
Contents
Neuroinflammation and Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches
www.ncbi.nlm.nih.gov/pmc/articles/PMC7037892/
オンラインで公開2020年1月21日
概要
成人の脳では、脳室下帯や歯状回で新しい神経細胞が成人になってからも生成される。成体神経発生の調節には、様々な内在的経路(シグナル伝達経路、エピジェネティック・遺伝子調節経路)や外在的経路(代謝成長因子調節、血管、免疫系経路)がある。
アルツハイマー病では、ヒトAD脳およびADモデルの両方で、ニューロン新生の変化が確認されている。アルツハイマー病における成体ニューロン新生の調節不全の正確なメカニズムは、まだ完全には解明されていない。
しかし、神経炎症が成体ニューロン新生を変化させることが明らかになっている。免疫細胞、サイトカイン、ケモカインなどの様々な炎症性成分の存在は、神経幹細胞の生存、増殖、成熟を制御する役割を果たしている。また、神経炎症は、アルツハイマー病の特徴的な神経病理学的特徴と考えられている。
この総説では、成人の神経形成、神経炎症、およびアルツハイマー病におけるこれら、2つの現象の関係について、最新の見解をまとめている。さらに、この分野で報告されている抗炎症および神経新生促進介入に焦点を当てて、治療アプローチの可能性について議論する。
キーワード
アルツハイマー病、ニューロン新生、神経炎症
1. はじめに
アルツハイマー病は、世界的に最も多い神経変性疾患であり、最近の記憶の早期障害を特徴とする。アルツハイマー型認知症は、初期の記憶障害を特徴とし、重症度が進むにつれ、言語能力、方向感覚、実行機能などの広範な症状が現れ、セルフケア能力の低下を招く。アルツハイマー病の特徴的な病態は、βアミロイド(アミロイドβ)の沈着と、リン酸化されたタウ(P-tau)を含む神経原線維のもつれ(NFT)である。アルツハイマー病の確定診断には、脳組織の剖検が必要である。しかし、最近では、脳脊髄液(脳脊髄液)のバイオマーカー(脳脊髄液中のアミロイドβ40,アミロイドβ42またはその比率、総タウまたは高リン酸化タウ)やポジトロン・エミッション・トモグラフィー(アミロイドPETまたはタウPET)と臨床的な基準を組み合わせることで、生きている人の診断の助けになることが提唱されている[1]。
アミロイドβに関する混乱は、過剰生産またはクリアランスの低下の結果である[2]。また、加齢や遺伝的・環境的要因により、アミロイド前駆体タンパク質(APP)の代謝がアミロイド生成経路にシフトすることが知られている[3]。アミロイドβペプチドは、不溶性のオリゴマーやプロトフィブリルに凝集し、さらにアミロイドβ線維種を誘発し、老人斑や神経斑に蓄積していく。アミロイドβの過剰産生に加えて、脳からのアミロイドβのクリアランスが低下すると、細胞外にアミロイドβが蓄積し、その後、P-tauの沈着、細胞骨格の変化、神経細胞の機能障害、神経細胞死の連鎖が生じる[4]。しかし、APPの生理的機能はいまだに解明されておらず、アミロイドβプラークは、観察可能なアルツハイマー病症状の発症や診断の10年以上前から観察されている[5]。
微小管関連タンパク質であるタウタンパク質は、微小管を安定化させる働きがある。タウタンパク質は、リン酸化やグリコシル化が異常に亢進しているが、アルツハイマー病ではその生物学的機能が損なわれている。P-tauのフォールディング異常は、アルツハイマー病の主要な神経病理学的特徴であるペアヘリカルフィラメント(PHF)やNFTの生成につながる[6,7]。PHFは、細胞内のタウタンパク質をアグレッソームに集積させ[6]、神経細胞から細胞外に放出されたPHFは、アルツハイマー病患者のさまざまな脳領域にタウの病理を伝播させる[8,9]。タウ蓄積の重症度は、アルツハイマー病における認知症の重症度および神経変性プロセスと密接に関連している[10,11]。
多くの研究が、アミロイド沈着とタウタンパク質が、アルツハイマー病の原因となる2つの中核的な神経病理であることを示しているが、直接的なアミロイド免疫療法またはタウ免疫療法によってこれら、2つの病理タンパク質を調節した臨床試験の結果は残念なものであった[12,13,14,15]。アルツハイマー病の病態解明には、まだかなりのギャップがある。例えば、外傷性脳損傷、脳卒中、多発性硬化症、筋萎縮性側索硬化症、低酸素性虚血性脳症など、アルツハイマー病以外の多くの神経疾患においても、脳内アミロイドβレベルは上昇することが知られている。アミロイドβ濃度が上昇するメカニズムはまだ明らかになっていないが、神経細胞への障害や損傷に対する反応である可能性がある。したがって、アルツハイマー病では、アミロイドβの過剰産生または蓄積は、最初の神経細胞の損傷を修復し、正常な脳機能を維持しようとする試みである可能性がある[16]。上流の病態生理学的プロセスは、神経炎症またはミクログリアの異常な活性化[17,18,19,20]、病因にかかわらず代謝不全[21]、酸化ストレス[20]、またはコレステロールに関連した持続的な神経細胞の苦痛[22]である。さらに、最近では、早期発症のアルツハイマー病や後期発症の散発性アルツハイマー病の存在を説明するために、「ストレス閾値の状態変化」モデルが提案されている[23]。現在までのところ、アルツハイマー病の神経病理と臨床症状の発症と進行に関しては、まだかなりのギャップがある。
ここ数十年の間に、アルツハイマー病の第三の中核的な神経病理学的特徴が現れてきた。アミロイドβやNFTに加えて、アルツハイマー病患者の脳では神経炎症が見られる[18,19,24,25,26,27,28,29]。このようなアルツハイマー病における神経炎症性の変化は、動物モデル[30,31]やヒトの死後脳[32,33,34]で観察されており、また、炎症プロセスを検出するための分子イメージングによっても観察されている[35,36]。多くの研究が、神経炎症の制御におけるミクログリア遺伝子の潜在的な役割と、ミクログリア遺伝子と遅発性散発性アルツハイマー病との関係を論じている[29,37,38,39,40,41]。この神経炎症プロセスは、アルツハイマー病における神経細胞の損失や、病的なタンパク質の凝集の存在に反応する。しかし、多くの研究では、持続的な神経炎症がアルツハイマー病の疾患初期段階で認められることも示唆されている。疾患の進行に伴い、このような初期の炎症は、アミロイドβやNFTの生成を促進・悪化させ、さらに神経細胞の毒性と死をもたらす[24,42,43,44,45]。
神経新生は、神経幹細胞(神経幹細胞)の分裂、神経前駆細胞(海馬前駆細胞)の成熟、そして神経細胞への移動と成熟を経て起こる[46]。このプロセスは出生後に停止すると考えられてたが,予想に反して,哺乳類の脳の一生を通じて,成人期にも検出された [47].哺乳類の脳における成人期の神経発生は,海馬の脳室下帯(SVZ)と顆粒下帯(SGZ)という限られた領域でのみ検出されている[48].これらの領域は,神経幹細胞のニッチを維持している。脳室下のニューロン新生は、嗅覚と嗅覚神経回路に貢献している[49]。SGZでは、成熟した顆粒細胞が複数の発達段階を経る。
ステージ1(増殖)は、神経幹細胞s、すなわちグリア線維酸性タンパク質(GFAP)ネスチン、性決定領域Y-box 2(Sox2)を発現するタイプ1放射状グリア様細胞によって示される。
ステージ2(分化)では、ダブルコルチン(DCX)やポリシアル化神経細胞接着分子(PSA-NCAM)を発現する中間前駆細胞(タイプ2細胞)が代表的である。
その後、タイプ2細胞はステージ3(移動)の神経芽細胞(タイプ3細胞)またはDCX、PSA-NCAM、および未熟な神経細胞のマーカー(Tuj-1bまたはNeuroD)を発現する神経細胞の系統に属する細胞を生み出す。
ステージ4(軸索と樹状突起のターゲティング)とステージ5(シナプスの統合)では、成熟した神経細胞がカルレティニンを発現するほか、ステージ4ではNeuN、ステージ5ではカルビンディンを発現する[50]。
成人海馬の神経新生(AHN)は、学習や気分を含む様々な海馬依存の機能に重要な役割を果たしている[51,52,53,54]。パーキンソン病、アルツハイマー病、ハンチントン病など、さまざまな神経変性疾患において、成人のニューロン新生が変化したり、調節されなかったりすることがわかっている[54,55,56,57,58]。これまでの研究では,遺伝的,環境的,薬理学的な要因が神経新生の制御に役割を果たしていることが明らかになっている[46,55]。神経変性疾患でよく見られる全身性の炎症や神経性の炎症も、神経新生に関連した影響を及ぼしている[49,54,59]。アルツハイマー病における神経炎症と神経変性は相互に関連しているため、本レビューでは、神経新生に対する神経炎症の影響とアルツハイマー病との関連性に関する現在の証拠をまとめた。さらに、この問題に関する潜在的な治療戦略についても論じている。
2. 成人の神経形成とアルツハイマー病におけるその変化
2.1. ヒトの成人脳におけるニューロン新生
ヒトにおける成人の神経発生の概念は,1998年に癌患者の死後組織に含まれるブロモデオキシウリジン(BrdU)+細胞を調べることで初めて発表された[60]。2010年には,免疫組織化学の手法を用いて,ヒトの死後の脳組織サンプル中のニューロン新生マーカーを同定することで,ニューロン新生の証拠が報告された[61]。Sanaiらによる吻側遊走路(RMS)の発見[62]は、初歩的な嗅覚神経発生の継続を示唆するものであった。胎児期のヒトの脳ではRMSの存在は活発であるが、成人の脳では論争の的となっている[50]。また、成人の脳では海馬のニューロン新生が検出されるという矛盾した知見があり、議論が続いていた。ある報告では、若いニューロンは生後1年目にしか検出されなかったと述べている。そして,若いニューロンの加入はその後も続かず,成人の脳では極めて稀でさえあった[63]。しかし,ヒトにおける神経新生の存在を裏付ける証拠が増えてきている[47,64]。Moreno-Jimeńezetalらは、成人になっても新しいニューロンの加入が起こり、検出されることがあると指摘している。死後の経過時間が短く、固定方法も十分に確立されていることから、中年期から第9期までの神経学的に健康なヒト被験者の歯状回において、数千個の新しいニューロンまたは未熟なニューロンが検出された[47]。研究プロトコルと組織保存技術の向上により,ヒトの脳における新しいニューロンの加入が検出されるようになり,したがって,成人期における神経新生の存在が支持されるようになった。
神経新生の役割を完全に明らかにするには十分な証拠がないかもしれないが、ほとんどの研究者は、神経新生が成人期の脳の可塑性に寄与することを示唆している。成人期の神経新生の潜在的な機能としては、ストレスに対する回復力の向上[65]、パターン分離[66]や記憶・学習の形成[67,68]、定着した記憶や古い記憶の消失の促進[69]などが挙げられる。したがって、神経新生は海馬の可塑性において役割を果たしており[66,70]、歯状回にこれらの新しいニューロンが継続的に追加されることで、海馬の回路が再構築される。
多くの行動因子が成人のニューロン新生を制御していることを示す証拠が増えている。ランニングや豊かな環境刺激は、ニューロン新生を誘導することができる[71,72,73]。一方,慢性的なストレスは,神経幹細胞sの増殖を抑制することがある[74,75].また、ニューロン新生の量は、個人の年齢にも依存する。加齢に伴い、神経幹細胞sとその前駆細胞は、細胞増殖と神経細胞の産生を低下させた[76,77,78]。最近の報告では,43歳のドナーの脳では約42,000個の未熟なニューロン/mm2が検出されたが,90歳近くになった長老のドナーの脳では未熟なニューロンの数が約30%減少していた[79]。アルツハイマー病のような加齢に伴う神経変性疾患は,神経幹細胞の維持,増殖,生存,機能統合にさまざまな影響を及ぼす。神経変性疾患における神経新生の障害は、生きた神経細胞が持続的に失われ、再生能力が低下する原因となる。このことは、これらの神経変性疾患の病態生理学的メカニズムに影響を与える可能性がある。
2.2. アルツハイマー病における成体ニューロン新生の変化に関するエビデンス
成体ニューロン新生は、PSEN1,PSEN2,APPの単独変異や組み合わせによって、実験条件が大きく異なる様々なアルツハイマー病のトランスジェニック動物モデルで研究されていた。さらに、これらの研究では、BrdU処理後のレジメン、投与量、および分析の時点も異なっていた。しかし,異なる条件下で,歯状回のSVZとSGZの両方で,ニューロン新生の変化と機能不全が認められた[80].このような神経新生の低下は、特徴的な病変や神経細胞の喪失の発症に先立って起こる可能性がある[80]。PSEN1の単一変異を持つADトランスジェニックモデルでは、ほとんどの場合、成人のニューロン新生が損なわれている[81,82,83]。PDGFプロモーターを使用したAPPトランスジェニックの単一変異(PDAPP)も、特にアミロイド沈着後の老化状態において、海馬前駆細胞の増殖と生存に悪影響を与えた[84]。しかし、APPの2重または3重の変異(APP751swe、APPswe、Ind.)は、新しいニューロンの増殖と分化の増加をもたらした[85,86,87,88]。ニューロジェネシスの増加は、病的なタンパク質の蓄積に対する代償反応として説明できるだろう[85]。別の報告では、生後8か月のAPP/PS1マウスを用いて、静止期のネスチン陽性のアストロサイト様幹細胞が減少し、一過性に増幅する前駆細胞の数は維持されていた。しかし、どちらの細胞もアミロイド沈着時には形態的な異常を示した[89]。これらの知見は,アミロイド生成環境が海馬前駆細胞の機能障害を引き起こし,神経新生がアルツハイマー病の様々な動物モデルで異なって変化していることを示す証拠となる。トランスジェニックのADモデルに加えて、低用量および糖尿病誘発性のストレプトゾトシン(STZ)の脳室内(ICV)注射は、ヒトの散発性アルツハイマー病を模倣する新しい非トランスジェニック動物モデルであることが実証されている[90]。STZのICV注射は,アルツハイマー病を模倣する酸化ストレス,神経炎症,脳内コリン作動性障害,認知障害を誘発した。この非トランスジェニックAD動物モデルでは、STZのICV注射は、長期期間(3ヶ月)における未熟および成熟ニューロンの生成に悪影響を及ぼした[91]。
ヒトのアルツハイマー病脳でも、成体ニューロン新生における様々な知見が観察された。ある報告では、アルツハイマー病脳ではDCX、PSA-NCAM、TOアルツハイマー病-64/Ulip/CRMP(TUC-4)NeuroDの発現が健常者の脳よりも高いことが示されており、ヒトAD脳では神経新生が亢進していることが示唆された[85]。別の報告では,前兆のあるヒトAD脳のCA1-3領域でKi-67+細胞の数が増加しており,これはグリアや血管系に関連する変化の増加を反映しているが,歯状回では神経新生に変化は見られなかった[92]。しかし,矛盾したデータも存在した。ヒトAD脳の歯状回では、成熟した神経細胞マーカーである高分子量微小管関連タンパク質(MAP)アイソフォームMAP2aの発現が劇的に減少しており、海馬における神経細胞の成熟が失敗していることを示していた[93]。また、別の研究では、アルツハイマー病の海馬の歯状回において、DCX+細胞とSox2+細胞が、失明していない対照例と比較して減少していることが報告されている[94]。最近、Moreno-Jimeńezetalら[79]は、厳重に管理された条件と最新の組織処理法を用いて、52歳から97歳の45人のアルツハイマー病患者の神経新生を研究した。DCX+細胞の数は、アルツハイマー病の神経病理学的ステージが進むにつれて、顕著に減少した。さらに、年齢に関係なく、アルツハイマー病患者は神経学的に健康な対照群よりもDCX+細胞の数が一貫して少なかった。彼らは、NFTが脳の横脳領域に限定されているBraakステージIまたはIIであっても、アルツハイマー病の初期段階で神経新生が変化していることを発見した。これらのデータは、アルツハイマー病の神経新生が、生理的な加齢による神経新生の変化とは異なることを示す証拠となる。疾患の初期段階であっても、いくつかの独立した病態生理学的メカニズムがアルツハイマー病における神経新生の障害に寄与している可能性がある。
2.3. 成体ニューロン新生の既知の調節因子と成人ニューロン新生に対するアルツハイマー病の影響
複数の内在性および外在性因子が成人の神経新生を制御することが報告されている。内在的な調節因子としては,
- シグナル伝達経路(Wntシグナル,Notchシグナル,Sonic hedgehogシグナル(Shh),Eph: ephrinシグナル) [95,96,97,98,99,100,101,102]
- メチル-CPG結合ドメイン(MBD)-1 [103]
- メチル-CPG結合タンパク質2(MeCP2) [104]
- DNA損傷誘導タンパク質45β(Gadd45b) [105]
- ヒストンアセチル化(HDAC3,HDAC5,HDAC7) [106]
- マイクロRNA(Let-7b、miR-9,miR-34a、miR184) [106]などのエピジェネティックモジュレーター
- RE-1サイレンシング転写因子遺伝子(REST)[107]
- G-coupled protein receptor adenosine receptor A2A(ADORA2A)[108]などの遺伝子変異がある。
外因性モジュレーターとしては、
- VEFG
- BDNF
- IGF-1
- FGF-2
- IGF
- PDGF
などの代謝性成長因子があり、神経幹細胞sや海馬前駆細胞sの増殖、移動、細胞運命決定、成熟に寄与する役割を担っている[109,110,111,112,113]。
もう一つの外因性モジュレーターには、
- 血管系
- 血管新生
があり、SVZとSGZの血管床が神経発生をサポートし、内皮細胞が神経幹細胞sの増殖、分化、生存を促進することを示唆する報告もある[114,115,116]。
免疫系もまた、成体ニューロン新生の重要な調節因子である。様々な研究により、炎症が成人のニューロン新生に影響を与えることが明らかになっている。効果が増強されるか阻害されるかは、
- ミクログリア
- マクロファージ
- アストロサイト
をどのように活性化するかと、
- 炎症の持続時間
に依存する[117,118]。以前の研究では、安静時のミクログリアが、アポトーシスを起こした新生神経芽細胞を食作用によって除去することが示され、ミクログリアが歯状回における神経新生カスケードのホメオスタシス維持に貢献していることが示された[119]。また、別の試験管内試験の研究では、ミクログリアが神経細胞の分化に指導的な役割を果たしているというデータも得られている[120]。しかし、活性化されたミクログリア、特に古典的に活性化された炎症誘発性(M1)表現型は、炎症を促進し、神経芽細胞の生存率を低下させることで海馬のニューロン新生を抑制する役割を果たしている[121,122,123]。2003年に発表されたある画期的な動物実験では、リポ多糖(LPS)を末梢に注射すると、歯状回のDCX+細胞の数が減少し、ミクログリアの数が増加した。このLPSによる神経新生の減衰は,非ステロイド系抗炎症薬によって回復した[122]。さらに,成体マウスの末梢に28日間,長期にわたってLPSを注射すると,神経新生に長期的な悪影響を及ぼすことがわかった[121]。ミクログリアの影響や炎症の持続時間に加えて、IL-1α、IL-6,IL-10,CX3CL1,CXCL1,CXCL12などのいくつかのサイトカインやケモカインは、神経新生を積極的に制御する上で重要な役割を持っている[124,125,126,127,128,129,130]。
IL-1β、TNF-α、IL-18,IFN-γなど、活性化したミクログリアやアストロサイトから放出される炎症性サイトカインや、CCL11などの一部のケモカインは、神経幹細胞sの増殖や分化にマイナスの影響を与える[131,132,133,134]。また、これらのサイトカインやケモカインの曝露期間によっても、神経発生に対する影響が異なる。例えば,成体ラット海馬神経幹細胞sと共培養したIL-6を6~7日間急性暴露すると,神経幹細胞sの分化が有意に誘導されたが[124],脳をIL-6に長期暴露すると,成体のニューロン新生が阻害された[135].つまり、神経炎症は、脳のホメオスタシスを維持するための生理的反応としては有益であるが、慢性的なプロセスであることが判明すると、成体ニューロン新生に悪影響を及ぼす可能性があるのである。
アルツハイマー病が成体ニューロン新生にどのような影響を与えるかはまだ議論の余地があるが、アルツハイマー病の中心的な分子の中には、成体ニューロン新生を調節する役割を果たすものがいくつかあることが指摘されている。アスパルチルプロテアーゼであるγ-セクリースの触媒コアであるPSEN1は,EGFRとWnt/β-カテニンのシグナルを介して,成体脳における海馬前駆細胞の分化を制御している[136]。海馬の海馬前駆細胞においてPSEN1をダウンレギュレートすると,樹状突起の枝分かれや樹状突起の棘が減少するという形態的変化が生じ,行動テストにおけるパターン分離や新規性探索に障害が生じることがわかった[137]。APPの代謝物の一つである可溶性APP(sAPP)は、α-セクレターゼによるAPPの切断後の切断産物であり、海馬前駆細胞から分化した神経細胞の神経突起伸長を促進した[138]。sAPP-α、sAPP-β、アミロイドβペプチド、APP細胞内C末端ドメイン(AICD)などの他のAPP代謝物は、神経幹細胞sにおいて、増殖、神経新生、グリオジェネシス、または細胞死などの様々な機能を調節しているようである[139]。Scopaら[140]は、TG2576トランスジェニックマウスの前駆無症候期(生後1.5ヶ月)における神経発生障害を報告しており、神経幹細胞sの増殖が著しく低下し、神経幹細胞sが成熟した神経細胞に分化できなかった。この減少は、神経幹細胞sにおける細胞内のアミロイドβオリゴマーの形成と蓄積に依存しており、アミロイドβオリゴマーが神経新生に悪影響を及ぼすことを示していた。AICDは,Fe65およびTip60と転写活性複合体を形成し,PTCH1(ptch1,Shhリガンドが存在しない場合にShhシグナルを抑制する受容体)[141]やSox2(胚発生および細胞運命決定を制御する転写因子)[142]など,いくつかの遺伝子の発現を制御しており,先に述べたように,神経発生に関与している。Disintegrin-metalloproteinase (ADAM)は、生体内でαセクレターゼ活性を持ち、APPのα部位のタンパク質分解を行うことが知られている。ADAM10ノックアウトマウスでは、SGZにおける海馬前駆細胞の数が顕著に減少し、ADAM欠損の海馬組織ではNotch-1とその下流の標的遺伝子の活性化が損なわれていた[143]。ADAM17は、NotchおよびEGFRリガンドを含む多くの基質を処理し、神経細胞中間前駆細胞の多極出射および放射状移動に不可欠である[144]。ADAM 21は、成体のSVZ細胞に発現し、神経芽細胞の移動と分化に関連していることが示された[145]。タウタンパク質のリン酸化亢進は、神経発生に影響を与える。若齢のAPPswe/PS1ΔE9マウスでは、SVZにおいてニューロン新生が損なわれ、特に海馬前駆細胞の増殖と初期分化が損なわれてた[146]。この領域では、アミロイドβは比較的低かったが、タウの過リン酸化は生後2ヶ月という早い時期に有意に増加していた[80,146]。さらに,アルツハイマー病でタウをリン酸化する主要なキナーゼであるグリコーゲン合成酵素キナーゼ3β(GSK-3β)も,神経新生に影響を与えている[147,148]。GSK-3βの過剰活性化は、成熟した神経細胞のアポトーシスを増加させ、神経芽細胞の分化を損ない、歯状回内の増殖性クラスターの数を減少させる[149,150]。歯状回におけるGSK-3β活性の変化は、脳のこれらの神経原性ニッチにおけるタウリン酸化の変化の背景にあるかもしれない[150]。
2.4. 神経新生の変化とアルツハイマー病の病理との関連について
ニューロン新生の変化とアルツハイマー病複合体の病理との因果関係を疑問視する人がいるかもしれない。上述の証拠は、多くの病理学的代謝物(例えば、APP代謝物)またはアルツハイマー病プロセスに関与する重要な調節因子(例えば、PSEN1またはGSK-3β)が、神経新生に影響を与える役割を果たしていることを示していた。ニューロン新生の変化がアルツハイマー病の病態を媒介する可能性はあるのであろうか?Choiら[151]は、最近、5x家族性ADマウスを用いて、ニューロン新生とアルツハイマー病の病態との関係を研究している。彼らは、焦点照射、DNAアンキル化剤(テモゾロミド、TMZ)Wntのドミナントネガティブ型を発現させたレンチウイルス(LV-dnWnt)の3つのモデルを用いて神経新生をブロックした。その結果、5x家族性ADマウスでは、ごく初期の病期(6~8週間)に神経新生を高度にブロックしても、アミロイドβの沈着レベルやグリオシスの重症度には影響がなく、後期の病期(5カ月)になると成熟した神経細胞の死を開始し、認知機能障害を悪化させることがわかった。このような現象は、野生型マウスでは見られなかった。このことから、アルツハイマー病初期のニューロン新生の変化は、アミロイドβ病理には影響を与えないが、アルツハイマー病初期に海馬のニューロン新生が損なわれると、海馬の神経細胞の脆弱性が高まり、脳環境が悪化するアルツハイマー病後期に認知機能障害の悪化や神経細胞死の増加につながる可能性があると考えられた。
3. アルツハイマー病の神経炎症と成人の神経新生への影響
3.1. アルツハイマー病の神経炎症
神経炎症は、アミロイドβやNFTに次ぐ、アルツハイマー病脳における第3の神経病理学的特徴である。活性化したアストロサイトやミクログリアが、神経細胞やプラークの周囲に特徴的に見られる。また、いくつかの炎症性サイトカインや炎症マーカーの発現が増加していることも指摘されている[152,153]。このような炎症反応は、アミロイドβプラークやNFTの進行した蓄積に対する反応であると提案された。これらの炎症プロセスの慢性的または非制御的な活性化は、神経細胞の損傷または死を誘発することによって有害である[154]。以下のセクションでは、アルツハイマー病の神経炎症プロセスを構成する多くの細胞成分とメディエーターについて説明する。
ミクログリアは、中枢神経系の常駐免疫細胞である。ミクログリアは、末梢の単球と同様に、貪食、抗原提示を行い、免疫メディエーターを産生する。アルツハイマー病では、ミクログリアは、スカベンジャー受容体(SCARA-1,MARCO、SCARB-1,CD36,RAGE)Gタンパク質共役受容体(FPR2,CMKLR1)トール様受容体(TLR2,TLR4,TLR6,TLR9)CD47,α6β1インテグリン、TERM2などを介して、アミロイドβオリゴマーやフィブリルと相互作用する[18,155,156]。最初の認識後、ミクログリアは、NF-κB経路を介して活性化される[157]。アミロイドβの持続的な遭遇と、フィブリル状のアミロイドβの非効率的な貪食クリアランスのため[158]、ミクログリアは持続的に活性化される。アミロイドβによって慢性的に活性化されたミクログリアは、炎症性メディエーターを産生し、これが貪食能力の低下を招き、神経炎症を長引かせる[159]。アストロサイトは、神経細胞への栄養補給、老廃物の除去、血液脳関門の維持などに関与する多機能グリア細胞である。アルツハイマー病では、GFAPの増加によって示されるアストロサイトの活性化[160]、アストログリア症、およびアストロサイトの萎縮[161]が、アミロイドプラークの形成前であっても、疾患経過の早い段階で起こる可能性がある。また、アミロイドβは、おそらくNF-κB経路を介して、アストロサイトを活性化する[162]。活性化されたアストロサイトは、アミロイドβ自体を分解し、ApoEの脂質化によってミクログリアの貪食を増加させる[163]。しかし,活性化されたアストロサイトは,炎症性メディエーターも産生し,神経炎症プロセスに寄与する[164]。オリゴデンドロサイトは、中枢神経系におけるミエリンの供給源である。アルツハイマー病の神経炎症におけるオリゴデンドロサイトの役割は、まだほとんどわかっていない。試験管内試験の研究では、オリゴデンドロサイトは補体を合成する能力があるため、神経炎症のプロセスに寄与する可能性があるとされている[165]。
サイトカインは、アルツハイマー病の神経炎症において異なる役割を果たしている。TNF-αは、β-およびγ-セクレターゼを介してAPPを形成するアミロイドβ生成を増加させる[166]。IL-1は、APPの合成と分泌を増加させ、アミロイドβの生成を増加させる[166]。IL-1はまた、p38-MAPK経路を介してタウタンパク質のリン酸化を増加させる[167]。IL-6は、APPの発現を増加させ[168]、また、cdk5/p35経路を介してタウタンパク質のリン酸化を増加させる[169]。ケモカインは,中枢神経系におけるミクログリアの移動を制御する.アミロイドβは,ヒト単核細胞におけるCXCR8,CCL2,CCL3,CCL4の産生レベルを上昇させた[170].CX3CR1/CX3CL1は,ミクログリアの安静状態維持やシナプスの成熟と機能の維持に重要である[171]。動物モデルでは,CX3CR1/CX3CL1がアミロイド沈着や認知機能低下に関係していることが示された[172]。中枢神経系における補体系成分は、ミクログリア、アストロサイト、オリゴデンドロサイト、およびニューロンによって産生される[173]。アルツハイマー病では、補体系の役割はまだ明らかにされていない。C1qはアミロイドβに結合して代替補体経路を活性化し,アミロイドβの貪食作用や神経炎症の増強につながる[174]。動物実験では,C3の欠損は,アミロイドの貪食作用の低下とアミロイドβの沈着増加に関連していた[175].ゲノムワイド関連解析では、補体阻害因子であるアポリポタンパク質Jと補体受容体1との関連が明らかにされ、補体系がアルツハイマー病に寄与する可能性がさらに示された[176]。
3.2. アルツハイマー病における神経炎症と神経新生の変化に関する証拠
神経炎症は、アルツハイマー病の重要な特徴であるため、アルツハイマー病の神経新生に顕著な影響を与える可能性がある。アミロイドβタンパク質の沈着は、ミクログリアやアストロサイトを活性化し、この活性化は、IL-1β、IL-6,TNF-αなどの炎症性サイトカインの分泌を伴う。これらのサイトカインは、前述したように、神経幹細胞の増殖や生存に悪影響を及ぼすなど、神経新生に悪影響を及ぼすことが証明されている。ヒトAD脳における神経炎症の神経新生への影響に関する直接的な証拠はまだないが、AD動物モデルを用いたいくつかの研究では、神経炎症と神経新生の関連性が示されている。Bassaniらは,アルツハイマー病の動物モデルであるSTZをICV注射すると,グリアマーカーであるIba-1とGFAPの免疫反応が増加し,海馬のSVZと歯状回における細胞増殖と未熟な神経細胞(Ki-67+細胞とDCX+細胞)の数が減少したと報告している[177]。その後、同じグループは、このアルツハイマー病のラットモデルを用いて、STZがSVZと海馬背で急性かつ持続的な神経炎症反応を引き起こし、短期および長期の空間記憶を障害し、新生ニューロンの生存、分化、成熟を低下させることを実証し、神経炎症が神経新生に悪影響を及ぼすという仮説を支持した[178]。また、Misharaら[179]は、STZのICV注射により、注射後11日目および18日目のSVZおよび歯状回におけるBrdU+細胞、BrdU+/Nestin+細胞、DCX+細胞の数が有意に減少したことを報告しており、神経幹細胞sの増殖および新生神経細胞の移動に悪影響を及ぼしていることを示している。また、SVZと海馬では、GFAPやNF-κBなどの神経炎症マーカーの有意な増加が同時に認められた。この増加は、この種のAD動物モデルにおいて、神経新生の減少と神経炎症の亢進が関連していることを示唆している。Kiyotaら[127]は、トランスジェニックAD動物モデルにおいて、アデノ随伴ウイルス(AAV)を用いた遺伝子導入法により、APP/PS1トランスジェニックマウスの海馬で抗炎症性サイトカインであるIL-10の発現を増強させた。IL-10の発現により、プラーク周辺の反応性グリオシスやミクログリアの蓄積が抑制され、海馬のSGZにおけるBrdU+/NeuN+細胞やDCX+細胞の数が有意に増加することで神経新生が促進された。さらに試験管内試験のミクログリア/神経幹細胞s共培養系では、IL-10はミクログリアを刺激することで神経幹細胞sの増殖を促進したが、IL-10単独では促進しなかった。この結果は、神経新生の機能を制御する上でのミクログリアの役割を支持するものである。Ghosalら[180]は、アルツハイマー病の別の動物モデルにおいて、AICDトランスジェニックマウス(59残基の長いAICDフラグメントとFe65を共発現し、神経新生の過程に関与するいくつかの遺伝子を制御する転写活性複合体を形成している)では、SGZにおけるBrdU+細胞とDCX+細胞の数が生後3カ月から少なくとも12カ月まで減少し、海馬の神経新生が低下していることを報告した。また、12週齢のAICDトランスジェニックマウスでは、CD45+ミクログリアが劇的に増加し、炎症性サイトカインの発現が増加していた。非ステロイド性抗炎症薬(NSAIDs)であるイブプロフェンとナプロキセンを3週齢から9週間経口投与したところ、SGZにおけるBrdU+細胞とDCX+細胞の増加が認められたことから、ADトランスジェニックマウスにおいて、神経炎症の抑制とニューロン新生の促進の関連性が示された。Valeroら[181]は、野生型および3重トランスジェニックADモデルマウス(3xTg-AD)において、LPSの腹腔内注射によって誘発された単一の全身性炎症が成体のニューロン新生に及ぼす影響を調べた。生後4カ月の3xTg-ADマウスにLPSを単回全身に注射すると、LPS注射から7週間後に海馬依存性の長期空間記憶障害が悪化した。3xTg-ADマウスでは、DCX+細胞の数が野生型マウスに比べて有意に減少し、LPS注射によって3xTG-ADマウスのDCX+細胞の樹状突起にあるシナプスパンクタの数も減少した。このことは、ADトランスジェニック動物モデルの新生神経細胞におけるシナプス特殊化の形成にも、単一の全身性炎症イベントが長期的な障害をもたらすことを示している。
ヒトのアルツハイマー病脳における神経炎症とニューロン新生に関する直接的な証拠はないが、上述の知見は、ADモデルにおける神経炎症とニューロン新生の変化の関連性を支持するものである。この2つの現象の正確な因果関係を検証するためには、さらなる研究が必要であると考えられる。以上のような、ADモデル動物における神経炎症とニューロン新生の変化に関する知見を表1にまとめた。
表1 ADモデル動物におけるニューロン新生と炎症または抗炎症の変化に関するエビデンス
| 参照 | モデル | 観察期間 | 神経新生(場所) | 炎症/抗炎症 | 動作 |
|---|---|---|---|---|---|
| Bassani etal。(2017)[ 177 ] | ICV-STZ(ウィスターラット) | 4週間 |  Ki-67 +DCX + Ki-67 +DCX +(SVZ、DG) |
 GFAPIba-1 GFAPIba-1(SVZ、DG) |
短期空間記憶(物体位置テストとY迷路)と短期認識記憶(物体位置テスト)の障害 |
| Bassani etal。(2018)[ 178 ] | ICV-STZ(ウィスターラット) | 急性、7日 遅れ、30日 |
急性期のKi-67 +細胞(DGおよびSVZ)後期のBrdU + NeuN +細胞(DG)後期のDCX +細胞(DGおよびSVZ) |
GFAP伊庭-1 急性および後期で(CA1とCA3、DG) |
短期空間記憶と長期空間記憶の障害(物体位置テストとY迷路) |
| ミシュラ等。(2018)[ 179 ] | ICV-STZ(Sprague Dawleyラット) | 11日目と18日目 | 11日目と18日目のBrdU + Nestin +セルBrdU + NeuN +セルDCX +セル (DGおよびSVZ) |
18日目のGFAPNF-κB (DGおよびSVZ) |
学習と記憶の障害(モリス水迷路テスト)(18日目) |
| 清田ほか (2012)[ 127] | APP + PS1Tgマウス+ AAVを介したIL-10の発現 | 3週間 | DCX +セルBrdU + NeuN +セル (DGのSGZ) |
抗炎症のためのIL-10発現は、プラーク周辺の神経膠症とミクログリアの蓄積を減少させました | 該当なし |
| Ghosal etal。(2010)[ 180 ] | AICDTgマウス | 6週間 | BrdU + DCX +セル(DGのSGZ) |
経口NSAID(イブプロフェンおよびナプロキセン)による抗炎症 | 該当なし |
| Volero etal。(2014)[ 181 ] | 3xTG-AD | 7週間 | DCX +細胞とDCX +細胞の樹状突起のシナプス涙点 |
LPS注射による全身性炎症 | 空間記憶の障害(モリス水迷路テスト) |
下向き矢印:レベル低下、上向き矢印:レベル上昇、N/A:該当なし。ICVは脳室内、STZはストレプトゾトシン、DCXはダブルコルチン、BrdUはブロモデオキシウリジン、GFAPはグリア線維酸性タンパク質、Iba-1はイオン化カルシウム結合アダプター分子1,SVZは脳室下帯、DGは歯状回、Tgはトランスジェニック、NSAIDsは非ステロイド系抗炎症薬。
4. 成人の神経形成と炎症を標的とした治療法の可能性
成体ニューロン新生を正常なレベルに回復させることが,アルツハイマー病の進行を遅らせたり逆転させたりするための潜在的な治療法になるかどうかは不明である。Choiら[151]は,5x家族性ADマウスにおいて,遺伝学的手法(WNT3タンパク質であるLV-Wnt3を発現させたレンチウイルスをマウスに投与し,海馬前駆細胞の増殖を促進する)と薬理学的手法(海馬前駆細胞の生存率を高める化合物であるP7C3)を用いて,神経新生を促進した。その結果、ニューロン新生を誘導するだけでは、ADトランスジェニックマウスの認知機能を改善する効果は限定的であることがわかった。しかし、BDNFのレベルを薬理学的に高める方法(AMP-activated protein kinase agonist 5-aminoimidazole-4-carboxamide riboside)と、LV-Wnt3またはP7C3による神経新生の活性化を組み合わせることで、このADモデルの認知機能障害が改善された。
カロリー摂取量,食事の頻度,食感,食事内容などの食行動は,神経新生に影響を与えることが報告されている[182].動物モデルでは,カロリー制限[183],硬い食感の食事[184],多価不飽和脂肪酸(PUFA,オメガ3脂肪酸など)[185],ポリフェノール[186](ブルーベリー[187],クルクミン[188]),ビタミンD[189]などの栄養成分が神経新生を積極的に調節し,行動障害を回復させる可能性があることが示された。
カロリー制限は、PS1とPS2のダブルノックアウトマウスにおいて、神経発生関連遺伝子の発現を増加させ、炎症関連遺伝子の発現を減少させることで、アルツハイマー病の神経保護的役割を果たしている[190]。また、カロリー制限は、海馬で発現し、正常な学習と記憶に重要なSIRT1の発現を高める。SIRT1は、細胞の生存と神経細胞の分化の促進に関与している[191,192]。
神経新生の促進に加えて、一部の栄養成分は抗炎症作用を発揮する。PUFAは、アルツハイマー病患者の血液単核白血球の炎症プロセスに関わる遺伝子制御に影響を与えることで、抗炎症作用を示した[193]。クルクミン[194]、ケルセチン[195]、およびカフェイン/コーヒー[196,197]は、抗炎症作用を有することが示されており、したがって、神経新生とアルツハイマー病に対する保護の両方に有益である可能性がある[198]。
食事による介入に加えて、腸内細菌叢の組成と機能を調整する有益な細菌であるプロバイオティクスは、AD動物モデルにおいて神経炎症を軽減し、認知障害を回復することが示されている[199]。また、プロバイオティクスの補給は、海馬におけるBDNF mRNAを増加させる[200]ことから、神経新生を積極的に制御する可能性がある。しかし、プロバイオティクスの補給がアルツハイマー病患者の抗炎症や認知機能に及ぼす影響は、これまであまり期待できなかった[201,202]。今後の研究では、食事の介入によるproneurogenic(神経新生促進)効果や抗炎症効果、腸内細菌叢の調節やADモデルにおける治療効果などの分野に焦点を当てるべきである[198,203]。
成体ニューロン新生のもう一つのポジティブな制御因子である環境エンリッチメントは、アミロイドβレベルやアミロイド沈着を減少させるだけでなく、APPswe/PS1DE9マウスやAPP23マウスにおける成体ニューロン新生の障害や認知機能障害を回復させることが実証されている[88,204]。しかし,家族性アルツハイマー病関連のPS1変異を保有するトランスジェニックマウスにおける成体ニューロン新生に対する環境エンリッチメントの効果については,否定的な結果が報告されている[83]。
過去数十年の間に、運動が海馬の神経新生に繰り返しかつ確実に有益な効果を及ぼすことが証明された。Praagらは、自発的なランニングが歯状回における新しいニューロンの増殖と生存を増加させ、長期的なホイールランニングが海馬のニューロン新生と空間学習能力の両方を増加させることを報告した[205,206]。このようなランニングによる脳形成促進効果と記憶課題のパフォーマンス向上は、野生型のネズミモデルでは異なる実験パラダイムで実証されており[207,208]、アルツハイマー病のネズミモデルでも実証されている[151,209,210]。ヒトを対象とした研究では、運動と神経新生の促進との関係を示す直接的な証拠はない。しかし、健常者[211]やパーキンソン病患者[212]やアルツハイマー病患者[213]などの疾患者において、異なる運動トレーニングプロトコルにより、運動が記憶課題を改善することが示されている。
最近では、自発的な運動が脳内の抗炎症作用を持つ可能性を示唆する報告もある[214,215]。また、動物実験では、運動が、神経炎症によって引き起こされる神経新生の障害レベルを回復させることが証明されている[216]。しかし、アルツハイマー病の動物モデルにおける神経炎症や神経新生に対する運動の影響に関する直接的な証拠はまだない。Parachikovaら[217]は、Tg2576老齢マウスに自発的な運動をさせると、空間学習能力が向上するだけでなく、神経新生促進効果があると報告されているCXCL1とCXCL12の海馬での発現が増加することを報告した[129,130]。Nicholら[218,219]も、運動によって老化したTg2576マウスの海馬におけるTNF-αとIL-1βの発現が減少したことを報告している。また、ケージ内ランニングホイールを3週間使用することで、トランスジェニックマウスの可溶性アミロイドβと可溶性線維性アミロイドβのレベルが低下した。しかし、運動には神経炎症を調節する効果がないという矛盾した結果が示されている[219]。いくつかの異なるデータが報告されているが、運動は、アルツハイマー病関連の神経炎症や神経新生の障害に対して可能性があると考えられる。今後の方向性としては、ADモデルにおける運動の神経新生促進効果と抗炎症効果、そしてその治療効果に注目する必要がある。
神経炎症とそれに関連するサイトカインやケモカインが成人の神経新生に悪影響を及ぼすことから、炎症細胞や炎症分子を標的にして神経新生を操作する治療法がいくつか報告されている。Ghosalの報告では,AICDトランスジェニックマウスのニューロン新生障害が,ブプロフェンとナプロキセンの経口投与によって予防された[180]。しかし、アルツハイマー病におけるNSAIDs[220]やグルココルチコイド・ステロイド[221]に焦点を当てた他の研究では、現在のところあまり期待できない。アルツハイマー病の疾患修飾療法として、神経新生の変化に対する抗炎症薬について確かな結論を出すには、今後の研究が必要である。
図1は、上記のセクションで挙げた潜在的な治療アプローチをまとめたものである。
図1 アルツハイマー病における成人の神経形成と炎症を標的とした治療アプローチの可能性
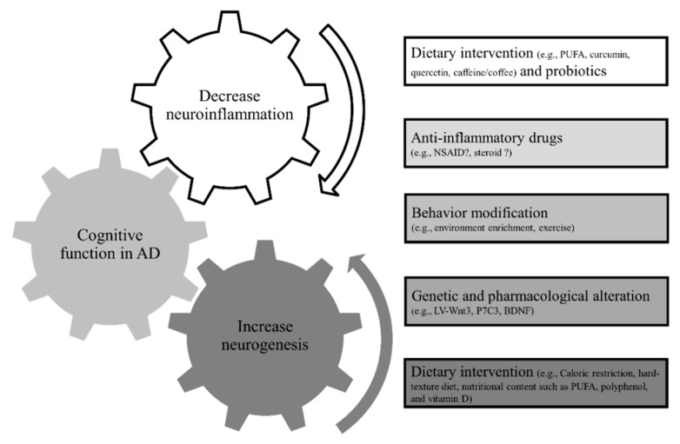
PUFAは多価不飽和脂肪酸、NSAIDは非ステロイド系抗炎症薬、LV-Wnt3はWNT3タンパク質を発現させたレンチウイルス。
5. おわりに
加齢したヒトの脳には神経新生が存在し、そのレベルは健常対照者に比べてヒトAD脳では確かに低下している。動物モデルでは、ニューロン新生の初期の障害が、アルツハイマー病の厳しい脳環境における神経細胞の脆弱性を増大させることが支持された。炎症は、アルツハイマー病の神経新生の変化に影響を与える中心的な病因の1つである。アルツハイマー病の神経変性プロセスを改善するためには、ニューロン新生の変化と神経炎症の相互作用をより深く理解する必要がある。