
SIRT1 as a therapeutic target for Alzheimer’s disease
https://pubmed.ncbi.nlm.nih.gov/27497424/
2016年12月1日
要旨
アルツハイマー病は、世界的に高齢化が進む中で認知症の最も一般的な原因となっている。SIRT1 は、ヒストンや転写因子の脱アセチル化により、複数の神経細胞や非神経細胞を標的とし、ストレス応答、エネルギー代謝、細胞の老化/死の経路を調節している。以上のことから、SIRT1 活性は、股関節-小脳および皮質ニューロンの機能や生存に様々な面で影響を与え、疾患の発症や進行を修飾する可能性があると考えられる。本レビューでは、アルツハイマー病に対するSIRT1の作用機序と治療標的としての可能性について考察する。
キーワード:βアミロイド、アルツハイマー病 (AD)レスベラ-コントロール、SIRT1,タウ
はじめに
現代における人間の平均寿命の増加(Kinsella, 1992; Olshansky, 2015)は、特に65歳以上の高齢者における加齢に伴う障害の症例を効果的に増加させている。認知障害および認知症は、深刻な社会的・経済的負担を構成し(滝沢 et al 2015a)高齢者における認知症の有力な原因は、アルツハイマー病である(アルツハイマー協会 2014; Scheltens et al 2016)。アルツハイマー病脳は、組織学的に細胞外アミロイド斑[Aβ(Aβ)ペプチドを含む]および脳内神経原線維のもつれ(高リン酸化タウを含む)によって特徴づけられる(Ittner and Götz, 2011; Bloom, 2014)。Aβペプチドは、逐次的なβ-セクレターゼおよびγ-セクレターゼ切断によってアミロイド前駆体タンパク質(APP)から生成され(De Strooper et al 2012)Aβオリゴマーおよび不溶性アグレゲートの神経細胞毒性は、アルツハイマー病らのアミロイドカスケード仮説を根底から支えている(Karran et al 2011)。一方、タウの病理学はアルツハイマー病の病因において重要な役割を果たしており、Aβとタウは神経細胞毒性の個別モードと相乗モードの両方を発揮するようである(Nisbet et al 2015)。高齢者の間のアルツハイマー病症例の大部分は散発性であるが、この疾患に対する遺伝的素因は知られている。APPとγ-セクレターゼ複合体のタンパク質分解成分であるプレセニリンの両方における突然変異(De Strooper et al 2012;Zhang et al 2014)は、必ずAβ産生を増加させ、疾患の早期発症を促進する(Tanzi 2012)。AβペプチドへのAPPのタンパク質分解処理は、それ自体およびセクレターゼの膜トラフィッキング遍歴に影響される(Tang 2009a;Bhalla et al 2012;Choy et al 2012;Jianga et al 2014,2014)。2014)一連の膜タンパク質調節因子(TangおよびLiou 2007)およびジシンインテグリンおよびメタロプロテアーゼ(ADAM)ファミリー(VingtdeuxおよびMarambaud 2012)のメンバーであるα-セクレターゼによる事前の切断が、β-セクレターゼの作用およびアミロイド生成を妨げるという事実。後者は、α-セクレターゼ活性を高めることが、アミロイド負荷を低下させ、アルツハイマー病の発症を遅らせる可能性があることを意味するであろう。
サーチュインまたはクラスIIIヒストン脱アセチラーゼは、ニコチンアミド・アデニン・ジヌクレオチド+(NAD+)依存性タンパク質脱アセチラーゼであり(Blander and Guarente, 2004)、広い範囲の細胞標的を持ち、細胞および組織の生理学、病理学、ならびに生物学的老化の多面的な側面に影響を与えている(Haigis and Sinclair, 2010; Sebastián et al 2012; Chang and Guarente, 2004)。2012;ChangおよびGuarente 2014;HerskovitsおよびGuarente 2014;Ng et al 2015)。) 出芽酵母Saccharomyces cerevisiaeにおける単一のSirtuinオルソログ、Sir2pは、酵母の交尾座、テロメアおよびリボソームDNA座からの転写を抑制し(Imai et al 2000)、そのrDNAサークル形成の抑制(Sinclair and Guarente, 1997)は、酵母の複製寿命の延長に積極的な役割を有する(Kaeberlein et al 1999; Fabrizio et al 2005)。elegansやDrosophila melanogasterなどの無脊椎動物のSIR2オルソログ(Tissenbaum and Guarente, 2001; Rogina and Helfand, 2004; Wood et al 2004; Burnett et al 2011; Viswanathan and Guarente, 2011)にも寿命延長活性が認められている。哺乳類では、Sir2と最も相同性の高いSIRT1パラログが寿命と健康寿命の向上の観点から広く研究されている。糖尿病マウスや肥満マウスでSIRT1をトランスジェニックに過剰発現させたり、薬理学的に活性化させたりすると、生涯の後半で代謝パラメータが改善された(Baur et al 2006; Alcendor et al 2007; Milne et al 2007; Herranz et al 2010)。SIRT1を脳特異的にトランスジェニックに過剰発現させたマウスは、SIRT1特異的活性化剤SRT1720を給与したマウスと同様に、寿命の測定可能な増加を示した(Satoh et al 2013)。他の哺乳類のサーチュインもまた、長寿に関与していることが示唆されている。例えば、SIRT6トランスジェニック過剰発現は、マウスにおいて性別に依存した長寿効果を有することが示され(Kanfi et al 2012)SIRT2は、分裂チェックポイントキナーゼBubR1に対して低モルフィックである短命マウスの寿命を延ばすことが見出された(North et al 2014)。初期の研究では、Sir2p/SIRT1が介在する寿命延長が、ある意味で食事制限またはカロリー制限(CR)の効果を模倣していることが明らかになった(Lin et al 2000;Anderson et al 2003;Cohen et al 2004)。栄養不良を引き起こすことなく食物摂取量を減少させるCRレジームは、ほとんどすべての動物モデルで寿命を延ばすことが確実に知られている(Chung et al 2013)。CR は SIRT1 の活性を増加させるが、SIRT1 が欠損したマウスは CR に反応しなかった (Boily et al 2008; Schenk et al 2011)。このように、CR 誘導長寿が SIRT1 を介して効果を発揮するという事実は、魅力的な考え方である。激しく議論されており、まだ結論が出ていないが(Kaeberlein and Powers, 2007; Cantó and Auwerx, 2009a,b; Guarente, 2013; Park et al 2013)CR-サーチュインのリンクは、サーチュイン、特にSIRT1を標的とした老化および老化関連疾患の治療的介入に大きな関心を呼び起こしている(Tang, 2011; Baur et al 2012; Chakraborty and Doss, 2013)。
健康寿命の延長の重要な側面は、認知機能の維持である。代謝性疾患以外にも、SIRT1は脳神経細胞で高度に発現しており、神経脱生成に対する一般的な役割が広く帰属している(Srivastava and Haigis, 2011; Herskovits and Guarente, 2014; Ng et al 2015)。また、SIRT1は、最近明らかになったシナプス可塑性および学習/記憶プロセスに内在する役割を持っている。脳特異的なSIRT1の過剰発現が寿命を延ばす可能性があるという事実(Satoh et al 2013)は、SIRT1が脳組織での活動を通じて老化に影響を与える可能性を示唆している。本レビューでは、SIRT1 と アルツハイマー病 との関連性を検討し、直接的および間接的なメカニズムについて議論する。また、SIRT1をベースにした、あるいは関連する認知症の治療研究についても簡単に見ていく。
SIRT1レベル/活動とアルツハイマー病の関連性
脳のSIRT1レベル/活性は加齢とアルツハイマー病で影響を受ける
SIRT1 は、すべての細胞/組織タイプで発現しており、齧歯類の成体およびヒトの脳では、その発現が顕著であり(Ramadori et al 2008; Michán et al 2010; Zakhary et al 2010)海馬や前頭前野などの アルツハイマー病 病理に関連する脳領域に遍在的に存在している(Michán et al 2010; Zakhary et al 2010)。げっ歯類の脳における In situ ハイブリダイゼーション研究では、SIRT1 トランスクリプトが基本的にすべての構造に存在することが示された。アルツハイマー病に関連する海馬領域では、CA1,CA2,CA3,歯状回のすべてが強いハイブリダイゼーションシグナルを示した(Ramadori et al 2008)。免疫細胞化学的解析により、齧歯類およびヒトの様々な脳領域における SIRT1 の局在が示された (Michán et al 2010)。可視化されやすい主なSIRT1発現細胞は神経細胞であり、SIRT1シグナルはパルバルブミンまたはチロシン水酸化酵素陽性のソーマにおいて大部分が核内に存在している(Michán er al)。 SIRT1の発現は、アストログリアやオリゴデンドログリアでも認められている(Lutz er al)。 海馬ニューロンで高発現しているようであるが、SIRT1の遺伝子欠失は、少なくともマウスでは、海馬領域や脳の総解剖学的な異常は観察されなかった(Zakhary et al 2010)。
脳内SIRT1レベルは代謝的に調節されており、身体活動(Falone et al 2012;Revilla et al 2014)およびCR(Satoh et al 2010;Quintas et al 2012;Gräff et al 2013)によって増加するが、高糖質/脂肪食によっては抑制される(Wu et al 2006;Heyward et al 2012)。CRは、マウスの脳内のSIRT1レベルに異なる影響を与えることが示されており、アルツハイマー病に関連する海馬および大脳皮質領域ではそのレベルを増加させ、小脳および中脳ではそのレベルを減少させる(Chen et al 2008)。重要なことに、SIRT1レベルは、一般に、老化したニューロンにおいて減少し(Quintas et al 2012)中枢神経系(CNS)SIRT1の減少は、しばしば、様々な神経病理学的状態と関連している(Pallàs et al 2008; Valle et al 2014)。動物実験モデルはさておき、SIRT1レベルは、ヒトAD脳において減少しているようであり(Julien et al 2009; Lutz et al 2014)、この減少は、アルツハイマー病の初期から後期への進行中に影響を受ける脳領域に対応している(Lutz et al 2014)。一方、脳組織におけるSIRT1転写産物の発現については、健常高齢者とアルツハイマー病患者群の死後サンプル間では有意差は認められなかった(Furuya er al)。 ほとんどの場合、SIRT1レベルと活性度がアルツハイマー病の発症/進行と相関しているだけなのか、それともSIRT1欠乏と疾患との間に本当に因果関係があるのかを立証することは難しいと思われる。
SIRT1 は学習や記憶に本来の役割を持っている。
宣言的記憶形成の障害は、アルツハイマー病の神経学的特徴である(Stern, 2012)。SIRT1が学習と記憶に役割を持つ可能性があるという事実が最近報告された。Michánらによって生成されたSIRT1ノックアウトマウスは、樹状突起の分岐と密度の低下を示し、マウスの海馬のシャッファー側副経路は、長期増強(LTP)の欠陥を示した。また、Sirt1欠損マウスでは、短期記憶、長期連想記憶、空間学習の障害が確認された。Gao er al)。 (2010)によるネスチン-クレアを用いたSIRT1の条件付きノックアウトマウス脳内でのノックアウトでは、股関節後頭CA1ニューロンの樹状突起密度の低下と海馬CA1のLTPの低下が認められた。興味深いことに、SIRT1欠損マウスの脳ではmiR-134のレベルが上昇しており、これがcAMP応答エレメント結合タンパク質(CREB)のトランスラーションを減少させ、結果的に神経発達とシナプス機能を制御する脳由来の神経栄養因子の産生を減少させていることがわかった。いずれの研究も、学習や記憶に関するSIRT1の神経発達とシナプス可塑性の役割を指摘している。記憶の形成と強化におけるCREBのよく知られた役割(Kida and Serita, 2014)を考えると、加齢とアルツハイマー病におけるSIRT1レベルの低下は、疾患の病理学の他の側面と相加的または相乗的な方法で認知機能を損なう可能性が高い。興味深いことに、認知障害は、タウ(Ahmed et al 2014; Lei et al 2014; Ma et al 2014)およびAPP欠損(Dawson et al 1999; Senechal et al 2008; Lee et al 2010)の両方について、老年期に報告されている。後述するように、SIRT1は、タウおよびAPPの両方と機能的に相互作用し、病理学的文脈および非病理学的文脈の両方において、これらのモルキュールを介して学習および記憶に影響を与える可能性がある。
動物モデルにおける SIRT1 の CR 活性化または過剰発現は、アルツハイマー病 に関連する病理を抑制した。
長年にわたる多くの研究により、CR による SIRT1 レベルまたは活性の上昇が動物モデルにおける アルツハイマー病 関連病理を抑制することが示されている(Wang er al)。 多くの報告では、メタボリックシンドローム/タイプII 糖尿病およびアルツハイマー病(Park 2011;Dar et al 2014)。パシネッティの研究室は、食事誘発性インスリン抵抗性が、アルツハイマー病のトランスジェニックマウスモデル(Tg2576,変異APP)においてアミロイド形成を促進することを示した(Ho et al 2004)。CRはトランスジェニックマウス(Wang et al 2005)と霊長類モデル(Qin et al 2006a,b)の両方でアルツハイマー病型脳アミロイドーシスを減衰させた。著者らは、CRがマウスの脳内でSIRT1とNAD+レベルを増加させ、培養ニューロンでSIRT1を外因性に発現させると、Rho依存性キナーゼ1-依存的な方法で非アミロイド性APPの処理を促進することで、Aβ産生が減少することを示した(Qin et al 2006b)。他の研究では、CRが変異APP(Mouton et al 2009; Schafer et al 2015)または変異タウ(Brownlow et al 2014)トランスジェニックマウスにおいて、Aβおよびアミロイド病理を減少させることも示されているが、これらの研究では、SIRT1の変化は明らかに抑止されていなかった。アルツハイマー病/タウ症のトランスジェニックp25(coactivator of cyclin-dependent kinase 5)モデルでは、実際にSIRT1がアップレギュレートされ、res-veratrolによるSIRT1の活性化は、海馬CA1領域へのレンチウイルス注入によるin situ SIRT1過剰発現と同様に、神経変性に対して保護されることが示された(Kim et al 2007)。このp25トランスジェニックモデルでは、CRは、シナプス可塑性と記憶を維持するだけでなく、神経細胞の損失と脳病理を救済した。著者らは、CRがヒップポカンプのSIRT1レベルと活性を増加させ、SIRT1の低分子アクチベーターであるSRT3657がCRの効果を本質的に再現できることを示した(Gräff et al 2013)。まとめると、これらの結果は、動物モデルにおけるアルツハイマー病の発症および進行とSIRT1レベル/活性との間の明確な相関関係を提供した。
複数の薬剤による動物モデルにおけるアルツハイマー病またはアルツハイマー病関連病態の抑制は、SIRT1の活性化に起因している。
一連の化合物は、実験動物における アルツハイマー病-関連または アルツハイマー病 関連の病理学を改善することが示されている。まず第一に、レスベラトロールは、酵母のレプリカ寿命を増加させ(Howitz et al 2003)動物のCRを模倣することが示されている(Wood et al 2004)最初のSIRT1活性化因子である。レスベラトロールは複数の細胞標的を有しており(Dasgupta and Milbrandt, 2007; Park er al 2012; Takizawa er al 2015a,b)それが真のSIRT1活性化因子であるかどうかは議論の的となっている(Pacholec er al 2010)。より最近の研究では、レスベラトロールおよび関連化合物が、特定のSIRT1基質上のアロステリック結合部位を介してSIRT1を活性化する可能性があることが示唆された(Hubbard et al 2013)。相関性のあるSIRT1エレバシオンを有するアルツハイマー病関連病理におけるレスベラトロールおよび他の化合物の有益な効果を文書化した多くの研究がある(Chen et al 2005;Feng et al 2013;Lee et al 2014a,b;Marwarha et al 2014;Sun et al 2014)。これらの研究は、培養中の神経細胞または一次ニューロンのいずれかを用いて行われた。これらの化合物を使用して動物モデルで実施された多くの研究は、SIRT1レベルまたは活性の付随的な上昇を明確に示さなかった。SIRT1 レベルまたは活性の相関を示した動物ベースの研究を表1
にまとめた。
上述したように、ヒトと動物の両方のデータから得られた SIRT1 と アルツハイマー病 の間の関連性は相当なものである。次に、Aβとタウに対するより直接的な効果からより間接的な効果へと進行しながら、SIRT1活性がアルツハイマー病の発症と進行に有益である可能性があるメカニズムと細胞/分子経路を見ていく。
SIRT1によるアルツハイマー病病態の抑制-直接的および間接的なメカニズム
非アミロイド性APP切断の誘導とAβレベルの低下
APPのα-セクレターゼ切断がAPPのその後のアミロイド生成処理を妨げるように、アミロイド産生と負荷の減少は、直接アルツハイマー病の発症と進行に影響を与えるだろう(唐 2005)。Pasinettiグループによる研究では、野生型SIRT1の過剰発現がαセクレターゼ活性を増強し(一方、ドミナントネガティブ変異体は減少し)対応するAβの減少とαセクレターゼ切断産物sAPPαのレベルの増加を伴うことが示されている(Qin et al 2006b)。より最近の結果は、この知見を裏付けるものである。SIRT1の過剰発現は、sAPPα分泌におけるApoE4媒介の減少を救済することが示された(Theendakara et al 2013)。ホスホジエステラーゼ阻害剤シロスタゾールは、SIRT1共役型レチノイン酸受容体β活性のアップレギュレーションを介してα分泌酵素ADAM10を活性化することが示された(Lee et al 2014a,b)。他の研究でも、APP切断におけるADAM10活性の上昇の役割が示唆されており(Endres and Fahrenholz, 2010)α-セクレターゼのSIRT1の転写制御は、したがって、Aβレベルを低下させ、アルツハイマー病病理を抑制する上で重要なメカニズムである可能性がある。
タウレベルと蓄積に影響を与えるSirt1活性
タウまたはその高ホスホス化によって媒介される病理学的損傷は、アルツハイマー病の進行において、下流またはAβと並行して作用し(Bloom, 2014; Nisbet er al)。 したがって、タウリン酸化またはそのレベルの抑制は、アルツハイマー病において有益であろう。Ganの研究室は、タウはヒストンアセチルトランスフェラーゼp300によってアセチル化され、SIRT1活性によって脱アセチル化され得ることを示した(Min et al 2010)。したがって、SIRT1を阻害すると、タウのアセチル化が著しく増加し、病的に高ホスホス化したタウが蓄積された(Min et al 2010)。タウのLys174アセチル化は、アルツハイマー病脳における初期症状であるようであり、タウ症の病因学的に重要である(Min et al 2015)。また、Lys280でのタウのアセチル化は、アルツハイマー病関連タウ症の病理組織学的マーカーとして関連性があることが示されている(Irwin et al 2013)。このアセチル化の薬理学的な減少は、おそらくその分解を促進することによって、タウ誘発性記憶障害および腰部ポカンプ萎縮を減少させる可能性がある(Min et al 2015)。したがって、加齢脳におけるSIRT1の減少は、リン酸化されたタウの安定性を促進し、したがって、アルツハイマー病の進行を助長する可能性があると考えられる。
神経炎症と老化表現型のSirt1抑制
現在、脳内のニューロン細胞とグリア細胞の両方における神経炎症プロプロセスが、アルツハイマー病の進行において極めて重要な役割を果たすことがますます明らかになってきている(Avila-Muñoz and Arias, 2014; Heneka et al 2015; De Strooper and Karran, 2016)。SIRT1は、NF-κBのRelA/p65を脱アセチル化する(Yeung et al 2004)ので、神経炎症を抑制する役割を持つであろう。いくつかの報告は、ミクログリアの増殖およびミクログリア依存性神経炎症の活性化を抑制するSIRT1の役割を明確に示唆しており(Chen et al 2005;Cho et al 2015;Li et al 2015b)これはアルツハイマー病の進行に対抗する可能性が高い。アストロサイト、特に傷害および神経炎症によって活性化されたものはまた、Aβの生成に影響を与えるためにニューロンと相互作用する可能性があり(Avila-MuñozおよびArias 2014)SIRT1の活性化は、抗酸化酵素のアップレギュレーションを介してアストロサイトにおける酸化ストレスを減衰させることが知られている(Cheng et al 2014)。脳内の一次免疫細胞以外にも、慢性炎症は、Senescence-Associated secretory phenotype (SASP)と呼ばれるプロ炎症性因子の分泌を介して、他のすべての老化および老化老化細胞によって非細胞的に誘導される可能性がある(Coppé et al 2010; Ovadya and Krizhanovsky, 2014)。最近、SIRT1がそのプロモーター領域でのヒストン脱アセチル化を介してSASP遺伝子の発現を抑制することが示された(Hayakawa et al 2015)。したがって、慢性的な神経炎症のSIRT1の抑制は、疾患の病態、特に散発的で遅発性のアルツハイマー病の形態を抑制する上で重要な役割を果たす可能性がある。
SIRT1活性は、一般的に神経保護的である可能性がある
SIRT1は一般的に神経保護的であり、この考えは、神経細胞p53の脱アセチル化(Luo et al 2001)に大きく起因しており、様々な文脈で再確認されている(Hegawa and Yoshikawa, 2008; Hernández-Jiménez et al 2013; Xiong et al 2015)。一般に、p53の脱アセチル化はアポトーシス誘導を抑制する。急性神経損傷については、活性化化合物によるSIRT1活性化(Raval et al 2006;Hernández-Jiménez et al 2013)またはSIRT1過剰発現(Hernández-Jiménez et al 2013)の両方が、虚血性損傷を減少させ(KoronowskiおよびPerez-Pinzon 2015)外傷性脳損傷における神経細胞死を減衰させることが示されている(Zhao et al 2012)。また、SIRT1活性化は、アルツハイマー病以外の慢性神経変性疾患の動物モデルにおいても保護効果があることが示されている。例えば、レスベラトロールで活性化されたSIRT1は、変異型ハンチンチンの細胞毒性からニューロンを保護した(Parker et al 2005年)マウスでのSIRT1ノックアウト/オーバー発現は、ハンチントン病の病態に相互に影響を与えた(Jeong et al 2011;Jiang et al 2011)。また、SIRT1は、筋萎縮性側索硬化症(Kim er al 2007; Song er al 2014)やパーキンソン病(van Ham er al 2008; Mudò er al 2012)の動物モデルにおいても、神経細胞の衰弱を抑制した。しかしながら、SIRT1の神経保護効果は文脈に依存する可能性があることに留意すべきであり(Tang, 2009a,b; Ng and Tang, 2013a)、そしていくつかの報告は、誘導ではなくSIRT1の阻害が、様々な症例において神経保護効果を示すことを示している(Li et al 2008; Liu et al 2009; Sansone et al 2013; Smith et al 2014)。
SIRT1の代謝調節効果はアルツハイマー病の進行をカウンターする
SIRT1 活性の NAD+ への依存性は、SIRT1 に複数の組織における栄養および酸化還元センシング能力を付与する。末梢組織におけるエネルギー代謝の調節におけるSIRT1の役割はよく知られている(Cantó and Auwerx, 2009a,b; Chang and Guarente, 2014)。哺乳類における代謝調節は、脳の視床下部の神経ペプチドを発現する視床下部ニューロンを介して中枢的に調節されており、これらのニューロンにおけるSIRT1活性は、この神経内分泌調節に重要である(Chang and Guarente, 2014; Toorie and Nillni, 2014; Ng er al 2015)。老化した視床下部ニューロンにおけるSIRT1活性の低下または変化は、代謝機能障害をもたらすことが示され(Ramadori et al 2010,2011;佐々木 et al 2014)インスリン抵抗性および糖尿病などの老化に関連した代謝症候群は、全身性SIRT1欠損性と関連している。脂質代謝およびグルコース代謝の異常は、βアミロイド産生/クリアランスとタウリン酸化の両方に影響を及ぼす(佐藤および森下 2015)。2型糖尿病(2型糖尿病)とアルツハイマー病との間の病態生理学的な関連は実際によく知られており(Park, 2011; Dar et al 2014; Mittal and Katare, 2016)アルツハイマー病の進行は糖尿病患者において明らかに加速される。動物モデルにおける2型糖尿病表現型の抑制におけるSIRT1活性化の結果(Baur et al 2006;Milne et al 2007;Herranz et al 2010)は、おそらくアルツハイマー病に対しても同様の利益をもたらすであろう。2型糖尿病とアルツハイマー病の間のもう一つの重要な関連は、インスリン分解酵素(IDE)である(Qiu and Folstein, 2006; Haque and Nazir, 2014)。これについては、Aβクリアランスに関するで議論する。
他のもっともらしいメカニズム
SIRT1はAβとタウのクリアランス機構を制御/促進する
SIRT1の活性化がアルツハイマー病の発症と進行を変化させる可能性のある他の考えられる方法がある。その一つは、Aβとタウのクリアランスの調節や促進である。細胞内の毒性凝集体は、オートファジー-リソソソーム経路またはユビキチン-プロ-テアソーム経路のいずれかを介してクリアされる可能性がある。これに関して、SIRT1活性は、マクロオートファジー(NgおよびTang 2013a,b;Chang et al 2015)およびマイトファジー(Guedes-DiasおよびOliveira 2013;Tang 2016)を調節することが知られており、おそらくオートファジー構成要素の直接的な脱アセチル化(Lee et al 2008)およびmTORシグナル伝達の阻害(Ghosh et al 2010;Liu et al 2010)を介してであろう。神経細胞に特異的な SIRT1 の過剰発現は、栄養飢餓状態での神経細胞の成長と生存性を高め、Aβ1-42 によって誘導される細胞死に対する抵抗力を高め、これは mTOR レベルとそのリン酸化のダウンレギュレーションと相関していることが示された(Guo er al)。 SIRT1は、プリオン病における毒性凝集体誘発神経変性への関与が十分に文書化されており、SIRT1活性化は、オートファジーの誘導を含むいくつかの方法でプリオンタンパク質に対して神経保護的である(Seo et al 2012; Jeong et al 2013)。しかしながら、AβおよびタウのクリアランスにおけるSIRT1の活性の関与についての良好な証拠はまばらである。上述したように、タウの場合、SIRT1の脱アセチル化をブロックすると、アセチル化されたタウおよびリン酸化タウの持続性が増強されることが知られている(Min et al 2010)。しかし、別の報告では、広範なサーチュイン阻害剤ニコチンアミドがSIRT2を阻害することにより、アセチル化α-チューブリンと微小管の安定性を増加させ、その結果、プロテアソーム分解による可能性が高いリン酸化タウの減少も効果的にもたらしたことが示されている(Green et al 2008)。Aβに関しては、シロスタゾールによるSIRT1活性化がオートファジーのアップレギュレーションを介してAβを減少させることが最近の報告で示唆されている(Lee er al)。
Aβに対するSIRT1の影響について、興味深いがよく理解されていない可能性のある側面として、Aβの細胞外クリアランスがある。Aβは、分泌された多くのメタロプロテアーゼによって分解される可能性があり(Miners et al 2008年)これらの中で顕著なものは、IDEとネプリリシンである。IDEは、インスリン、アミリンおよびAβペプチドを分解し(Farris et al 2003)したがって、2型糖尿病でしばしば起こる高インスリン血症は、インスリンとAβとの酵素の競合によってAβレベルを上昇させる可能性がある(Qiu and Folstein 2006;Haque and Nazir 2014)。したがって、SIRT1活性を介した2型糖尿病におけるインスリン抵抗性の逆転は、Aβクリアランスを高める可能性がある。レスベラトロールはマウスのネプリライシンレベルを上昇させることが示されているが、SIRT1の関与は明確に実証されていない(El-Sayed and Bayan, 2015)。
他のアルツハイマー病修飾転写/エピジェネティック因子との相互作用の可能性
SIRT1は転写複合体に存在し、転写因子やヒストンの脱アセチル化を介して遺伝子発現に影響を与える(Oberdoerffer et al 2008)。これに関連して、それは、アルツハイマー病の他の転写/エピジェネティック修飾因子と相互作用するであろう(Gjoneska et al 2015)。例えば、転写・エピジェネティック調節因子であるリプレッサーエレメント1-サイレンシング転写因子(REST)(Bithell, 2011)は、最近、アルツハイマー病に関与していることが示唆されている。RESTのレベルは、軽度認知障害(MCI)やアルツハイマー病を有するヒトの脳では低く、ストレス応答遺伝子を誘導しながら、神経細胞死やアルツハイマー病の病態を促進する遺伝子を抑制している(Lu er al)。 RESTのミスセンス変異体(rs3796529)は最近、MCIにおける海馬萎縮に対する保護変異体であることが示され(Nho et al 2015)別の解析では、この変異体がアルツハイマー病リスクの低下と関連していることも示された(Li et al 2015a,b)。クラスIおよびIIのヒストン脱アセチル化酵素(HDAC)の阻害剤(一般的にはサーチュインに有意な影響を与えない)は、記憶および認知機能低下のモデルにおいて有益性を実証しており(Benito et al 2015)SIRT1と他のHDACのエピジェネティックな活性との関連は、さらなる調査に値するであろう。
アルツハイマー病におけるSIRT1活性化因子と治療試験
SIRT1 活性化が無数の加齢関連疾患に有益である可能性があることが約束されていることを考えると、SIRT1 活性化化合物を用いたヒト試験の実施には多くの衝動がある。レスベラトロールは、アルツハイマー病患者を対象とした無作為化二重盲検プラセボ対照試験が発表されている唯一のSIRT1活性化剤です(Turner et al 2015)。この第2相試験は、主要な臨床転帰の差を検出するのに十分なパワーを有していない可能性が高いため、神経学的な有益性は認められなかった。しかし、それはレスベラトロールの大量投与が長期にわたってよく忍容されていることを再確認し、化合物とその代謝物は確かにささやかに アルツハイマー病 バイオマーカーのいくつかの軌道を変更するには、血液脳関門を貫通することができた。後者の中で、著者らは2つのパラメータのセットを強調した。第一に、脳脊髄液と血漿中のAβ40はレスベラトロール投与群ではよりゆっくりと低下し、その差は試験終了時には統計的に有意になった。第二に、MRIによる脳体積の変化では、レスベラトロール投与群はプラセボ投与群に比べて脳体積の減少が大きいことが示された。いずれの変化も、化合物またはその代謝物の中枢神経系への影響を示している。Aβ レベルは通常 アルツハイマー病 の進行と脳のボリュームが増加する可能性がある神経炎症とその結果浮腫、レスベラトロール治療から生じる両方の変更は楽観的にやや肯定的な結果として見られる可能性がある。
レスベラトロールは、低毒性ではあるが、バイオアベイラビリティーが限られている。最近報告された代謝機能に関する試験では、混合した結果が得られている(Timmers et al 2011; Yoshino et al 2012)。レスベラトロールは幅広い標的を持っているため、よりSIRT1に特異的な結果を得るには、マウスの寿命を緩やかに延長することが示されたSRT 2014(Mitchell et al 2014)など(Hubbard and Sinclair 2014)のように、より高度なSIRT1標的特異性を持つ化合物を用いた試験が待たれる。アルツハイマー病治療薬の開発および臨床試験(Mangialasche et al 2010)は、失敗の点で、また、動物モデルとヒト被験者における利益の間の明らかに大きな不一致のために、あまり気にしていなかった。最近の挫折には、ヒト化抗Aβモノクローナル抗体であるバピヌズマブを用いた待望の第3相試験が含まれる(Salloway et al 2014)。SIRT1活性化単独ではまだ効果があるとは言えないが、これらがアルツハイマー病バイオマーカーの軌跡を変える能力を持ち、長期間の忍容性が良好であれば、補助療法として有用である可能性は十分にあるだろう。
エピローグ
上記の議論では、SIRT1 の活性と アルツハイマー病 関連または関連する病理との間の関連性、および解読されたり暗示されたりしている多数のニューロン細胞の自律/非自律メカニズムの概要を簡単に説明した(図 1)。SIRT1 が アルツハイマー病 病理に対抗する可能性があるこのような多様な可能性を考えると、SIRT1 が アルツハイマー病 の治療標的として有用であることへの期待は高いと言える。特異的で強力な SIRT1 活性化剤を用いたヒト試験が既に実施されている。SIRT1活性化化合物(Sinclair and Guarente, 2014)またはNAD+生成化合物(Yoshino et al 2011; de Picciotto et al 2016)を用いたSIRT1の医薬的活性化は重要な治療戦略であり続けるだろうが、継続的な研究は、SIRT1活性化の他の方法を探索する必要がある。これは、miR-34a(Yam-akuchi et al 2008)miR-133b(Tian et al 2016)および最近報告されたmiR23a/bおよびmiR-212/132クラスター(Weinberg et al 2015)のようなSIRT1転写のリプレッサーの局所的なサイレンシング、またはDBC1(Zhao et al 2008)のような内因性SIRT1インヒビターのサイレンシングを含むかもしれない。臨床的な利点はさておき、これらのヒトベースの実験から多くの科学的洞察が得られる可能性がある。
図 1:SIRT1 が アルツハイマー病 の発症と進行に対抗する主なメカニズムを示す模式図
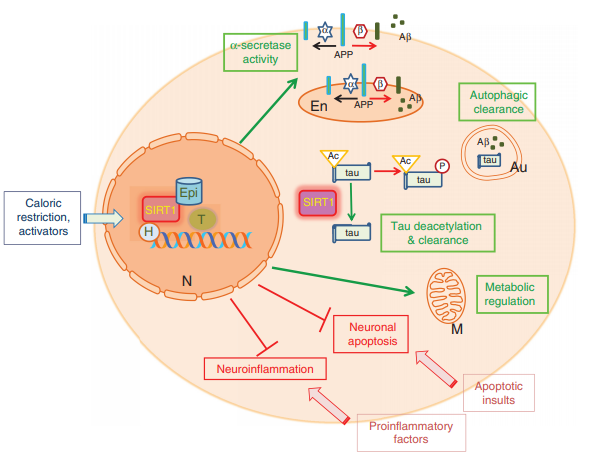
SIRT1は大部分が核内に存在するが、核内-細胞質間のシャトルリングを示し、その活性は複数の細胞サブコンパートメントに浸透している可能性がある。
SIRT1は、CRや様々な活性化化合物などの代謝状態によって活性化される。ヒストン(H)、転写因子(T)、および他のエピジェネティック因子(Epi)との潜在的な相互作用により、α-セクレターゼおよび非アミロイド性APP処理を増強する転写変化を誘発し、内因性および外因性ソースからの神経炎症を抑制する。また、オートファジーの調節により細胞内のAβやタウのクリアランスを促進し、タウの脱アセチル化によりタウの分解を促進する。また、SIRT1はp53の活性を調節することで神経細胞死を抑制する。詳細は本文を参照。Auはオートファゴソーム、Enはエンドソーム、Mはミトコンドリア、Nは核。