Contents
概要
アルツハイマー病は、神経細胞やグリア細胞の内外に誤って蓄積されたタンパク質の異常蓄積により、細胞内のタンパク質の恒常性が失われ、記憶力の低下が進行することを特徴とする神経変性疾患である。
現在、アルツハイマー病の進行を阻止したり遅らせたりする治療法はなく、そのメカニズムは完全には解明されていない。オートファジーは、損傷した小器官や病原体、不要なタンパク質の集合体を除去することで、細胞の恒常性を維持するために重要な細胞内分解経路である。近年、オートファジー機能障害は、アルツハイマー病やその他の神経変性疾患において、最終的に神経細胞死の原因となる誤ったタンパク質の蓄積と関連していることから、注目を集めている。興味深いことに、オートファジー活性化化合物もまた、臨床試験や前臨床試験において有望な結果を示している。
本総説では、オートファジー機能障害に関する現在の知見をアルツハイマー病の病態生理の観点からまとめ、アルツハイマー病が媒介するオートファジーのフラックス障害に関する最近のメカニズムを明らかにし、アルツハイマー病治療のためにオートファジーを標的とした治療の可能性と新規治療機会を明らかにすることを目的としている。
キーワード
アルツハイマー病、動物モデル、オートファジー、臨床研究、プロテオスタシス、治療薬
アルツハイマー病は、脳の神経細胞や経路の不可逆的な損傷による認知障害や記憶喪失を特徴とする進行性の神経変性疾患である(1)。今日では、アルツハイマー病の進行をブロックしたり、遅らせたりする治療法はなく、病気の原因となる正確なメカニズムは謎に包まれたままだ(1)。
アルツハイマー病は、アミロイド-b(アミロイドβ)やタウなどの誤ったタンパク質の異常蓄積が特徴で、細胞の恒常性の喪失につながり、最終的には細胞死をもたらする(2,3)。構造的に正常なタンパク質は、生きている細胞の基本的な高分子として機能しており、その恒常性は生物の生存に不可欠である。
タンパク質の恒常性の制御は、合成または分解のいずれかの制御を通じて行われる。オートファジーは、細胞質の内容物がオートファゴソームと呼ばれる二重膜結合小胞に取り込まれ、消化のためにリゾソームに運ばれる細胞内分解経路である(4,5)。
このため、いくつかの報告では、アルツハイマー病ではオートファジーが制御不能であることが示されており、オートファジーの活性化が新たな治療法と考えられている(6,7)。いくつかの薬理学的オートファジー活性化薬で有望な結果が得られている(8,9)。しかし、この薬理学的アプローチが本当に病気を改善する治療戦略になる可能性があるかどうかは、まだ調査中である。
アルツハイマー病 概要
現在、570万人のアメリカ人と5,000万人の世界の人々がアルツハイマー病と共に生活している。リバスチグミン、ガランタミン、ドネペジル、メマンチン、メマンチン-ドネペジル配合剤、タクリンの6つの薬が、長年にわたって食品医薬品局によってアルツハイマー病治療薬として承認されている。
これらの化合物の作用機序は、グルタミン酸N-メチル-D-アスパラギン酸受容体の遮断、アセチルコリンエステラーゼの阻害、またはその両方の組み合わせである。しかし、これらの化合物は、神経病理の進行をうまく止めることなく、一時的に症状を緩和するだけである(1)。
家族性または若年性アルツハイマー病と散発性または遅発性アルツハイマー病(10):本疾患は、2つの主要な形態に分類することができる。家族性または早期発症型は、アミロイド前駆体タンパク質(APP)プレセニリン 1,プレセニリン 2 をコードする遺伝子の特定の常染色体優勢変異の結果として発症する。
この疾患のこの型は、30~40 歳頃から症状が始まる アルツハイマー病 人口のわずか 5%にしか影響を与えない(11)。対照的に、散発性または遅発型は、総アルツハイマー病患者の95%に影響を与え、典型的には65歳以降に症状が現れる。
今日では、後者の変異は素因性リスク対立遺伝子と環境リスク因子への曝露によるものであるというコンセンサスがある(1)。加齢とともに有病率が劇的に増加するため、年齢は散発性アルツハイマー病の最も強い危険因子である(12)。この病気の家族歴は散発性アルツハイマー病のもう一つの危険因子であり、この病気に罹患した母親、父親、または兄弟を持つことは、この病気のリスクを有意に増加させる(13-16)。
さらに、APOE遺伝子のε4対立遺伝子を持っていることも、人生の後半でのアルツハイマー病発症の遺伝的危険因子である。APOEはコレステロールトランスポータータンパク質をコードする遺伝子で、3つのバリアントがある。ε4変異体は散発性アルツハイマー病を発症するリスクの増加と関連しているが、ε2変異体はそれよりも低いリスクと関連している(17)。
* *
研究では、疾患の素因に重要な役割を果たすいくつかの修正可能な危険因子の存在が実証されている。特に、喫煙、中年期の肥満、高血圧、2型糖尿病、高コレステロール血症などの心血管疾患の危険因子はすべて、遅発性アルツハイマー病の強い素因効果を持っている(18-21)。教育レベルと外傷性脳損傷の既往歴もまた危険因子と考えられており、今日では晩年のアルツハイマー病感受性の調節因子として十分に受け入れられている(22,23)。
アルツハイマー病:脳病理学
アルツハイマー病は、アミロイドβプラークを形成するアミロイドβペプチドや、脳実質に神経原線維のもつれを形成する高リン酸化タウタンパク質など、誤ったタンパク質が蓄積することが特徴である。これらのタンパク質の凝集体は非常に毒性が強く、神経細胞死につながる。
これらの有害な凝集体を除去しようとすると、ミクログリアやアストロサイトの慢性的な活性化がいくつかの炎症性経路を促進し、結果として神経細胞の生存に不利な環境を作り出する(24,25)。アミロイドβペプチドは、特定のプロテアーゼによる大きな膜貫通タンパク質であるAPPのタンパク質分解的切断の結果として生成される。
特に、APPは、健康な脳では平衡状態にあると考えられているアミロイド原性または非アミロイド原性経路のいずれかによって切断することができる(図1)。しかし、アルツハイマー病脳では、アミロイド原性経路は、APPをアミロイドβペプチドに切断するb-セクレターゼ1とg-セクレターゼなどの特定のタンパク質分解酵素の活性の増加のために普及するようになる。
非アミロイド原性経路では、APPはa-セクレターゼおよびg-セクレターゼによって順に切断され、アミロイドβの形成を妨げる。アミロイド原性経路によって産生されるアミロイドβペプチドは、36〜43個のアミノ酸モノマーからなる(26,27)。
一旦形成されると、アミロイドβのモノマーは、フィブリルに集合したオリゴマーに凝集し、その後、b-プレアチン化されたシートになり、最終的にアミロイド斑を形成することができる(図1)。今日では、アミロイドβオリゴマーはより細胞毒性の高い種と考えられている(28,29)。
アミロイドβは、インスリン分解酵素であるネプリルリシンのような特異的なプロ溶血酵素によってクリアされる(30,31)。さらに、アポリポ蛋白質Eは、脳実質内のアミロイドβを輸送し、血液脳関門を介して実質からアミロイドβの転送を調節し、シャペロンとして機能することができるリポ蛋白質である(32)。アルツハイマー病では、クリアランスとアミロイドβペプチドの産生の間のバランスは、進行性のアミロイドβ蓄積につながる、制御不能と考えられている(27)。
* *
タウは神経細胞の軸索コンパートメントに豊富に存在する微小管関連タンパク質であり、そのリン酸化状態に基づいて微小管の安定性を制御している(33)。タウをリン酸化するキナーゼとしては、サイクリン依存性キナーゼ5,グリコーゲン合成酵素キナーゼ3β、p38マイトジェン活性化プロテインキナーゼ、ストレス活性化プロテインキナーゼ/Junアミノ末端キナーゼなどが知られている(34)。
タウの翻訳後修飾に関与する主要なホスファターゼは、プロテインホスファターゼ2Aである(35)。アルツハイマー病では、タウキナーゼとホスファターゼのバランスは、タウのリン酸化に極端に偏っている(図2)。一旦高リン酸化されると、タウは微小管に対する親和性を失い、タウオリゴマーを形成し、それはその後、ペアらせん状のフィラメントに凝集し、最終的には神経原線維のもつれ(33)(図2)になる。アミロイドβに関しては、タウオリゴマーはタウ神経原線維性のもつれよりも毒性が強い(36)。
微小管の不安定性とタウオリゴマーの蓄積は、軸索輸送の著しい障害をもたらし、その結果
* *
神経細胞死(36)。進行性のアミロイドβとタウオリゴマーの蓄積の結果として、脳のシナプスは、その完全性を失い、最終的にはアルツハイマー病脳の親球内で失われる。この概念のサポートでは、このようなシナプトフィシンとシナプス後密度タンパク質95などの事前および事後シナプスタンパク質のレベルは、健康なマッチしたコントロール被験者と比較した場合、アルツハイマー病脳で有意に減少しているという観察である(37,38)。さらに、ミクログリアやAS-trocytesのような炎症性細胞は、アミロイドβフィブリルとタウオリゴマーによって生成された毒性環境に応答して過剰に活性化される(39-41)。
オートファジー:概要
オートファジーとは、細胞質の不要な内容物がオートファゴソームと呼ばれる二重膜結合小胞に取り込まれ、リソソソームに運ばれて消化される細胞内分解経路のことである。オートファジーには、マクロオートファジー、ミクロオートファジー、シャペロンを介したオートファジーの3つのタイプがある。
しかし、「オートファジー」という用語は、特に明記されていない限り、このレビューの焦点であるマクロオートファジーを指すのが一般的である(4)。オートファジーは伝統的に、栄養不足時に細胞質内容物を非特異的に分解して再利用するプロセスと考えられてきた。
今日では、飢餓に対する適応応答に加えて、オートファジーは、飢餓状態にない細胞でも活性化し、選択的オートファジー受容体を介して侵入病原体やタンパク質凝集体を分解し、細胞の健康状態に関する情報を報告することで、細胞の恒常性を維持していることが知られている(5)。
さらに、オートファジーは、ミトコンドリアなどの損傷した小器官の除去にも関与しており、ミトファジーとしても知られているマクロオートファジーの特異的かつ特殊な変種である(42)。
* *
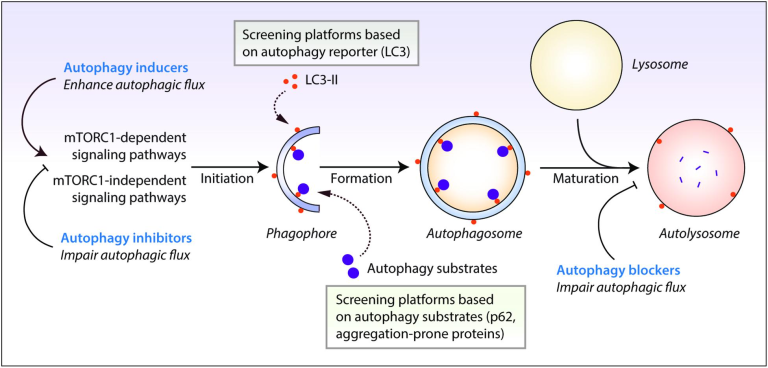
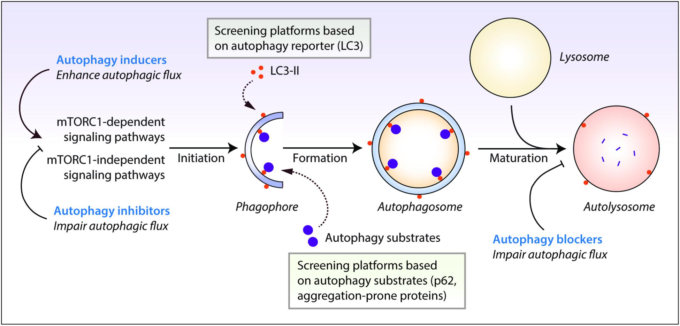
オートファジーは高度に制御されたプロセスで、開始、小胞の伸長、リソソソームの融合と分解の3つのフェーズに分解することができる(図3)。オートファジーの開始は、主に哺乳類のラパマイシン複合体1(mTORC1)とAMP活性化プロテインキナーゼ(AMPK)によるオートファジー活性化キナーゼ1(ULK1)のようなunc-51の反対側のリン酸化によって制御されている(43-45)。
また、栄養が不足している時には、オートファジー関連タンパク質ATG13,ATG101,200kDaのFAKファミリーキナーゼ相互作用タンパク質と複合体を形成しているULK1のセリン757をリン酸化することで、オートファジー誘導を抑制する(44,46,47)。
細胞のエネルギー状態が低い場合、AMPKはAMPの結合により活性化され、ULK1のセリン317およびセリン377を直接リン酸化し、オートファジーを開始することができる(48)。
さらに、細胞飢餓または細胞ストレスシグナルの他の条件の間、mTORC1は不活性化され、mTORC1による抑制されたリン酸化は、ULK1の自己リン酸化を可能にし、これは、順番に、ATG13および200kDaのFAKファミリーキナーゼ相互作用タンパク質をリン酸化する。
ULK1は次に、クラスIIIのホスファチジルイノシトール3-キナーゼとして作用し、ホスファチジルイノシトール3-キナーゼを産生し、ホスファチジルイノシトール3-キナーゼ結合タンパク質のリクルートおよびファゴフォアの核生成を可能にする液胞タンパク質ソーティング34複合体(ベクリン-1,液胞タンパク質ソーティング34,ホスファチジルイノシトール3-キナーゼ調節サブユニット4,およびATG14Lを含む)を活性化する(図3)(49,50)。
当初は小胞体がファゴフォア核生成の主要な膜源であると考えられてたが、今日では、ファゴフォア形成時にエンドソーム、ミトコンドリア、形質膜、ゴルジ体も膜ドナーとして機能することが示唆されている(51,52)。ファゴフォアのエロン化と拡張は、いくつかのユビキチン様共役系に依存している。まず、E1ユビキチンリガーゼATG7およびE2ユビキチンリガーゼATG10は、ATG12をATG5に共役させる。次に、ATG12-ATG5はATG16L1に結合し、結果として生じる複合体は、微小管関連タンパク質1A/1B-軽鎖3(LC3)のリクルートのためのプライミングのためにファゴソーム膜と結合する(53)。LC3をATG4で切断してLC3-I、ATG7,ATG3,およびATG12-ATG5-ATG16L1複合体を形成した後、LC3-IIを生成する(54)。完全に処理されたLC3-IIは、その後、ファゴソーム膜に結合することができ、成熟したオートファゴソームを形成するために、その脂質化を介してファゴフォアの伸長および閉鎖を促進するのに役立つ(図3)。他にもATGタンパク質がファゴフォアの伸長と閉鎖を助けると考えられているが、その役割はまだ明確に定義されていない。成熟したオートファゴソームが形成されると、ライソソソームと融合してオートリソソームになるか、エンドソームと融合してアンフィソームと呼ばれる中間体を形成し、リソソームと融合する。融合後、リソソームは液胞アデノシン三リン酸酵素を利用してオートリソソームのコンパートメントを酸性化し、オートファジーのサブストラクトを消化するリソソームヒドロラーゼを活性化する。消化された貨物から生成された代謝物は、その後、細胞内で再利用できるようになり、細胞内シグナル伝達経路に影響を与えるために放出される可能性がある(図3)(55)。
アルツハイマー病における自律神経失調症
複数の証拠のラインは、オートファジー機能不全がアルツハイマー病の病態生理に重要な役割を果たしているという仮説を支持している(6)。免疫金標識と電子 顕微鏡では、Nixonら(7)は、前頭頂皮質では、アルツハイマー病患者は対照被験者と比較して有意に高いオートファゴソームの蓄積を表示することを実証することに成功した。
その後の研究では、オートファジー制御タンパク質ベクリン-1のタンパク質とメッセンジャーRNAレベルがアルツハイマー病の脳組織で減少していることが報告された(56)。遺伝学的研究では、オートファジーの調節に関与する他の潜在的な候補が同定されている。
カテプシンDは、アミロイドβペプチドとタウタンパク質の両方のクリアランスに関与するリソソソームペプチダーゼである。リソソーム脱顆粒活性に影響を与えるこの酵素の遺伝的変異は、アルツハイマー病のリスクの増加と関連している(57)。
機能の喪失がオートファジーに悪影響を及ぼすアルツハイマー病感受性遺伝子であるホスファチジルイノシトール結合型クラスリン・アズアセンブリータンパク質のレベルもアルツハイマー病では変化する(58-60)。アルツハイマー病におけるオートファジーは主にリソソームのタンパク質分解不全のために欠損しており、その結果、脳内のオートファジー空胞の脳内蓄積が再び起こるという研究が支持されている(58)。
ホスファチジルイノシトール結合性クラスリン結合蛋白質は、アルツハイマー病で異常に切断され、その結果、完全長蛋白質のレベルが低下し、オートファゴソームの合成と成熟を混乱させる(61,62)。
データは、オートファジー障害がアルツハイマー病の両方の主要な特徴に影響を与えうることを示している。アミロイドβペプチドとタウタンパク質の蓄積(8)。オートファジーはアミロイドβの産生とクリアランスの両方の重要なモジュレーターであるため、オートファジーとアミロイドβの間の関係は複雑である。Kurtishiら(63)は、APPは通常細胞質に分布しているが、オートファゴソームにもAPPが含まれており、g-セクレターゼ複合体の4つの構成要素に非常に濃縮されていることを示した。オートファゴソームの液胞では、これらの酵素は完全に活性化されており、アミロイドβを産生する能力があり、アルツハイマー病におけるアミロイドβ産生の重要な追加的な供給源である可能性を示唆している(45,63)。興味深いことに、オートファジーの誘導とともにアミロイドβの産生が増加することが示されている(63)。同時に、細胞外空間でのアミロイドβペプチドの放出とそれに続くプラーク形成もオートファジーに依存している(64)。アミロイドβ分泌に対するオートファジーの影響を研究するために、Nilssonら(64)はATG7をノックダウンすることにより、オートファジー欠損APPトランスジェニックマウスモデルを作製した。驚くべきことに、これらのオートファジー欠損動物では、アブの分泌が90%減少したが、オートファジーを回復させると正常なアミロイドβの分泌が得られた(65)。さらに、mTOR阻害剤ラパマイシンによるオートファジー誘導は、アルツハイマー病のマウスモデルでは、アミロイドβクリアランスを促進し、認知障害を改善する(66)。
* *
アミロイドβに加えて、オートファジーはまた、タウクリアランスの制御における重要なプレーヤーである。タウタンパク質はユビキチン-プロテアソーム系によって分解されるが、オートファジー-リソソーム系の機能不全はタウオリゴマーの形成と不溶性タウ種の蓄積を促進することが示唆されている(67)。
死後の研究では、高ホスホリル化タウ免疫反応性が、アルツハイマー病患者や関連するタウ症患者の脳内でLC3-およびユビキチン結合タンパク質p62陽性のオートファゴソームと関連していることが実証されているが、対照者ではそうではない(68,69)。
オートファジーもまた、タウのリン酸化状態に直接影響を与えるようである。実際、オートファジー欠損マウスはタウのリン酸化が亢進している;対照的に、同じマウスでオートファジーが回復すると、リン酸化されたタウは減少する(68,69)。
これらの観察結果は、オートファジーもリン酸化タウクリアランスの代替経路として考慮されるべきであるという仮説を支持している(70)。一貫して、ラパマイシンを介したmTORシグナル伝達の薬理学的阻害は、試験管内試験ではタウのリン酸化を減少させ、生体内試験では神経原線維のもつれの形成を停止させる(69)。
さらに、トレハロースを用いてP301Sマウスモデルでオートファジーを促進すると、不溶性およびリン酸化されたタウが減少し、これは神経細胞の生存率の増加と相関していた(71)。
逆に、別の論文では、オートファジーの阻害はタウのリン酸化を増加させることが示されている(72)。興味深いことに、タウはオートファジー-リソソソーム系にも作用し、リソソーム膜に結合してその完全性を変化させることが示されている(8,73)。さらに、最近の報告では、タウタンパク質が細胞内に蓄積すると、直接的な相互作用によってヒストン脱アセチル化酵素6の活性を阻害し、結果としてオートファジー機能不全を引き起こすことがわかった(9)。
アルツハイマー病の治療標的としてのオートファジー
アルツハイマー病におけるミスフォールドされたタンパク質の重要な役割と、アルツハイマー病脳におけるオートファジーフラックスの機能不全が確立されていることを考えると、今日では、オートファジーの恒常性が理想的な治療法の候補であるというコンセンサスがある。
したがって、オートファジーを活性化および/または促進して不要なタンパク質凝集のクリアランスを刺激することは、理論的にはアルツハイマー病に対する有望な治療アプローチである可能性がある。
興味深いことに、このアプローチは、特定の分子(b-セクレターゼ1のようなセクレターゼやサイクリン依存性キナーゼ5のようなキナーゼ)を典型的に標的としたり、アミロイドβ凝集やタウの神経毒性を中和することを目的とした現在の治療法とは全く異なる新規で実行可能な治療法である可能性がある。
したがって、これらのアプローチとは対照的に、細胞生物学全体の欠陥を標的としたオートファジーフラックスの薬理学的活性化は、神経変性の文脈で共通の細胞メカニズムを調節するであろうし、この理由から、潜在的には、その病因の核心にあるアルツハイマー病のタールゲットのためのより望ましい、信頼性の高い戦略であり得る。
* *
いくつかのオートファジー刺激性化合物が開発され、提案されており、それらのいくつかは有望な結果を示しているが、他の化合物はそうではない(表1)。興味深いことに、これらの化合物は通常、mTORまたはAMPK経路を介して作用する。以下に、これらの薬剤のいくつかと、前臨床試験と臨床試験の両方で利用可能なデータについて説明する。
* *
N-メチル-D-アスパラギン酸受容体拮抗薬メマンチンは、中等度から重度のアルツハイマー病治療薬として食品医薬品局(FDA)に承認されている数少ない化合物の一つである。この薬剤を用いた臨床試験のメタ分析では、一般的に忍容性が高く、アルツハイマー病においてある程度の有効性があることが示されている(74)。
さらに、メマンチンは、記憶力の低下、脳アミロイドーシス、および アルツハイマー病 マウスのシナプス機能にいくつかの有益な効果を示している (75)。この薬剤がアミロイドーシスを減少させる正確なメカニズムはまだ完全には解明されていないが、最近の報告では、メマンチンがアルツハイマー病の細胞モデルにおいてオートファジーとアポトーシスの両方を阻害することが示唆されている(76)。しかし、メマンチンのオートファジーに対する効果が脳病理に影響を与えるかどうかは、まだ解明されていない。
* *
抗てんかん薬のカルバマゼピンは、アルツハイマー病患者、特に攻撃的行動の改善に治療の可能性を示している(77,78)。また、3xTgマウスモデルにおいて、mTOR依存性およびmTOR非依存性の両方のメカニズムを介したオートファジー誘導により、記憶機能障害および脳アミロイドーシスを改善することが研究で示されている(79,80)。
抗ヒスタミン薬であるラトレピルジンは、アルツハイマー病とハンチントン病性絨毛を対象とした臨床試験において、いくつかの相反する結果が示されている(81-83)。対照的に、別の報告では、アルツハイマー病のTgCRND8マウスモデルにおいて、mTORおよびATG5依存的なオートファジーの活性化を介して認知機能障害および脳アミロイドーシスを減少させることが示されている(84)。さらに、同じ化合物はミトコンドリア機能に有益な効果を示した(85)。
* *
気分安定剤のリチウムは、アルツハイマー病の臨床試験で相反する結果を示している。特に、いくつかの臨床試験では、アルツハイマー病患者における毒性を伴わない認知機能の改善が再報告されている(86)が、他の臨床試験では限定的な効果が報告されている(87)。
生体内試験研究からのデータは、リチウム投与がオートファジーを介して3xTgマウスのタウ病理を減少させる可能性があることを示している(88)が、これにはイノシトール一リン酸経路が関与している可能性がある(89)。
* *
ニコチンアミド(ビタミンB3/PP)は、アルツハイマー病患者を対象とした臨床試験では有効性は限られているが、安全性は良好であることが示されている(90)。対照的に、ADマウスモデルの生体内試験研究では、いくつかの有望な結果が示されている。
特に、酸化ニコチンアミド・アデニン・ジヌクレオチド(NAD1)の前駆体として、ニコチンアミド治療は、サーチュイン1活性とプロトン勾配の調節を通じて記憶力の低下、アミロイドーシス、およびタウの病理を減少させ、これは最終的に3xTgマウスにおけるオートファゴリーソームの酸性化を促進する(91,92)。
興味深いことに、ウロリスチンAやアクチノニンと組み合わせたNAD1前駆体は、マイトファジーを改善することで、ADモデルにおけるアミロイドβとタウの病理を阻害し、認知障害を逆転させる(93)。最後に、ニコチンアミドの有益な効果も一部でエネルギー代謝を改善する既知の作用に起因する可能性がある(94)。
* *
抗糖尿病薬のメトホルミンは、アルツハイマー病患者の記憶力低下を改善することで臨床試験で有望な結果を示している(95)。一方、ADマウスを用いた研究では、メトホルミンがプロテインホスファターゼ2Aの活性化を介してタウのリン酸化を緩和する効果が示されている(96)。
さらに、メトホルミンによるAMPKを介したオートファジーの活性化は、オートファゴソームへのAPPの蓄積とg-セクレターゼによるアミロイドβへの処理を促進することが報告されている(97)。
* *
mTOR阻害剤であるラパマイシンは、最近、哺乳類の寿命を延ばすことが報告され(98)心臓の老化に対する臨床試験で肯定的な効果を示し(99)トランスジェニックマウスのアルツハイマー病様病態に対して有益な効果を示すことが報告されている。
特に、ラパマイシンは、認知機能の低下、脳 また、3xTgマウスとAPPトランスジェニックマウスの両方において、オートファジー活性化を介して、アミロイドーシス、タウ病理が改善された(66,100)。さらに、ラパマイシンの投与は、アポリポタンパクE ε4トランスジェニックマウスの記憶・認知機能、代謝パラメータ、血管機能障害を改善した(101)。
* *
ADマウスモデルでは、ブドウ由来のポリフェノールであるレスベラトロールは、AMPKを介したオートファジー活性化を介して、アルツハイマー病様病理に対する保護効果を示した(102)。
興味深いことに、同じ薬剤はまた、サーチュイン1の活性化剤として作用し、酸化/還元NADの上昇を誘導し、mTOR依存性およびmTOR非依存性の方法で異常なタンパク質のクリアランスを強化し、神経細胞の生存を促進することができる(103)。しかし、安全性の高い同化合物の投与により、アルツハイマー病患者を対象とした第II相臨床試験では相反するデータが得られている(104,105)。
* *
これらの薬剤とその有効性については相反する結果があるが、オートファジーを標的とした治療法の研究は重要な領域である。異なる作用機序、薬物動態、オートファジーフラックス内の特定の標的を持つ薬剤を比較することは困難であるが、これらの研究を解釈し、結果を正当化することも一般的には困難である。
さらに、オートファジーの複雑さや、既知と未知の両方の異なる関与者を考慮すると、おそらくこのメカニズムの核心をターゲットにする立場にはまだないことは驚くべきことではない。それにもかかわらず、前臨床試験に関しては、異なるモデル、様々な薬物濃度、使用された実験デザインの違い(例えば、治療期間や投与経路)が、これまでに得られた一貫性のない結果の一部を説明することができると考えられる。
一方、臨床試験については、試験デザイン、母集団の募集、登録時の診断の割合などが大きく異なっていたことが、相反する結果の一因になっている可能性がある。最後に、前臨床試験の結果を臨床の場で翻訳するという一般的な課題を忘れてはならない。
12/15-リポキシゲナーゼ:オートファジーの内因性モジュレーター
最近、リポキシゲナーゼ(LOX)とオートファジーとの意外な関係が発見された。LOXは、遊離脂肪酸やエステル化した多価不飽和脂肪酸に分子状の酸素を挿入する触媒作用を持つユビキタス酵素の一群である。酸素化された炭素原子の位置に基づいて、これらの酵素は、5-LOX(位置5の炭素)や12/15-LOX(位置12と15の炭素)などの様々なアイソフォームに分類されている(106)(図4)。
この反応を触媒することにより、LOXは、ヒドロペルオキシ脂肪酸、ロイコトリエン、およびリポキシンなどのシグナリング分子として作用するほとんどの異なる脂質に脂肪酸を変換する。
* *
興味深いことに、12/15-LOXは、試験管内試験および生体内試験でミトコンドリアおよび小胞体膜などの様々な細胞小器官の膜に直接結合し、酸化することが示されている(107-109)。また、12/15-LOXが正常ラット肝臓のペルオキシソームやミトコンドリアの生理的分解に関与していることが示されている(110-112)。
これらの観察結果は、van Leyenら(113)が当初提案したように、12/15-LOXがオルガネラのプログラム分解に関与していることを裏付けている。これらの知見に沿って、最近の論文では、12/15-LOXの遺伝子欠損は、マウスの脳と肝臓の両方でLC3タンパク質レベルを増加させることにより、オートファジーが強化されることが示された(図5)。同様の結果は、12/15-LOX活性を薬理学的に阻害した後の培養肝細胞HepG2およびSH-SY5Y神経芽腫細胞でも再報告されている。
同じ研究では、結果としてオートファジーが増加し、損傷したミトコンドリアや細胞要素の蓄積を予防することが示されている(114)。別の研究では、12/15-LOXが異なる細胞タイプでオートファジーの内因性調節因子として作用することも実証された。
この研究では、野生型と12/15-LOX欠損マウスのマクロファージを用いて、膜の超構造、LC3発現、脂質化の違いを調べ、酸化リン脂質がLC3とAGT8の脂質化の基質として作用する能力を調べた。彼らは、12/15-LOXの活性化により生成した酸化リン脂質が また、この酵素を持たない細胞では、オートファジー機能不全の生化学的・細胞学的徴候が明らかに見られた(115)。
先行研究との明らかな矛盾は、異なる細胞タイプ(すなわち、肝細胞とマクロファージ)に起因する可能性があるにもかかわらず、これらの研究は、12/15-LOXが異なる細胞タイプでオートファジーの内因性調節因子として作用するという新しい概念の支持を提供したので、重要である(図5)。
近年、証拠は、12/15-LOXが機能的なプレーヤーであり、アルツハイマー病の病態生理に貢献していることを示唆している。研究は、そのタンパク質レベルと酵素活性がアルツハイマー病患者や軽度認知障害の臨床診断を受けた被験者で有意に上昇していることを示しており、アルツハイマー病の病態形成におけるこの経路の早期関与を示唆している(116)。
さらに、12/15-LOX酵素活性化の遺伝的欠失または早期の薬理学的阻害は、本疾患の異なるトランスジェニックマウスモデルにおいて、認知障害およびアルツハイマー病様神経病理の発症を予防する(117-119)。高齢化した3xTgマウスに12/15-LOX阻害剤PD146176を投与すると、認知機能の低下、脳アミロイドーシス、タウ病変が逆転し、この経路が疾患発症の後期にも役割を果たすことを初めて証明した(120)。
興味深いことに、同じ研究では、対照動物と比較して、治療マウスではATG5-12とLC3B2/1の定常状態レベルが有意に増加したことを示すことで、この効果が神経細胞のオートファージックフラックスの活性化と関連していることが実験的に裏付けられている。
観察された現象の機序的関係の裏付けとして、12/15-LOX欠損マウスの脳にも同様の生化学的サイン(ATG5-12とLC3B2/1比の上昇)が見られることが示された。このことは、神経細胞と構造的に異なる2種類の12/15-LOX阻害剤を用いた一連の試験管内試験試験でさらに確認された。
これらの研究では、これらのブロッカーで処理された神経細胞は、オートファジーマーカーの上昇だけでなく、最も重要なことに、タウ蓄積の有意な減少を示した(120)。最近では、12/15-LOXを過剰発現させた3xTgマウスでは、アミロイドβとタウの両方のタンパク質の不溶性および凝集性画分が有意に増加し、ネプリリジンやインスリン分解酵素などの古典的な分解経路の変化とは無関係であることが、遺伝学的アプローチを用いて示された(121)。
対照的に、これらのマウスの中枢神経系における12/15-LOXの過剰発現は、AGT7,ATG5-12,およびLC3B1/2比の有意な減少をもたらし、この酵素経路がオートファジー活性化に直接影響を与え、それによって細胞内のアミロイドβおよびタウレベルを調節するという概念を支持するものであった(121)。
結論
全体的に、オートファジーの機能不全がアルツハイマー病における誤ったタンパク質凝集体の蓄積の原因因子である可能性があるという考えを支持する文献がある。したがって、オートファジーを活性化してそれらのクリアランスを刺激するという概念は、有望な治療アプローチであると考えられている。
そのため、オートファジーの活性化が認知機能の低下やアルツハイマー病の脳病理を遅らせる可能性があることが実験的に示されている。しかし、いくつかの前臨床研究は有望であるが、これまでのところ、臨床試験では、アルツハイマー病患者におけるこれらの活性化剤のための効果は中程度または全くないと報告されている。
* *
この矛盾を考慮すると、アルツハイマー病のオートファジー仮説は有効ではないか、あるいはオートファジーはアルツハイマー病の病態形成における二次的なイベントに過ぎないと結論づけることができる。我々は、利用可能なデータに基づいて、これらの結論のいずれかを支持するのに十分な情報がないと考えている。
前臨床試験と臨床試験の間で、また臨床試験自体でも相反する結果が出ているという事実は、明らかに一つのことを示唆している:アルツハイマー病のオートファジー仮説を再考する必要がある。単純な解釈では、オートファジーを回復させることができる薬剤は有益であるはずだと予測されていたが、現実はこのシナリオよりもはるかに複雑であり、前進するためには、すべての証拠を批判的に検討する必要がある。
そのためには、いくつかの教訓がある。我々は文献から学んだこと:薬物の有効性だけでなく、作用機序を理解することを目的とした前臨床研究が必要であること、関心のある薬物の標的関与のためのサロゲートマーカーを開発する必要があること、研究対象となる薬物の薬物動態に関する知識が必要であること、より大きなサンプルサイズの臨床試験が必要であること、患者の登録(軽度の認知障害から完全なアルツハイマー病まで)と病期分類に関して広い異質性を減らす必要があること、そして信頼性の高い認知測定とバイオマーカーを得る必要があること。
* *
結論として、アルツハイマー病のオートファジー仮説はまだ有効であるが、もっと多くの研究が必要であると考えられる。将来的に計画されたより良い前臨床研究や臨床研究、バイオマーカーの分野における技術的進歩、より良いマウスモデルや試験管内試験細胞系、そして新たな創薬努力とともに、より選択性の高い薬剤を生み出すことができれば、最終的にはアルツハイマー病におけるオートファジーの役割を解き明かし、アルツハイマー病の新規治療薬としてのオートファジー活性因子の実現可能性と有効性を実証することができると期待される。
