ぐ。AD患者においては、フィブリノゲンの血中濃度が上昇し、大きな無秩序な血栓を形成して内皮機能障害を誘発する(E. B.Smith,1986;van Oijen,Witteman,Hofman,Koudstaal,&Breteler,2005)。
試験管内試験の研究データでは、フィブリノゲンがF-アクチン繊維の形成を開始し、これが内皮間結合の拡大とアルブミン漏出につながることが示されている((Patibandlaら 2009;Tyagi,Roberts,Dean,Tyagi,&Lominadze,2008)。
脳実質では、フィブリノゲンはアルブミンと不溶性の複合体を形成し、分解に抵抗性があり、免疫化学的に検出することができる(Adams,Passino,Sachs,Nuriel,&Akassoglou,2004;Chung et al,2016;Cortes-Canteli et al.,2015;Cullen,Kocsi and Stone,2005,Cullen,Kocsi and Stone,2006;Lipinski&Sajdel-Sulkowska,2006;Viggars et al.,2011)。
Fialaら(2002)は、AD患者(n=3)の側頭葉皮質スライスにおいて、非発症者(n=4)のものと比較して、フィブリノーゲン免疫反応性が7倍高い(p<0.05)ことを報告した。Ryu and McLarnon(2009)は、AD患者(n=8)の内嗅皮質スライスにおいて、非痴呆者(n=7)のスライスと比較してフィブリノーゲン免疫反応性が4.5倍(p=0.003)高かったと報告している。
毛細血管CAAを有するアルツハイマー病患者(n=8)の白質組織において、Magakiら(2018)は、非認知症者の組織(n=10)と比較して2倍のフィブリノーゲン免疫反応性の上昇(p<0.05)を見いだした。他の研究者は、AD患者の楔前部サンプルにおいて、非認知症者(n=37)のものと比較して、フィブリノゲン蛋白質レベルが1.2倍高い(p=0.0026;n=49)ことを発見した(Minersら、2018)。
*
血管外フィブリノゲンは、脳血管障害の重症度と相関がある。Cullenら(Cullen,Kocsi and Stone,2005,Cullen,Kocsi and Stone,2006)は、AD患者(n=11)の脳スライスにおいて、非発達者(n=10)と比較して、細動脈、静脈、毛細血管の分岐点に多くあるAβ沈着とヘムに富む沈着に隣接したフィブリノーゲン免疫反応性を見いだした。
Minersら(2018)は、重度のCAA病理を有するアルツハイマー病患者の脳組織サンプルにおいて、CAA病理を有さないアルツハイマー病患者のサンプルと比較して、フィブリノゲン蛋白質レベルが1.2倍高く(p<0.01)、フィブリノゲンレベルと血管Aβ沈着の関連を示唆することを見いだした。
また、血管外フィブリノゲンは、重度の脳灌流低下と直接相関することが明らかとなった。フィブリノゲン合成は損傷した脳で起こることが知られているため、フィブリノゲンを漏出指標として使用する場合は慎重な解釈が必要である(Golanovら、2019)。
*
フィブリノーゲンに加えて、フィブリンも脳内に蓄積し、主に前頭葉皮質、海馬、内嗅皮質、海馬形成、海馬傍回に蓄積する。Cortes-Canteliら(2015)は、AD患者(n=29)の皮質脳組織サンプルでは、非認知症者(n=8)のサンプルで検出されたレベルと比較して、フィブリンタンパク量100倍(p<0.05)であることを示している。
AD患者の海馬のサンプルでは、非認知症の人のサンプルと比較して、フィブリンレベルが20倍高かった(p<0.01)。血管外へのフィブリン沈着は、毛細血管周辺の「血管周囲雲」としても存在した。
*
これらの研究により、AD患者の死後脳組織サンプルにおいて、血管外層のフィブリノゲンとフィブリンがバリア漏出の代用マーカーであることが確認された。
5.アルツハイマー病におけるバリアリーケージの分子機構
ADにおけるバリアー漏出の分子メカニズムは、まだ十分に解明されていない。このセクションでは、試験管内試験の脳内皮細胞株、AD患者の死後脳組織サンプル、あるいは生体内試験の動物モデルでこれまでに同定された最も顕著なメカニズムを要約する(図5)。
*

図5 アルツハイマー病における血液脳関門漏出のメカニズム
A) invitroあるいは生体内試験で血液脳関門漏出を誘発することが以前に示された神経血管ユニットの膜貫通型受容体とそのリガンドの概略図。
B)これらのリガンドと受容体のペアの多くは、PI3K/Akt、MAPK、RhoA/ROCKなどの一つ以上の下流シグナル伝達経路を引き起こし、毛細血管内皮細胞内および周辺に生理的変化を引き起こす。
C)これらの変化の中には、タイトジャンクションタンパク質の発現低下、タイトジャンクションタンパク質の内皮間接合部からの非局在化、タイトジャンクションタンパク質の分解、または内皮細胞の細胞骨格構造の変化などがある。
5.1.低酸素によるバリアー漏出
低酸素は、内皮細胞におけるバリアー漏出の引き金となる(Brown&Davis,2005;Lochhead et al.)低酸素におけるシグナル伝達経路の一つは、転写因子である核因子赤血球2関連因子2(Nrf2)の活性化を伴う。
この経路では、低酸素がNrf2を活性化し、核内に移動してヘムオキシゲナーゼ-1(HO-1)やNAD(P)Hデヒドロゲナーゼ(NQO1)などの標的遺伝子のプロモーター領域の抗酸化応答要素(ARE)に結合して、抗酸化物質の転写と翻訳を開始し活性酸素種(ROS)生成を抑制する(Kensler,Wakabayashi,&Biswal,2007;)。
Biswal,2007;McSweeney,Warabi,&Siow,2016;Tebay et al.,2015).しかし、この経路は、Aβが媒介するROSレベルの増加により、ADにおいて制御不能となる(Butterfield,2018;Fao,Mota,&Rego,2019)。
ROSは、血液脳関門のZO-1、クローディン-5、およびオクルディンを変化させ、TJ複合体を分解する(Carranoら、2011;Hyeon、Lee、Yang、&Jeong. 2013;Wan、Chen、&Li. 2014)。
活性酸素の発生を介してバリアー漏出を引き起こすもう一つのシグナル伝達経路は、ブラジキニンシグナル伝達である。ADでは、tPA活性の上昇によりブラジキニン経路がアップレギュレートされる(Medinaら 2005;Singh、Chen、Ghosh、Strickland、&Norris. 2020)(図5A)。
メカニズム的には、tPAはプラスミノーゲンをプラスミンに変換してブラジキニンを生成し、内皮細胞上のブラジキニン受容体を活性化する(Gauberti,Potzeha,Vivien,&Martinez de Lizarrondo,2018;Marcos-Contreras et al,2016;Medina et al,2005;Singh et al.)ブラジキニン受容体を活性化すると、動脈/細動脈が拡張し、静脈/細静脈が収縮して、脳低灌流、血管損傷、バリアリークをもたらす(Marcos-Contreras et al.、2016)。
脳低灌流はさらにROSを発生させ、TJタンパク質を破壊し、IgGの血管外遊出を増加させるが、これらはすべてAPP23マウスで観察される現象である(Shangら、2019;S.Shangら、2016;Shiら、2019)。
第3のシグナル伝達経路は、ADで発現が増加するアンジオテンシンを含み、5xFADマウスのROSおよびIgG脳内濃度の上昇をもたらす(Takane et al.、2017)。ROSに加えて、一酸化窒素はバリアリーケージの誘導に関与する重要なプレーヤーである。Logsdonらによる最近の研究では、一酸化窒素合成酵素が、外傷性脳損傷の72時間後まで、マウスの血液脳関門のクローディン-5を一過性に破壊することが確認された(Logsdonら、2018)。
これらの効果は、一酸化窒素合成酵素阻害剤であるN(G)-ニトロ-L-アルギニンメチルエステル(L-NAME)で処理したマウスで軽減された。ADにおけるバリアリークに対する一酸化窒素合成酵素の影響については、AD動物モデルでまだ研究されていない。
5.2.細胞骨格の変化とバリアリーケージ
細胞骨格タンパク質は、TJタンパク質複合体を安定化させることによって、バリアーの完全性を制御している(Gonzalez-Mariscal,Betanzos,Nava,&Jaramillo,2003)。この点に関して、細胞骨格の変化は、アクチン重合、アクチン-ミオシンストレスファイバーの形成、およびカルシウムの調節異常につながり、最終的にTJタンパク質複合体を不安定にする(Carman,Mills,Krenz,Kim,&Bynoe,2011;M. K.Choi,Liu,Wu,Yang,&Zhang,2020)(図5C)。
TJタンパク質複合体を不安定化する経路の1つに、アデノシンシグナル伝達がある。簡単に説明すると、脳損傷は、アクチン結合タンパク質のリン酸化、ストレスファイバーの形成、および内皮間接合部からのTJタンパク質の剥離(図5A)を誘導するアデノシン・シグナル伝達を活性化する(Carman et al,2011;Garcia-Ponce,Citalan-Madrid,Velazquez-Avila,Vargas-Robles,&Schnoor,2015;C.Guo et al.,2020;Hicks,O’Neil,Dubinsky,&Brown,2010;Koss et al.,2006;Kuhlmann et al.,2007)。
生体内試験研究のデータによると、損傷によってアデノシンレベルが上昇し、その結果、APP/PS1マウスの10kDaおよび70kDaデキストランの血液脳関門伝染性が増加する(Carmanら、2011)。
もう一つのシグナル伝達経路はカルシウムによるもので、正常な状態ではカルシウムはE-カドヘリンとオクルディンに結合し、脳内皮細胞株の細胞間接着を安定化させる。しかし、カルシウムが過剰になると、内皮細胞間接合部において細胞骨格からタイトジャンクション複合体が解離する(Pokutta,Herrenknecht,Kemler,&Engel,1994;Ye,Tsukamoto,Sun,&Nigam,1999)。
In vitroの研究により、Aβ42の高度糖化最終生成物受容体(RAGE)の活性化が、bEnd.3細胞におけるカルシウムのホメオスタシスを調節することが明らかになった(Ahnら、2018;Kookら、2012;Kookら、2014)(図5A)。
RAGEに結合するAβ42はまた、哺乳類ラパマイシン標的(mTOR)またはカルモジュリン依存性タンパク質キナーゼ-β/AMP活性化タンパク質キナーゼシグナルを活性化することによってアドヘレンス接合タンパク質を破壊する(Chen,Li,Wang,&Liu,2020;Van Skike et al.,2018)(図5A、図5B)。
障壁漏出を駆動する別のメカニズムは、ADにおけるいくつかのマイトジェン活性化タンパク質(MAP)キナーゼのアップレギュレーションを含む(Rodriguez-Perdigon,Solas,&Ramirez,2016;S.Shang,Yang,et al,2016;Zhu et al,2001)(Fig.5B)。
周皮細胞における細胞骨格の再編成もバリアー完全性に影響を与え、これらの変化はRhoA/Rho-associated protein kinase(RhoA/ROCK)シグナルに起因するものである。この点に関して、Parkら(2017)は、5xFADマウスでは、野生型マウスと比較して、周皮細胞が低レベルのアネキシンA1を分泌することを示した。アネキシンA1は内因性のRhoA/ROCKシグナル阻害剤および抗炎症タンパク質であり、Parkら(2017)は、低レベルのアネキシンA1レベルが障壁漏出を開始させることを示した(図5C)。
5.3.成長因子を介したバリアー漏出
VEGF、トランスフォーミング増殖因子β(TGFβ)、顆粒球マクロファージコロニー刺激因子(GM-CSF)の3つの成長因子がバリアリークに影響を与えることが示されている。
VEGFやTGFβは、様々なシグナル伝達経路によって神経細胞の生存や神経細胞/グリア細胞の増殖を促進する一方で、内皮細胞において接着タンパク質をリン酸化しTJタンパク質を分解するSrcキナーゼを活性化することによってバリアリークを誘発する(Croll&Wiegand,2001;Garcia-Barroso et al,2013;R.Paul et al.,2001;Ruiz de Almodovar,Lambrechts,Mazzone,&Carmeliet,2009;Weis&Cheresh,2005)がある。
脳毛細血管内皮では、VEGFは、MMP-9タンパク質レベルをアップレギュレートし、ZO-1を再分配し、タイトジャンクション複合体からクラウディン-5とオクルディンを呼び寄せるPI3キナーゼ/Aktシグナルを活性化する(Kilicら 2006;Miaoら、2014;Valableら 2005)。
一方、TGFβは、アストロサイトのRGMa-CDC42-PAK-1シグナル伝達経路を活性化してグリア痕形成を引き起こすか、毛細血管周囲の周皮細胞および壁細胞の被覆を減少させて漏出を誘導し、小血管症を引き起こす(Kato et al.2020;佐藤、タブノキ、石田、サイトウ、&有馬,2013)。
GM-CSFは、ユビキチン-プロテアソーム経路内皮細胞を介してZO-1およびclaudin-5タンパク質の発現低下を誘導する(Boydら、2010;Liら、2018;Murphy Jr.,Yang,&Cordell,1998;Ridwan,Bauer,Frauenknecht,von Pein,&Sommer,2012;Satoら、2013;J.Shang,Yamashita,et al,2016)。
5.4.サイトカインによるバリアリーケージ
インターロイキン-1β(IL-1β)、インターロイキン-6(IL-6)、腫瘍壊死因子αサブユニット(TNF-α)などの炎症性サイトカインは、複数のシグナル伝達経路を介してバリアリークを誘発する。例えば、IL-1βはアストロサイトのソニックヘッジホッグシグナルを抑制し、それが試験管内試験の内皮細胞のTJをダウンレギュレートする(Y.Wang et al.、2014)。
IL-1βはまた、細胞外シグナル制御キナーゼ(ERK)シグナル伝達を活性化し、MMP-9をアップレギュレートしてTJタンパク質を破壊する(Gu&Wiley,2006;Mori,Kondo,Ohshima,Ishida,&Mukaida,2002;Ralay Ranaivo,Zunich,Choi,Hodge,&Wainwright,2011;Yang,Zhao,Zhang,&Xu,2016)。
TNF-αは、TJタンパク質を分解するMMPをアップレギュレートするc-Jun N-terminal kinases(JNK)を活性化する(Machida et al.、2017;Zhan et al.、2015)。さらに、Guglielmottoら(2019)は、TNF-αおよびIL-6がともに、5xFADマウスにおけるクローディン-5の発現を乱す活性化B細胞の核因子カッパ-ライトチェーン-エンハンサー(NF-kB)およびマイトジェン-活性化プロテインキナーゼ(MAPK)を活性化することを示している。
もう一つの炎症性サイトカインであるインターフェロンγ(IFNγ)は、PDGFRβの内在化を開始することにより、内皮細胞の周皮被覆を減少させる。これは、周皮細胞の剥離と分解をもたらす重要なシグナル伝達ステップである。この周皮細胞の損失は、最終的にバリア漏出の一因となる(Heldin&Westermark,1999;Jansson et al.)
*
これらの研究を総合すると、いくつかのシグナル伝達経路がTJタンパク質の破壊と周皮細胞の被覆の喪失に関与し、ADモデルにおけるバリアー漏出の一因となっていることが明らかになった。しかし、AD患者においてこれらのシグナル伝達経路を確認するためには、さらなる研究が必要である。
6.アルツハイマー病におけるバリアリーケージの臨床的関連性
ADにおけるバリアー漏出が患者の認知機能低下に寄与していることを示す新たな証拠が得られており、したがって臨床的意義がある。このセクションでは、バリアリーケージと認知機能障害の関係を定義する文献を要約し、併存疾患の重要性を強調し、ADにおけるバリアリーケージを修復するための治療介入の機会を指摘する。
障壁漏出は一般的に加齢とともに増加するが(Montagneら、2015;Verheggenら、2020)、漏出指数に対する年齢効果は、AD患者と非認知症患者で大きく異なっている。これらの変動は、このセクションでは説明しないが、患者の年齢、遺伝的要因、ライフスタイルの選択に一部起因する。
6.1.バリアリーフと認知機能障害との関連性
ADにおける障壁の漏出は40年以上前に初めて観察されたが、障壁の漏出が認知障害に寄与するという概念は比較的新しいものである(Glenner,1979;Iadecola,2013)。初期の知見は、死亡したアルツハイマー病患者の脳で血管外タンパクが検出されたことに基づいており、バリアの漏れを示唆していた(Blennowら、1990;Wada、1998)。
しかし、当時はすべての研究者がバリアー漏出を検出したわけではなく、この分野では数十年にわたり論争が続いた(Alafuzoff et al.)さらに、他の研究者は、AD患者における障壁漏出マーカー(Qalb値)と認知スコア(MMSE)との間の関連を観察しなかった(Janelidzeら、2017;松本ら 2007、松本ら 2008;Skillbackら、2017)。
神経画像における最近の進歩により、研究者は、バリア漏出の信頼性の高い検出を可能にする毛細血管マーカーと組み合わせた新しいMRI技術を使用して、これらの研究間の不一致に慎重に対処することが可能になった。2015年、Montagneら(Montagne et al.,2015)は、MCI患者において、老化した海馬における障壁漏出が認知障害と関連しているという生体内試験証拠を提供した。
さらに、van de Haar,Burgmans,et al.は(2016)、AD患者の脳では対照者と比較してガドブトロール漏出が5倍増加することを示した(p<0.05)。このデータは、バリアリーケージがグローバルであり、早期AD患者の認知機能低下と相関していることも示した。このデータに基づき、著者らは「BBBの障害は、最終的に認知機能の低下と認知症につながる病理学的事象のカスケードの一部である可能性がある」と仮定した(van de Haar,Burgmans,et al.、2016)。
同様の結果は、最近、他のグループでも得られている(Montagneら、2016;Shamsら、2015)。具体的には、Montagneら(2016)は、MCIを持つ人(n=20)が年齢をマッチさせた非認知症者(n=18)と比較して1.5倍高いKtrans値(p<0.001)を有することを実証した;男女両方の被験者が含まれている。さらに、MCI患者(n=17)では、Ktransの値が、非認知症患者(n=14)のQalbの値の1.5倍高い値と直接関連していた(p<0.01)。
同様に、Shamsら(2015)は、高齢の認知症患者(n=1,504)において、脳微小出血の有病率は低い認知スコア(p<0.006)と関連していることを示している。また、重度の脳血管障害(CMB>8)の場合、CMBはアルツハイマー病患者(n=21)のMMSEスコアの低さと相関することがわかった(Goosら 2009)。バリア漏出と認知機能の低下は、広範なAD病理を有しないMCIの個人にも生じる(p<0.001)(Nationら、2019)。
これらの研究を合わせると、バリアリーケージと認知機能の低下の間の相関の証拠が得られる。したがって、新たな証拠は、AD患者において、障壁漏出と記憶の低下が潜在的に機構的に関連していることを示すが、この関連を定義するためにはより多くの研究が必要である。
6.2.アルツハイマー病における合併症のバリアー漏出への影響
様々な臨床ケア環境にわたって検査されたアルツハイマー病患者の61%近くが、ADの重症度上昇につながる3つ以上の併存疾患を有する(Doraiswamy,Leon,Cummings,Marin,&Neumann,2002)。AD患者は、てんかん、腎機能障害、糖尿病および/または心血管疾患を発症するリスクが著しく高い(Fordら、2018;Rojasら、2021;Songら、2021)。本セクションでは、これらの併存疾患に焦点を当て、それぞれがADにおけるバリアリークにどのように寄与するかを議論する。
6.2.1.癲癇(てんかん)
てんかんは、ADにおけるよく知られた併存疾患である(Forstlら、1992;Volicer、Smith、&Volicer、1995)。実際、一般的なアルツハイマー病患者集団の25%以上が発作を発症している(Palop&Mucke. 2016;Vossel et al.、2013;Vossel et al.、2016)。
新たな証拠は、早期AD患者が発作を経験することを示しており、最新の臨床研究からのデータは、65%ものアルツハイマー病患者が発作を有することを示している(Horvath,Kiss,Szucs,&Kamondi,2019;Lam et al,2020;Vossel,Tartaglia,Nygaard,Zeman,&Miller,2017)。
これらのデータは、ADにおけるてんかん発症率は長い間過小評価されており、当初報告されたよりもはるかに大きいことを示唆している。血液脳関門の漏出は、両疾患に共通する病理学的特徴の一つです(Vossel et al.、2016)。
てんかん患者は、バリア機能障害、神経血管の炎症、および白血球の蓄積を発症し、バリアリーケージにつながる可能性がある(Rempe et al.)障壁漏出自体は、てんかんの進行を促進する悪質な正のフィードバックループを通じて、これらの患者における発作の発生に寄与する(Weissberg,Reichert,Heinemann,&Friedman,2011)。このフィードバックループにおいて、発作は、神経炎症とその後の発作負荷を悪化させるバリアリークを駆動する(Vezzani&Janigro,2009;Weissbergら,2015)。
AD患者においては、臨床研究のデータがてんかんの影響を最もよく強調している:てんかんを有するアルツハイマー病患者では、認知機能の低下が5〜7年早く始まり、寿命は発作のないアルツハイマー病患者と比較してさらに短くなる(Amatniekら 2006;Cretinら、2016;Forstlら、1992;Irizarryら、,2012;McAreavey,Ballinger,&Fenton,1992;M.Mendez&Lim,2003;M. F.Mendez,Catanzaro,Doss,Arguello,and Frey,1994;Picco et al.,2011;Rao,Dove,Cascino,&Petersen,2009;Romanelli,Morris,Ashkin,&Coben,1990;Sarkis et al,2015;Volicer et al,1995;Vossel et al.,2013)が挙げられる。
*
臨床的な関連性が高いにもかかわらず、研究者はてんかん発作がADに果たす役割をほとんど無視しており、臨床医はこれらの患者に対する治療の選択肢を制限されたままにしている。てんかんを伴うADのバリアリーケージは十分に理解されておらず、効果的な治療法もない。したがって、バリアリークを悪化させる根本的なメカニズムを明らかにし、てんかんを有するADのバリア機能障害の修復を助ける介入戦略を確立するために、さらなる研究が必要である。
6.2.2.腎機能障害
慢性腎臓病(CKD)は、クレアチニン血清レベルの低下と糸球体濾過不良を示す高い尿アルブミンレベルによって特徴付けられる腎機能の漸減であり、したがって、腎臓障害(Deckers et al,2017)である。
メカニズム的には、CKDは、血中へのサイトカインの放出に続いて、CNSへのサイトカインの浸潤を引き起こし、既存の血液脳関門漏出を悪化させる(Helmerら、2011;Miranda,Cordeiro,Soares,Ferreira,&Simoes,2017;Miwaら、2014;Zammit,Katz,Bitzer,&Lipton,2016)。
軽度のCKDは内皮機能障害およびADと関連しているが、重度のCKDはラクナ梗塞、白質高濃度、そして最終的には血管性認知症を引き起こす高度の内皮機能障害と関連している(Helmerら,2011;Miwaら,2014;Zammitら,2016)。
6.2.3.糖尿病
糖尿病は、体内の血糖値が持続的に高くなる代謝異常である。Starrら(2003)による症例対照DCE-MRI研究のデータでは、糖尿病患者は非糖尿病患者と比較してガドリニウム脳内取り込みが増加し、特に皮質領域と比較して脳深部領域でバリアリークを示す可能性が高いことが示されている。さらに、Janelidzeら(2017)は最近、糖尿病患者が非糖尿病患者と比較してQalb値が上昇することを確認した。
ADにおける糖尿病によるバリアリーケージの相対的な寄与はまだ明らかにされていないが、Salamehらによる生体内試験研究では、酸化ストレスが糖尿病マウスにおける周皮細胞の損失とバリアリーケージを誘発することが示されている(Salamehら、2016)。糖尿病性ADと糖尿病性非認知症患者におけるバリアリークに対する酸化ストレスの原因的役割を検証するために、さらなる調査が必要である。
6.2.4.心血管系の併存疾患
高血圧、心房細動、一過性虚血発作などの心血管共存症は、AD患者、MCI患者、および非発達者におけるガドベンテートジメグルミンの高いKtrans値と相関する(Blennowら、1990;Bowmanら 2007;Nationら、2019)。他の研究はまた、CMBがアルツハイマー病患者の高血圧と相関することを強調している(Benedictusら、2013;Nagasawaら、2014;Olazaranら、2014)。
さらに、Bridgesら(2014)は、高血圧患者の皮質下脳領域において、非高血圧患者と比較してフィブリノゲン濃度の上昇を示した(n=84;Braakステージ:0-II)。これらの研究を総合すると、AD患者におけるバリアリークと心血管状態の相関が示唆される。
*
以上のことから、てんかん、腎機能障害、糖尿病、心血管疾患などの併存疾患は、これらの疾患によって引き起こされる病態や認知障害の重症度とは無関係に、AD患者における障壁漏出の発症と程度を増加させることが示唆された。
6.3.ADにおけるバリアリーケージに対する治療戦略:機会と課題
現在FDAが承認しているAD治療薬は、記憶喪失を遅らせることによって臨床的な認知症症状を治療するように設計されている。しかし、これらの薬剤は、認知機能に対する効果はわずかであり、ADの進行を止めることはできず、すべての患者に有効というわけではなく、6~12カ月後に効果がなくなる。
したがって、AD患者をうまく治療するための新たな疾患修飾的アプローチが緊急に必要とされている。この点で、バリアー漏出を修復する治療戦略は、治療の新しい可能性を開くものである。以下では、バリア特性を変える現在のアプローチを紹介し、前臨床試験で得られた成功と臨床で直面する課題を対比する(図6)。

図6 アルツハイマー病における血液脳関門漏出に対する治療戦略
トランスジェニックADマウスおよびラットにおける関門漏出に対する治療戦略の関連性を示す選択した研究の要約。ここに描かれた治療法は、血液脳関門開口効果(赤)、血液脳関門封鎖効果(青)、または血液脳関門への効果なし(濃い灰色)のいずれかを有する。
各四角の中の数字は、各治療法とモデルの組み合わせについて表4に示した発表済みの研究を示している。なお、白抜きの数は、未検討・未知の戦略・モデルの組み合わせを示す。
6.3.1.抗凝固剤、抗血小板剤、線溶療法剤
抗凝固剤、抗血小板剤、線溶剤は、血栓形成を抑制することで血流を調節する(Hogg&Weitz,2017)。
これらの薬剤は、ADの血液脳関門に対して差動的に作用する。抗凝固剤のうち、ヘパリンやワルファリンなどのビタミンK拮抗薬は、ビタミンK依存性凝固因子を減少させるが、AD患者において関門漏出および微小出血を誘発する(W.Cheng,Liu,Li,&Li,2018;Harter,Levine,&Henderson,2015)。
別の抗凝固剤カテゴリーは、年齢を合わせた未処置のTgCRND8マウスと比較して、7〜14カ月齢のTgCRND8マウスの肥大した周皮細胞および血管周囲のアストロサイトの数を減少させるトロンビン阻害剤です(Cortes-Canteli et al.、2019)。
ジピリダモール、シロスタゾール、およびタダラフィルなどの抗血小板薬は、それぞれTg2576、TgSwDI、およびJ20マウスの微小出血の悪化を防ぐ(Fisherら、2011;Garcia-Barrosoら、2013;Hattoriら、2016;Maki、ら、2014)。
組み換え活性化プロテインC(3K3-APC)は、凝固を遅延させ、高血圧ラットのIgG漏出および微小出血を低減する、抗凝固および抗血小板特性を有する別の治療薬である(Lazicら、2019;Marottoliら、2017)。
スピノシンや遺伝子組み換えtPAなどの線溶薬は、血栓中のフィブリンを分解するが、5xFADマウスやAPP23マウスでは微小出血を誘発することが知られている(Cai et al.、2020)。要約すると、非ビタミンK拮抗薬、経口抗凝固薬、抗血小板薬、および3K3-APCはバリアシーリング効果を有するが、ビタミンK拮抗薬および線溶薬は前臨床ADモデルにおいて微量出血を増加させる。
6.3.2.アンジオテンシン受容体拮抗薬
アンジオテンシン受容体拮抗薬は、高齢者集団におけるAD発症リスクの上昇と相関する併存疾患である高血圧症に対して処方される(Kivipelto et al. 2001)。アンジオテンシン受容体拮抗薬は、脳血流を増加させる血中アンジオテンシンII濃度の上昇に対応して処方される(Fleegal-DeMotta,Doghu,&Banks,2009)。
逆に、オルメサルタンなどのアンジオテンシンII受容体拮抗薬は、APP23マウスや5xFADマウスにおいてタイトジャンクションmRNAレベルを上昇させたり、酸化ストレスを制限することでバリアリークを防ぐ(中川ら、2017;中川、長谷川、上川、&金光山、2017;Pelischら、2011;武田ら 2009)。この効果は、ヒドラジンやニフェジピンなどの非アンジオテンシン受容体遮断薬では観察されなかった(Takeda et al. 2009)。
これらの前臨床試験を総合すると、アンジオテンシンII受容体拮抗薬は、前臨床ADモデルにおいてバリアリークを減少させるが、患者における治療効果を確認するためには、さらなる試験が必要であることが示された。
6.3.3.非ステロイド性抗炎症薬(NSAIDs)
NSAIDは、シクロオキシゲナーゼ酵素COX-1およびCOX-2を阻害することによって炎症性サイトカインの産生を抑制し、生体内のバリア破壊を防止する(Banks et al,2015;Candelario-Jalil et al,2007;Grosser,Ricciotti,&FitzGerald,2017)。最近の研究で、Elfakhri、Abdallah、Brannen、およびKaddoumi(2019)は、NSAIDエトドラクと抗酸化物質a-トコフェロールの組み合わせが、5xFADマウスにおけるIgG外遊を減少させたことを示した。
この併用療法は、この研究でもAPPプロセッシングを非アミロイド生成経路の方にシフトさせた。Qosaら(Qosa et al.,2015;Qosa et al.,2015)およびAl Rihaniら(Al Rihani,Darakjian,&Kaddoumi,2019;Al Rihani,Lan,&Kaddoumi,2019)も、オレカンタールとエクストラバージンオリーブオイルの組み合わせがTgSwDIマウスのIgG extravasationを低減したと報告している。
これらの前臨床研究は、NSAIDsがADにおけるバリアリークを減少させることを示しているが、「AD Anti-inflammatory Prevention Trial」の結果は、NSAIDsがAD病態の重症度を高めることを示している(Breitnerら、2011)。このように、NSAIDsの臨床的有用性には疑問があり、さらなる研究が必要である。
6.3.4.受動的Aβ免疫療法
受動的Aβ免疫療法は、非特異的トランスサイトーシスを介して脳実質に入ると考えられている異なるAβコンフォメーションに対して上昇させたモノクローナル抗体を含む。過去数十年にわたり、受動的Aβ免疫療法療法療法は、臨床試験で広く研究されてきた(van Dyck,2018)。
しかし、これらの臨床試験において、抗体治療群に登録されたアルツハイマー病患者において、プラセボ群のアルツハイマー病患者と比較して認知機能の改善は観察されていない(van Dyck,2018)。
この点で、受動的Aβ免疫療法を用いたすべての臨床試験は、AD患者の認知機能を改善することができなかった。受動的Aβ免疫療法群におけるアルツハイマー病患者の悪い認知アウトカムは、1)抗体の脳内浸透性が低い(0.1%未満)、2)進行したアルツハイマー病患者に焦点を当てたり、Aβ沈着が確認されていない患者を含むことによって誤った患者集団を試験したことに起因する(Jack Jr. 他,2013;van Dyck,2018)とされてきた。
モノクローナル抗体を投与された患者の脳MRIスキャンは、受動的Aβ免疫療法が脳浮腫(ARIA-E)および微小出血(ARIA-H)などのAβ関連画像異常(ARIA)を引き起こし、これらの患者における障壁漏れを示している(van Dyck、2018)。
AD患者におけるARIA-EまたはARIA-Hの発現の重症度は、検査したモノクローナルAβ抗体によって異なる。bapineuzumab(IgG1:Aβ1-5残基に対して)、gantenerumab(IgG1:Aβ13-24およびAβ18-27に対して)、およびaducanumab(IgG1:Aβ36に対して)などのIgG1抗体では、登録患者集団において高い(>3%)ARIA発生率(Rinne&Nagren,2010;Salloway et al,2009;Salloway et al.,2014;Sevigny et al.,2016;van Dyck,2018)。
一方、ソラネズマブ(IgG1:Aβ16-26に対して)およびクレネズマブ(IgG4:Aβ13-24に対して)などの中間Aβ残基に対するヒト化抗体は、すべての形態のAβ種を認識し、低い(1%未満)ARIA発生率になる(Cummingsら、2019;Doodyら、2014;Farlowら、2012;van Dyck,2018)。
場合によっては、抗体はARIA発生を引き起こさない。そのような例の1つは、Aβ30-40残基に対して上昇したIgG2であるponezumabであり、これは単量体のみを認識し、患者においてARIA発生を引き起こさなかった(Landenら、2013;van Dyck. 2018)。
Aβプロトフィブリルに対して上昇させたIgG1であるBAN2401に関する研究は進行中であるが、これまでの報告によると、BAN2401を投与された患者とプラセボとの間でARIAの発生数は同程度である(Landen et al,2013;Logovinsky et al,2016;van Dyck,2018)。
*
Aducanumabは、Aβ(N末端3-6)の可溶性オリゴマーと不溶性ファイバーに特異性を持つ高親和性の完全ヒト型モノクローナルIgG1抗体で、AD治療薬探索の希望を蘇らせた。この抗体は、血管内Aβではなく、実質内Aβの凝集体に結合するよう特異的に設計されている。
Ferreroらが主導した単回投与試験(Ferreroら、2016)により、投与量≦30mg/kgのアデュカヌマブはヒトにおいて線形薬物動態をたどり、許容できる安全性と忍容性のプロファイルを有することが示された。
この報告に続いて、Sevignyら(2016)は、前駆期または軽度のアルツハイマー病患者において、1年間毎月投与されたaducanumabが用量および時間依存的にAβ脳内濃度を低下させることを示した。
重要なことは、本試験のデータから、アデュカヌマブ投与により、Clinical Dementia Rating-Sum of BoxesおよびMini-Mental State Examinationスコアで測定した認知機能の低下も遅くなることが示されたことである。
2021年6月、aducanumab(Aduhelm™)は、AD治療のためのFDAの加速承認パスウェイの下で承認を受けた。したがって、aducanumabは、Aβレベルを低下させる最初のFDA承認薬となる。しかし、早期承認には、承認後の試験で薬剤の臨床的有用性を確認することが義務付けられている。
Aducanumabがこの承認後の試験で臨床的有用性を示さない場合、FDAの承認は撤回される可能性がある。最近発表された報告書では、aducanumab治療の有用性に疑問が呈されている。例えば、早期認知症患者において、aducanumab投与群とプラセボ投与群の間で主要評価項目に有意な変化は認められなかった(Angelo&Ward,2021)。
さらに、アデュカヌマブを投与された患者やAPOE4の遺伝子変異を有する患者は、プラセボ投与患者に比べてARIA-EおよびARIA-Hの発生率が高く、慢性的であると報告している(Salloway&Cummings,2021;VandeVrede et al,2020)。副作用と比較的低い有効性プロファイルに加えて、高い患者関連コストが患者におけるアデュカヌマブの使用を制限する可能性がある(Anderson,Ayanian,Souza,&Landon,2021)。
*
つまり、受動的免疫療法は、最近の有望な進歩であるが、同時に課題もある。例えば、抗体がCSFから脳内に入り、生体内でAβを分解する仕組みはまだ不明である。さらに、抗体が効果を発揮する「治療濃度域」に関する情報が限られているため、受動免疫療法はアルツハイマー病患者においてバリアリークを引き起こす危険性があり、その結果、潜在的な治療効果が相殺される可能性がある。
6.3.5. mTOR阻害剤
mTOR阻害は、前臨床ADモデルにおいてバリアリーケージを減少させるために最近開発された戦略である(Van Skike et al.2018;Van Skike&Galvan,2019)。いくつかの研究は、AD、脳虚血、およびてんかんの前臨床モデルにおいて、mTOR阻害がバリアリーケージを低減することを示している(W.Guo,Feng,Miao,Liu,&Xu,2014;Van Skike et al,2018;van Vliet et al,2012)。
Linら(Lin et al.,2020;Lin,Parikh,Hoffman,&Ma,2017)は、ラパマイシン(mTOR阻害剤)がシクロフィリンA依存性の炎症性経路を阻害することによってAPOE4トランスジェニックマウスの障壁漏出を防ぐことを示すデータを提示した。AD患者におけるバリアリークを減少させるmTOR阻害剤の可能性を評価するために、さらなる研究が必要である。
*
これらの研究を総合すると、前臨床ADモデルにおけるバリアー漏出を修復するための潜在的な薬理学的介入の効果が概説され、これらの戦略をアルツハイマー病患者の治療に応用することの難しさが浮き彫りにされた。
7.今後の展望
このレビューで要約された研究は、バリアー漏出がAD病理の本質的な部分であることを強調し、バリアー漏出を修復することで認知機能の低下を遅らせることができることを示唆している。しかし、効果的な疾患修飾療法は今のところない。バリアー漏出を引き起こす誘因を特定し、そのメカニズムを解明し、ADにおけるバリアー漏出の根底にあるシグナル伝達経路を明らかにするためには、さらなる研究が必要である。
前臨床および臨床レベルでは、生化学的手法と新しいイメージング技術を組み合わせた縦断的研究が、バリアー漏出の発症とその程度を検出し可視化するのに役立つだろう。動物モデルを用いて、バリアー漏出の根本的なメカニズムを明らかにし、ADにおけるこの漏出を修復する新しい治療法を検証することが可能である。さらに、バリアー漏出と認知機能の低下との関連は十分に理解されておらず、この関連性をさらに明確にするためにさらなる研究が必要である。
さらに、バリアー漏出は、ADおよびAD関連認知症における認知障害への血管の寄与(VCID)の早期バイオマーカーとして機能する可能性がある。この点で、認知機能検査と組み合わせた大規模な縦断的神経画像臨床研究は、この関連について重要な洞察を与える可能性がある。
ある患者ではバリアリーケージが重要な特徴であるが、他の患者ではそうでないかもしれないことを認識することは、VCID、ADおよびAD関連認知症の定義の形成にさらに役立ち、治療介入のための新しい領域を切り開くことになるであろう。また、バリアー漏出は、臨床試験において重要な「チェックポイント」となり、漏出の発生しやすさに基づいた患者のプロファイリングに役立ち、より良い、的を絞った介入戦略を可能にすることが期待される。
*
まとめると、バリア機能障害全体とバリア漏出は、ADの早期発見と、AD患者において初めて疾患修飾効果を発揮する新しい治療戦略の開発につながる、まだ十分に研究されていない領域であることがわかる。
資金調達先
このプロジェクトは、National Institute on Agingの助成金番号2R01AG039621(A.M.S.H.)およびR01AG075583(A.M.S.H.とB.B.)によって支援されている。内容はあくまで著者の責任であり、必ずしもNational Institute on AgingまたはNational Institutes of Healthの公式見解を示すものではない。
競合する利害関係の宣言
著者らは、利益相反がないことを宣言する。