
Brain Kynurenine and BH4 Pathways: Relevance to the Pathophysiology and Treatment of Inflammation-Driven Depressive Symptoms
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6095005/
要旨
うつ病は世界的に増加しており、特に抗うつ薬の有効性を高める薬剤の開発が停滞していること、従来の抗うつ薬では効果が得られない患者の割合が高いこと、また、慢性疾患の増加に伴いうつ病の併存率が高くなっていることなどから、うつ病の病態やメカニズムの解明が急務とされている。
したがって、標的とする治療戦略の開発に資するためには、うつ病の病態生理と慢性疾患におけるうつ病併発のメカニズムに関するより良い知識が緊急に必要とされている。本レビューでは、炎症過程がうつ病症状の病態生理と治療の鍵を握っていることを示唆するエビデンスを提示する。特に、特定の代謝経路、すなわちキヌレニンとテトラヒドロビオプテリン(BH4)経路における炎症による変化が、セロトニン、グルタミン酸、ドーパミンの代謝に大きな変化をもたらし、それが主要なうつ病症状の発現に寄与している可能性があることを示す前臨床および臨床所見を報告している。
したがって、キヌレニンおよびBH4経路を標的とした抗炎症性介入は、特にうつ病症状と炎症が併存している場合には、新規治療として、あるいはモノアミンに直接作用する従来の薬物のアジュバントとして有効であると考えられる。この考え方は、既知の抗うつ薬と炎症過程との密接な相互作用やその治療的意味合いを示す最近の知見に照らして論じられている。以上のことから、炎症に関連した抑うつ症状を治療するための、より適応の高い個別化された治療戦略に向けての貴重な知見が得られた。
キーワード
炎症、神経炎症、キヌレニン、テトラヒドロビオプテリン(BH4)モノアミン、抑うつ症状、抗うつ薬治療、抗炎症戦略
序論
うつ病は現在、世界的な公衆衛生上の懸念を表している。その有病率は世界的に着実に増加しているだけでなく、罹患率や死亡リスクの増加にも関係している(世界保健機関 2017)。さらに、うつ病治療の進歩にもかかわらず、うつ病患者の少なくとも3分の1は従来の薬物療法に反応しない(Rush et al 2006)。うつ病の世界的な負担にさらに貢献しているのは、代謝性疾患、自己免疫疾患および心血管疾患を含む、有病率も継続的に上昇している慢性疾患のほとんどが、うつ病のリスクの増加と関連していることである(Luppino et al 2010;Capuron et al 2017;Zuzarte et al 2018)。後者は、影響を受けた患者の生活の質を損ない、その後の医学的合併症の強力な貢献者として浮上する(Penninx et al 2013)。このように、うつ病の病態生理学的基盤に関するより良い知識と、治療の進歩に関連する新たなターゲットの同定は、まだ明らかに必要である。
異なる神経生物学的システムがうつ病の病態生理に関与している可能性が高いが、このレビューでは、炎症性プロセスのための主な役割を支持する証拠を要約する。最初に自然免疫系と脳との複雑な関係を解明することに専念したのは、精神神経免疫学の分野で行われた初期の研究で、炎症が行動や神経精神に及ぼす影響の根底にある分子メカニズムを明らかにし始めたからである(Dantzer et al 2008,Capuron and Miller 2011年を参照)。一方、臨床研究では、炎症性因子の循環レベルの上昇と気分変調の発症リスクの増加との関連性が報告されている(Capuron et al 2002;Evans et al 2005)。これらの知見により、サイトカインが標的とする神経生物学的経路をさらに特定し、その神経精神作用を媒介することを目的とした研究は、現在も継続中である。さらに、肥満やメタボリックシンドロームで報告されているような基礎炎症の亢進が、従来の抗うつ薬治療の治療効率を損なう一因となる可能性があることを示唆する最近のデータ(Kloiber er al)。 このレビューでは、うつ病の病態生理における炎症の役割についてのより深い機序的理解と、新規治療のための関連する洞察を提供する最近の知見に照らして、これらの問題について議論する。
うつ病の炎症性仮説
双極性障害、不安障害、統合失調症など、いくつかの精神疾患にも徐々に拡大してきているが(Potvin et al 2008; Hoge et al 2009; Barbosa et al 2013)神経精神症状の病態生理に炎症が関与している可能性があるという考えは、特にうつ病の文脈で研究されてきた(Raison et al 2006; Capuron and Miller, 2011)。この概念を強く支持する最初の研究では、健康な対照者と比較して、うつ病患者において、C反応性タンパク質(CRP)サイトカイン(特にインターロイキン-6,IL-6)および各種ケモカインを含む炎症性メディエーターの循環レベルが高いことが報告されている(Dowlati et al 2010;Leighton et al 2018)。さらに、縦断的な調査から、より高い炎症性プロファイルが抑うつ症状の発症を予測することが明らかになっている(Valkanova et al 2013;Smith et al 2018)。相関性から因果関係へと移行するために、慢性的なサイトカイン投与は、サイトカイン免疫療法を受けている医学的疾患患者の最大50%においてうつ病を誘発することが示されている(Musselman et al 2001;Capuron et al 2002;Kawase et al 2016)。同様に、健康なボランティアへのサイトカイン、またはリポ多糖(LPS)などのサイトカイン誘導剤の直接投与は、抑うつ症状を誘発する(Schedlowski et al 2014;Engler et al 2017)。臨床所見と一致するように、げっ歯類における急性または慢性の免疫チャレンジの両方が、持続的な抑うつ様行動および不安様行動を誘発する(Merali et al 2003; Frenois et al 2007; Moreau et al 2008; Klaus et al 2016)。逆に、抗炎症性化合物は、炎症性疾患の動物モデルにおいて、これらの行動を減少させる(Llorens-Martin et al 2014;Zaminelli et al 2014;Norden et al 2015)。同様に、特定の炎症性サイトカインを直接標的とすることは、ヒトおよびげっ歯類の両方において気分変化を減少させる(Tyring et al 2006;Kekow et al 2010;Haji et al 2012;Bayramgürler et al 2013;Fleming et al 2015)。
炎症誘発性の行動変化は、活性化された免疫細胞によって末梢的に放出される炎症性サイトカイン[例えば、IL-1β、IL-6,腫瘍壊死因子(TNF)-αなど]が、体液性、神経系および化学的経路を介して脳に到達し、脳の免疫細胞である活性化されたミクログリアによって局所的に脳サイトカインの産生を誘導することを可能にする、大規模なコミュニケーションネットワークに依存していることが示されている。これらのサイトカインは、神経伝達物質の代謝、神経内分泌機能、神経可塑性などの行動や気分の調節に関与する経路に影響を与える(Castanon er al)。 これらの神経免疫相互作用は、免疫応答を調整するだけでなく、病気行動と総称される適応的な行動変化の発生にも関与している。脱力感、倦怠感、倦怠感、食欲不振、疲労感などを含むこれらの変化は、身体が攻撃に対して積極的に戦うのを助けることで、回復に必要であることが示されている。炎症性の活性化は通常一過性で、抗炎症メカニズムによって制御され、病気の行動の時間制限と可逆性を保証する。逆に、持続的な炎症の発現を可能にするこれらの制御機構の障害は、炎症に関連した気分変化の主要な主要な原因であることが示されている(Dantzer et al 2008;CapuronおよびMiller 2011;Castanon et al 2014;CapuronおよびCastanon 2017)。実際、炎症が最初は感染を制御し、組織修復を促進するという保護機能を果たしていたとしても、長期的には脳の神経伝達を妨害し、組織の損傷を引き起こす可能性があり、最終的には持続的な行動や気分の変化を促進する一因となる。興味深いことに、説得力のある臨床研究により、サイトカイン免疫療法を受けている患者は、神経栄養学的レベル(例えば、疲労、調子および意欲の低下)から精神神経学的レベル(例えば、気分の落ち込み、不安、認知の変化)までの多次元にわたる症状を示すことが明らかにされた(Musselman et al 2001;CapuronおよびMiller 2011;CapuronおよびCastanon 2017)。重要なことに、これらの症状は、古典的な抗うつ薬、特にセロトニン神経伝達を標的とする薬による予防的治療に対する時間経過および反応性が異なる。特に、神経性症状は、サイトカイン免疫療法を開始してから早期に出現し、患者の大部分に見られる。逆に、神経症状は、サイトカイン投与の数週間後に進行的に発現し、患者の30〜50%にしか影響を与えない。これらの最後の症状は、この介入に反応しないか、または最小限にしか反応しない神経症状とは対照的に、抗うつ剤治療によって予防することができる(Musselman et al 2001; CapuronおよびMiller 2011)。これらの所見から、これらの所見は、異なる基礎となるメカニズムの関与を示唆している。
この仮説を強力に支持するのは、免疫刺激を受けた動物を対象とした研究で、免疫刺激後早期に発生した病気の行動と、長引く抑うつ様行動とを実験的に解離させることが可能であることである(Merali et al 2003; Frenois et al 2007; Moreau et al 2008; Klaus et al 2016)。それぞれの基礎となる神経生物学的メカニズムを評価する最初の試みは、同じ前臨床モデルに集中している。この文脈では、病気の行動を特徴づける行動変化のいくつか、特に運動鈍化が、運動反応に基づくパラダイムで評価した場合に、抑うつ症状に似た行動の測定を妨げる可能性があることに言及する価値がある。しかし、この潜在的な方法論的バイアスは、うつ病の異なる中核症状をモデル化した信頼性の高い相補的な行動テストをいくつか使用し、病気の行動から完全に回復した後にのみ、これらのテストでマウスをテストすることによって回避されている(Frenois et al 2007; Godbout et al 2008; O’Connor et al 2009c)。興味深いことに、LPSを負荷したマウスを用いたこの実験デザインは、LPSによって誘発された病的行動と抑うつ的行動の根底にある脳構造の間の神経解剖学的な解離をそれぞれ示すことができた(Frenois et al 2007)。文献の間にはいくつかの矛盾が存在し(Biesmans et al 2013年を参照)LPSによる急性免疫活性化のモデルが必ずしも臨床状況に関連していないことが議論されている。慢性炎症モデルで行われたいくつかの研究は、より適切であると主張することができるが、病気の行動はもはや検出できず、循環サイトカインのレベルも上昇していない一方で、持続的な抑うつ様行動の存在を確認した(Moreau et al 2008; O’Connor et al 2009a,b)。臨床研究と合わせて、これらすべてのモデルは、炎症に関連した抑うつ症状の神経生物学的基盤の同定に非常に有用であることが判明した。
炎症関連うつ症状の神経生物学的基盤
臨床所見および前臨床所見の積み重ねにより、我々および他の研究者は、多次元的な炎症関連症状は、炎症性サイトカインが重要な代謝経路、すなわちキヌレニンおよびテトラヒドロビオプテリン(BH4)経路を変化させる能力に依存している可能性があり、これにより、気分調節に関与するモノアミン、特にセロトニン、グルタミン酸およびドーパミンの神経伝達が障害される可能性があることを提案した(Dantzer et al 2008; CapuronおよびMiller 2011; CapuronおよびCastanon 2017; Haroon et al 2017)。2008;カプロンおよびミラー 2011;カプロンおよびカスタノン 2017;ハルーン et al 2017)。)
キヌレニン経路。炎症と気分の岐路に立つ
必須アミノ酸トリプトファンの代謝は、行動や気分、特にセロトニンにリンクされた主要な生理学的プロセスを調節することができる重要な要因の配列を生成する。キヌレニン経路(KP)に沿ったトリプトファン代謝は、タンパク質合成に使用されないトリプトファンの大部分を占め、最終的には3-ヒドロキシキヌレニン(3-HK)やキノリン酸(QA)など、いくつかの神経活性代謝物の生産につながる。は、N-メチル-D-アスパラギン酸(NMDA)グルタミン酸受容体を刺激し、酸化ストレスを促進することができる、そしてむしろ神経保護特性を示すキヌレン酸(KA)である(図11)(Schwarcz and Stone, 2017)。ミクログリアは優先的にQAを産生し、KAはアストロサイトによって合成されるので、免疫活性化の条件下では神経毒性が優勢である。したがって、血漿および/または脳脊髄液のQAレベルの増加は、気分症状の有病率の増加とともに、炎症性および神経変性損傷を包含する多くの状態で報告されている(Capuron et al 2011;Bay-Richter et al 2015;Lovelace et al 2017;O’FarrellおよびHarkin 2017;SchwarczおよびStone 2017)。より重要なことに、KPの活性化は、脳損傷の伸張および神経精神症状の重症度の両方と相関する(Capuron et al 2002;Raison et al 2010;Savitz et al 2015a,b)。これらの知見は、KPに沿ったトリプトファン代謝の第一段階および律速段階を触媒するインドールアミン2,3-ジオキシゲナーゼ(IDO)という酵素を皮切りに、これらの症状の誘発におけるKP酵素および代謝物の関与に対する関心の高まりを促した(Lestage et al 2002; Moreau et al 2005; André et al 2008)。例えば、Bacillus Calmette-Guerin(BCG)をマウスに接種すると、末梢および脳のIDO活性が慢性的に増加し、この活性化は持続的な抑うつ様行動の発達と平行していることが示された(Moreau et al 2008; O’Connor et al 2009a,b)。高齢マウス(Godbout et al 2008;Wynne et al 2010)または構成性ミクログリアの過剰活性化を示すマウス(Corona et al 2013)もまた、免疫チャレンジ後に持続的なサイトカイン産生を示し、脳内IDO発現の長期化と抑うつ様行動を示す(Godbout et al 2008;Wynne et al 2010;Corona et al 2013)。同様に、炎症に関連した脳内IDO活性化と抑うつ様行動との関連は、肥満のマウスモデルなどの他の慢性炎症性疾患においても報告されている(André et al 2014; Dinel et al 2014)。重要なことに、脳内でのKPの直接的な活性化は、げっ歯類の感情行動を変化させるのに十分であり、特に行動および気分のための重要な脳領域、特に海馬で発生する場合には十分である(Henry et al 2009; Fu et al 2010; Park et al 2011; Dobos et al 2012; Lawson et al 2013)。IDOを遺伝的または全身的に阻害し、免疫チャレンジに供したマウスを使用することにより、我々および他の研究者は、抑うつ様行動および不安様行動の誘導におけるIDO活性化の因果的役割を報告した(Godbout et al 2008;O’Connor et al 2009a,b,c;Salazar et al 2012;Corona et al 2013;Gibney et al 2013;Xie et al 2014;Castanon et al 2015)。この誘導は、トリプトファンのバイオアベイラビリティーを減少させることにより、IDOがセロトニン合成に悪影響を与える可能性と関連している。しかしながら、免疫活性化が、IDO活性化(O’Connor et al 2009c)とは関係なく、実際にセロトニンターンオーバーの増加と関連しているという事実(Godbout et al 2008; O’Connor et al 2009c; Gibney et al 2013; Parrott et al 2016a)は、この仮説を弱めている。これらの知見は、むしろ、セロトニントランスポーターおよび受容体の調節を含む、セロトニン神経伝達における他のサイトカイン誘発性変化の介入を示唆している可能性がある(Zhu et al 2006)。したがって、セロトニン関連プロセスの時間的・空間的パターンに対するKPの活性化の影響を深く研究するためのフォローアップ研究が必要である。
図1 炎症状態におけるキヌレニン経路の活性化
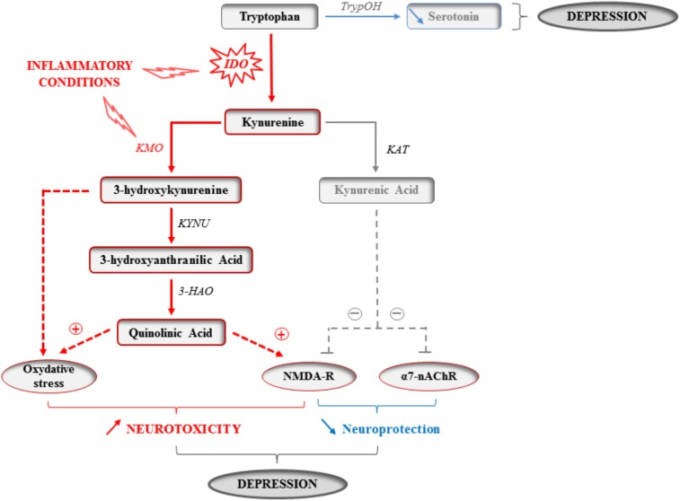
炎症状態では、活性化された単球、マクロファージ、脳ミクログリアの炎症性サイトカインにより、インドールアミン2,3-ジオキシゲナーゼ(IDO)の酵素活性が上昇する。したがって、必須アミノ酸トリプトファンは、キヌレニンの合成に使用され、これは、その前駆体および制限因子トリプトファンの利用可能性に直接依存するモノアミンセロトニンの合成を犠牲にしている。したがって、IDOの活性化はセロトニンの神経伝達を損なう可能性がある。キヌレニンは、その後、グルタミン酸NMDAおよびα7-ニコチンアセチルコリン(α7-nAChR)受容体の両方に作用して神経保護作用を示すキヌレニン酸、および酸化ストレスの促進および/またはNMDA受容体の活性化によって神経毒性を示す3-ヒドロキシキヌレニンおよびキノリン酸を含む、異なる神経活性を有するグルタミン酸代謝物を産生するために使用される。3-ヒドロキシキヌレニンを合成するキヌレニンモノオキシゲナーゼ(KMO)の活性が炎症性サイトカインによって活性化されたミクログリアにおいて増加するように、キヌレニンの産生の増加は、最終的には、神経毒性の増加に向けたキヌレニン代謝バランスのスキューイングと関連している。セロトニンの神経伝達を障害し、神経細胞の損傷を促進することにより、サイトカイン誘発性のキヌレニン経路の活性化は、炎症に起因する抑うつ症状の発症に寄与する可能性がある。TrypOH、トリプトファン水酸化酵素、KAT、キヌレニンアミノトランスフェラーゼ、KYNU、キヌレニナーゼ、3-HAO、3-ヒドロキシアントラニル酸オキシゲナーゼ。
炎症に関連した気分症状におけるKP活性化の関与についての代替的な説明は、神経毒性のあるグルタミン酸性キヌレニン代謝物の生成であり、それらは気分変化の重症度と臨床的に関連している(Capuron et al 2011;Bay-Richter et al 2015;Haroon et al 2017;SchwarczおよびStone 2017)。神経変性疾患の臨床的転帰を微妙に変化させるニューロンの完全性および/または機能の変化の確立された役割に沿って、神経細胞の損傷もまた、炎症誘発性の抑うつ症状の発現に関連している(DantzerおよびWalker 2014)。これは特にNMDA受容体が関与している可能性があり、最近うつ病研究の分野で注目されている(Dantzer and Walker, 2014; Haroon et al 2017)。この考え方をさらに支持する前臨床研究では、LPSチャレンジ(Parrott et al 2016a,b)または神経障害性疼痛(Laumet et al 2017)に関連して誘発される抑うつ様行動は、神経毒性代謝物の産生に向けたキヌレニン代謝バランスのスキューイングと関連しており、これは海馬が特に影響を受けている部位特異的な方法であることが報告されている。さらに、キヌレニン(O’Connor et al 2009c; Salazar et al 2012; Agudelo et al 2014)または3-HK(Parrott et al 2016a,b)の全身投与は、用量依存的に情動行動を障害するが、IDO欠損マウスはNMDA受容体介在性興奮毒性から保護される(Mazarei et al 2013)。注目すべきことに、NMDA受容体遮断は、最終的にNMDA受容体アゴニストQAを合成する下流KP酵素、すなわちキヌレニン3-モノオキシゲナーゼ(KMO)または3-ヒドロキシアントラニル酸ジオキシゲナーゼ(HAAO)の選択的阻害とともに、炎症誘発性抑うつ様行動を消失させる(Walker et al 2013)(Parrott et al 2016b; Laumet et al 2017)。興味深いことに、これは、特に、臨床的な行動絶望に関連する抑うつ様行動(すなわち、尾部懸垂試験での不動)を改善する一方で、行動変化にはあまり効率的ではなく、むしろ無気力(すなわち、ショ糖嗜好)をモデル化している(Parrott et al 2016b)。これらの結果は、それぞれ、異なる炎症に関連した抑うつ症状の根底にある神経生物学的メカニズムを特定するのに特に関連している。それぞれの行動/症状は、離散的な脳領域における選択的なニューロン経路の優先的な活性化によって機能的に支配されているので、このような次元の解離は、これらの実験研究で報告されたKP活性化の地域差に依存している可能性がある(Parrott et al 2016a,b; Laumet et al 2017)。サイトカインのLPS誘発脳内発現は、大部分が領域に依存しないが、3-HK/KA比は海馬で有意に増加しているが、中枢扁桃体または後頭核では増加していない(Parrott et al 2016a)。これらの前臨床所見と一致するように、うつ病患者の死後の研究は、重度のうつ病と選択的な皮質小領域におけるミクログリアQA検出の増加とを関連づけている(Steiner et al 2011)。逆に、KA/QA比の上昇によって反映される高い神経保護指数は、臨床的にはうつ病であっても薬を服用していない患者の海馬体積と正の相関がある(Savitz et al 2015a)。そうでなければ、KPの活性化と特定の抑うつ症状の発症との間の上記の解離は、むしろ他の代謝経路および/または神経伝達系の関与を反映している可能性がある(Capuron and Castanon, 2017)。この仮説は、LPS処理されたIDO欠損マウスではモチベーション駆動行動の炎症誘発性障害が持続するため、IDO活性化とは無関係であることを示した最近の研究によってうまく支持されている(Vichaya et al 2018)。炎症に関連した意欲性障害に関しては、最終的にドーパミン神経伝達を障害する可能性のあるBH4経路が、可能性のある候補として現れる(Felger and Treadway, 2017)。これは、ドーパミン関連の行動変容を示すBH4欠損マウスで得られたデータによって裏付けられている(Choi and Tarazi, 2010)。KAは、α7-ニコチン性アセチルコリン受容体に作用して線条体のドーパミン作動性トーンを調節することが示されており(Wu et al 2007)これらの受容体の薬理学的活性化は慢性ストレスのマウスモデルにおいて無気力症を緩和する(Zhao et al 2017)ので、両方の選択肢が必ずしも切り離されていないわけではない。さらに、臨床データは、循環KA/QA比とうつ病患者における無気力症の程度との間の逆相関を報告している(Savitz et al 2015a)。
BH4経路 うつ病症状の神経生物学的調節におけるキープレイヤー
BH4 は一酸化窒素合成酵素アイソフォーム(NOS)と 3 つの芳香族アミノ酸水酸化酵素(フェニルアラニン水酸化酵素(PAH)トリプトファン水酸化酵素(trypOH)チロシン水酸化酵素(TH)の最適な機能のための重要な補酵素(Thöny et al 2000; Sumi-Ichinose et al 2001)。したがって、必須アミノ酸であるチロシン、トリプトファン、アルギニンからそれぞれドーパミン、セロトニン、一酸化窒素の合成に必要とされている(Sumi-Ichinose er al)。 BH4 の新規合成には 3 つの酵素が順次活性化されるが、最初の酵素である GTP-シクロヒドロラーゼ 1 (GCH1) が律速段階 (Thöny et al 2000) (図 22)。モノアミン含有細胞と共局在するこの酵素の活性は、脳の部位によって異なることが示されている(Sawada er al)。 1986; Nagatsu er al)。 1995; Dassesse er al)。 1997)。IL-1β、インターフェロン-γおよびTNF-αを含む炎症性サイトカインは、GCH1の発現および活性の両方を誘導することができ、したがってBH4合成を増加させる(Shi et al 2004)。翻訳後レベルでは、GCH1は、シクロヒドロラーゼフィードバック調節タンパク質(GFRP)との複合体形成を介して、BH4によって阻害され、フェニルアラニンによって刺激される(Neurauter er al)。 このプロセスは、体内のBH4レベルを生理的範囲内にしっかりと保つことを保証する。BH4はまた、ジヒドロ葉酸還元酵素(DHFR)によってBH4に逆に変換されることができるBH2に酸化される(Harada et al 1993)。つまり、BH4の正味の細胞内バイオアベイラビリティは、de novo合成、BH2への酸化、DHFRによる再生のバランスに起因していると考えられる。
図2 炎症関連 BH4 経路
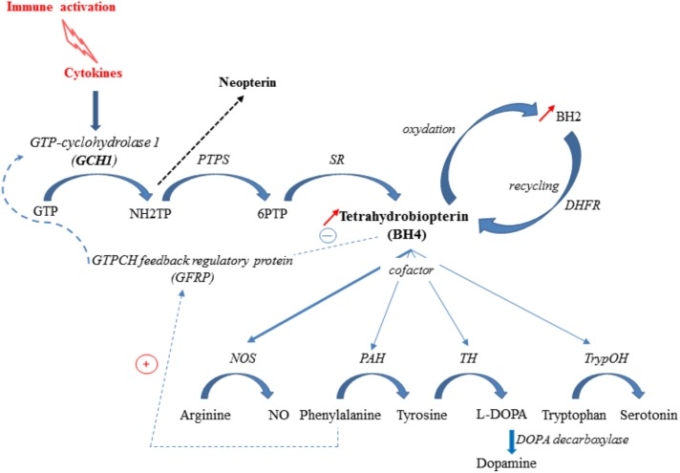
免疫課題に応答して誘発されるサイトカイン産生は、GTP-シクロヒドロラーゼ-1(GCH1)6-ピルボイル-テトラヒドロプテリン合成酵素(PTPS)セピアプテリン還元酵素(SR)の活性化(de novo synthesis pathway)を誘導し、テトラヒドロビオプテリン(BH4)合成を誘導する。GCH1の活性は、GTP-シクロヒドロラーゼ・フィードバック・プロテイン(GFRP)とエフェクター分子であるBH4とフェニルアラニンの相互作用によって調節される。BH4は急速にBH2に酸化され、その後、酵素DHFRによってBH4に還元され、サルベージ経路を表している。ヒトでは、PTPSは炎症性の挑戦の下で律速になり、BH4の不利になるようにネオテリンの形成を促進する。炎症性条件下では、BH4は、一酸化窒素(NO)の合成のためのNOS(一酸化窒素合成酵素)活性のための補酵素として優先的に使用され、PAH(フェニルアラニン水酸化酵素)TH(チロシン水酸化酵素)およびTrypOH(トリプトファン水酸化酵素)活性のためのより低い利用可能性およびモノアミンの減少した合成につながる。NH2TP:7,8ジヒドロネオプテリン三リン酸;6PTP:6ピルボイル-テトラヒドロビオプテリン。
いくつかの臨床症状はBH4経路の欠陥と関連しており、主に異なる酵素ステップに影響を与える変化に関連している。GCH1の変異はBH4レベルの大幅な低下をもたらし、これはドーパミン反応性ジストニアなどの神経疾患を引き起こすことが示されている(一ノ瀬 et al 1994; Müller et al 2002)。BH4を遺伝的に欠損したマウス(hph1モデル)は、ドーパミンレベルの低下および抑うつ様行動の増加を示す(Nasser et al 2014)。前臨床所見を裏付けるように、GCH1欠損症の患者は、うつ病および不安を含む精神疾患の頻度の増加を示すことが示されている(Van Hove et al 2006; Trender-Gerhard et al 2009)。さらに、最近、GCH1活性のマーカーの変動が、高齢者の神経性症状および炎症性因子と相関することが報告されている(Capuron et al 2011)。また、重度のうつ病の既往歴のある被験者の死後の脳では、BH4の低値が発見されている(Blair et al 1984)。逆に、うつ病患者では尿中および血漿中の総ビオプテリン濃度の上昇が測定されており(Duch et al 1984;Garbutt et al 1985;Knapp et al 1989;Hashimoto et al 1994;アミロイドβou-Saleh et al 1995)BH4代謝の障害が示唆されている。さらに、電気けいれん療法に反応するうつ病患者において、フェニルアラニン/チロシン比の増加によって評価されるGCH1活性の低下が報告されている(Anderson et al 1994)。このフェニルアラニン/チロシン比はBH4の利用可能性とPAH活性の指標としても使用され、したがって、ドーパミンとノルエピネフリン合成の間接的なバイオマーカーとして機能する可能性がある(Neurauter et al 2008; Capuron et al 2011)。
ヒトでは、炎症などのGCH1刺激条件下では、低PTPS活性はBH4を犠牲にしてネオテリンの産生を誘導する。したがって、ネオテリンは、炎症状態における細胞介在性免疫のマーカーとして考えられている(Murr et al 2002)。フェニルアラニン濃度およびフェニルアラニン/チロシン比の増加は、慢性炎症状態に苦しむ患者において報告されており、この増加はネオテリン濃度と相関している(Neurauter et al 2008;Ploder et al 2008;Zangerle et al 2010;Murr et al 2014;平山 et al 2016;Ormstad et al 2016)。炎症性刺激が誘導性NOSを活性化し、それによって最適な酵素活性のためのBH4の使用が大幅に増加し、大量の酸素ラジカルの形成を誘導し、それによってBH4の酸化的損失に寄与することがよく知られている(Werner et al 2003)。慢性炎症状態によるBH4の使用量の増加と損失の両方が相乗的に作用してBH4依存性酵素の機能を変化させ、モノアミンの生合成を阻害し、気分障害の発症に寄与する可能性がある(Neurauter et al 2008; Felger et al 2013a,b)。
前臨床研究では、免疫刺激後の脳内ドーパミンとセロトニン、および/またはそれぞれの代謝物レベルの変化が示されている(Kamata et al 2000; Kumai et al 2000; Kitagami et al 2003; Sato et al 2006)。興味深いことに、酸化的損傷の誘導を伴う可能性のあるドーパミン神経伝達の炎症誘発性障害は、LPSを負荷したマウスで報告された動機付け変化の根底にある潜在的なメカニズムとして提案されている(Vichaya et al 2018)。臨床研究もまた、意欲の障害および運動鈍化と関連して、ドーパミン性報酬系のいくつかの機能的変化を明らかにしている(Capuron et al 2007; Eisenberger et al 2010; Capuron et al 2012)。報酬に対する神経反応の同様の鈍化は、ドーパミン合成のためのアミノ酸前駆体が食事で枯渇している状態で観察されている(Bjork et al 2014)。ドーパミン合成および放出の減少は、炎症性条件下のサルおよびマウスのマイクロダイアリシスを用いて確認されている(Felger and Treadway, 2017 for review)。また、いくつかの証拠は、炎症性サイトカインが、小胞性モノアミントランスポーター(VMAT2)の発現および機能の変化を通じて、シナプス前のドーパミン貯蔵を変化させる可能性があることを示している(Kazumori et al 2004)。さらに、ドーパミン再取り込みポンプであるDATの機能変化は、HIV感染または出生前LPSチャレンジに関連した神経炎症状態において示唆されている(Gelman et al 2006; Tien et al 2013)。そうすると、シナプス前小胞貯蔵の減少および/またはドーパミンのDAT再取り込みの変化は、炎症状態でのドーパミンターンオーバーの減少につながる可能性がある。
これらのデータに基づいて、BH4経路は、炎症性メディエーターの過剰生産に関連する多くの症状および病態の重要な調節因子として浮上している。さらに、先に述べたように、最近のデータではKPとBH4経路の関連性が示唆されており、炎症状態で相乗的に作用してモノアミン合成を低下させる可能性があるとされている。したがって、3-HKの代謝物であるキサントウレン酸の増加は、セピアプテリン還元酵素を阻害することによってBH4生合成を直接低下させることが示されている(Haruki et al 2016)。同様に、キヌレニンの同時アップレギュレーションとBH2産生の同時アップレギュレーションは、(キヌレニンによる)NOSの活性のアップレギュレーションと、NOS補因子としてのBH4の利用可能性の低下の組み合わせにつながる可能性が示唆されている(Oxenkrug, 2011)。このような組み合わせは、NOSのアンカップリングをもたらし、その結果、さらなる酸化的なBH4の損失を引き起こす「発火性」活性酸素種(ROS)に有利なNOの産生を減少させる。これらのデータは、炎症性関連のうつ病症状の発症におけるキヌレニンとBH4の両方の経路の役割を強く支持している。
治療への影響
うつ病性障害に対処する際の大きな問題の一つは、標準的な抗うつ薬治療に対する抵抗性の高まりである(Rush et al 2006)。そのような文脈において、臨床転帰を改善するために、炎症性プロセスを代替的および/または併用的に標的化する可能性が、最近特に注目されている(Schmidt et al 2016;Colpo et al 2018;Kappelmann et al 2018;Shariq et al 2018;Zuzarte et al 2018)。モノアミン神経伝達に一次的に作用することが知られているほとんどの古典的な抗うつ薬[すなわち、選択的セロトニン再取り込みブロッカー。SSRI、ノルエピネフリン再取り込みに作用する三環系抗うつ薬(TCA)およびセロトニンおよびノルエピネフリン再取り込みの両方に作用する抗うつ薬。SNRI]は、それらが末梢および脳内の両方で抗炎症性を示すので、炎症にも作用する可能性がある(Lanquillon et al 2000;Tynan et al 2012;Strawbridge et al 2015;Wiêdłocha et al 2018)。重要なことに、それらは循環サイトカインレベルを減少させるだけでなく、KPの下流活性化も減少させ(AraおよびBano 2012;Zoga et al 2014;Reus et al 2015)最終的には神経保護性および神経毒性のキヌレニン代謝物の間の不均衡を修正する(Kocki et al 2012;Eskelund et al 2017)。したがって、うつ病のげっ歯類モデルにおける持続的なSSRI治療は、気分調節に関与することが知られている異なる脳領域におけるQAレベルを低下させる(Eskelund et al 2017)。さらに、IDOおよびGCH1をコードする遺伝子の変異は、うつ病患者におけるSSRI治療の転帰を予測することが示されており(Cutler et al 2012;Kishi et al 2012)治療反応における両経路の役割をさらに支持している。注目すべきは、抗うつ薬の免疫調節作用はそのクラスによって異なり、SSRIおよびSNRI薬はほとんどが抗炎症性であるのに対し、TCAはむしろいくつかの研究では抗炎症性を示していることである(Hamer et al 2011;Vogelzangs et al 2012;Nazimek et al 2016;Chen et al 2018)。抗うつ薬と炎症との関係の複雑さをさらに強調すると、与えられた抗うつ薬は、どのような免疫パラメータ(例えば、サイトカイン放出または特定の細胞内シグナル伝達経路の活性化)を評価するかに応じて、プロまたは抗炎症性の両方の特性を示し得る(Horowitz et al 2014)。治療抵抗性は、特定の遺伝子変異(Baune et al 2010)選択的な細胞内経路の活性化(Horowitz et al 2014)または慢性炎症性疾患/疾患の存在(Luppino et al 2010;Vogelzangs et al 2012;HughesおよびKumari 2017)に関連しているかどうかにかかわらず、炎症の上昇によって予測される可能性があることを示唆する証拠が数多く存在する。この概念を支持するために、慢性的な低悪性度の炎症状態とうつ病の高い有病率を特徴とする過体重または肥満のような状態は、従来の抗うつ薬に反応しないより大きなリスクと関連していることが最近示されている(Kloiber et al 2007; Toups et al 2013; Woo et al 2016; Jantaratnotai et al 2017)。興味深いことに、これらの条件において、全身性炎症は、循環中のキヌレニンレベルの増加および肥満被験者の脂肪組織におけるKP酵素-特に神経毒性代謝物-の発現によって明らかにされるように、KP活性化と関連しており(Favennec et al 2015;Alemán et al 2017年)および気分症状(Capuron et al 2008;Daly 2013)。同様の結果は、肥満の前臨床モデルにおいて得られた(Dinel et al 2011,2014;André et al 2014;Boitard et al 2014;Castanon et al 2015;Almeida-Suhett et al 2017;de Cossio et al 2017)。興味深いことに、炎症およびKP活性化を減少させる減量(CancelloおよびClement 2006;Alemán et al 2017)は、有意な気分の改善と相関する(mery et al 2007;Capuron et al 2011)。KP活性化の他に、肥満におけるネオテリンレベルの増加を報告する研究(Brandacher et al 2006;Oxenkrug et al 2011;Mangge et al 2014)もまた、肥満に関連した抑うつ性合併症におけるBH4経路の潜在的な関与を示唆しており、肥満で報告されたドーパミン神経伝達の障害と一致している(Sharma and Fulton 2013;Krishna et al 2015)。
説得力のある証拠は、治療された患者において炎症と抑うつ症状が共存している限り、抗炎症性介入が新規抗うつ薬または従来の抗うつ薬のアジュバントとして有効であり得るという考え方を支持している(Strawbridge et al 2015;Kappelmann et al 2018;Köhler et al 2016;Schmidt et al 2016;Jantaratnotai et al 2017;JhaおよびTrivedi 2018)(表11)。例えば、非ステロイド性抗炎症薬(NSAIDs)は、うつ病性障害患者における抗うつ治療の転帰を改善する(Müller et al 2006; Akhondzadeh et al 2009)。同様に、NSAIDsの投与は、癌(Norden et al 2015年)アルツハイマー病(Llorens-Martin et al 2014年)およびパーキンソン病(Zaminelli et al 2014)などの炎症性疾患のいくつかの動物モデルにおいて、情動変化の重症度を減少させる。テトラサイクリン系抗生物質ミノサイクリンの有意な抗うつ効果も、プラセボと比較してうつ病患者において報告されている(レビューはRosenblatおよびMcIntyre 2018)。また、ω-3多価不飽和脂肪酸(PUFA)などの天然の抗炎症剤も、特に従来の抗うつ薬とのアドオン療法として、気分に関して有望な結果を示していることにも言及する価値がある(レビューLayé et al 2018)。肥満とうつ病の間のリンクを支持して、この戦略は、体格指数(BMI)の上昇にリンクされている低悪性度の基底性炎症(Rapaport et al 2016)を持つうつ病患者に特に効果的であることが明らかになった。興味深いことに、肥満の被験者におけるω-3 PUFAsの補充はまた、低カロリーの食事によって誘導される体重減少を強調する(Kunesová et al 2006)肥満の文脈での体重減少は、前述のように-炎症の減少(Lasselin et al 2014)および改善された抑うつ症状(Emery et al 2007; Capuron et al 2011)と関連している。神経精神症状を促進する特定の炎症性経路の暗示に関する現在の知識はまた、標的化された抗炎症性介入の可能性を提供している。したがって、IL-6,IL-17,およびTNF-αに対するモノクローナル抗体は、有意な抑うつ症状を有する慢性炎症患者、および基底性低悪性度炎症を有する抑うつ患者において抗うつ効果を示す(Traki et al 2014;Gossec et al 2015;Griffiths et al 2017;JhaおよびTrivedi 2018;Kappelmann et al 2018)。豊富な文献はまた、エタネルセプトまたはインフリキシマブなどの抗TNF-α薬の抗うつ効果を臨床試験(Kekow et al 2010;Arisoy et al 2013;Raison et al 2013;Fleming et al 2015)および炎症性疾患のマウスモデル(Haji et al 2012;Bayramgürler et al 2013)で報告している。特筆すべきは、治療抵抗性のうつ病患者におけるTNF-αの標的化は、特にベースラインの炎症が高い患者(Raison et al 2013年)すなわちBMIが高い傾向にある患者において、うつ病の症状を改善することが明らかになったことである。また、慢性的なストレス誘発性の抑うつ様行動は、IDO活性化の低下を介してTNF-α拮抗作用により軽減される(Fu et al 2016)。
表1 炎症、キヌレニンまたはBH4経路を標的とした抗うつ薬の介入を検討している研究
| 人口/モデル | 処理 | 主な成果 | |
|---|---|---|---|
| 臨床試験 | |||
| Akhondzadeh et al。、2009 | n = 40うつ病患者 | COX-2阻害剤(セレコキシブ) | 抗うつ治療の改善 |
| ミュラー他、2006年 | n = 40うつ病患者 | COX-2阻害剤(セレコキシブ) | 抗うつ治療の改善 |
| Kappelmann et al。、2018(レビューとメタアナリシス) | 慢性炎症状態 | 抗サイトカイン | ↙うつ症状 |
| Arisoy et al。、2013 | n = 9人の患者強直性脊椎炎 | TNFαブロッカー(インフリキシマブ) | ↙うつ病と不安のスコア |
| Fleming et al。、2015(データベースレビュー) | n = 464人の患者乾癬 | TNFαブロッカー | ↙うつ症状 |
| Raison et al。、2013 | n = 60うつ病患者 | TNFαブロッカー(インフリキシマブ) | ↙うつ症状 |
| エメリー他、2007年 | n = 13人の肥満女性 | 減量(胃バイパス) | ↙うつ症状 |
| ↙炎症性血液マーカー(CRPレベル) | |||
| Pan et al。、2011 | 症例報告(治療抵抗性自殺念慮) | サプロプテリン | 気分の改善 |
| Curtius et al。、1983 | 症例報告 | BH4 | 気分の改善 |
| Woggon et al。、1984 | 症例報告 | BH4 | 気分の変化なし |
| 前臨床試験 | |||
| Norden et al。、2015 | マウス(がん関連の倦怠感) | イブプロフェン | ↙うつ病のような行動と倦怠感のような(FST /ホイールランニング) |
| ↙海馬のIL-1bとIL-6 | |||
| Zaminelli et al。、2014 | ラット(ロテノンパーキンソンモデル) | イブプロフェン | ↙うつ病様行動(FST) |
| Fu et al。、2016 | ストレスラット(UCMS) | TNFαブロッカー(インフリキシマブ) | ↙うつ病のような行動 |
| ↙脳IDOおよびHAAOmRNA発現 | |||
| Bayramgürleretal。、2013 | ラット | TNFαブロッカー(エタネルセプト) | ↙うつ病様行動(FST) |
| Laumet et al。、2017 | マウス(神経損傷を温存) | (IL-1RA KMO阻害剤(Ro 61-8048) | ↙うつ病様行動(FST) |
| ↙脳KMOmRNA発現 | |||
| ↙うつ病様行動(FST) | |||
| Dobos et al。、2012 | マウス(LPS) | 1-MT | ↙うつ病のような行動 |
| コロナ他、2013年 | マウス(LPS) | 1-MT | ↙うつ病様行動(TST) |
| Xie et al。、2014 | ラット(ピロカルピン) | 1-MT | ↙うつ病様行動(FST) |
| ↙脳IDOmRNAの発現と活性 | |||
| O’Connor et al。、2009c | マウス | 1-MT | ↙うつ病のような行動 |
| ギブニー他、2014年 | (LPS) | ミノサイクリン | (TST / FST) |
| ラット(拘束ストレス) | アロプリノール | 脳IDOmRNAの発現と活性 | |
| ↙うつ病様行動(FST) | |||
| ↙循環kyn / tryp | |||
COX-2, cyclo-oxygenase-2; UCMS, unpredictable chronic mild stress; FST, forced swim test; TST, tail suspension test; IL-1RA, interleukin-1 receptor antagonist; KMO, kynurenine 3-hydroxylase; 1-MT, 1-methyl tryptophan (IDO inhibitor); LPS, lipopolysaccharide; IDO, indolamine 2,3-dioxygenase; HAAO, hydroxyanthranilic acid oxygenase.
炎症そのものではなく、KPのような炎症の脳神経生物学的標的に作用する可能性もまた、特にグローバルな抗炎症戦略は重要な副作用を伴うことが多いため、注目されている。したがって、説得力のある前臨床研究は、KPの活性化を減少させることが抗うつ効果を促進することを報告している(Reus et al 2015;RemusおよびDantzer 2016;JeonおよびKim 2017;Lovelace et al 2017;O’FarrellおよびHarkin 2017)。最初の有望なオントライアル戦略は、KP酵素、特にIDOおよびKMOの遮断を含むが、その効率は、しかしながら、どのような症状ドメインを評価したかによって異なる抗うつ薬様の特性を示すことが示されている(Toledo-Sherman et al 2015; Laumet et al 2017; Lovelace et al 2017; Tashiro et al 2017)。KP酵素を直接標的とするのではなく、他の研究は、むしろ、脳キヌレニンの大部分が末梢から来るので、血液脳関門を横切る末梢キヌレニン輸送を減少させる可能性を調査してきた。この手順は、活性化されたミクログリアによるQAの産生を防止することが実際に示されている(Carrillo-Mora et al 2010)だけでなく、LPSによって誘発された抑うつ様行動(RemusおよびDantzer 2016)。あるいは、神経保護的なキヌレニン代謝物であるKAの合成/利用可能性を高めることも価値があるかもしれない(Vécsei et al 2013)。QAや他のNMDA受容体アゴニストの神経毒性効果を打ち消すことにより、KAの生成を増加させることで、神経細胞の損傷やそれに伴う実験的に誘発された発作を減少させることが示されている(Russi et al 1992; Silva-Adaya et al 2011)。炎症に関連した抑うつ症状に関しても、同様の有益な効果を確認する必要があるが、これらのデータはすでに非常に心強いものとなっている。最後に、グルタミン酸を気分障害と関連させ、ケタミンなどのNMDA受容体拮抗薬の抗うつ効果を強調している文献と一致し(レビューについてはHaroon et al 2017を参照)いくつかの興味深い研究は、グルタミン酸活性を標的とするか、またはQAがNMDA受容体を活性化するのを阻止することも、さらなる治療の機会を提供することを示唆している(DantzerおよびWalker 2014;Reus et al 2015)。慢性ストレス誘発性抑うつ様行動(Li et al 2011)に対して保護することが既に示されているこれらの受容体の活性化をブロックすることは、LPS誘発性行動変容に関して同様に有効である(Walker et al 2013)。さらに、IDO欠損マウスはQA誘導性の神経細胞障害に対して感受性が低い(Mazarei et al 2013)。炎症関連うつ症状を改善するための潜在的な治療標的としてのNMDA受容体活性化の役割をさらに支持するために、抗うつ剤治療は、最近、KPのストレス誘発性活性化およびNMDA受容体発現の変化の両方を減少させることが示された(Martín-Hernández et al 2018)。同様に、脳のKYNAレベルを増強することによってNMDA受容体の活性化を減少させることは、アミロイドβ関連の神経変性を打ち消す有望な方法として提案されている(Carrillo-Mora et al 2010)。
BH4経路を標的とした潜在的な抗うつ効果、例えば経口的なBH4補充による効果は、これまでのところあまり研究されておらず、一貫性のない結果しか得られていない(Curtius et al 1983; Woggon et al 1984)としても、合成BH4の補充はすでにフェニルケトン尿症に苦しむ患者の治療に使用されている(Blau, 2013)。さらに、興味深い症例報告では、BH4置換タンパク質を投与すると、大うつ病に苦しんでいる患者の抑うつ症状が改善されることが示されている(Pan et al 2011)。前臨床データはまた、その末梢投与によって脳のBH4レベルを増加させる可能性を強調しており(大橋 et al 2016)その結果、セロトニンおよびドーパミン代謝(Brand et al 1996)THタンパク質含有量(Homma et al 2013)ドーパミンレベルおよび脳内の神経細胞活動(Koshimura et al 2000)を変化させている。BH4によるドーパミンとノルエピネフリンの神経伝達の変化が期待される行動改善、特にうつ病に関連した無気力症状の背景にあるのかどうかを検証するためには、さらなる研究が必要である。
これらの所見は、炎症に起因する抑うつ症状の背景にある炎症および/または神経生物学的メディエーターを直接または間接的に標的にすることが、有望な新しい治療戦略であることを明確に示している(図33)。これらの戦略は、抗うつ薬として処方されている現在入手可能な薬剤を用いた薬理学的アプローチだけでなく、まだ発見・検証されていない薬剤の使用にも依存している可能性がある。KPに関して、KP酵素阻害剤の潜在的な有益な効果は、現在、他の医療分野、特に腫瘍学において検討されている(レビューについては、Prendergast et al 2018を参照のこと)。したがって、炎症に起因する抑うつ症状の文脈で実施された研究は、他の分野で報告された知見から利益を得る可能性が高いはずである。薬理学的アプローチの他に、栄養学的介入などの非薬理学的治療戦略は、合併症のリスクと経済的コストの低減に関連した有望な代替手段である可能性がある。これには、特に、ω-3 PUFAs、アミノ酸または抗酸化物質、すなわち、最終的に炎症、酸化ストレスおよび/または他の神経毒性の障害から保護することが知られている化合物のような天然の薬剤を含む食事の補充が含まれる。重要なことは、これらの異なる治療戦略を組み合わせたり、むしろ治療すべき抑うつ症状のタイプに応じてそれらのうちの一つを好んだりする機会は、現在使用されている抗うつ剤の介入の作用スペクトルを広げ、したがって、炎症に関連する抑うつ症状の管理および/または治療を改善するのに役立つはずであるということである。
図3 炎症状態における抑うつ症状の誘導に関与するメカニズム
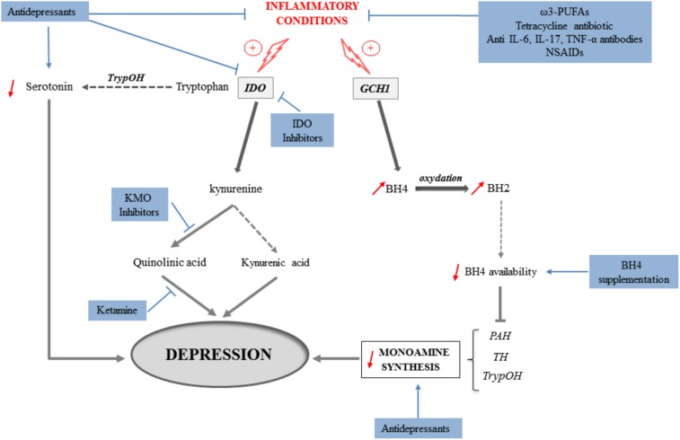
(灰色の矢印)と薬理学的または栄養学的介入(青矢印)に続く潜在的な治療上の意味合い。炎症状態におけるインドレアミン2,3-ジオキシゲナーゼ(IDO)の活性化は、セロトニン産生を犠牲にしてトリプトファンからキヌレニンを産生することにつながる。セロトニンおよび他のモノアミンの合成もまた、サイトカインによる GTP-シクロハイドロレース-1 (GCH1) の誘導に起因するテトラヒドロビオプテリン (BH4) のバイオアベイラビリティーの低下のために障害される。キヌレニンモノオキシゲナーゼ(KMO)を同時に活性化することにより、炎症は神経毒性代謝物であるキノリン酸の産生を促進し、キヌレニン酸の合成はむしろ神経保護的に減少する。炎症によるモノアミン産生の減少と神経細胞の損傷の増加は、最終的にはうつ病症状の発症に寄与する。このような背景から、これらのメカニズムを標的としたさまざまな治療戦略が同定されている。それらには、モノアミン合成を増加させることを目的とするが、炎症やキヌレニン経路の活性化を減少させることもできる古典的な抗うつ薬、抗生物質、ω3-PUFAs、抗サイトカイン抗体またはNSAIDsによる抗炎症介入、IDOまたはKMO阻害薬、ケタミンのようなNMDA受容体の拮抗薬、およびBH4の補充などがある。PAH、フェニルアラニン水酸化酵素;TH、チロシン水酸化酵素;TrypOH、トリプトファン水酸化酵素。
結論
うつ病と炎症を基盤とした慢性疾患の有病率が着実に上昇しており、それらの組み合わせが他の重篤な疾患の病因に悪影響を及ぼしていること、そして従来の抗うつ薬に対する抵抗性が高まっていることを考えると、炎症を基盤としたうつ病症状の病態生理学的メカニズムをよりよく理解することは、新たな効果的な治療戦略を同定するために緊急に必要であると考えられる。以上の結果から、炎症性プロセスを標的とした治療法は、うつ病の病態生理において極めて重要な役割を果たし、気分の調節に関与するモノアミンの代謝に大きな影響を与えることから、特に有望である可能性があることが示された。過去10年間で重要な進歩を遂げたにもかかわらず、炎症性経路内で標的とすべき最適な候補の同定から、最適な治療プロトコルの決定まで、いくつかの課題が残されている(すなわち、治療としての抗炎症戦略、または従来の抗うつ薬との併用治療など)。これらの問題を解決することは、患者さんの個性に依存することが明らかであり、抗うつ薬の処方をパーソナライズすることが可能になり、ひいては精神医学における精密医療の発展に貢献することができるという意味で、今後の重要な課題であると考える。