
Contents
Pharmacological aspects of the neuroprotective effects of irreversible MAO‑B inhibitors, selegiline and rasagiline, in Parkinson’s disease
pubmed.ncbi.nlm.nih.gov/29417334/
Received: 2018年1月4日 / Accepted: 2018年1月31日

要旨
MAO-B阻害剤の時代は50年以上前にさかのぼる。それは、Kálmán Magyarによる選択的阻害剤であるセレギリンの優れた発見から始まった。この化合物は今でもMAO-B阻害剤のゴールドスタンダードとみなされているが、新しい薬剤もこの分野に導入されている。MAO-Bを選択的に、しかも不可逆的に阻害することで、非選択的MAO阻害剤の重篤な副作用であるチラミンの増強、いわゆる「チーズ効果」が起こらないことが早くから明らかにされていた。MAO-Bは主にドーパミンの分解に関与しているため、この阻害剤には抗うつ作用がないが、ドーパミンを節約する作用があることから、パーキンソン病治療の第一選択薬となった。セレギリンの広範な研究により、MAO-B非依存的なメカニズムが関与する複雑な薬理活性プロファイルが示された。神経保護作用や抗アポトーシス作用などの有益な作用の一部は、プロパルギルアミンの構造に関連していた。パーキンソン病の治療薬として承認されている2番目のMAO-B阻害剤であるrasagilineも、この構造要素を持ち、同様の薬理学的特性を示している。本総説では,セレギリンとラサギリンの前臨床試験の概要を紹介する。
キーワード MAO-B阻害,セレギリン,ラサギリン,神経保護
はじめに
カテコラミンの分解酵素であるモノアミン酸化酵素(MAO)の阻害剤は、古くから治療に用いられてきた。デプレニルは、精神的な活力剤として最初に合成され(Varga and Tringer 1967)、抗うつ剤として試験された後、1960年代に不可逆的なMAO阻害剤として特徴づけられた(Knoll er al)。 1965)。デプレニルのR-(-)-異性体は、後にセレギリンと呼ばれ、より強力なMAO阻害剤として同定された(Magyar er al)。 1967)。1968年、Johnstonは、この酵素が不均一であることを発見し、クロルギリンによってより強力に阻害されるアイソフォームをMAO-Aと名付けた(Johnston 1968)。その後、マジャールとノールは、セレギリンも選択的阻害剤であり、MAO-Bと呼ばれるもう一つのアイソフォームに優先的に作用することを発見した(Knoll and In memory of Professor Kálmán Magyar (1933-2017) (Magyar 1972)。この2つのアイソザイムは、基質特異性や組織分布が異なるため、その生理的役割も大きく異なる。これまで使用されてきた非選択的MAO阻害剤のよく知られた抗うつ効果は、MAO-Aの活性を阻害した結果であることがわかった。しかし、MAO-Aを不可逆的に阻害することは、チラミンを多く含む食事を摂取した後に起こる生命を脅かす高血圧性危機という重篤な副作用の原因でもある。これは、チーズ製品にチラミンが多く含まれていることから、「チーズ効果」と呼ばれている。セレギリンは、MAO-Bを選択的に阻害するため、このような危険な副作用はないが(Knoll et al 1968年)抗うつ作用はない。セレギリンは中枢神経系におけるドーパミンの分解を優先的に減少させるため(Riederer et al 1978年)1975年以降、パーキンソン病の治療において重要性を増した(Birkmayer et al 1975,1977年、Parkinson Study Group 1993)。
脳内におけるMAOアイソザイムの分布は、Riedererらによって死後組織で広範囲に研究された。MAO-Bは主にグリアに存在し(Konradi er al)。 1989)、その活性は高齢の患者で上昇していた(Kornhuber er al)。 1989)。死後脳の研究では、セレギリンを投与されたパーキンソン病患者の黒質線条体系において、ドーパミン(Riederer and Youdim 1986)特にフェニルエチルアミン(Riederer er al)。1984)の増加が認められた。これらの知見に基づき、フェニルエチルアミンの上昇によるカテコラミン放出作用がセレギリンのドーパミン免役作用に寄与している可能性が示唆された(Reynolds et al 1978年、Youdim and Riederer 1993)。
セレギリンは酸化ストレスを軽減し(Cohen and Spina 1989)神経毒である1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine(MPTP)によるドーパミン枯渇を防ぐことができることがわかったので(Langston et al 1984; Cohen et al 1984)その薬理学的特性を明らかにするためにさらなる研究が開始された。その結果、MAO-B阻害作用に依存しない神経保護作用、抗アポトーシス作用、神経救済作用の可能性が示され、これらの作用におけるプロパルギルアミン部位の重要性が指摘された。その約30年後には、プロパルギルアミン構造を持つもう一つのMAO-B阻害剤であるラサギリンがパーキンソン病の治療に導入された。本稿では,これら2つの薬剤の神経保護作用,抗腫瘍作用,神経救済作用を理解することを目的とした薬理学的研究をレビューし,議論する。
神経毒に対するセレギリンの保護作用
ドパミン作動性神経毒であるMPTPは、自社で合成したペチジン類似物質を使用した薬物中毒者に急速に発症した重度のパーキンソン症候群が偶然発見されたものである。その後、この化合物による黒質線条体の損傷も確認された(Davis et al 1979)。その後、MAO-B阻害剤であるパルジリンやセレギリンがMPTPの毒性を抑制することがわかり、MPTPの活性型である1-methyl-4-phenyl-pyridinium (MPP+)への変換にMAO-Bが関与していることが示唆された(Langston et al 1984年、Cohen et al 1984)。続いて、MAO-Bによって生成されたMPP+がドーパミン神経細胞に侵入して損傷を与え、MPTP毒性の原因となることも明らかになった。MAO阻害剤の他に、ドーパミン取り込み阻害剤も保護作用を示し、このトランスポーターが毒素の選択的蓄積に重要な役割を果たしていることを示した(Fuller and Hemrick-Luecke 1985)。
以下の研究では、セレギリンの活性毒素MPP+に対する有効性を様々な実験デザインで解析した。まず,MytilineouとCohenは,セレギリンの前処理によって,ラット胚中脳神経細胞培養におけるMPP+によるドーパミン枯渇が抑制されることを報告した(Mytili- neou and Cohen 1985)。一方,Vagliniらの研究(1996)では,この効果は確認されなかった。生体内での動物実験の結果も議論の余地があり、セレギリンによる枯渇の失敗、部分的効果、防止が同様に報告されている(Bradbury et al 1985年、Mihatsch et al 1988年、Vizuete et al 1993)。これらの研究では,MPP+による毒性の後に線条体のドーパミンが枯渇したが,これは必ずしもドーパミン作動性細胞の損失とは一致しなかった。
MPP+はドーパミン作動性ニューロンのミトコンドリア毒素として同定された(Nicklas er al)。 1985; Heikkila er al)。 1985)。MPP+は電子伝達系の複合体Iの活性を阻害し(Nicklas et al 1987)、その結果、エネルギー生産が低下し(Mizuno et al 1987)、酸化ストレスが増大する(Cleeter et al 1992)。同様の変化は、死後のパーキンソン病脳で見られる複合体Iの発現低下(水野 et al 1989年、Schapira et al 1989年、Reichmann and Riederer 1989)や鉄分の増加(Sian-Hülsmann et al 2011)によっても誘発され、著しい酸化的損傷を引き起こす。さらに、酸素フリーラジカルの発生を促進する原因として、毒素による細胞損傷後に放出される膨大な量のドーパミンの自動酸化が挙げられる。MPTP毒性に対するMAO-B阻害剤の保護効果の構成要素を図1にまとめた。
セレギリンの保護効果についてさらに情報を得るために、その示唆された抗酸化特性(Cohen and Spina 1989; Gotz et al 1995)の関与を調べた。MAO-Bの阻害自体が過酸化水素の生成を減少させるが、抗酸化活性の他の構成要素も仮説として考えられた。古くは1989年にKnollが、老齢ラットにセレギリンを慢性的に投与すると、線条体のスーパーオキシドディスムターゼの活性が高まることを報告している(Knoll 1989)。これは後にCarilloらによって若い動物でも確認された。さらに、スーパーオキサイドディスムターゼに加えてカタラーゼ活性の上昇も認められた(Carrilloら 1991)。別の研究では、セレギリン投与後の抗酸化酵素活性の増加は、高齢ラットの線条体でのみ測定できた。抗酸化酵素の他に、グルタチオンのレベルも上昇していることがわかった。若い動物の大脳皮質や海馬などの脳領域では、影響は検出されなかった(Takahata er al 2006)。試験管内試験の細胞培養実験では、神経成長因子(NGF)と同様に、セレギリンの投与によりスーパーオキシドディスムターゼのmRNAの発現が用量依存的に誘導されることが示された。また、セレギリンはNGFの効果を増強したことから、スーパーオキシドディスムターゼmRNAの誘導に対するセレギリンの作用はNGFに依存しないことが示唆された(Li er al)。1998)。これは、セレギリンが単離されたミトコンドリアの酸素消費を阻害することを発見し、この障害がスーパーオキシドディスムターゼ活性の適応的な増加を誘発することを示唆したThiffaultらの知見と一致する(Thiffault et al 1997)。しかし、抗酸化酵素の誘導による抗酸化作用は、慢性投与後にゆっくりと発現するため、セレギリンの急速な神経保護作用を十分に説明することはできない。MPP+によるヒドロキシルラジカル生成に対するセレギリンの急性効果は、ドーパミンのオーバーフローには大きな影響を与えないことが報告されており、直接的な抗酸化作用が重要な役割を果たしていることが示唆されている(Wu er al)。 このことは、他のヒドロキシルラジカル捕捉剤や抗酸化剤でも同様の効果が得られていることからも支持される(Wu er al)。 1996; Khaldy er al 2000)。MPP+損傷を受けた神経細胞をセレギリンで処理すると、低酸素症のマーカーである乳酸の放出が減少することも、セレギリンの保護効果を示している(Matsubara er al 2001)。また、セレギリンは、ロテノンや鉄分過多のような他のタイプの酸化ストレス誘発剤からも、神経細胞を保護することができ(Saravanan er al 2006; Budni er al 2007; de Lima er al 2005)セレギリンの抗酸化特性の重要性がさらに裏付けられた。
セレギリンは、他のモノアミンを枯渇させる神経毒に対しても保護作用があることがわかった。DSP-4(N-(2-クロロエチル)-N-エチル-2-ブロモベンジルアミン塩酸塩)は、ノルアドレナリン神経末端に選択的に蓄積されるため、脳のいくつかの領域でノルアドレナリン濃度を特異的に低下させる。さらに、脳内のドーパミン-β-水酸化酵素活性を低下させ、ノルアドレナリン作動性神経細胞の変性と一致する(Ross 1976)。DSP-4によるノルアドレナリン減少に対して、MAO-B阻害剤のセレギリンと非セレギリン阻害剤のパルギリンには保護効果があり、MAO-A阻害剤のクロルギリンには効果がないことが報告された(Gibson 1987)。このことから、保護にはMAO-Bの阻害が関与していることが示唆された。しかしその後、構造的には無関係で選択的なMAO-B阻害剤であるMDL 72974がDSP-4の毒性を防ぐことができず、セレギリンの保護効果には別のメカニズムがあることが示された(Finnegan et al 1990)。DSP-4はノルアドレナリン末端に取り込み輸送体によって蓄積されるため、セレギリンおよび/またはその代謝物による阻害も提案されていた(Magyar er al)。 1996, 1998; Magyar and Haberle 1999)。しかし、代謝物であるR-(-)-メタンフェタミンおよびR-(-)-アンフェタミンは、より強い取り込み阻害活性を有するが、DSP-4による海馬のノルアドレナリン減少に対して、セレギリンに比べて弱い保護作用を示した。このことから、取り込み阻害が保護効果の唯一のメカニズムであるとは言えない(Haberle er al 2001)。同様に、セレギリンは6-ヒドロキシドーパミンや3,4-メチレンジオキシメタンフェットアミンなど、生体アミンの枯渇を引き起こす他の毒素に対しても保護作用を示すことがわかっているが、正確な作用機序はまだ解明されていない(Salonen er al)。 1996; Sprague and Nichols 1995)。
これらの毒素は、パーキンソン病のある側面をモデル化するのに非常に有用であるが、その効果はパーキンソン病の病態生理からは程遠いものである。その後、セレギリンの保護効果を調べるために、おそらく神経変性疾患の病態とより密接に関連する他の神経細胞傷害を用いた。
神経細胞変性モデルにおけるセレギリンの保護効果
NMDA受容体を介した興奮毒性は、神経細胞の死に関与するメカニズムの一つである。セレギリンとその代謝物であるデスメチルセレギリンは、ドーパミン系中脳の培養神経細胞におけるNMDA誘発神経毒性を予防した。デスメチルセレギリンはこの試験において親化合物よりも強力であることが実際に確認され、セレギリンの神経保護効果にこの代謝物が寄与していることが示唆された(Mytilineou et al 1997a、b)。また、海馬スライスにおいてもNMDA毒性に対する保護作用が示され、同時に、NMDA依存性の長期増強過程を阻害しないことも示された(Niittykoski er al 2003)。生体内試験の実験では、intravitreal NMDA注入によって網膜の損傷が誘発された。局所的ではなく非経口的にセレギリンを投与したところ、保護効果が認められ、局所的にデスメチルセレギリンも有効であった。この結果は、神経保護効果に代謝物であるデスメチルセレギリンが寄与している可能性を示している(Takahata er al 2003)。
また、セレギリンがプロオキシダントによる損傷からミトコンドリアを保護することも示された。膨潤、膜電位の崩壊、シトクロムcの放出を防止した。ミトコンドリアの伝染性遷移孔のいくつかの構成要素との直接的な相互作用と阻害が提案された(De Marchi er al 2003)。
タンパク質の凝集と沈着物の形成は、いくつかの神経変性疾患の特徴である。パーキンソン病では、特徴的なレビー小体の主成分はαシヌクレインである。試験管内試験の実験では、セレギリンが凝集過程を修飾し、無害な大きな凝集体を形成することが示された(Braga er al 2011)。セレギリンが結合したα-シヌクレインの構造は、おそらくよりコンパクトになっていると考えられる。また、その代謝物であるメタンフェタミンの同様の作用も報告されている(Kakish er al 2015)。マウスの研究では、セレギリンは、急性および亜慢性の投与スケジュールで、アミロイドβペプチドによって誘発された認知機能障害を回復させた(Pazini er al 2013)。
虚血再灌流は、脳卒中の脳梗塞に最も近い、神経細胞の損傷と変性の確立されたモデルである。慢性的なセレギリンの前処置は、一過性脳虚血後の梗塞サイズを効果的に減少させた(Knollema er al)。1995)。梗塞サイズ以外にも、アポトーシスのマーカーである切断されたカスパーゼやDNAの断片化も有意に減少した(Unal er al 2001)。虚血による脂質過酸化や記憶障害も緩和された(Maia er al 2004)。セレギリンとイチョウ葉エキスの併用療法は、虚血による神経細胞の減少を防ぐのにより効果的であった(Kwon er al 2004)。しかし、全体的な前脳虚血の直前または直後に適用された前処理は、細胞死を防ぐのに効果的ではなかった(Ballabriga er al)。 1997)。
セレギリンの神経救済作用
Tattonらの報告によると、セレギリンは遅延投与により死にかけている神経細胞を効果的に救済するということで、セレギリンの研究に新たな弾みがついた。マウスにMPTPを投与すると、20日間で黒質のチロシン水酸化酵素陽性細胞数が約40%減少した。投与後72時間後にセレギリンを投与すると、細胞の減少は約15%に抑えられた(Tatton and Greenwood 1991)。セレギリンのレスキュー効果は、中脳の初代ニューロンとグリアの共培養でも観察された。セレギリンをMPP+による傷害の24時間後に投与したところ、培養液中のドーパミン濃度、TH陽性細胞数、アストロサイト数が増加した(Koutsilieri er al)。 1994, 1996)。また、顔面運動ニューロンの軸切除後の非ドーパミン系ニューロンに対しても同様にレスキュー効果が認められた。著者らは、セレギリンが標的由来の栄養サポートの損失を補う可能性を示唆した(Salo and Tatton 1992)。遺伝性の運動ニューロン変性を有する変異マウスを用いたさらなる詳細な研究では、セレギリンは保護作用を示すことができなかった(Oh er al)。 1994)。神経救済効果は、外傷性脳損傷のラットモデルでも観察され、セレギリンは、運動機能は変わらないものの、認知機能を改善した。対症療法的な効果と一致して、傷害による海馬のDBH陽性細胞の消失が減少した(Zhu er al)。 また、カイナイトによる海馬の細胞減少とそれに伴う行動変化も、セレギリンの投与によって影響を受けた。セレギリン投与を中止しても改善が持続したことから、対症療法ではなくニューロレスキュー効果が確認された(Gelowitz and Paterson 1999)。
トロフィックファクターの産生に対するセレギリンの影響は広範囲に検討された。最初の報告の一つは、セレギリンを長期間投与することにより、ラットの新線条体における塩基性線維芽細胞成長因子(bFGF)およびグリア線維酸性タンパク質(GFAP)の産生が、病変により増強されることを示した(Biagini er al)。1994)。ラット皮質アストロサイトにおけるcAMP誘発のbFGF mRNA上昇の増強も示された(Riva er al)。 また、高用量のセレギリンおよびデスメチルセレギリンを投与した24時間後に、マウス培養アストロサイトにおいて、NGF、脳由来向神経性因子(BDNF)グリア由来向神経性因子(GDNF)などの他の向神経性因子の産生が増加することが示された(Mizuta er al 2000)。アストロサイトからの向神経性因子の放出を増加させることに加えて、活性化したミクログリアに関連する神経細胞の損傷に対する効果の可能性も提案され、モデルシステムで研究された。神経芽細胞腫細胞を、活性化した単球性THP-1細胞の条件付き培地で処理した。セレギリンを単球に投与すると、神経芽細胞ではなく、単球の損傷を用量依存的に防止した。他のMAO阻害剤では効果がなく、MAO非依存性のメカニズムが示唆された(Klegeris and McGeer 2000)。セレギリンによるNGF産生の促進と、その結果としての神経細胞の外因性および虚血性障害に対する保護について、詳細な研究が報告された。試験管内試験の細胞培養実験では、かなり低い濃度(10pM-1nM)のセレギリンに反応してNGFの産生が増加した。また、ラットの大脳皮質においても、虚血傷害に対する保護を伴うセレギリン腹腔内投与によるNGF産生の増加が認められた(Semkova er al)。1996)。
さらに、培養ドーパミン神経細胞では、セレギリンとBDNFによって神経突起長が同様に増加することも示された。ただし、セレギリンは神経突起の平均的な枝の長さを増加させるが、BDNFでは新しい枝の形成が増加する(Kontkanen and Castren 1999)。向神経性因子の誘導および/または模倣効果と一致して、セレギリンは様々な幹細胞を神経細胞の表現型に分化させた。これらの試験管内試験実験において、セレギリンはMAO-B活性をかなり阻害できない低濃度(約10-8 M)で有効であることが一般的に認められた。向神経性因子の発現増加とニューロンへの分化は、セレギリン処理後の胚性幹細胞(Esmaeili et al 2006年)骨髄間質細胞(Ghorbanian et al 2010年)胚性癌幹細胞(Bakhshalizadeh et al 2011年)および神経幹細胞(Hassanzadeh et al 2015)で実証された。
セレギリンの抗アポトーシス活性
セレギリンが向神経性因子や抗酸化酵素の合成を誘導することを明らかにした後、その神経保護・神経救済効果は、細胞生存機構の調節に関わる複合的な作用に基づいていることが示唆された(Tatton and Chalmers-Redman 1996; Magyar er al 2006)。分化したPC12細胞において、セレギリンおよびその代謝物であるデスメチルセレギリンは、血清およびNGF休薬により誘発されるいくつかの遺伝子の発現を修飾した。これまでに報告されている抗酸化酵素であるスーパーオキシドディスムターゼとグルタチオンペルオキシダーゼの増加に加えて、他のアポトーシス関連タンパク質の発現の変化も観察された。c-JUNとGAPDHの上昇、BCL-2レベルの低下、さらにはBAXのミトコンドリアへの転位が阻止された。これらの変化はすべて、ミトコンドリアの膜面電位を維持し、伝染性遷移孔の開口部を防ぐことで、抗アポトーシス作用をもたらすと考えられる(Tatton and Chalmers-Redman 1996)。小脳顆粒細胞において、セレギリンと他のプロパルギル化合物は、低カリウムによるアポトーシス細胞死に影響を与えることなく、アポトーシスに関連するミトコンドリア機能の低下を防止した。これらの結果から、セレギリンはアポトーシスのミトコンドリアルートを選択的に阻害することがさらに確認された(Paterson er al)。1998)。セレギリンおよび関連するプロパルギルアミンの効果は用量に依存し、10-9Mの濃度で最大の抗アポトーシス効果が発現し、さらに濃度を上げると効果が低下した(Tatton er al)。 この低濃度のセレギリンは、培養海馬細胞、小脳顆粒細胞、Neuro-2a神経芽細胞において、オカダイン酸によるアポトーシス反応を防ぐのに有効であることがわかっている。しかし、セレギリンはこれらのモデル細胞において、血清欠乏に対する保護作用を発揮しなかった(Suuronen er al 2000)。この知見とは逆に、サブマイクロモル濃度のセレギリンは、培養網膜神経細胞において、血清欠乏または低酸素のいずれかによって誘発されるアポトーシス細胞死を防止した(Xu er al)。1999)。別の研究では、同様にセレギリンを低用量投与すると、神経外胚葉由来の2つのメラノーマ細胞株において、血清剥奪によって誘発される過剰なアポトーシスを2日間遅らせることができたが、そのエナンチオマー対であるS-(+)-デプレニルは効果がなかった。この研究では、セレギリンおよびデスメチルセレギリンの高用量(ミリモル)は、血清を除去しなくてもアポトーシスによる細胞死を増加させることが示された(Szende er al 2000)。これらの結果から、抗アポトーシス作用のベル型の用量依存性が確認され、注意深い実験計画とセレギリンの投与スケジュールの再検討が重要であることが示された(Magyar er al 2004; Magyar 2011)。
セレギリンおよび関連するプロパルギル化合物の抗アポトーシス効果は、試験管内試験の細胞培養実験で様々な神経毒に対しても示された。MPP+で処理したSK-N-SH神経芽腫細胞では、シトクロムcの放出やカスパーゼ3の活性化など、毒素によるアポトーシスマーカーの変化が抑制され、部分的に保護された(Sharma er al)。 SH-SY5Y神経芽細胞腫細胞では、セレギリンを前処理することで、仮説的内因性毒素であるN-methyl-R-salsolinolのアポトーシス効果から細胞が保護され、DNA断片化が減少した。毒素に加えて、反応性酸素種および窒素種によって誘発されるアポトーシスも研究された(Naoi er al)。 神経芽細胞腫細胞を用いたこれらの実験では、セレギリンの有効濃度はマイクロモル領域とはるかに高かったことは特筆に値する。この矛盾は、分化した細胞と未分化な細胞の性質が異なることで説明できるかもしれない。
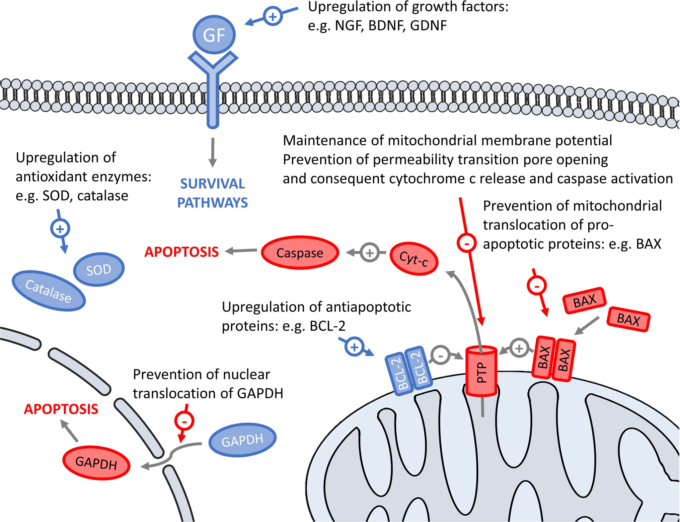
最近の研究では、セレギリンのアポトーシス防止効果がさまざまな動物実験で報告されている。セレギリンは、3-nitropropi-onic acidによる線条体および皮質のアポトーシスを抑制し、カスパーゼ3の活性化やBCL-2/BAX比の変化を示した。さらに、毒素によって誘発される行動変化も改善された(Wahdan er al)。 マウスのMPTP病変後にセレギリンを投与すると、栄養因子の産生が増加したほか、BCL-2およびBAXの発現が抗アポトーシス的に変化した。同時に、この薬剤は毒素による歩行障害を救済した(Zhao er al 2013)。局所脳虚血後にセレギリンを投与すると、梗塞サイズが縮小し、抗アポトーシス遺伝子が誘導され、アストロサイトのNOTCH-JAGGEDシグナルが促進された。これは、脳の微小循環が維持されたためと考えられ、梗塞周囲の浮腫の減少を伴っていた(Nardai er al 2015)。セレギリンの抗アポトーシス活性に寄与するメカニズムとして提案されているのが、図2である。
セレギリンの細胞保護作用、抗酸化作用、抗アポトーシス作用は、損傷した神経細胞に限らない。
図2 セレギリンおよびラサギリンのMAO-B酵素阻害とは独立した作用に関与する標的の推定値
培養気道上皮細胞において、タバコ煙抽出物による酸化ストレスおよび炎症は、セレギリンによって10-8-10-6Mの範囲で用量依存的に回復した。著者らは、本剤の抗酸化特性が、炎症に関連する転写因子Nrf2およびNF-κBの活性化を防ぐことによる抗炎症作用にも関与していることを示唆した(Cui er al 2017)。慢性的な低用量のセレギリンは、血漿の総スカベンジャー容量を増加させ、脂肪分の多い食事で誘発される肝脂肪量の増加を防ぐことが示された(Bekesi er al 2012)。また、動物実験では、腎細胞が低酸素誘導によるアポトーシス死から救われることが示された(Toronyi er al 2002)。ウサギの心不全モデルでは、セレギリンを8週間投与すると、心筋細胞がアポトーシスから救出され、左心室機能が改善された(Qin er al 2003)。
老化に対するセレギリンの有益な効果も、Birkmayerら(1985)が最初に発表して以来、いくつかの論文で報告されている。セレギリンを慢性的に投与すると、ラットの寿命が大幅に延び、性行為も改善された(Knoll.)
また、セレギリンを慢性的に投与することで、ラットの寿命が大幅に延び、高齢動物の性行為(Knoll 1988; Knoll er al)。 長寿誘導の潜在的なメカニズムとしては、抗酸化作用、細胞保護作用、神経活動促進作用、いわゆるモノアミン系伝達のエンハンサー効果が示唆されている(Knoll and Miklya 1994; Kaur er al 2003; Singh er al 2012; Kitani er al 2002)。
セレギリンの神経保護作用に関する研究の中で、そのファーマコフォアにはプロパルギルアミン部分が含まれていることが明らかになった。これは、そのデスメチル代謝物の活性が観察されたことで裏付けられた。しかし、デスメチルセレギリンは、投与量の1%以下のわずかな代謝物であることが示されている(Szökő er al 2004b)。MAO阻害活性を持つ、あるいは持たない他の芳香族および脂肪族プロパルギル化合物についても検討した。試験管内試験および生体内試験での抗アポトーシスおよび神経保護試験におけるそれらの効果は、セレギリンと同様であった(Paterson et al 1998;Waldmeier et al 2000a、b)。
ラサギリンの神経保護・神経救済効果
セレギリン以外にも,いくつかのプロパルギル化合物がMAO阻害作用を目的として1970年代に合成され,研究された。その中でもアミノインデン構造のJ-508は、MAO-Aに対する阻害作用よりもMAO-Bに対する選択性がセレギリンよりも劣るものの、MAO-Bに対する阻害作用が約10倍強いことが明らかになった(Magyar et al 1979; Magyar 1994)。この化合物のデスメチル誘導体であるrasagilineは、パーキンソン病の治療薬として承認された2番目のMAO-B阻害剤として開発された。この化合物の神経保護作用と神経救済作用についても広く研究された。
rasagilineの神経保護作用、抗酸化作用、抗腫瘍作用、ニューロレスキュー作用を明らかにするために、セレギリンと同様に試験管内試験および生体内試験の試験が行われた。セレギリンと同様に、様々な神経毒に対して有効であることがわかった。黒質のドーパミン神経細胞の生存率を高め、6-ヒドロキシドーパミンで誘発されるステレオタイプの行動を改善した(Blandini er al 2004)。SH-SY5Y細胞を用いた試験管内試験の実験では、保護効果のメカニズムとして、毒素によるミトコンドリア障害に関連したアポトーシスの抑制が実証された。同様の結果は、ペルオキシナイトライトを供与するSIN-1やN-methyl-(R)-salsolinolを用いて神経細胞障害を誘発した場合にも報告されている(Maruyama er al)。 Rasagilineは、MPTP後に病変を起こしたマウスに慢性的に投与することで、神経救済作用を示した。ラサギリンは、チロシンキナーゼ受容体(Trk)-ファチジルイノシトール3(PI3)キナーゼ-Akt経路のアップレギュレーションにより、中脳におけるドーパミン神経細胞の生存率を高めた(Weinreb er al 2006; Sagi er al 2007)。また、自然発症した高血圧ラットの視床下部室傍核の神経細胞の変性を用量依存的に抑制した。この効果は、血圧の低下を伴うものであった。降圧剤であるカプトプリルやヒドラジンは神経変性の予防に効果がなく、ラサギリンによる直接的な神経保護が主な作用であることが示唆された(Eliash er al 2005)。
ラサギリンの慢性投与によるカタラーゼおよびスーパーオキシドジスムターゼ(抗酸化酵素)活性の誘導は、海馬ではなく黒質および線条体で確認された。興味深いことに、末梢組織、腎臓、心臓でも抗酸化酵素の活性が上昇していた(Car- rillo er al)。 セレギリンと同様に、虚血性障害に対しても有効であることが証明された。NGFで分化させたPC12細胞では、酸素-グルコース欠乏による細胞死を防ぐことができた(Abu-Raya er al)。 動物実験では、中大脳動脈閉塞後にNGFを投与すると、梗塞サイズが縮小し、神経機能と認知機能が改善された(Speiser er al)。 α-シヌクレインを過剰発現させた細胞培養モデルでは、パラコートの使用により著しい酸化ストレスとカスパーゼ3の活性化が引き起こされたが、ラサギリンにより阻止された(Chau er al 2010)。別の試験管内試験モデルでは、ラサギリンは、ミクログリアの活性化に伴う酸化ストレスや炎症性サイトカインの放出を抑制し、神経保護作用に寄与していると考えられている(Trudler er al)。
ラサギリンの神経保護-抗アポトーシス作用のメカニズムは、主にSH-SY5Yドーパミン神経芽細胞腫細胞を用いた細胞培養実験で広く研究されている。その結果、GDNFを中心とした向神経性因子の誘導(Maruyama er al 2002a, 2004)や、抗アポトーシス作用を持つBCLタンパク質ファミリーの誘導(Akao er al 2002b; Inaba-Hasegawa er al 2012)が報告された。おそらく、これらはミトコンドリアの完全性の保護と、同じく実証されたミトコンドリアのパーミビリティー転移孔の開口部の防止に貢献していると思われる(Maruyama er al)。 抗アポトーシス効果におけるミトコンドリア保護の重要性は、小脳顆粒細胞においても確認されており、ラサギリンはシトシンβ-d-アラビノフラノシド、l-ブチオニン(S,R)-スルホキシミン、またはグルタミン酸を防ぐが、低カリウムや血清剥奪によるアポトーシスは防げなかった(Bonneh-Barkay er al 2005)。
また、単離されたミトコンドリアを用いて、ラサギリンによるミトコンドリアの脱分極および伝染性遷移孔の開口の防止が報告されており、ミトコンドリアを直接標的としていることが示唆された(Akao er al)。 また、GAPDHの核内移行を阻害することが示され、GAPDHもプロパルギルアミン化合物の標的となりうることが示唆された(Maruyama er al 2001a; Ou er al 2009; Waldmeier er al 2000b)。ラサギリンを慢性的に投与すると、プロテインキナーゼCの発現と活性化が増加し、同時にアミロイド処理に有益な影響を与えることが示された(Bar-Am et al 2004)。細胞培養実験では、この効果は、αセクレターゼ活性、プロテインキナーゼCおよびERKキナーゼのシグナルに依存することが示された。また、非MAO阻害剤であるラサギリンのS-エナンチオマーにも同様の効果が認められ、この作用はMAO-B阻害とは関係なく、プロパルギルアミンの構造に依存することが示された(Yogev-Falach er al)。
可逆的なMAO-B阻害剤であるサフィナミド
最近、第3のMAO-B阻害剤であるサフィナミドも治療に導入された。サフィナミドは、セレギリンやラサギリンとは異なる化学構造と薬理学的特性を有している。ラット脳の単離ミトコンドリアにおいて、サフィナミドはMAO-BをIC50値98 nMで強力かつ可逆的に阻害するが、MAO-Aに対する作用はその約5000倍である(Caccia er al 2006)。これらの試験管内試験での結果と同様に,本薬は0.6 mg/kgの単回投与で,ヒトのMAO-A活性に影響を与えることなく,MAO-Bを完全に阻害する(Marzo er al)。 また,MAO-B阻害作用に加えて,はるかに高濃度ではあるが,電位依存性のナトリウムおよびN型カルシウムチャネルを阻害することがわかった。試験管内試験の実験では,チャンネル阻害のIC50値とMAO-B阻害のIC50値の差は2~3桁であった。また、イオンチャネルに対する作用の結果として、グルタミン酸の放出も抑制する(Caccia er al 2006; Fariello 2007)。
サフィナミドによる神経保護についても研究された。サフィナミドは、MAO-B阻害作用と一致してMPTP誘発毒性を防止したが、弱いドーパミン取り込み阻害作用(Fariello 2007)も寄与していると考えられる。MPTPの4時間後には、ドーパミンの減少には影響を与えなかったが、ドーパミン神経系のニューロンには若干のスペア効果が認められた。また、前処理では、カイネイトで誘発されたニューロンの損失を軽減した。虚血によって引き起こされる神経変性も、前処理と後処理の両方で軽減された(Caccia er al 2006)。これらの保護作用は、サフィナミドの高用量で報告されており、サフィナミドのチャネル遮断特性と、その結果としてのグルタミン酸放出の抑制が関与している可能性を示している。非可逆的MAO-B阻害剤に加え、サフィナミドの臨床特性についても最近レビューされている(Dezsi and Vecsei 2017)。
可逆的MAO-B阻害剤の研究には継続的な関心がある。サフィナミドに加えて、いくつかの他の天然および合成化合物が可逆的MAO-B阻害剤として報告され、前臨床および初期臨床研究で検討された。サフィナミドと構造的に関連のある化合物であるセンブラジリンは、高選択的なMAO-B阻害剤であり、効果が長く持続する(Borroni er al 2017)。最近、中等度のアルツハイマー病に対する有効性が第2相臨床試験で評価されているが、決定的な結果は得られていない(Nave er al)。 前臨床試験では、さらなるビアリール化合物(Yeon er al 2018)だけでなく、フラノカルコン(Suresh er al 2018)のような新しい化学構造も研究されている。天然化合物の中でもクリシンは、セレギリンやラサギリンに似たいくつかの薬理作用の他に、MAO-B阻害活性を有することが報告されている(Guo er al)。
セレギリンとラサギリンの効果の比較
ラサギリンの報告された効果の大部分は、これまでにセレギリンで発表された効果と類似している。また、いくつかの直接的な比較試験も行われた。MPTPによる神経毒性の予防をサルで比較したところ、生化学的、組織学的、行動学的に同等の効果が認められた。MAO-Bの阻害がMPTP毒性の軽減に大きく寄与していることから、この結果は予想された(Kupsch et al 2001)。チロシンヒドロキシラーゼ陽性の神経細胞の試験管内試験での生存率は,MAO-B阻害剤の両方によって,1〜10μMという比較的高い濃度で選択的に改善された。MAO-A阻害剤であるclorgylineとR-(-)-methamphetamineは、この濃度範囲では細胞生存率に影響を及ぼさなかった。GABA作動性ニューロンの生存率は、調べたどの化合物にも影響されなかった(Finberg et al 1998;Goggi et al 2000)。ラサギリンおよびセレギリンも同様に、ミトコンドリアの伝染性遷移孔が開いた後のミトコンドリアからのCa2+流出を抑制し、スーパーオキシドの生成を減衰させた(Wu er al 2015)。海馬スライスを用いた研究では、ラサギリンとセレギリンの両方でグルタミン酸を介した興奮毒性が減弱することが観察されたが、より詳細な分析により、異なるメカニズム、すなわち異なるグルタミン酸受容体の反応を調節することが明らかになった(Dimpfel and Hoffmann 2011)。局所脳虚血を対象とした生体内試験において,rasagilineは,1 mg/kgではなく3 mg/kgを投与することで,交感神経の回復を促進したが,最終的な神経学的スコアは改善しなかった。同時に、梗塞サイズを縮小させた。同じ用量のセレギリンはこれらの効果を示さなかった。より高用量の投与は検討されていないので、効果がなかったのは、2つの薬剤のよく知られた効力の違いによるものかもしれない(Speiser er al)。1999)。ユビキチン・プロテアソーム系阻害剤であるラクタシスティンを用いた別の生体内試験神経変性モデルでは、セレギリンとラサギリンの前処理は同様に有効であった。しかし、傷害後に治療を開始した場合、セレギリンの効果は低いことがわかった(Zhu et al 2008)。
いくつかの研究では、保護効果に対する代謝物の影響を評価することも目的としていた。セレギリンの代謝物の中には、プロパルギルアミンの構造を維持しているものがある。デスメチルセレギリンの神経保護効果は、NMDA毒性に対する有益な効果(Mytilineou et al 1997b;Takahata et al 2003)向神経性因子(Mizuta et al 2000)および抗アポトーシスタンパク質(Tat-ton and Chalmers-Redman 1996)の産生を誘導することなど、様々な試験で観察された。そのN-オキシド代謝物は、それまで使用されていた分析方法であるガスクロマトグラフィーに問題があったため、後になってようやく同定された(Katagi er al)。 DSP-4によるノルアドレナリン減少に対するセレギリン-N-オキシドの神経保護効果は、親化合物のそれと比較して高いことがわかった(Szökő er al)。 セレギリンの主な非プロパルギル代謝物はR-(-)-メタンフェタミンとR-(-)-アンフェタミンであり(Szoko er al)。 1999; Tabi er al 2003; Shin 1997)、ラサギリンは1-R-アミノインダンに変換される(Siddiqui and Plosker 2005)。アンフェタミンのS-エナンチオマーはかなりの精神運動刺激作用と神経毒性を持つため、セレギリンの代謝物がその神経保護作用を打ち消すのではないかと考えられた。しかし、R-アンフェタミンは、カテコールアミンを放出する作用が非常に弱く、セレギリンが代謝される際に低濃度で生成される(Magyar and Tothfalusi 1984; Magyar er al 2004)。In vitroの細胞培養実験では、高濃度のメチルアンフェタミンは、血清やNGFの放出によって誘発されるPC12細胞の死を防ぐことができず、セレギリンやラサギリンによる保護効果も低下した(Bar Am er al 2004)。しかし、セレギリンの治療的使用中には、はるかに低濃度のアンフェタミンが生成されるため(Heinonen er al 1989)セレギリン効果の相殺はあまり考えられない。1-R-アミノインダンの効果に関するデータも議論の余地がある。1-R-アミノインダンは、1μMというかなりの高濃度で、血清およびNGFを用いたPC12細胞死モデル(Bar Am et al 2004)およびデキサメタゾンによるSH-SY5Y神経芽細胞および1242-MG膠芽細胞のアポトーシスモデル(Tazik et al 2009)に有効であることがわかっている。しかし、小脳顆粒細胞を用いた別の試験では、広い濃度範囲で様々な細胞傷害に対して有効性が認められなかった(Bonneh-Barkay er al 2005)。これらの結果から、MAO-B阻害剤の神経保護作用におけるプロパルギルアミン構造の重要性がむしろ認められている。このことは,N-プロパルギルアミン自身が,PC12細胞の血清休薬によるアポトーシスに対して神経保護作用を示すという結果からも裏付けられる(Weinreb er al 2005)。
臨床試験における神経保護作用の評価
パーキンソン病におけるMAO-B阻害剤の疾患修飾効果、神経保護効果を評価するために計画された臨床試験がいくつかあったが、最終的な結論は出ていない(レビューと詳細な議論はRiederer and Laux 2011を参照)。DATATOP試験では、推定される抗酸化作用にしたがって、他の抗パーキンソン治療を受けていない病初期の患者を対象に、セレギリンとトコフェロールの効果を評価した。運動スコア(UPDRS)と日常生活動作(ADL)を測定し、症状コントロールのためにレボドパを開始する必要性を登録した。セレギリン投与はスコアを改善し、レボドパ治療の開始を約9カ月遅らせることができた。トコフェロールはプラセボよりも効果がなかった。セレギリン休薬2ヵ月後には運動症状が低下し、神経保護効果は証明できなかった(Parkinson Study Group 1993)。ラサギリンの場合は、異なるデザインの研究が行われ、早期に開始したラサギリン治療の効果が、治療継続期間中も維持されることが明らかになった。しかし、有意差が認められたのはラサギリン1mg/日投与の治療法のみで、高用量である2mg/日投与の治療法では有意差が認められなかった(Parkinson Study Group 2004)。臨床試験データの再評価とポストホック解析を何度か行ったが、両薬剤の症状改善効果と疾患修飾効果を明確に区別することはできなかった。
結論
両非可逆的MAO-B阻害剤の神経保護効果を支持するいくつかの薬理学的実験データが発表されたが,試験管内試験実験の結果を臨床効果に結びつけることは困難であった。前臨床試験のデータでも,薬剤の有効濃度や代謝物の役割については一貫性がなく,試験管内試験と生体内試験の結果が一致しない原因になっていると考えられる。前臨床試験の結果の中には、セレギリンとラサギリンの保護効果の違いを示唆するものもある。しかし、最近の無作為化臨床試験のメタアナリシス(Marconi and Zwingers 2014)セレギリンまたはラサギリン治療中の患者のレボドパ処方を遅らせる薬物利用分析(Peretz er al 2016)ヘッド・ツー・ヘッドの3年間のレトロスペクティブケースコントロール研究(Cereda er al 2017)では、2つの薬剤の有効性と神経保護の可能性は同様であると結論づけられた。
