Contents
pubmed.ncbi.nlm.nih.gov/33157162/
Therapeutic resistance of pancreatic cancer: Roadmap to its reversal
ジャーナル・プリプルーフ
セン・ユー, チャン・チュンユー, 謝克萍
PII: S0304-419X(20)30180-3Reference: BBACAN 188461
受理日:2020年8月6日
改訂日:2020年10月20日
受理日:2020年10月24日
本論文は、受理後に表紙やメタデータの追加、読みやすさのための書式設定などの修正を行った論文のPDFファイルであるが、まだ確定版ではない。このバージョンは、最終的な形で出版される前に、さらにコピー編集、組版、校閲が行われる予定であるが、このバージョンを提供することで、論文を早期に確認していただけるようにしている。また、ジャーナルに適用されるすべての法的免責事項が適用される。
膵臓癌の治療抵抗性:その逆転へのロードマップ
セン・ユー1、チャン・チュンユー1、謝克萍2,3*(敬称略)
2テキサス大学MDアンダーソンがんセンター消化器内科、ヒューストン、テキサス州77030
通信: Dr. Keping Xie, The Precision Institutes of Medicine and Oncology, 10700 Richmond Avenue, Unit-143, Houston, Texas 77042, USA. 電話:832-398-3103; Email: kepingxie@gmail.com
要旨
膵癌は致死的な疾患であり、診断が遅く転移が早いため、治癒のための第一選択として切除可能な手術の機会は限られている。膵癌の非形成性間質と細胞の遺伝的あるいはエピジェネティックな変化は、化学療法、放射線療法、標的療法、免疫療法などの有効な治療法に対して物理的、生物学的な障壁を課している。ここでは、膵癌に対する現在の治療選択肢と、この致命的な疾患の特徴である治療抵抗性の根底にあるメカニズムと逆転の可能性について概説する。
キーワード:膵癌、化学療法、放射線療法、免疫療法、治療抵抗性
はじめに
膵臓がんは、最も致死率の高いヒトのがんのひとつであり、5年生存率は10%未満と低い[1]。2018年には、世界中で458,918人の膵臓がんの新規症例が発生し、432,242人が死亡した[2]。膵臓がん患者は通常、遠隔転移を伴う末期段階で診断され、治癒可能な手術の機会はほとんど残されていない[3,4]。したがって、膵臓がんの効果的な治療のために、非外科的治療アプローチを開発することが急務である。しかしながら、治療抵抗性は膵癌の治療効果や予後を左右する根強い課題である。治療抵抗性のメカニズムを理解し、新たな治療アプローチを探索することが不可欠である。
膵管腺癌に対する現在の治療選択肢
膵癌全体の約90%が膵管腺癌(PDA)である。 [5] 切除可能性により、膵管腺癌の臨床病期分類は4つのタイプに分類される: 切除可能:腫瘍が血管に浸潤することなく手術で切除できる;境界切除可能:腫瘍が血管または他の局所構造に浸潤し、おそらく1~3個の局所リンパ節に転移を伴うが手術で切除できる;局所進行:腫瘍が局所浸潤し、4個以上の局所リンパ節に転移を伴うが手術で切除できない;転移性:腫瘍が原発膵管腺癌を超えて他の臓器に転移し、手術で切除できない。 [6]
切除可能な膵管腺癌に対しては、手術が第一選択であり、その後に補助化学療法が行われる。[7]。歴史的に、補助ゲムシタビン単剤療法 [8] または補助5-FU単剤療法 [9] は、手術単独療法と比較して、外科的切除後6カ月の全生存期間(OS)を改善する。しかし、FOLFIRINOX(オキサリプラチン、フォリン酸/ロイコボリン、イリノテカン、5-FU)化学療法[10]やゲムシタビンとカペシタビンの併用療法[11]など、ゲムシタビン単剤療法と比較して全生存期間中央値を増加させることができる新しい術後補助併用療法に多くの臨床研究が焦点を当てている。近年、ネオアジュバント療法(手術前に行われる化学療法と放射線療法の併用または非併用)が登場し、切除断端が脅かされる可能性のある縮小を達成するために最初に使用されている。いくつかの第III相試験では、術後補助療法として5-FUベースの放射線療法が全生存期間に有益であることが示されている。[13-15]。
切除不能な膵管腺癌に対しては、標的治療を併用する、または併用しない化学療法が第一選択である。切除不能な膵管腺癌に対しては、FOLFIRINOX [16]とゲムシタビンベースの化学療法も適している。しかし、ゲムシタビンとナノ粒子-アルブミン結合パクリタキセル(nab-パクリタキセル)[17]、ゲムシタビンとエルロチニブ[18]の併用などのゲムシタビンベースのレジメンは、切除可能な膵管腺癌のものとは異なる。上皮成長因子受容体(EGFR)の発現が増加しているため、[19]、EGFRに対する標的薬であるエルロチニブがゲムシタビンと併用され、ゲムシタビン単独と比較して中央値および1年生存率の改善を示す。 [18] さらに、ゲムシタビンベースの化学療法後の2次治療として、5-FUとフォリン酸を併用する、または併用しないナノリポソームイリノテカン [20]、OFF(5-FU、フォリン酸、オキサリプラチン) [21]、FOLFOX(フォリン酸、5-FU、オキサリプラチン) [22]などのレジメンが利用されている。
免疫療法は多くの種類のがんの治療で一定の成功を収めているが、膵管腺癌の免疫療法ではほとんど成功していない。免疫チェックポイント阻害薬、キメラ抗原受容体T(CAR-T)細胞、免疫調節薬、ワクチンによる免疫療法の治療成績のほとんどは理想的なものではない。しかしながら、免疫療法の可能性を完全に否定することはできず、膵管腺癌の免疫学と免疫療法抵抗性のメカニズムを完全に理解することに希望がある。ある臨床試験では、ワクチンGVAXとCTLA-4(細胞傷害性Tリンパ球関連蛋白4)の免疫チェックポイント阻害薬であるイピリムマブの併用療法が、膵管腺癌患者の全生存期間中央値を向上させることが示されている。 [25] さらに、GVAX、抗PD-1/PD-L1(プログラム死蛋白1/プログラム死蛋白リガンド1)免疫チェックポイント阻害薬、および化学療法薬シクロホスファミドの併用療法を受け入れた患者は、対照群と比較して生存期間が最も長く、治癒率も高かった[26]。したがって、免疫療法と化学放射線療法との併用療法は、膵管腺癌治療のための免疫療法応用の将来的な方向性の1つとなる可能性がある。
膵管腺癌治療抵抗性のメカニズム
手術は膵管腺癌患者の長期生存を期待させるが、ほとんどの患者が末期と診断されるため、手術が適用されるのは15%~20%の症例に限られる。[27]。さらに、手術を受けた患者のほとんどが1年以内に再発する。[27]。それにもかかわらず、膵管腺癌の間質は腫瘍全体の90%を占めており、[28]、緻密な脱形成性間質は従来の薬剤の浸透を妨げる物理的障壁となっている。[29]。効果的な治療レジメンをデザインするためには、治療抵抗性の潜在的メカニズムを理解することが不可欠である。ここでは、膵管腺癌の化学療法、放射線療法、免疫療法の耐性機序について概説する(図1)。
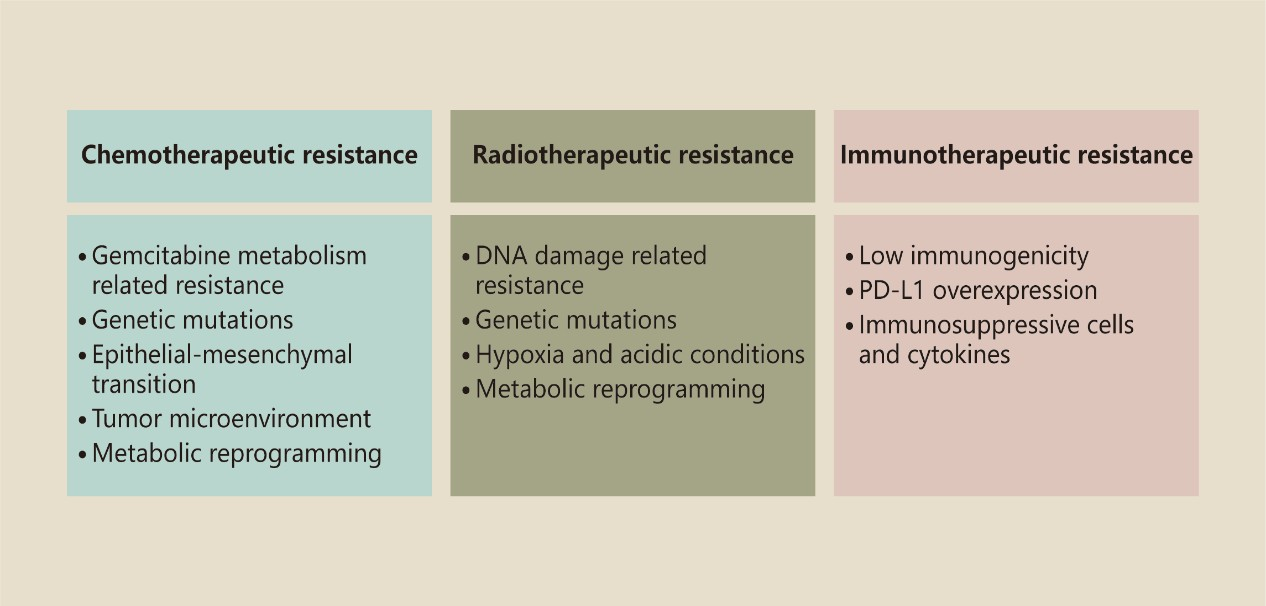
図1 膵管腺癌治療抵抗性のメカニズム
A. 化学療法抵抗性
ゲムシタビンは膵管腺癌に対する第一選択の化学療法薬であり、その耐性は膵管腺癌で一般的であり、ほとんどの研究の焦点となっている。リン酸化により活性化され、DNA合成を阻害し、[31, 32]、細胞増殖を抑制し、細胞周期を阻害する。[33]。膵管腺癌治療の第一選択薬としてのゲムシタビンの臨床的重要性を考慮し、膵管腺癌におけるゲムシタビン耐性の主要な基礎メカニズムに関する総説に焦点を当てる。
ゲムシタビン代謝に関連した耐性
ゲムシタビンは細胞膜を通過して細胞内でその機能を発揮する。この過程において、ゲムシタビンの細胞内への取り込みは、ナトリウム依存性(濃縮型ヌクレオシドトランスポーターhCNT)およびナトリウム非依存性(平衡型ヌクレオシドトランスポーターhENT)トランスポーターを含むヌクレオシドトランスポーター(NT)によって主に媒介される。 [ZIP4 は転写因子ZEB1の発現を増加させ、次いでインテグリンサブユニットITGA3とITGB1の発現を促進し、インテグリンα3β1シグナルの活性化につながる。活性化されたシグナル伝達は、c-Jun-N末端キナーゼ(JNK)を介してhENT1の発現を阻害し、ゲムシタビン耐性をもたらす。hENT1が高発現している膵管腺癌患者は全生存期間が長いことから、hENT1はゲムシタビンベースの化学療法を受けた切除膵管腺癌患者の予後バイオマーカーであることが示唆される。[37, 38]。さらに、hCNT1の発現は腫瘍細胞で常に低下しており、[39, 40]、がんの進行とゲムシタビン抵抗性と一致している。hCNT1の減少は、細胞周期の停止、細胞のアポトーシス、細胞遊走の阻害を引き起こすが[39, 40]、同時にゲムシタビンの取り込みを減少させ、治療抵抗性をもたらす。
NTに加えて、ATP結合カセット(ABC)トランスポーターもまた、化学療法抵抗性に関係している。例えば、ABCC5は5-FUまたはゲムシタビンの排出に関与しており、その発現レベルは膵管腺癌の化学療法抵抗性と正の相関がある。 [43] 膵管腺癌で過剰発現している転写因子である特異性蛋白質1(Sp1)は、グルコース調節蛋白質78(GRP78)の発現を増加させ、小胞体(ER)の恒常性を高めることにより、ABCトランスポーターの排除活性を高め、結果としてゲムシタビン耐性を高める。 [残念なことに、ABCトランスポーターを標的としたいくつかの臨床試験は、高い毒性と過剰な血中薬物量のために失敗に終わっている。
ゲムシタビンは細胞に取り込まれた後、デオキシシチジンキナーゼ(dCK)によってゲムシタビン一リン酸(dFdCMP)にリン酸化され、ゲムシタビンの活性型であるゲムシタビン二リン酸(dFdCDP)および三リン酸(dFdCTP)に変換される。 [42] 研究は、dCK阻害が腫瘍細胞におけるゲムシタビン化学療法抵抗性を引き起こす一方、ゲムシタビン抵抗性細胞におけるdCKレベルの回復がゲムシタビン化学療法感受性をもたらすことを示している[47, 48] dCKレベルは、RNA結合タンパク質であるHu抗原R(HuR)によって転写後制御されており、HuRレベルと正の相関がある[49-51] HuR過剰発現のがん細胞は、ゲムシタビン療法に対して高い感受性を示す。逆に、細胞内のゲムシタビンは、dFdCをdFdUに変換する酵素であるシチジンデアミナーゼ(CDA)によって不活性化される[52]。腫瘍細胞では、CDAの抑制によってdFdCTPが大幅に増加し、dFdUが減少し、ゲムシタビンの化学感受性が低下する。 [53] CDAレベルは、膵管腺癌微小環境に多く存在する腫瘍関連マクロファージ(TAM)によってアップレギュレートされ、TAM阻害剤GW2580はマウスモデルにおいてゲムシタビン効果を増強することができる[54]。 さらに、ゲムシタビン耐性乳がんではmiR-484の減少の結果としてCDAがアップレギュレートされる[55]。
リボヌクレオチド還元酵素(RR)は、リボヌクレオチドをdNTPに変換することができ、DNA合成において重要な役割を果たしている。dFdCDPによるRRの阻害は、ゲムシタビン活性を増強する最も重要なメカニズムである。[31, 33]。dNTPsプールを拡大することにより、RRの過剰発現はdNTPsアナログであるdFdCTPのDNAへの取り込みを減少させ、ゲムシタビン細胞毒性を減弱させる。 [56, 57] DNA損傷応答(DDR)に重要な役割を果たすATR(ataxia telangiectasia and Rad3-related)やCHK1(checkpoint kinase 1)が、ともにRRの発現を増強することを示す研究もある。したがって、RR過剰発現を伴うゲムシタビン抵抗性膵管腺癌に対しては、DDR過程を標的とする薬剤が有望な選択肢となる。同様に、dUMPをdTMPに変換する酵素であるチミジル酸シンターゼは、dNTPsプールの調節を通じてゲムシタビン活性の低下を誘導することができる[60]。
上皮間葉転換
上皮間葉転換(EMT)は、上皮細胞が運動性の間葉細胞に分化する過程である。EMTは、発生、創傷治癒、幹細胞の挙動において起こり、線維化や癌の進行に寄与している[61]。EMTは、ジンクフィンガーE-ボックス結合(ZEB)、SNAIL、TWIST、トランスフォーミング増殖因子β(TGF-β)、受容体チロシンキナーゼ(RTKs)などの様々な主要タンパク質や、PI3K-AKT、ERK、NOTCH、NF-κBなどの主要シグナル伝達経路によって制御されている[61]。
EMTは膵管腺癌ゲムシタビン抵抗性に関与しているようである(図2)。ゲムシタビンベースの化学療法は腫瘍の出芽(TB)と関連しており、これは形態学的にEMTの過程を反映している。[62]。TB細胞は、ビメンチン [63]、ZEB1、ZEB2 [64]などの間葉系マーカーの発現が増加し、E-カドヘリンやサイトケラチンなどの上皮系マーカーの発現が減少または限局していることが特徴である。
リン酸化ERK1/2によって誘導されるZEB1発現の増加は、上皮性マーカーであるE-カドヘリンとオクルディンの発現を減少させ、間葉性マーカーであるビメンチンを増加させ、EMTとゲムシタビン耐性を増強するが、ZEB1またはリン酸化ERK1/2の阻害はその逆を行う[62]。
加えて、EMTに寄与する2つの重要な転写因子であるSNAILとTWISTの阻害は、hENT1とhCNT3の発現を増加させ、結果として腫瘍細胞におけるゲムシタビン感受性を増強する[66]。
NOTCH-2レセプターの過剰発現は、EMTを起こすゲムシタビン抵抗性膵管腺癌細胞で観察される[67]。さらなる研究は、EMTのプロセスが、ヘパリン結合性成長因子であるミッドカイン(MK)の増加によって促進されることを発見した。MKはNOTCH-2受容体と相互作用し、NOTCHシグナル伝達経路とそれに続くNF-κBシグナル伝達経路を活性化し、EMTを引き起こし、アポトーシスを減少させ、腫瘍細胞のゲムシタビン耐性を増強する。さらに、MKはNOTCH-1誘導性のEMTを増強することにより、胆道がんのゲムシタビン耐性を促進する[68]。
また、RNAシークエンシング研究により、グルタチオンペルオキシダーゼ-1(GPx1)の欠損によるAKT/GSK3β/SNAILシグナルの活性化と活性酸素種(R全生存期間)レベルの増加が、EMTとゲムシタビン抵抗性の過程に関与していることが示唆されており、GPx1がゲムシタビン治療膵管腺癌患者における潜在的なバイオマーカーであることが示唆されている[69]。
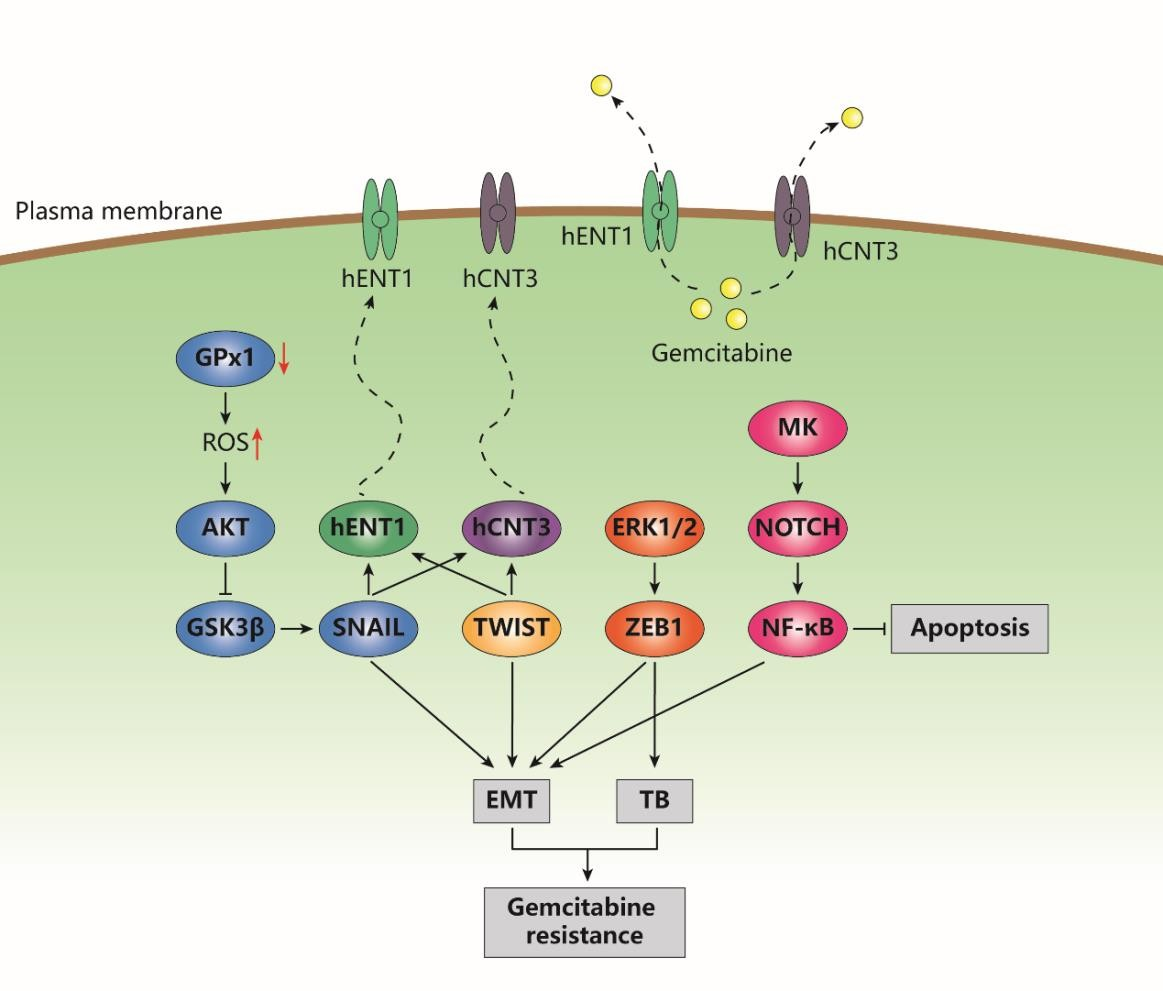
図2 膵管腺癌におけるEMTとゲムシタビン耐性の分子シグナル伝達
上皮間葉転換(EMT)は、主に上皮マーカー遺伝子を抑制し、間葉マーカー遺伝子を活性化するSNAIL、TWIST、zinc-finger E-box-binding(ZEB)転写因子によって駆動される。ゲムシタビン抵抗性膵管腺癌細胞では、SNAILとTWISTはともにヒト平衡型ヌクレオシドトランスポーター1(hENT1)とヒト濃縮型ヌクレオシドトランスポーター3(hCNT3)の遺伝子発現を活性化し、その結果ゲムシタビンの排出が促進される。SNAIL活性は、AKT/グリコーゲン合成酵素キナーゼ-3β(GSK3β)/SNAILシグナルの活性化により、グルタチオンペルオキシダーゼ-1(GPx1)の阻害と活性酸素(R全生存期間)レベルの上昇によって制御されている。ZEB1の発現上昇は、細胞外シグナル制御キナーゼ1/2(ERK1/2)を介し、細胞のEMTと腫瘍の出芽(TB)を誘発する。これは、間葉系マーカーの発現上昇と上皮系マーカーの発現低下または局所的な発現を特徴とするEMTの過程を形態学的に反映する。さらに、ヘパリン結合性成長因子であるミッドカイン(MK)の発現増加は、NOTCHとそれに続く核因子κB(NF-κB)シグナル伝達経路を活性化し、EMTを促進しアポトーシスを減少させる。総合すると、EMTは膵管腺癌におけるゲムシタビン耐性を積極的に制御している。
腫瘍微小環境
腫瘍微小環境(TME)は、線維芽細胞、内皮細胞、免疫細胞を含む様々なタイプの細胞、および成長因子、サイトカイン、ホルモン、細胞外マトリックスを含む細胞外成分から構成され、これらは腫瘍細胞を取り囲み、血管網によって栄養されている。 [70] TMEは、腫瘍の発生、進行、および転移の過程において極めて重要な役割を果たすだけでなく、治療効果にも大きな影響を及ぼす。
膵星状細胞(PSCs)は膵TMEの主要な構成要素であり、[71]、脱形成反応中にラミニン、コラーゲン、フィブロネクチンなどの細胞外マトリックスの要素を大量に排出する。[72]。TGF-βはPSCsにおいてCYR61の発現を誘導し、CYR61はゲムシタビンの取り込みを抑制するヌクレオシドトランスポーターhENT1およびhCNT3を負に制御し、ゲムシタビン耐性を誘発する。 [73] さらに、PSCsはNOTCHシグナル伝達経路を介してがん細胞における転写因子HES1の発現を促進し、ゲムシタビン耐性をもたらす。
TMEのもう一つの支配的な構成要素は、がん関連線維芽細胞(CAF)であり、その大部分は常在線維芽細胞に由来する。CAFsは腫瘍の進行と化学療法抵抗性を促進する可能性がある。 [75] CAFsから分泌されるタンパク質SDF-1は、腫瘍細胞におけるタンパク質SATB-1の発現を刺激し、CAFsの特性を維持することで、相互フィードバックループを形成する。さらに、α-SMA陽性CAFでは、mTOR/4E-BP1経路の活性化を通じて化学療法抵抗性が誘導される。この経路の阻害剤SOM230は、ソマトスタチン受容体sst1を活性化し、IL-6の合成を阻害することにより、ゲムシタビンと相乗的に腫瘍増殖と線維化を抑制する。また、CAFではIL1受容体関連キナーゼ4(IRAK4)がIL1βによって活性化され、次にNF-κBを活性化して腫瘍の線維化、増殖、生存およびゲムシタビン耐性を増強する。
阻害剤IPI-926によるHhシグナル伝達経路の阻害は、α-平滑筋アクチン(SMA)とマトリックスの減少を引き起こし、その結果、腫瘍血管密度が一過性に増加し、血液潅流とゲムシタビンの腫瘍内濃度が上昇し、細胞増殖と転移が阻害される。 [80] さらに、Hhシグナル伝達経路のもう一つの阻害剤であるシクロパミンは、ABCトランスポーターABCG2の発現をダウンレギュレートすることにより、膵管腺癌細胞の増殖を低下させ、ゲムシタビン耐性を誘導する。
TMEの免疫細胞は、自然免疫細胞(マクロファージ、マスト細胞、好中球、樹状細胞、骨髄由来抑制細胞、ナチュラルキラー細胞)および適応免疫細胞(TおよびBリンパ球)から構成され、腫瘍細胞と相互作用し、腫瘍の進行および治療に対する反応性に影響を及ぼす。特筆すべきことに、TMEの構成要素は、抗腫瘍M1マクロファージと親腫瘍M2マクロファージという、異なる表現型を持つ2種類のマクロファージの形成を制御していることが観察される。[42, 82] 腫瘍関連マクロファージ(TAM)は、TMEにおける治療応答の主要な制御因子である。[70]。TAMは、薬物の取り込みと代謝の際にゲムシタビンと競合するデオキシシチジンなどのピリミジン種を放出することができ、そのためゲムシタビン耐性が増強される。 [83] また、TAMは膵管腺癌細胞のEMTを刺激することにより、腹膜転移とゲムシタビン耐性をもたらす。 [84] さらに、TAMはtoll-like receptor 4(TLR-4)とcyclase-associated protein 1(CAP-1)のリガンドであるレジスチンを産生し、TLR-4とCAP-1を活性化することができ、細胞増殖、遊走、浸潤、ゲムシタビン耐性の増加につながる。
B. 放射線治療抵抗性
細胞が放射線に曝された後、高エネルギー光子は電子と相互作用し、そのエネルギーは電子に伝達される。この電子は、DNAらせん内の原子をイオン化することによって直接的に、あるいは水などの酸素含有分子をイオン化して活性酸素を生成することによって間接的に、DNA損傷を誘発する。 [86] 膵管腺癌の治療には、放射線療法と化学療法の併用が臨床で行われているが、治療成績はごく一部の症例で軽度の成功を収めている一方で、大多数の患者は放射線抵抗性である。
DNA損傷に関連した抵抗性
放射線によってDNA損傷が起こると、まずDDRタンパク質がそれを感知し修復する。[86]。同時に、細胞周期チェックポイントが活性化され、細胞周期を停止させることによってDNA修復のための時間を確保する[88]。放射線治療後の細胞は、DNA損傷のレベルや状況に応じて、生存、老化、アポトーシス、壊死、オートファジーなど、いくつかの異なる結果を得る可能性がある[86]。
二重鎖切断(DSB)はDNA損傷の一種であり、DSBの認識はATM(ataxia-telangiectasia mutated)とATR(ataxia telangiectasia and Rad3-related)キナーゼに依存している。[89]。ATM阻害剤であるAZD8055の使用は、癌細胞に対する放射線の有効性を高めることができ、この阻害剤は固形癌の放射線感作に利用されている。 [90]。DSBは、相同組換え(HR)と非相同末端結合(NHEJ)の2つのメカニズムで修復される[91]。NHEJの過程で重要なタンパク質DNA-PKcsを阻害すると、膵管腺癌細胞を放射線感作することができ[92]、放射線抵抗性にDDRが重要であることを示している。さらに、膵管腺癌患者の4-8%には、HRの過程でDNA修復機能を発揮するBRCA1またはBRCA2の生殖細胞系列変異が存在する[93, 94]。
14-3-3σは、リン酸化CDC25と結合して細胞周期をG2/Mチェックポイントで停止させることができる細胞周期チェックポイントタンパク質であるが、多くの膵管腺癌症例で過剰発現しており、放射線抵抗性に関係している[97-100]。
アポトーシスはプログラムされた細胞死の一形態であり、アポトーシスシグナル伝達経路の調節異常は、侵攻性のがんに頻繁にみられる。抗アポトーシス蛋白質IGF1Rは、放射線抵抗性の膵管腺癌、黒色腫および乳がん細胞で発現が上昇している。加えて、もう一つの抗アポトーシス蛋白であるREG4は、患者の血清中に高値を示し、放射線治療後の局所再発と正の相関があることから、膵管腺癌の放射線感受性の有望な予測因子である。[105]。
オートファジーは、DNA損傷、栄養欠乏、低酸素、熱ストレスなどのさまざまな条件に応じて細胞を保護または破壊する重要なプロセスである。HOTAIRは、オートファジー関連遺伝子7(ATG7)の発現をダウンレギュレートすることができるlncRNAであり、腫瘍細胞ではその発現が増加する[108]。HOTAIRのノックダウンまたはラパマイシンの使用は、いずれもオートファジーのプロセスを活性化し、細胞増殖を促進して放射線感受性を高める。さらに、放射線抵抗性と腫瘍細胞におけるmiR-23bの発現レベルとの間には正の相関がある。しかしながら、オートファジー機能の亢進も放射線抵抗性に寄与する。例えば、SMAD4は膵管腺癌の進行における一般的な変異抑制遺伝子であり、放射線抵抗性は腫瘍細胞におけるSMAD4の発現に関係しているようである。[110]。SMAD4を欠失させた細胞は、活性酸素の蓄積と放射線誘発性オートファジーのレベルを増加させるが、SMAD4を欠失させた細胞に野生型SMAD4を過剰発現させると放射線感受性が回復する。さらに、膵管腺癌において発現低下したmiR-216aは、ベクリン-1(BECN1)を介したオートファジーを促進することにより、放射線に対する細胞の感受性を低下させる。膠芽腫細胞における放射線療法は、オートファジーの亢進を誘導することが示されている[111]。放射線とオートファジー阻害剤であるクロロキン(CQ)の併用療法は、細胞のアポトーシスと放射線感受性を促進する。その結果、膵管腺癌放射線抵抗性におけるオートファジーの役割は複雑であり、オートファジー蛋白を標的とすることは、放射線抵抗性膵管腺癌患者に対する代替アプローチとなりうる。
低酸素と酸性条件
理論的には、低酸素症はほとんどの固形がんにおいて、血管系の異常による酸素供給不足から存在し、がんの進行に基本的な影響を与える[112]。
放射線抵抗性膵管腺癌細胞では、熱ショックタンパク質90(HSP90)の発現が増加し、HSP90はHIF-1αをアップレギュレートする。HSP90の阻害剤であるガネテスピブは、HIF-1αの発現を抑制し、放射線抵抗性を減弱させることができる。[114]。HIFsの分解を制御する律速酵素であるPHD3は、腫瘍細胞において発現が低下している。[115]。
正常酸素下または低酸素下でPHD3を過剰発現させると、HIF-1αの発現が減少し、正常酸素下よりも低酸素下の方が放射線誘発アポトーシスが大きくなり、その結果放射線感受性が上昇する。さらに、フコイダンは褐藻由来の多糖類であり、抗血管新生作用がある。フコイダンでコーティングされた二酸化マンガンナノ粒子(Fuco-MnO₂-NPs)は、低酸素で放射線抵抗性の膵管腺癌を放射線感受性の高い状態にすることができる。Fuco-MnO₂-NPsは、H₂O₂の存在下で効率的に酸素を産生し、HIF-1αの発現をダウンレギュレートし、低酸素条件下での放射線感受性を高める[116]。低酸素は、がん細胞のアポトーシス、代謝、オートファジーを制御することにより、放射線抵抗性と関連することが示されているが[117-119]、膵管腺癌放射線抵抗性のメカニズムにおけるその役割は、依然として不明である。
C. 化学療法抵抗性と放射線療法抵抗性の共通機序
遺伝子変異
膵管腺癌では、がん遺伝子KRASおよび抑制遺伝子TP53、SMAD4、CDKN2Aの変異を含むいくつかの遺伝子変異が、がんの進行および予後の悪化に寄与している。
KRAS、TP53、SMAD4およびCDKN2Aの変異は一般的であるが、遺伝子変異のメカニズムやゲムシタビン耐性との関連性に関する研究は限られており、KRAS変異による耐性のみが研究されている。癌遺伝子KRASは、肺癌、結腸直腸癌、膵癌において最も高い変異頻度を有しており、これらは米国において癌死亡率のトップ3を占めている[121]。がん細胞の増殖に加えて、KRASは化学療法の有効性にも影響を及ぼし、甲状腺乳頭がん、肺がん、大腸がんでKRAS変異と化学療法耐性の増強の関係が報告されている。[122-125]。
癌抑制遺伝子FBW7の変異または発現低下は多くのヒトがんで観察されており、その発現は変異KRASによって抑制される。FBW7の減少は、リソソーム機能を活性化することによりhENT1をタンパク質レベルでダウンレギュレートするため、ゲムシタビン抵抗性につながる。 [126] KRAS/ERKシグナル伝達経路の下流タンパク質であるRF6は、膵管腺癌で過剰発現しており、hENT1およびdCKの発現を阻害してゲムシタビン抵抗性につながる。 [127] さらに、膵管腺癌でしばしば発現が上昇するDCLK1が過剰発現すると、KRASが活性化され、ゲムシタビン感受性が低下する。KRASを直接標的とする理想的な薬剤は存在しないが、そのエフェクターであるPI3KまたはMAPKの阻害剤がゲムシタビン/ナブパクリタキセルの化学療法反応を増強するのに有効であることが示されている。[129, 130]。
KRAS変異とSMAD4変異は、それぞれ化学療法抵抗性と放射線抵抗性と関連していることは以前にも述べた。さらに、KRAS、TP53、CDKN2A変異は肺がんの放射線抵抗性と関連している。[131, 132]。放射線療法を受けた肺がんの亜集団では、放射線抵抗性の表現型である有糸分裂様凝縮クロマチン(MLCC)が観察され、骨ポンチンとEGFRによって制御されている。MLCC陽性がん細胞はKRAS、TP53、CDKN2A変異によって特徴づけられ、MLCCは放射線誘発性DSBから細胞を保護することができる[131]。
KRAS変異を有するPDC細胞は、試験管内試験および生体内試験でKRAS標的siRNAによる治療を受けると放射線感受性を回復する[133]。TGF-βⅡ型受容体の発現は、変異KRASによって調節異常となり、放射線抵抗性と関連する。ファルネシルトランスフェラーゼ阻害剤(FTI)を用いると、KRASタンパク質のファルネシル化と機能を抑制することができ、その下流タンパク質であるDNAメチルトランスフェラーゼ1(DNMT1)の発現を低下させる。DNMT1のダウンレギュレーションは、TGF-βⅡ型受容体のプロモーターメチル化を減少させ、その発現と放射線感受性を促進する。しかしながら、FTIはすべての膵管腺癌細胞株で機能するわけではなく、FTIの臨床応用にはさらなる研究が必要である[134]。さらに、KRAS遺伝子変異を有する肺がん細胞において、EGFRチロシンキナーゼ阻害剤BIBX1382BS(BIBX)はEGFR依存性のPI3K-AKT経路を阻害することができ、DNA-PKcsの活性を減弱させ、DSBsの蓄積を増加させ、放射線感受性をもたらす[135]。
代謝の初期化
再プログラムされたがん細胞の代謝は、がんの特徴の一つである[136]。がんの進行と生存を促進するために、がん細胞は多くの代謝経路を変化させ、正常組織やTMEの細胞を含む非悪性細胞との代謝的相互作用に依存する[137, 138]。最近の研究では、代謝の再プログラム化が、エピジェネティック修飾を制御することにより、膵臓腫瘍の発生と転移を促進することが証明されている[139, 140]。
ゲムシタビンによる長期治療後の膵管腺癌細胞は、R全生存期間レベルの低下と好気的解糖の増加を示し、がん幹細胞(CSC)およびEMTの表現型を誘導することにより、化学療法抵抗性を惹起することができる[142]。さらに、R全生存期間の発生は栄養欠乏状態により亢進し、解糖は低酸素によるHIF-1α発現の亢進により亢進し、ゲムシタビン抵抗性をもたらすことができる[143, 144]。予想通り、HIF阻害剤ジゴキシンまたはYC1による治療は、ゲムシタビン感受性を改善する[145]。
膵管腺癌化学療法抵抗性は、脂質代謝異常によっても誘導される。例えば、アップレギュレートされた脂肪酸合成酵素(FASN)は、ピルビン酸キナーゼ筋アイソザイム2(PKM2)の発現を増加させ、解糖の増加とゲムシタビン抵抗性につながる。 [147] 脂肪酸の他に、コレステロールの取り込みもゲムシタビン抵抗性に関係する。低比重リポ蛋白受容体(LDLR)のサイレンシングによってコレステロールの取り込みを阻害すると、抵抗性が逆転する。
化学療法抵抗性に加えて、代謝のリプログラミングも放射線抵抗性膵管腺癌で観察される。Mucin1(MUC1)は、膵管腺癌を含む固形がんでしばしば過剰発現されるがん遺伝子であるが、解糖、ペントースリン酸経路、およびヌクレオチド生合成を増強することにより、放射線誘発性細胞毒性を減弱させることができる[149]。放射線抵抗性膵管腺癌細胞に解糖阻害剤2-DGを投与すると、放射線療法に対する感受性を回復させることができる[150]。
脂肪酸およびコレステロール合成を含む脂質代謝の異常もまた、膵管腺癌の放射線抵抗性に寄与している。 [151, 152] コレステロール合成経路で過剰発現したファルネシル二リン酸合成酵素は、ゾレドロン酸(ZOL)によって阻害され、放射線抵抗性を低下させる。しかしながら、代謝リプログラミングと膵管腺癌放射線抵抗性の関係に関する研究は限られており、さらなる研究が必要である。
D. 免疫療法抵抗性
免疫療法は、免疫系を活性化することで腫瘍細胞を死滅させることができる治療法である。しかし、免疫チェックポイント阻害剤、CAR-T細胞、免疫調節剤、ワクチンなどによる免疫療法の失敗を引き起こす主な理由は、膵管腺癌の低い免疫原性、免疫チェックポイントシグナルの増強、免疫抑制性微小環境である。
低い免疫原性
TMEにおけるネオアンチゲンの提示は、腫瘍浸潤リンパ球(TIL)をリクルートし、免疫反応と免疫チェックポイント阻害剤の有効性を高めることができる。 [153] しかしながら、ネオアンチゲンの分類は腫瘍細胞の体細胞変異率に依存するのに対し、膵管腺癌では1変異/メガベース(メラノーマや肺がんでは10変異/メガベース)に過ぎないため、膵管腺癌の免疫原性は低く、その後のT細胞の活性化を妨げる[154]。特に、体細胞変異を促進しうるミスマッチ修復欠損を有する膵管腺癌患者では条件が異なる可能性がある。第II相試験において、ミスマッチ修復欠損を有するか有さない進行性転移性がん患者41人が抗PD-1薬で治療され、ミスマッチ修復欠損を有する患者は免疫活性が高く、生存期間が延長した[155]。とはいえ、ミスマッチ修復欠損を有する膵臓がんの割合は約2%に過ぎず、さらなる研究が残されている[156]。
PD-L1の過剰発現
免疫チェックポイントは免疫系の調節因子であり、免疫系から無差別に攻撃されないように細胞を守る自己寛容にとって重要である。免疫系の制御因子の一つとして、PD-L1はがん細胞で頻繁に観察される[158]。PD-L1と、活性化T細胞上に存在するPD-L1の受容体であるPD-1との結合は、T細胞の抗腫瘍免疫能力を抑制する。
PD-L1は、ヒト膵管腺癌組織の60%~90%、およびヒト膵管腺癌細胞株10株のうち9株で発現しており、がん細胞の免疫回避を引き起こす[159]。メカニズム的には、PD-L1プロモーターはH3K4トリメチル化(H3K4me3)を濃縮し、タンパク質MLL1はプロモーターに結合してPD-L1の転写を亢進する。抗PD-L1または抗PD-1抗体免疫療法と組み合わせてMLL1をサイレンシングすると、細胞増殖の抑制に成功する。また、PD-L1のアップレギュレーションは、膵管腺癌においてJAK-STATシグナル伝達経路によって媒介される可能性がある。活性化されたJAK-STATシグナル伝達経路は、細胞傷害性Tリンパ球(CTL)を不活性化するために、腫瘍細胞の免疫抑制性サイトカインの分泌を促進する。これらの結果は、免疫チェックポイント阻害免疫療法の失敗の一端を説明するかもしれない。さらに、PD-L1の高発現は膵管腺癌幹細胞の高比率と密接に関連しており、免疫療法の効果に影響を及ぼす可能性があり、CD8+ T細胞の浸潤が多い患者は予後が良好である。[161]。
デュルバルマブ、ペムブロリズマブ、アテゾリズマブなど、いくつかの抗PD-L1薬が臨床試験で検証されているが、[162]、これらの薬剤の単剤療法の効果はいずれも期待外れであり、他の免疫療法薬や化学療法薬との併用療法ではほとんど効果が得られない治療成績もある。[162]。したがって、抗PD-L1薬の単剤療法は膵管腺癌にとって理想的ではない可能性があり、おそらく併用療法に重点を置くべきである。
免疫抑制細胞およびサイトカイン
免疫抑制細胞とは、TMEにおける抗腫瘍免疫を阻害しうる細胞を指し、例えばTAM、骨髄由来抑制細胞(MDSC)、B細胞などが挙げられる。免疫抑制性サイトカインは、インターロイキン(IL)、インターフェロン(IFN)、コロニー刺激因子(CSF)など、腫瘍細胞や間質細胞によって産生される。これらは免疫抑制細胞の増殖と蓄積を促進し、免疫エフェクター細胞を抑制する。
TAMは免疫療法抵抗性において重要な役割を担っている。例えば、TAM上の骨髄成長因子受容体CSF1Rによって媒介されるシグナル伝達を阻害すると、膵管腺癌におけるTAMの抗原提示能と抗腫瘍T細胞応答を減弱させることができる。
MDSCsは、細胞表面上のタンパク質CD11bの高発現レベルを特徴とし、腫瘍に浸潤し、T細胞の活性化を阻害することができる[165]。MDSCsの一つである樹状細胞(DC)およびTAMsの活性低下は、膵管腺癌における免疫療法に対する抵抗性を引き起こす[166]。低分子アゴニストADH-503を介したCD11b活性化は、TAMsを再分極させ、DC応答を増強し、MDSCs数を減少させ、抗腫瘍T細胞免疫を改善する。さらに、膵管腺癌のTMEにおけるIL-10レベルの増加は、樹状細胞のIL-12産生を抑制し、抗腫瘍T細胞およびナチュラルキラー細胞(NK)の活性を制限する[167]。
168] B細胞はIL-35を産生し、CD8+T細胞の転写因子STAT3を活性化する。STAT3は、IFN-γ、CXCR3およびCCR5の発現を阻害することにより、CD8+ T細胞の腫瘍内浸潤および機能を抑制し、免疫療法抵抗性をもたらす。
治療抵抗性を克服するための潜在的アプローチ
様々なメカニズムが膵管腺癌の治療抵抗性を引き起こすが、TMEは不変のメカニズムであり、腫瘍間質の割合が大きいことから予測可能である。したがって、間質によって生じる物理的・生物学的障壁に取り組むことが不可欠である。膵管腺癌の治療抵抗性を解消するために、多くの試みがなされている(図3)。現在、この問題を解決するための2つの新しいアプローチが推定されている:冗長な間質を除去することと、ナノ粒子(NPs)を介して薬剤を送達することである。
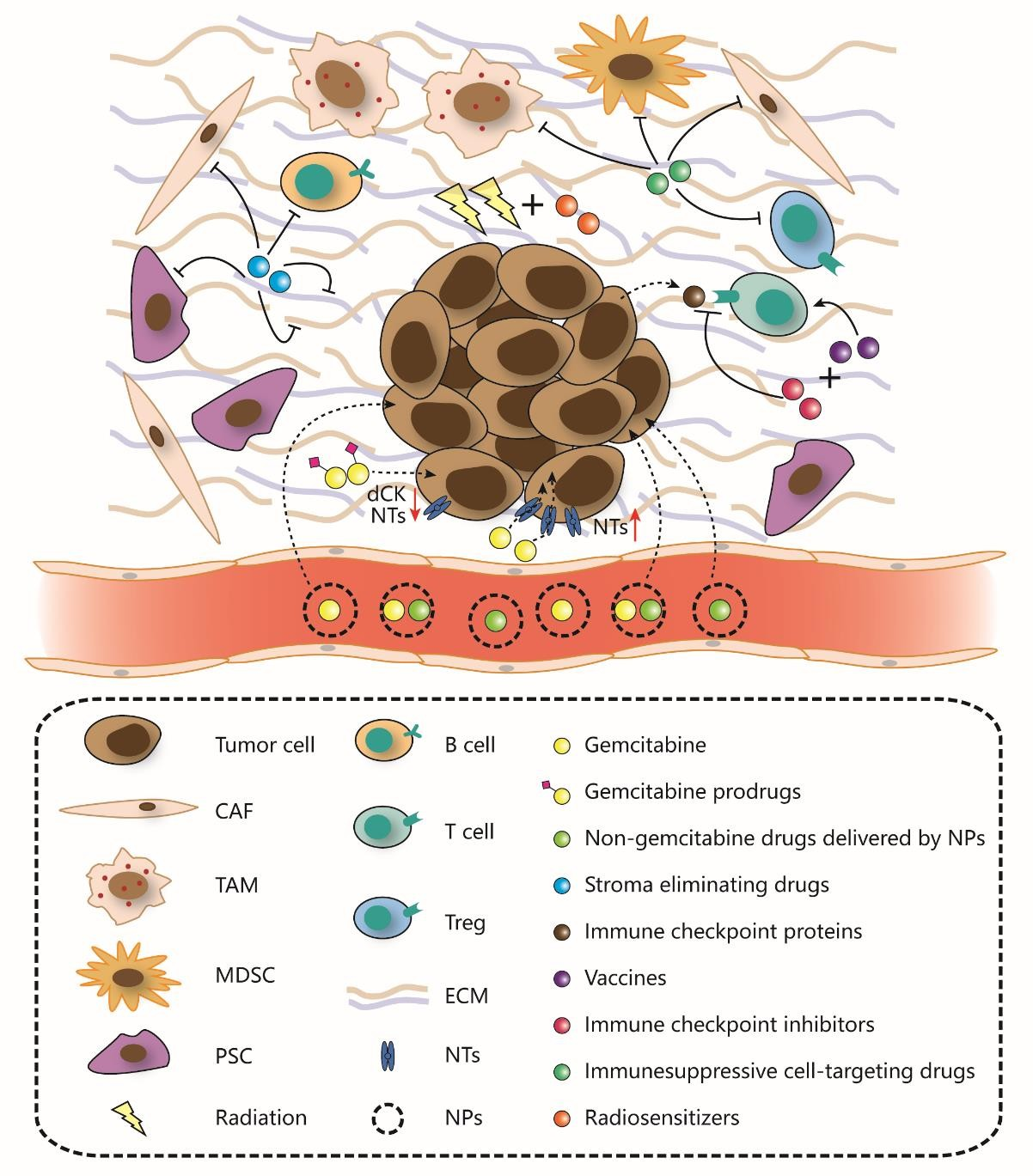
図3 膵管腺癌の治療抵抗性を解決する試み
膵管腺癌は腫瘍細胞と、膵星状細胞(PSCs)、がん関連線維芽細胞(CAFs)、腫瘍関連マクロファージ(TAMs)、骨髄由来抑制細胞(MDSCs)、B細胞、T細胞、制御性T細胞(Tregs)、細胞外マトリックス(ECM)、血管系などを含む腫瘍間質の大部分から構成されている。明瞭な腫瘍微小環境(TME)は、ほとんどすべての治療アプローチに影響を及ぼすため、いくつかの間質細胞、サイトカイン、ECMを標的とする様々な間質除去薬が膵管腺癌治療を促進するために試みられている。化学療法抵抗性に関しては、ゲムシタビンの取り込みはヌクレオシドトランスポーター(NT)のアップレギュレーションによって促進される。さらに、ゲムシタビンプロドラッグは、ゲムシタビンを活性化しうる欠損したNTとデオキシシチジンキナーゼ(dCK)を迂回することにより腫瘍細胞を死滅させるように設計されている。ナノ粒子(NPs)は近年、新しいタイプの薬物キャリアとして台頭してきた。NPsは血管壁や細胞膜を容易に通過することができ、薬物の分解を回避することで薬物の循環時間を延長し、薬物の吸収と有効性を高める。放射線治療抵抗性に関しては、放射線とヌクレオシドアナログや酸素代替物のような放射線増感剤の併用が試みられている。免疫療法抵抗性に関しては、ワクチンと免疫チェックポイント阻害剤の併用が、T細胞活性を効果的に増強する有望なアプローチである。さらに、T細胞活性化の制限を緩和するために、いくつかの免疫抑制細胞やサイトカインが標的とされている。
A. 冗長な間質を除去する
現在、間質の除去に関連する最も重要な薬剤はナブパクリタキセルであり、これは腫瘍細胞と間質細胞の両方を標的とし、[169]、転移性膵管腺癌患者の第一選択治療としてゲムシタビンとの併用で臨床応用されている。 [169] ゲムシタビン+ナブパクリタキセルで治療された転移性膵管腺癌患者は、ゲムシタビン単独療法を受けた患者と比較して、全生存期間、無増悪生存期間(PFS)、客観的奏効率(ORR)の中央値が高いことが第III相試験で示されている[17]。さらに、第I/II相臨床試験では、ゲムシタビンの細胞内濃度がゲムシタビン単独療法と比較して約2.8倍に増加した[170]。
ヒアルロン酸とI型コラーゲン
ヒアルロン酸(HA)とI型コラーゲンは、膵管腺癌間質内で物理的バリアとして働く2つの主要な要素である。したがって、HAの除去は腫瘍血管の柵状突起と血管間接合ギャップを誘導し[171]、腫瘍細胞への薬物送達を促進することができる[172]。事実、HAの除去は膵管腺癌ストーマを方向づける最もエキサイティングで期待されるアプローチの一つである[169]。 [172, 173] ゲムシタビンに続いてPEGPH20を投与すると、腫瘍内ゲムシタビン濃度を増加させることができる[171] さらに、薬物送達を強化した治療は一貫して腫瘍応答を達成し、マウスの生存期間を延長する[171, 172] 第II相試験では、HA高転移性膵管腺癌においてGEMとnab-パクリタキセル+PEGPH20の効果が評価された。PFS中央値はPEGPH20群で9.2カ月、対照群で5.2カ月であった(NCT01839487)。この有意な結果は、第III相臨床試験(NCT02715804)の継続を支持するものである。残念ながら、第III相臨床試験は中止されたが、その理由は不明である。
I型コラーゲンの架橋はリシルオキシダーゼ(LOX)ファミリーによって制御されており、線維性コラーゲンの増加はゲムシタビンの腫瘍内濃度を抑制する。CAFsを標的とすると、膀胱がんや膵管腺癌を含む脱形成がんにおいて、腫瘍の硬さと総コラーゲン含量が減少し、薬物送達が促進される。 [176] しかしながら、CAFsの枯渇は、血管伝染性や灌流、またはゲムシタビンに対する腫瘍細胞の感受性を増加させることはできず、その機序は不明である。 [172, 177] また、コラーゲン産生は、B細胞、DC、線維芽細胞に発現し、T細胞活性化のために抗原提示細胞を活性化しうる腫瘍壊死因子(TNF)の受容体であるCD40によって制御されている。 [179] 第I相臨床試験において、進行した膵管腺癌患者におけるCD40作動薬とゲムシタビンのORRは19%であり、一過性の免疫活性化が観察された。 [181] マウスモデルでは、選択的FAK阻害剤VS-4718とゲムシタビンの併用は生存期間中央値を増加させ、VS-4718と養子T細胞療法の併用は腫瘍組織におけるT細胞浸潤を約4.7倍増加させる。
成長因子および対応する受容体
膵間質の形成は、多数の分子およびシグナル伝達経路によって制御されている。TGF-β、EGFR、血小板由来成長因子(PDGF)および結合組織成長因子(CTGF)は、膵管腺癌の脱形成反応を促進しうる分子であり、すべて潜在的な治療標的である。[182]。TGF-β阻害剤galunisertibによるTGF-βの阻害は、化学療法の送達を改善し、転移を抑制し、CSCsの更新を阻止する。 [185] さらに、前述のように、ゲムシタビン+エルロチニブは、第III相臨床試験において進行性または転移性膵管腺癌患者の生存期間を改善しうる。しかしながら、別の第III相臨床試験では、セツキシマブを含むEGFR阻害剤は末期の膵管腺癌には無効であることが示されており、このような結果となった理由は依然として不明である。 186] さらに、PDGF受容体(PDGFR)-IgGは、腫瘍細胞と間質線維芽細胞間のPDGFRシグナル伝達を阻害することにより、膵管腺癌細胞の増殖を低下させる。さらに、CTGFは前臨床膵管腺癌モデルで過剰発現しており、[188]、新規ヒトモノクローナル抗体であるパムレブルマブとゲムシタビンの併用によるその阻害は、生体内試験での動物の生存期間を延長させる。[189]。
Hhシグナル伝達経路
Hhシグナル伝達経路は、造血幹細胞や線維芽細胞の分化や運動性など、膵管腺癌の脱形成間質の形成に関与している。ゲムシタビンとの併用によるHhシグナル伝達経路の阻害は、間質容積を減少させ、化学療法の有効性を増加させるが、その結果は一過性のものである可能性がある。マウスモデルにおける腫瘍容積は1-2週間以内に減少し、その後腫瘍の増殖が再開し、血管レベルが低下することから、薬剤に対する治療抵抗性が示唆される。 [80] Ib/II相試験では、別のHhシグナル伝達経路阻害剤であるvismodegibは、転移性膵管腺癌においてゲムシタビンに追加しても臨床的有用性を示さなかった[190]。したがって、Hhシグナル伝達経路が膵管腺癌における他の薬剤の治療効果に影響を及ぼすメカニズムについては、さらなる研究が必要である。
ブルトン型チロシンキナーゼ
ブルトン型チロシンキナーゼ(BTK)は、B細胞受容体シグナル伝達経路の非受容体型チロシンキナーゼである[191]。CD20+ B細胞は、膵管腺癌のTMEにおいて正常膵細胞と比較して増加する[191]。血液悪性腫瘍の治療に用いられるBTK阻害剤イブルチニブ [192] は、TAMを抗腫瘍M1マクロファージに変化させることができる。 [193] しかしながら、第III相試験では、ゲムシタビンおよびナブパクリタキセルと併用したイブルチニブは、進行膵管腺癌患者において全生存期間またはPFSの有益性を示さなかった[82]。
まとめると、間質除去剤による膵管腺癌の治療成績のすべてが有益であるわけではなく、その大部分は議論の余地があり、説明のつかないものである。重大な問題は、前臨床試験の結果の多くが臨床試験で再現できないことである。転移の失敗は、おそらく研究に用いる膵管腺癌モデルの選択が不適切であったためであろう。間質がほとんど形成されない異種移植膵管腺癌モデルでは、ゲムシタビンの細胞内投与量が多く、腫瘍の増殖を効果的に抑制する[80]。一方、膵臓細胞に内因性の変異型KRASおよびTP53対立遺伝子を条件付きで発現させ、ヒトの膵管腺癌間質に類似した脱形成間質を形成するKPCマウスモデルでは、ゲムシタビンの細胞内投与量が少なく、ほとんど効果がない。従って、前臨床試験において、特に複雑な成分の間質に関する研究においては、膵管腺癌モデルを正しく選択することが特に重要である。さらに、最大限の治療効果を得るためには、薬剤の投与順序を最適化する必要がある。他の薬剤の送達や効果を抑制する可能性のある脱形成性間質をまず除去する必要があるかもしれない。さらに、膵管腺癌間質を標的とするほぼすべての薬剤は、一種類の分子や細胞のみに作用するが、間質成分は様々であり、そのうちの一つだけを標的にしても治療効果は限定的である。従って、複数の間質細胞によって特異的に産生される分子を研究することは、間質を広範囲に除去し、より良い治療成績を達成するための、将来の別の方向性かもしれない。現在、ストロマを標的とした治療薬の臨床試験がいくつか進行中である(表1)。
表1 膵管腺癌間質を標的とした治療薬を検討する進行中の臨床試験
【本文参照】
B. ナノ粒子による薬物送達
ナノ粒子(NPs)は通常、1~100 nmの大きさの粒子と定義される[194]。最近の研究では、NPsががん治療のための薬物キャリアとして使用できることが示唆されている。NPは物理的障壁を容易に通過することができ、薬物の分解を回避することで薬物の循環時間を延長し、薬物の吸収と有効性を高めることを示す証拠がある。 [注目すべきは、低分子薬物、抗体、siRNA、アンチセンスヌクレオチド、毒素などの部位特異的リガンド、あるいは特定の腫瘍微小環境で活性化されることにより、スマートNPsは腫瘍を標的とし、特定の部位での薬物放出を制御できることである。 [その後2008年には、ゲムシタビンとセツキシマブをEGFR標的薬として用いた膵管腺癌のNP標的療法が報告され、試験管内試験と生体内試験で効果を示した。 [200] さらに、最新の研究では、クルクミン-エルロチニブ結合体の自己組織化に基づくキャリアフリーのNPが、クルクミンとエルロチニブのフリーの組み合わせよりも膵管腺癌治療に優れた効果を示し、担癌マウスの生存期間中央値が長いことが示されている[201]。結果として、ナノテクノロジーは、高密度間質を特徴とする膵管腺癌の治療抵抗性を逆転させる有望なアプローチである。現在、膵管腺癌治療に対してポジティブな効果を示す有機または無機のNPは、主に4つのカテゴリーに分類される。
アルブミンは、栄養輸送のための最も豊富な血漿タンパク質であり、NPの製造に利用されている。現在、アブラキサン®とも呼ばれるナブパクリタキセルは、2013年にFDAから臨床応用の承認を受けたナノ医薬品のひとつである。 [202] 上述のように、ゲムシタビンと組み合わせたナノ粒子-アルブミン結合パクリタキセル(nab-パクリタキセル)は、ゲムシタビンの腫瘍内濃度を高め、全生存期間、PFS、ORRの中央値を促進する転移性膵管腺癌の治療に使用されている。 [17, 169, 170] さらに、パクリタキセルとクルクミン(クルクミンの非毒性アナログであり、膵臓に特異的に集積する)[203-205]は、ナノ粒子-アルブミン結合パクリタキセル/クルクミンの形で共送達され、試験管内試験でパクリタキセルのみを含むNPと比較して、効果的な薬物放出と71%低い半量最大阻害濃度(IC50)をもたらす[206]。
一般に、抗腫瘍薬は親油性が高いため、薬物の溶解性や吸収性が低い。しかし、この問題は、親水性ポリマーNPと薬物を結合させ、疎水性コアに薬物を含むコアシェル凝集体を形成することで解決される。最近の研究では、溶解性に乏しいイリノテカンの活性代謝物であるSN38と、Hhシグナル伝達経路の阻害剤であるvismodegibを親水性高分子NPに結合させた二重薬剤を用いた試験管内試験での効果的な膵管腺癌治療が示されている[208]。さらに、ゲムシタビンと親水性高分子NPとの結合は、試験管内試験において遊離の薬剤と比較して、ゲムシタビンの分解と細胞毒性の減少を示す[209]。別の研究では、ゲムシタビンと腫瘍抑制因子miR-205模倣体を含む親水性高分子NPが、試験管内試験において化学療法抵抗性、細胞浸潤および遊走を有意に減少させ、生体内試験において腫瘍増殖を阻害することが報告されている[210]。
次に、両親媒性ポリマーNPである。親水性高分子NPと比較して、両親媒性高分子NPは親水性と疎水性の両方の構造を持ち、水溶液中で自発的に凝集して高分子ミセルを形成する[195]。したがって、疎水性コアは親油性薬物の送達に利用される。試験管内試験の研究では、ゲムシタビンと塩酸ドキソルビシンを1セットの混合ミセルに結合させると、混合していない薬物に比べて腫瘍細胞毒性が増強されることが示された[211]。さらに、野生型TP53遺伝子と、腫瘍細胞で過剰発現しているアンジオテンシンII 1型受容体を標的とする薬物であるカンデサルタンを結合させた両親媒性高分子NPは、腫瘍関連の血管新生と腫瘍増殖を有意に抑制した[212]。 [両親媒性ポリマーNPに結合させた新規薬剤BNI膵管腺癌octは、異種移植マウスにおいて膵管腺癌細胞の増殖を阻害することができ、BNI膵管腺癌octの投与量は臨床的に使用されているゲムシタビンの8倍である。 [213]。ナノリポソームはリポソームのナノメートルバージョンであり、1つ以上の同心二重層に配置された両親媒性脂質からなる小さな球状構造体であり、疎水性二重層と親水性コアにそれぞれ疎水性薬物と親水性薬物の両方を運ぶことができる。 [214] 第III相臨床試験において、ナノリポソームイリノテカン+5-FUとフォリン酸の治療レジメンは、5-FUとフォリン酸のレジメンと比較して、ゲムシタビンベースの治療を受けたことのある転移性膵管腺癌患者の全生存期間中央値を延長した[20]。
無機NPは、炭素、酸化鉄、金などの無機材料で作製される。上記の有機NPsとは異なり、無機NPsは明確な磁気的、電気的、光学的、電気化学的特性を持っており[195]、特殊な目的への可能性を示唆している。例えば、カーボンナノチューブが膵管腺癌細胞に内在化され、808nmの照射を受けると、加熱効果が生じ、ミトコンドリア膜の脱分極をもたらし、フリーラジカルの流出を引き起こし、その結果、酸化ストレスの亢進によって誘発される細胞死を引き起こす可能性がある。 [ゲムシタビンと抗CD47抗体を結合させた酸化鉄NPは、複数の膵管腺癌細胞株と共培養しても細胞毒性を増加させることはできないが、腫瘍細胞へのゲムシタビンの取り込みは改善される。
現在、膵管腺癌におけるナノ粒子に関するほとんどの前臨床試験は、包括的な臨床試験には至っていない。なぜなら、ナノテクノロジーに関するいくつかの未解決の問題については、さらなる研究が必要だから: ナノ材料は健康に害を及ぼさないのか?ナノ材料の分解時間をどのように制御できるか?ナノ材料はどのようにして長期保存を実現し、物理的・化学的特性を維持できるのか?また、どのようにしてNPsの製造コストを削減できるのか?スマートNPは、腫瘍の標的化や特定部位での薬物放出の制御など、革新的な機能を持つように設計されているが、NPの複雑な構造や組成は、生体内での詳細な作用を研究する上でより多くの困難をもたらす。
結論と今後の方向性
過去数十年間、膵管腺癌治療のために数多くの治療レジメンが作成されてきたが、膵管腺癌患者の5年生存率は常に低いレベルにとどまっており、膵管腺癌は複雑な疾患であり、実質的な研究作業が緊急に必要であるという悲惨な事実を再確認している。膵管腺癌の病態、特に治療抵抗性のメカニズムを根本的に理解し、新薬を開発することが急務である。今後の研究は、3つの主要な分野に焦点を当てなければならない: 膵管腺癌のTMEは、ほとんどすべての治療過程に影響を及ぼすため、有望なターゲットである。早期診断、早期発見と診断のみが、膵管腺癌を最初から克服するための効果的なアプローチを設計することを保証する。精密治療、個々の特徴を持つ患者ごとに治療法を調整することで、腫瘍の不均一性がもたらす問題を軽減することができると考えられる。
リキッドバイオプシーは、循環腫瘍DNAを早期に検出するための侵襲性の低い方法である。しかしながら、循環腫瘍DNAは限局がん患者の約50%にしか存在しない。[218]。そのため、リキッドバイオプシーの感度および特異度が低く、早期診断には限界がある。CA19-9は、症候性膵管腺癌患者の診断に有効な血清バイオマーカーであり、その感度は約80%、特異度は約86%である。[219]。 [220-223] さらに、CA19-9濃度は、腫瘍切除後の切除可能な膵管腺癌の予後を予測することができ [219, 224]、切除不能な膵管腺癌の予後を反映することもある。[225, 226] しかしながら、CA19-9は症状のない膵管腺癌患者の早期診断には使用できず、胆道閉塞患者では濃度が変化するため、膵管腺癌診断への応用には限界がある。 [227] 早期発見に用いられるこれらの分子因子に加えて、新規発症糖尿病(NOD)は散発性膵管腺癌の最も高い危険因子と考えられている。 229]さらに、NODと膵がんの関係は、異なる人種や民族にも当てはまる。しかしながら、膵管腺癌に関連した糖尿病を一般的な糖尿病と区別することは困難であるため、この問題を克服するためのより高度なアプローチを開発することが重要であり、膵管腺癌の早期発見のためのより多くの研究が必要である。
さらに、腫瘍の不均一性は、すべての癌の進行に存在する非常に困難な問題であり、これは、異なる腫瘍細胞が、細胞形態、遺伝子発現、代謝、運動性、血管新生、増殖、免疫原性、転移を含む、異なる表現型を持つ可能性があるという現象を指す[232]。がん幹細胞の存在とクローン進化は腫瘍の不均一性の原因として広く考えられており、[233]、高い可塑性を有するこれらの腫瘍細胞は治療抵抗性の一因となっている。 [234] 腫瘍の不均一性は、ゲノムまたはトランスクリプトームシークエンシングにより、患者間の複数の腫瘍サブタイプや1つの腫瘍内の細胞サブタイプに反映されうる。[235-239] Collissonらは先に3つの膵管腺癌サブタイプを発見し、異なる膵管腺癌サブタイプがそれぞれ化学療法薬ゲムシタビンおよびエルロチニブに対して異なる感受性を有することを示している。 [同様に、Baileyらは、変異遺伝子、シグナル伝達経路、遺伝子発現プロファイルなどの腫瘍組織の異なる特徴に従って、4つの膵管腺癌サブタイプをより正確に定義した:(1)扁平上皮、(2)膵前駆細胞、(3)免疫原性、(4)異常に分化した内分泌外分泌。今日では、単一細胞RNAシーケンシング(scRNA-seq)は、この問題を部分的に解決し、膵管腺癌の複雑なTME構成要素を研究する大きな機会を提供している。例えば、CAFが腫瘍細胞およびTMEに及ぼす影響については、浸潤性EMT移行、増殖促進、免疫応答調節など、scRNA-seqを通じてよく研究されている。膵管腺癌のサブタイプとその細胞に関するこれらの研究は、臨床において特異的な治療戦略を実行できるよう、精密医療への可能性を提供する。シークエンシングと解析法の進歩とともに、正確な分類と標的治療の組み合わせは、将来膵管腺癌患者に利益をもたらすかもしれない。
現在、いくつかの先端技術が登場しており、それらは膵管腺癌の治療効果に役立つ可能性がある。KRASは膵管腺癌で最も頻繁に変異する癌遺伝子であり、癌の進行を促進するために下流のタンパク質とシグナル伝達経路を活性化する。残念なことに、KRAS遺伝子はGTPに対する親和性が高く、薬剤がアクセス可能な結合ポケットがないため、KRAS遺伝子を標的とする有効な薬剤は設計されていない。 [243, 244] 研究者らは、ジフテリア毒素(DT)の受容体標的性と膜移行性を利用して、人工キメラ毒素を作製し、RRSPを標的とするがん細胞に導入した。遺伝子治療に加えて、静脈・動脈再建、ネオアジュバント化学療法、定位体放射線療法、切除放射線療法、免疫療法の併用、NPを用いた光触媒療法、間質除去療法などの他の治療分野での進歩も、最終的には膵管腺癌治療抵抗性の逆転と治療成績の向上に寄与するであろう。
利益相反
著者らは利益相反はないと宣言した。
謝辞
Leon Wang博士の批評的読解に感謝する。本総説に関連する研究の一部は、中国国家自然科学基金(82072632)および広州市科学技術局(中国広州市)の助成金により行われている。
