fizer and Moderna Analysis Re-do
rwmalonemd.substack.com/p/pfizer-and-moderna-analysis-re-do
ロバート・W・マローン医学博士、MS

今月初め、「無作為化試験におけるmRNAワクチン接種後の重大な有害事象」と題するプレプリント(未査読)論文が投稿され、最近ちょっとした騒ぎになっている。
この研究結果を表現するのに、「bombbshell study」(爆弾研究)という言葉が使われている。これはかなり重要なことで、確かに私の注意を引いた。BMJ誌のシニアエディターであるピーター・ドーシ博士がシニアオーサーであるから、あり得るかもしれませんね。Doshi博士は、不都合な真実を伝えることに定評がある。では、見てみよう。
以下は、アブストラクトからのヘッドラインの結果である。
- ファイザーとモデルナのmRNA COVID-19ワクチンは、特に関心のある重篤な有害事象のリスク増加と関連しており、プラセボのベースラインと比較して、1万人当たりの絶対リスク増加はそれぞれ17.6と42.2(95% CI -0.4~20.6 と -3.6~33.8 )と10.1 と15.1であった。
- mRNAワクチンの組み合わせは、1万人あたり12.5人(95%CI 2.1~22.9)の重篤な有害事象の絶対リスク増加と関連していた。
- 特別に関心のある重篤な有害事象の過剰リスクは、ファイザーとモデルナの両試験におけるプラセボ群に対するCOVID-19入院のリスク低減(参加者1万人あたりそれぞれ2.3人と6.4人)を上回った。
そして、要旨の考察部分。
「本研究で認められた重篤な有害事象の過剰リスクは、正式な有害事象-ベネフィット分析、特に入院や死亡などのCOVID-19の重篤な転帰のリスクに応じて層別化した分析の必要性を指摘している。」
この見出しは一見するとかなり深刻に見える。しかし、考察の項では、著者らが慎重であることを警告している。著者らは「火に油を注ぐ」ような知見を示しているわけではない。
本当に何が起こっているのだろうか?これを理解するには、カナダのCOVID Care Allianceによるファイザーの初期試験結果の素晴らしく明確で正確な要約から始めるのが良いだろう(この投稿が、昨年12月に私がツイッターとリンクトインから追い出され、約60万人のフォロワーから縁を切ることになった罪であるらしい)。
また、この分析と調査結果をまとめたPDFをこちらで見ることができる。
要するに、NIAID、FDA、CDCが緊急使用許可を正当化するために使用したファイザーの第3相臨床試験は、意図した追跡期間を満たす前に不適切に中止され、ワクチン接種に伴う有害事象の十分長い追跡分析も行われず、対照群が意図的に排除された、まさにジャンク臨床試験であるということだ。このため、ファイザー社のmRNA接種の真のリスクは何だったのか、その真相に迫る機会は基本的になくなってしまった。さらに小さなリスクについては、この研究では評価するための検出力がなかった(十分な大きさがなかった)。
このような状況の中、(主に)上級の学術研究者からなる勇敢なグループが名乗りを上げた。「天使が踏みつけるのを恐れるところへ愚か者が殺到する」という表現が思い浮かぶ。つまり、承認されたワクチンのシナリオに疑問を呈することは、いかなる学者にとっても非常に危険なことなのだ。しかし、この明らかに愚かでないグループは勇敢にも前に出た。
私の読みでは、彼らがこの分析と報告でとったアプローチは、モデルナとファイザーが行うべき第3相臨床試験(これらは製品の認可前の「大きな、最終」臨床試験であるはず)の分析を行うために誠実に努力することであったと思われる。基本的には、FDAが自ら行うべきであり、また、モデルナとファイザーに行わせるべきであった分析である。ホワイトハウス首席補佐官のMark MeadowsがFDAに圧力をかけなければ、おそらくFDAは正しいことをしただろう。しかし、FDAは明らかに屈服し、仕事をしなかったので、今ここにいるのである。
ここに問題がある。FDAは自分の仕事をしなかっただけでなく、FDAもモデルナもPfizerも主要データを公開しないので、他の誰もそれをすることができないのだ。この最近の分析の著者が考察で述べているように。
様々な人口統計学的サブグループにおける害と利益の問題に対処するために、個々の参加者データを用いた系統的レビューとメタ分析が行われるべきである。これらの疑問を適切に評価するためには、COVID-19ワクチンの臨床試験データの完全な透明性が必要である。残念ながら、COVID-19ワクチンの普及から1年以上経過しても、参加者レベルのデータにはアクセスできないままだ。
Doshiと同僚たちは、2つの出版物で繰り返し完全な情報開示を要求しているが、無駄だった。したがって、裁判所が義務づけたデータ公開にデータが含まれない限り、今回のプレプリントレポートで彼らが行った分析が、私たちが手に入れようとしているもののうちで最も良いものになるかもしれない。これについては、以下を参照してほしい。
Tanveer S, Rowhani-Farid A, Hong K, Jefferson T, Doshi P. Transparency of COVID-19 vaccine trials: decisions without data.(COVID-19ワクチン試験の透明性:データなしの意思決定)。BMJ Evid Based Med [インターネット]を参照してほしい。2021年8月9日
Doshi P, Godlee F, Abbasi K. COVID-19ワクチンと治療法:我々は今、生のデータを持たなければならない。BMJ [インターネット]。2022 Jan 19;376:o102.
Doshi博士らが適切に指摘しているように。
2013年、米国と欧州の業界団体は、臨床試験データの共有に関する共同声明を支持し、「患者、医療、経済の利益のために臨床試験データを共有することの重要性を認識する」という一連のコミットメントを行った6 2015年、米国医学研究所は、同様に臨床試験データの共有による利益を支持し、「研究者の主張の検証および再現」が科学プロセスにとって不可欠であると強調し、「患者、その医師、研究者はもちろん、医療の支払者を含む関係者にとって多くの利益がある」ことに言及した。
しかし、願いが馬なら、乞食は乗るだろう。ファイザー社、モデルナ社、FDAは、米国の裁判所がそうさせない限り、英国医学雑誌の上級編集者の訴えに耳を傾けるつもりはないことは明らかであり、その場合でも、できるだけ長く踵を返すつもりである。その理由は想像に難くない<皮肉>。
Doshi博士らがとったアプローチは、各企業(「スポンサー」)の学術出版物、FDAやカナダ保健省のウェブサイトをくまなく調べ、公開プレゼンテーションから得られた有害事象の表やリストを、できる限り「本物」の主要データに近い形に組み立てて、それらのデータセットを分析することによって、オリジナルと考えられるものを厳密に組み立てることだった。
雑誌の出版物に加え、FDA(諮問委員会の資料)とカナダ保健省(スポンサーが規制当局に提出する資料の一部)のウェブサイトを検索した。FDAのウェブサイトでは、FDAとスポンサーの両方によるプレゼンテーションを検討した。これらの資料の中から、特定のSAEタイプ別に情報を提示するSAE結果表を検索し、FDAが要求する投与後1ヶ月以上の安全性追跡調査期間(中央値)に対応する最新のSAE表を選択した。
SAEは、serious adverse event(重篤な有害事象)の略語だ。最後の行、2回目の投与から2ヶ月後に注目してほしい。昨年1月のCell誌の論文で、mRNAではない合成mRNAも、そのmRNAから生成されるスパイクタンパク質も、少なくとも60日間持続することが分かっているので、「薬」は投与2回目以降も少なくとも2カ月間は存在することになる。FDAがSAEのフォローアップを1カ月以上にするよう主張すれば、おそらくもっとよくなるはずである。しかし、トランプの首席補佐官が「早くやれ」と言っていたので、彼らは慌てていた。そこで、これである。原因と結果だ。
論文の内容に戻るが、Doshiらは、得られた有害事象のデータを分析するために、CEPIとBrighton Collaborationが作成し、WHOが承認した「有害事象リスト」(AESI: adverse events of special interest)を適用している。このリストは試験開始前に作成されたものである。今、振り返ってみると、ファイザー社の広範なAESI表があるが、これは緊急時使用許可が出た後に作成されたようで、著者らはそれを使うこともできたはずである。しかし、Doshiらは真のボーイスカウトであり、本来行われるべきことを遡及的に行おうとして、試験データが解析に利用可能となる前に存在したAESIリストのみを評価することを選択したのである。
この問題点は、彼らは実際に患者レベルのデータを入手できないため、これらの主要データについて、特にその数値/統計分布の観点から、いくつかの仮定をしなければならなかったということだ。
もう一つの限界は、個々の参加者のデータにアクセスできないことで、標準誤差を保守的に調整することを余儀なくされたことだ。そのため、標準誤差を保守的に調整せざるを得なかった。したがって、算出された95%CIは、どの患者が複数のイベントを発症したかが分からないため、あくまで概算に過ぎない。さらに、有効性のエンドポイント(COVID-19,COVID-19肺炎、「SARS-CoV-2検査陽性」と表示されたSAE)を解析から除外しようとしたにもかかわらず、COVID-19の重大な合併症(例えば、急性呼吸不全、心停止、急性腎障害)がよく起こるSAEを識別して除外できなかった…。
つまり、彼らはできる限りのことをしたが、いくつかの仮定を含めなければならなかった。
このような苦労の末に出来上がったのが、主要なデータ表だ。
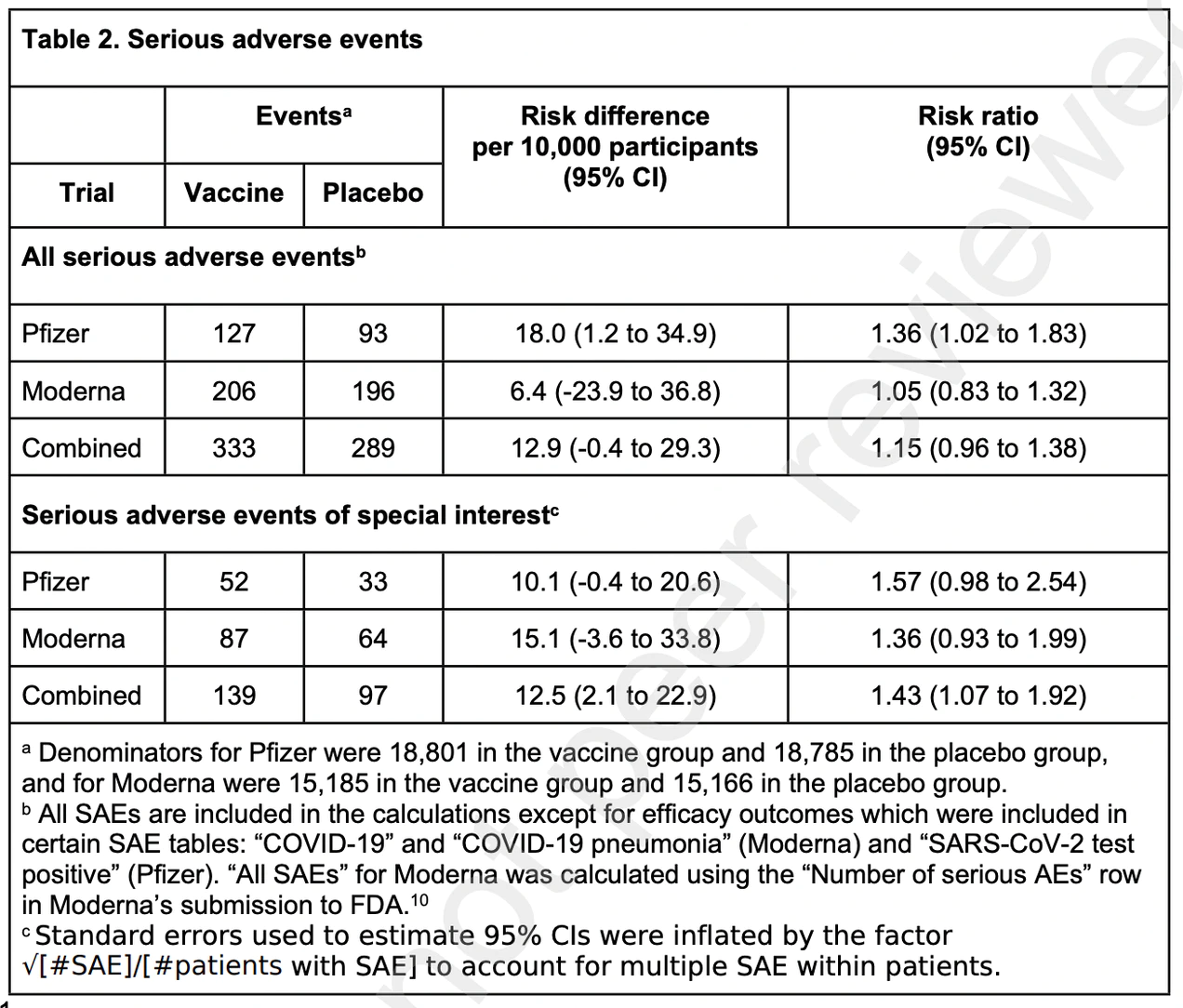
リスク比の列と、特に95%信頼区間(CIと略す)に注目してほしい。対照群と実験群が同等である場合のリスク比は、1.0となる。1.0より大きいと(この場合)ワクチンを接種した人に有害事象のリスクがより高いことを意味する。しかし、その数値の周辺には統計的な範囲がある(無作為に検定を行い、100回中95回はその範囲内に結果が収まるという統計検定の閾値を設定した場合)。であるから、信頼区間が1未満から1以上にわたる場合、対照群とワクチン投与群の結果に統計的な差があると結論づけることはできない。このような試験の多くがそうであるように。つまり、もしテストした患者の数がもっと多ければ、統計的に有意になる可能性があることを示唆している。しかし、これは第3相のワクチン試験としては控えめなサンプル数だ。繰り返しになるが、FDAはスポンサーにこれを許したが、これが入手可能なデータなのである。そして、この時点に戻ることはできない。なぜなら、現在、ほとんどすべての人がワクチンを接種しているか、感染しているかのどちらかだからである。
ワクチン研究では、研究サンプルサイズを推定するために、「3の法則」を適用している。1000人に1人の割合で発生する有害事象を確実に検出したいのであれば、ワクチン接種群から3000人の患者をテストする必要がある。つまり、ファイザーの試験では、(18,800/3)=6,266人に1人程度の割合で発生する有害事象を検出する検出力があることになる。モデルナは、(15,185/3)=5,061人。それ以下の頻度で発生する有害事象は、一般に統計学的に有意なレベルでは検出されないだろう。対照群にランダムに発生する有害事象の頻度を補正し、1万人当たりの事象数で正規化すると、表にまとめたデータになる。
また、「特に注目すべき重大な有害事象」については、統計的有意性を示す推定値を得るために、ファイザー社とモデルナ社の臨床試験のデータを組み合わせなければならないことに注意してほしい。この2つの製品は異なり、異なる製剤を含み、mRNAの投与量も全く異なるため、「現実の世界」では決して行われないことだ。
以上のことから、この分析が、著者の手持ちの資料の中では、かなり良いものであることがおわかりいただけると思う。しかし、なぜ彼らが(適切に)簡潔に調査結果を報告し(「我々が欲しいのは事実だけだ、奥さん」)そして適切に慎重な結論を導き出したのかも理解いただけると思う。
我々の研究で見つかった重篤な有害事象の過剰リスクは、正式な害と利益の分析、特に入院や死亡といったCOVID-19の重篤な転帰のリスクに応じて層別化した分析の必要性を示唆している。
これは、これらのmRNAワクチンの臨床研究史において、文字通り歴史の流れに影響を与える重要な決定がなされた時点に戻ろうとする英雄的な共同作業だった。その時の決断は急がれ、2つの研究の有用性は、研究を早期に中止し、対照群にワクチンを接種することで失われた(意図的に?今回の解析のやり直しの著者たちは、最善を尽くしている。しかし、Doshi博士らが繰り返し要求しているように、オリジナルのデータセットが公開されない限り、適切な解析はできない。
それまでは、ずっとパリのままだ。お前を見ているようだ
もう一回やってくれ、サム 「As time goes by 」を再生してほしい。
