Contents
- Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease
- 要旨
- 序論
- アルツハイマー病の進行期
- 古典的なアルツハイマー病病変
- アルツハイマー病の進行中のコリノトロフィー性基底前脳欠損
- アルツハイマー病の進行中の神経成長因子
- NGFとアルツハイマー病の進行
- アルツハイマー病の進行中のNGF受容体の発現
- アルツハイマー病の進行に伴う皮質proNGFレベル
- アルツハイマー病の進行中のNGF代謝経路
- アルツハイマー病の進行中に海馬のproNGFと下流経路
- アルツハイマー病進行中のコリノ栄養基底前脳ニューロン遺伝子発現
- アルツハイマー病の進行中のコリノトロフィー神経細胞TAUの病理学
- アルツハイマー病の進行中のコリノトロピックエピジェネティックな変化
- アルツハイマー病の進行のためのコリノトロフィーバイオマーカー
- アルツハイマー病の治療戦略としてのNGF療法
- アルツハイマー病治療のための低分子ニューロトロフィン化合物
- おわりに
Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease
www.ncbi.nlm.nih.gov/pmc/articles/PMC6613497/
要旨
今回のレビューでは、アルツハイマー病の進行中における神経成長因子(NGF)とその認知受容体の病態をまとめている。トランスクリプトおよびタンパク質の両方のデータから、コリノトロフィー性神経細胞の機能不全は、TrkAが媒介する生存シグナルとNGF前駆体(proNGF)/p75NTRが媒介するプロアポトーシスシグナルの間の不均衡に関連しており、NGFの代謝の変化に関連している可能性があることが示唆されている。
データは、メイナート基底核(NBM)内に位置するコリノトロフィー性神経細胞サブグループ内のタウ病理の進化に関連した変性の時空間的パターンを示している。これらの変性イベントにもかかわらず、コリノ栄養系は、疾患の前段階および後期の間、細胞の回復力および/または可塑性を発揮することができる。
ニューロトロフィン機能不全に加えて、研究は、エピジェネティックに制御されたタンパク質の変化は、アルツハイマー病の進行中にコリノトロフィー nbM ニューロン内で発生することを示し、転写産物の発現の変化の下にあるかもしれないメカニズムを示唆している。
proNGFの脳脊髄液レベルの上昇がMCIの発症とアルツハイマー病への移行を示すという知見は、このプロニューロトロフィンが潜在的な疾患バイオマーカーであることを示唆している。
NGF機能障害を治療するための新規治療法としては、NGF遺伝子治療や、アルツハイマー病治療のためのプロニューロトロフィン受容体TrkAに対する低分子アゴニストやパンニューロトロフィンp75NTR死レセプターに対するアンタゴニストの開発が挙げられる。
キーワード
アルツハイマー、神経成長因子、軽度認知障害、エピジェネティクス、ニューロトロフィン受容体、バイオマーカー
序論
アルツハイマー病は、記憶力の低下、日常生活動作の障害、神経精神症状、その他の行動障害を臨床的に特徴とする、進行性で致死的な加齢に伴う脳疾患である。有病率報告によると、世界では約1,800万人がアルツハイマー病を発症しており、米国では580万人以上がアルツハイマー病を発症しているとされている(アルツハイマー病協会 2019)。症例の割合は、年齢が約5年上昇するごとに2倍に増加し、60歳の個人の1%、85歳の個人の約30%が本疾患を呈することが示されている。大幅な介入がなければ、米国では今世紀半ば(2019)には症状のある人が1,380万人に増加すると予測されている。アルツハイマー病を持つ人々の介護にかかる費用は、年間1000億米ドルを超えることになる(Christensen, 2007; Wimo, 2007; Wimo et al 2017)。これらの憂慮すべき統計は、アルツハイマー病の初期または前駆段階で使用するための効果的な治療法を開発することの圧倒的な重要性を強調している。
アルツハイマー病の進行期
アルツハイマー病には広範囲の前臨床段階があり、早ければ臨床症状の発症の15~20年前にも発症する可能性がある(Sperling et al 2014)(図1)。軽度認知障害(MCI)は、現在では進行性アルツハイマー病と同義語となっているが、正常な脳の老化とフランクな認知症の中間段階であり、認知障害のない人(NCI)と比較して神経原線維のもつれ(NFT)やアミロイドβペプチド(アミロイドβ)病変が増加する(Guillozet et al 2003;Markesbery et al 2006;Markesbery 2010)。臨床的概念としてのMCIは、毎年認知状態の検査を受けた高齢者コホートの縦断的調査から、軽度の痴呆患者を評価していた記憶クリニックから発展した。そのような調査では、以前の軽度の認知機能低下を持つ多くの人がNINDS/薬物有害反応DA アルツハイマー病診断に必要な2つの認知領域の障害を示すことができなかったことが示された(Mckhann et al 1984)。これらの人々は、健忘性障害と定義され、健忘性MCI(aMCI)と呼ばれた(Petersen et al 1999)。記憶クリニックでは、aMCIがアルツハイマー病につながるMCIのより一般的なタイプであることが示唆されていたが、この実体は、この臨床分類のマイナーではあるが重要な側面を構成していることは明らかであった。全体的に、MCIの臨床診断は、単一領域のaMCIとして分類される孤立した記憶問題を持つ患者を含む異質な患者集団を包含し、一方で、記憶欠損と他の認知領域の障害を持つ患者はマルチドメインMCI(mdMCI)として分類される(Petersen, 2004; Johnson er al)。 無脳性MCIの症例は、アルツハイマー病を発症するリスクが高い(Petersen, 2004; Johnson et al 2010)。NCIまたはMCIと臨床的に診断された高齢者のかなりの割合が、アルツハイマー病に見られるのと同様のアミロイドプラークおよびNFT病理を示し、これらの病変だけで認知症の発症を早めるという病理学的に基づく概念に挑戦している(Mufson and Kordower, 1999; Price and Morris, 1999; Markesbery, 2010; Mufson et al 2016a,b)。
図1 アルツハイマー病の臨床的および病理学的進行の軌跡を描いた模式図
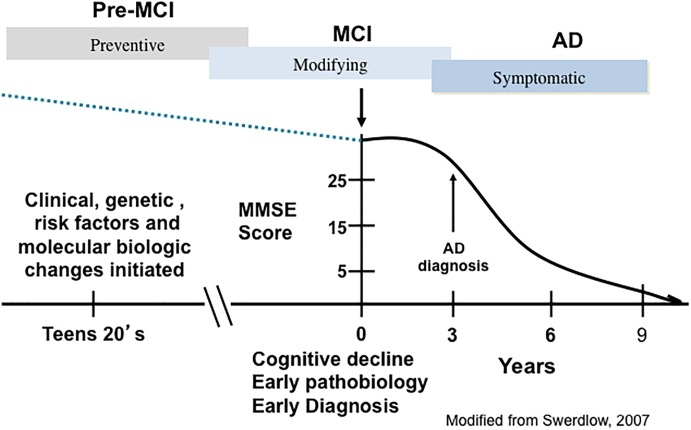
PreMCI、pre-mild cognitive impairment、MCI、mild cognitive impairment。
古典的なアルツハイマー病病変
20世紀になると、神経病理学的な調査により、認知症の高齢者から死後に採取された脳に異常な細胞外プラークが存在することが報告された(Blocq and Marinesco, 1892)。ドイツの精神科医アロイス・アルツハイマー博士は、被害妄想と記憶障害を呈し、診断から5年後に死亡した51歳の女性オーギュスト・デターを治療した。剖検では、彼女の脳は萎縮しており、神経細胞が失われており、NFTと老人性プラーク(SP)を含んでいた。エミール・クレーペリン博士は、この3つの特徴を “アルツハイマー病 “と呼んだ。細胞外マトリックスに存在するSPは、βサイトAPP切断酵素1(BACE1)と膜内γセクレターゼ複合体による連続的な切断によって、より大きな膜貫通アミロイドβ前駆体タンパク質(APP)から産生されるアミロイドβの不溶性フィブリルから構成される(Shoji er al)。 NFTsは、高リン酸化タウタンパク質の細胞内凝集体からなる(Trojanowski et al 1993; Yoshiyama et al 2013)。SPとNFTはアルツハイマー病の定義的な病理学的特徴と考えられているが、アルツハイマー博士は、「…プラークは老人性認知症の原因ではなく、中枢神経系の老人性侵襲の付随的な特徴に過ぎない」(アルツハイマー、1911)と書いている。この声明にもかかわらず、アルツハイマー病の研究分野は、「アミロイドカスケード仮説」(ハーディとSelkoe 2002)によって推進されており、治療戦略は、プラーク沈着を除去するための抗アミロイド薬の開発を中心に展開し続けている。しかし、事実上すべての抗アミロイド臨床試験は、主要なエンドポイントである認知の改善を達成していない(Hampel et al 2015,2018)。このような薬剤の有効性の欠如は、アミロイドがアルツハイマー病の初期のバイオマーカーである可能性はあるが、臨床的な衰えには必要ないという概念を支持するものである。より可能性が高いのは、アルツハイマー病は多面的な多遺伝子疾患であり、そのうちアミロイドはこの疾患の病態形成のパートナーであるということである。アミロイド仮説に反して、大規模な文献の体は、認知の損失は、大規模な皮質切断症候群につながる複数の神経伝達経路の選択的脆弱性を伴うことを示唆している。
アルツハイマー病の進行中のコリノトロフィー性基底前脳欠損
大脳皮質と海馬全体を支配するコリン作動性基底前脳(脳血流)ニューロン(図2A-D)の変性は、30年以上前から、アルツハイマー病治療のターゲットとなる可能性がある疾患の初期に影響を受ける重要な神経伝達物質系として研究されてきた(Hampel et al 2018)。アルツハイマー病の「コリン作動性仮説」(Bartus et al 1982)は、アセチルコリンエステラーゼ阻害剤(AChEI)がアルツハイマー病患者において有意な症状効果を有することを発見して勢いを増した(Summers et al 1986)。1986)これは、アセチルコリンエステラーゼ阻害剤(AChEI)のより大きなファミリーの開発につながった(Hampel et al 2018)(図2E-G)が、アルツハイマー病治療のためのFDA承認薬の数少ないクラスの1つであり続けている(Johannsen 2006;Mangialasche et al 2010;Hampel et al 2018)。例えば、AChEIであるドネペジルは、アルツハイマー病の前駆症状におけるメイナート基底核(nbM)および内側中隔/対角線帯内の前脳基底部の萎縮を減少させることが示されており、症状の緩和だけでなく、構造的な効果も実証されている(Cavedo et al 2017)。
図2
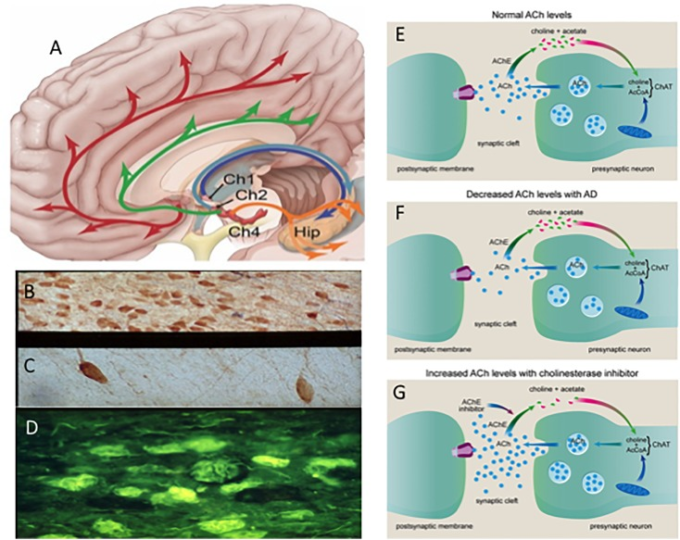
A)コリノ栄養皮質と海馬投影系の模式図。老化したコントロール
(B)アルツハイマー病(C)の減少とチオフラビン(黄色)のもつれベアリングp75NTRニューロン(紺色)(D)のコリノトロフィーニューロンの画像を示すフォトマイクログラフは、アルツハイマー病(D)。正常(E)とアルツハイマー病(F)とコリン作動性シナプス(G)でアセチルコリンエステラーゼ阻害剤の効果との間のアセチルコリン(ACh)の変化を示す漫画。Ch1,内側中隔コリン作動性細胞群;Ch2,対角線帯コリン作動性細胞群Ch4,基底核コリン作動性細胞群、Hip、海馬。赤と緑の矢印は両側のCh4の突起を示し、紺色と水色の矢印は海馬への中隔/対角帯の突起を示す。
最近、若年性認知症の分野で脳血流投影系への関心が復活している(Douchamps and Mathis, 2017; Hampel et al 2018)。イメージング研究は、認知機能の低下に関連するシグナリングにおける前脳基底回路の制御異常の重要性の証拠を提供している(Ballinger et al 2016)実行機能、エピソード記憶に重要なデフォルトモードネットワーク(DMN)の制御異常(Nair et al 2018)認知機能の伝播の重要性の証拠を提供している(Nair et al 2018)。2018)および疾患の進化の早い段階での皮質萎縮の伝播(Schmitz and Nathan Spreng, 2016)およびアルツハイマー病の前症候性バイオマーカーとして(Ho et al 2008; Grothe et al 2012)。アルツハイマー病発症時のDMNや他の皮質部位へのコリン作動性皮質投射ニューロンへの関心の高まりは、この投射系の機能不全の根底にあるメカニズム的要因を理解することの重要な必要性を強調している。
アルツハイマー病の進行中の神経成長因子
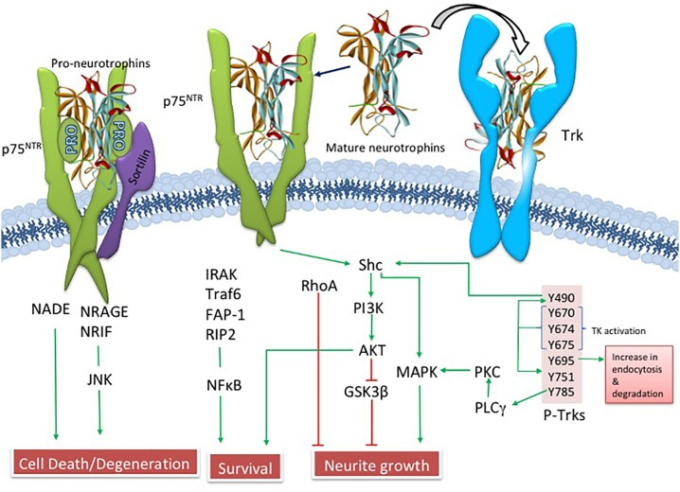
ラモン・イ・カハールが脳細胞が「特別な食物」を必要とすることを示唆して以来、研究者は神経細胞の生存に役割を果たす成長刺激剤を探してきた(Henry, 1998)。Levi-MontalciniとCohen (Levi-Montalcini, 2000)は、栄養物質である神経成長因子(NGF)が培養ニューロンの選択的生存の基盤となっていることを明らかにしたことでノーベル賞を受賞した。彼らは、神経細胞の生存に関する神経栄養学的仮説を提案した最初の研究者である。NGFは第1染色体上に見られる単一の遺伝子の産物であり、27キロダルトン(kDa)と35キロダルトン(kDa)のproNGF前駆体タンパク質(Francke et al 1983; Edwards et al 1988)を産生し、これらはタンパク質分解的に切断されて成熟した生物学的に活性なペプチドとなる(Edwards et al 1988; Lee et al 2001)。成熟したNGFではなく、ProNGFは、ヒトの脳で見られる主要な形態である(Fahnoshostork et al 2001)。NGFは、トロポミオシン関連キナーゼA(TrkA)受容体とp75パンニューロトロフィン受容体(p75NTR)に結合する(Ibanez, 2002; Chao, 2003; Kaplan and Miller, 2004)。NGFがTrkAに結合すると、Aktを活性化することにより下流の生存経路が活性化される(Ulrich et al 1998)一方、proNGFおよびp75NTRは、その共受容体であるsortilin(Nykjaer et al 2004)およびニューロトロフィン受容体ホモログ2(NRH2)(Murray et al 2004)とともに、細胞のアポトーシスに関連するc-Jun N末端プロテインキナーゼ(JNK)を活性化する(Nykjaer et al 2005)(図3)。臨床試験は、NGFがアルツハイマー病における脳血流の生存および神経可塑性を増強する治療的可能性を有することを示している(Tuszynski et al 1990年;TuszynskiおよびBlesch 2004;Tuszynski et al 2015)。
図3 NGFに関連する上流・下流の経路を示す図
NGFとアルツハイマー病の進行
長年にわたり、アルツハイマー病におけるNGFの損失により、コリノ栄養基底前脳皮質および海馬突起ニューロンが退化するという仮説が立てられていたが(Hefti and Mash, 1989; Tuszynski et al 1990; Smith er al)。 1991; Murase er al)。 1993; Jette er al)。 1994)減少(Hellweg er al)。 1998)または増加(Crutcher er al)。 1993; Scott er al)。 1995; Fahnestock er al)。 1996; Narisawa-Saito er al)。 1996; Hellweg er al)。 1998; Hock er al 2000)のNGFレベルが、重度アルツハイマー病患者の組織を用いて報告されている。しかし、MCI、軽度アルツハイマー病、重度アルツハイマー病の臨床診断を受けて剖検に来た人では、5つの皮質領域(上前頭頂皮質、上側頭頂皮質、中側頭頂皮質、前帯状皮質、下頭頂皮質)と海馬でNGFレベルが保存されていた(図4)(Mufson et al 2003)。対照的に、他の人は、体積減少が体重または体積あたりのNGF濃度の増加をもたらす可能性があるアルツハイマー病末期における皮質および海馬のNGF mRNAおよびタンパク質の増加を報告している(Crutcher et al 1993; Jette et al 1994; Scott et al 1995)。1994; Scott et al 1995; Fahnestock et al 1996; Narisawa-Saito et al 1996; Hellweg et al 1998; Hock et al 2000)またはNgfからコード化されたNGFタンパク質への翻訳が損なわれるか、またはアルツハイマー病症例間で発現レベルが異なる可能性がある。我々は、早期から後期のアルツハイマー病症例に及ぶコホートにおいて、広範囲のNGF活性を報告し、NGFの最高レベルと最低レベルのいくつかは、末期のアルツハイマー病症例で見られた(Scott et al 1995年)与えられたコホート内で、NGFレベルは、疾患発症時の年齢または疾患過程の違いによって異なる影響を受け得ることを示唆している。本研究では、アセチルコリン合成の律速酵素である皮質コリンアセチルトランスフェラーゼ(ChAT)活性とNGFのレベルとの間には、また、MCIと軽度アルツハイマー病におけるChAT-(Gilmor et al 1999)TrkA-(Mufson et al 2000)またはp75NTR-(Mufson et al 2002b)を含むニューロンの数の減少との間には、何の関係もなかった。さらに、アポリポ蛋白質ε4遺伝子型とNGFレベルとの間の相関の欠如は興味深いものであり、それは、アポEε3およびε4対立遺伝子が、アルツハイマー病末期におけるコリン作動性マーカーのより大きな減少と関連していることが報告されているからである(Poirier et al 1995)。Rush Religious Orders Study(RROS)から調査した重度のアルツハイマー病症例の90%以上が少なくとも1つのApoE ε4対立遺伝子を含んでいたが、NGFレベルは評価された臨床グループ間で差はなかった(Mufson et al 2003)。これらの観察から、ApoE ε4遺伝子型はNGFの代謝に直接影響を与えないことが示唆された。
図4 アルツハイマー病の進行中に大脳皮質と海馬でのNGFタンパク質レベルの安定性を示すボックスプロット
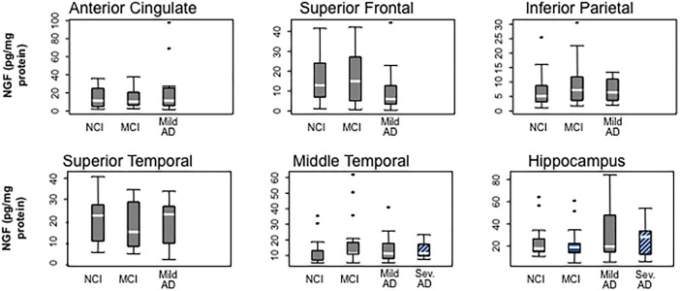
(Mufson et al 2003)から転載。
アルツハイマー病の進行中のNGF受容体の発現
コリン作動性基底部前脳ニューロンの機能は、NGFのそのコグナートレセプターTrkA、およびそのパン-ニューロトロフィンp75NTRへの結合に依存していることは、NGFとそのレセプターの調節障害がアルツハイマー病におけるコリン作動性ニューロンの機能不全の根底にあることを示唆する支持を貸す。TrkA受容体とp75NTRは、脳血流ニューロンのペリカリア内で産生され、NGF産生のスティーツである大脳皮質と海馬に前方に輸送される(Schwab et al 1979)。脳血流ニューロン内では、成熟したNGFはTrkA受容体に結合し、NGFによって誘導されるニューロンの生存を制御するシグナル伝達経路を活性化する(Kaplan and Miller, 2004)。しかしながら、p75NTRは、NGF/TrkA結合の正のモジュレーターであり(Kaplan and Miller, 2004)、アポトーシスまたは細胞死経路の刺激を含むいくつかの文脈依存的な機能を示す(Bamji et al 1998; Yoon et al 1998; Frade, 2000; Friedman, 2000; Lee et al 2001; Roux and Barker, 2002)。これに関して、p75NTRの特定の下流効果は、様々な受容体シャペロンとの相互作用に依存する(Mamidipudi and Wooten, 2002; Nykjaer et al 2004; Teng and Hempstead, 2004)。
NGF受容体を含む脳血流ニューロンの数がアルツハイマー病の進行の初期に変化するかどうかを評価するために、我々は臨床的にNCI、MCI、またはアルツハイマー病に分類されたRROS被験者の組織を調べた(Gilmor et al 1999)。興味深いことに、ChATを含むニューロンの数はMCIおよび軽度のアルツハイマー病で安定していたが、TrkAおよびp75NTR免疫反応性ニューロンはNCIと比較して有意に減少しており、MCIにおける直接的なニューロン変性ではなく、脳血流機能をサポートする受容体の表現型ダウンレギュレーションを示唆している(Gilmor et al 1999)(図5)。あからさまなコリン作動性細胞の喪失ではなく、萎縮によるコリン作動性マーカーの表現型の喪失は、フィムブリア-小角切断および興奮毒性による中隔コリン作動性ニューロン軸切りの動物モデル研究と一致している(Hefti, 1986; Williams et al 1986; GinsbergおよびMartin, 1998)。アルツハイマー病では、皮質TrkAレベルの低下はMini-Mental State Exam (MMSE)で評価される認知パフォーマンスの低下と正の相関を示した(Counts et al 2004)ことから、nbMと皮質NGF受容体タンパク質レベルの低下はアルツハイマー病の早期発症を示す可能性があることが示唆されている。
図5 アルツハイマー病の進行中のコリン作動性、TrkAおよびp75NTR免疫反応性ニューロンの差動的な減少を示すヒストグラム
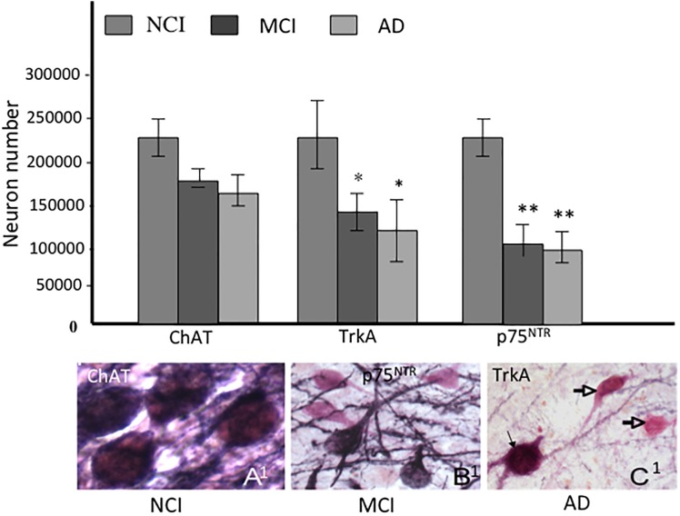
A1-C1。認知障害なし(NCI)軽度認知障害(MCI)とアルツハイマー病の間でChAT(ピンク)陽性ニューロンと比較してp75NTR(紺色)の表現型ダウンレギュレーションを示す二重標識された基底核ニューロンのフォトマイクログラフ。MCIとアルツハイマー病におけるChAT陽性細胞のp75NTR免疫反応性染色の消失に注意してほしい(開いた矢印)。黒矢印はアルツハイマー病(C1)の二重染色ニューロンを示し、**および**はそれぞれp<0.05および0.01を示す。
アルツハイマー病の進行に伴う皮質proNGFレベル
NGF前駆体タンパク質であるproNGFの変化は、アルツハイマー病の進行中の認知機能障害に寄与する前頭皮質、後帯状体、前帯状体、上側頭皮質(Perez et al 2015)および海馬(Mufson et al 2012b)を含む皮質DMNの構成要素の臨床病理学的調査において広範な研究を受けている(Sperling et al 2014)。アルツハイマー病皮質から単離されたProNGFは、受容体のγ-セクレターゼの脱落に依存するメカニズムを介してp75NTRと相互作用することにより、神経細胞培養物中でアポトーシスを誘導するが、対照脳から単離されたProNGFはアポトーシスを活性化しない(Pedraza et al 2005)。MCIまたは軽度アルツハイマー病の臨床診断を受けて死亡した患者の側頭頂皮質では、NCIの患者と比較してproNGFレベルが増加している(Peng et al 2004)。対照的に、前頭頂前野のproNGFレベルはアルツハイマー病末期まで安定しており(Perez et al 2015)前頭前野(Fahnestock et al 2001,2004;Podlesniy et al 2006)および海馬(Al-Shawi et al 2008;Mufson et al 2012b)と同様であり、これらはすべて、罹患脳におけるproNGFの変化を示唆している。ウェスタンブロッティングでは、臨床群をまたいでTrkA、p75NTR、および前楔前野(Perez et al 2015年)および海馬(Mufson et al 2010)内の共受容体であるソルチリンのレベルに変化は見られなかった。ProNGFは、より高い親和性でp75NTRに結合し、これは、アポトーシスを誘導するためにソルチリンの存在下で増強される(Lee et al 2001; Nykjaer et al 2004; Pedraza et al 2005; Al-Shawi et al 2008)。NGF受容体の恒常的な調節は、proNGFのTrkAへの結合(成熟NGFよりも親和性が低いとはいえ)と相まって、脳血流ニューロンの機能に関与する下流経路の活性化(Fahnestock et al 2001,2004)およびTrkA受容体への親和性が低い結合を介した神経栄養活性の誘導をもたらす(Fahnestock et al 2001,2004)。アルツハイマー病発症時にp75NTRレベルが楔前部および他の皮質領域で安定したままであるという知見(Counts et al 2004;Mufson et al 2012b)は、アルツハイマー病におけるp75NTR皮質ニューロンのde novo出現の実証に関連しているかもしれない(Mufson and Kordower、1992)。
p75NTRが媒介するproNGFシグナルのプロアポトーシス効果は、p75NTRとVps10pドメイントラフィッキングタンパク質であるソルチリンとの相互作用に依存しており、proNGF活性化細胞死を媒介するためにp75NTRとの細胞表面共受容体として作用する。この受容体ファミリーは、アルツハイマー病への潜在的な関与のために重要性を増している(Nyborg et al 2006)。ソルチリンは、proNGF処置に続くp75NTR誘導アポトーシスを活性化する(Nykjaer et al 2004年)細胞死における役割を示唆する(MamidipudiおよびWooten 2002;RouxおよびBarker 2002)。この結合イベントをブロックすることは、proNGFのp75NTRへの結合およびその後の細胞変性を妨げる(BronfmanおよびFainzilber 2004;KaplanおよびMiller 2004;Nykjaer et al 2004;Teng et al 2005)。プロニューロトロフィンに応答するp75NTRシグナル伝達は、結合した共受容体の同一性と有効性に依存している可能性がある。特筆すべきことに、ソルチリンの皮質レベルは、アルツハイマー病の進行中にp75NTRと同様に、安定したままである。おそらく、脳血流ニューロンにおけるプロ生存またはプロアポトーシスのシグナル伝達は、TrkA、p75NTR、選択された共受容体の利用可能性、およびアルツハイマー病の初期段階での異なる環境内でのproNGFの生理的役割の変化に依存している。これらの要因のバランスをシフトすることは、アルツハイマー病の進行中に脳血流ニューロン内でproNGF結合が活性化する反応を変化させる可能性がある(図6)。これらの相互作用を定義することが、認知症の神経栄養戦略の開発の鍵となる(Bruno et al 2004; Longo et al 2007)。proNGFが生体内試験でp75NTRと結合し、アポトーシスを誘導する場合(Lee et al 2001;Nykjaer et al 2004)proNGFのp75NTRへの結合をブロックする薬剤の開発が極めて重要になる。一方、proNGFがTrkAと結合して細胞の生存を誘導する場合(Fahnestock et al 2004年)この相互作用を強化する薬剤の開発は、アルツハイマー病における神経保護を提供する可能性がある。
図6 アルツハイマー病の進行中のNGF活性に関連した細胞生存から細胞死へのシフトを示す漫画
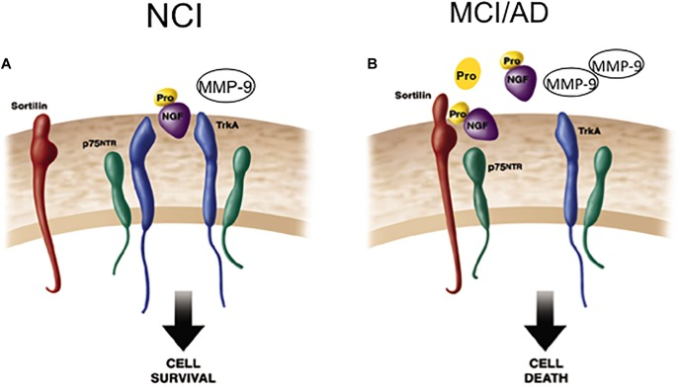
A)ProNGF/TrkA複合体は細胞生存機構を活性化し、健康な高齢者の脳では細胞表面にp75NTRが共発現することで促進される。また、ProNGFはproNGFの共受容体であるSortilinと同時に結合してシグナル伝達複合体を形成している。
B)アルツハイマー病では、減少したTrkAに直面して上昇した皮質proNGFは、p75NTR /ソルチリン複合体へのproNGFの結合を高める。ソルチリンは、p75NTR媒介プロアポトーシスシグナルを支配する分子スイッチとして機能するので、増加したproNGFは、減少したTrkAに直面して細胞死を誘発する。Mufson et al 2012a)より改変。
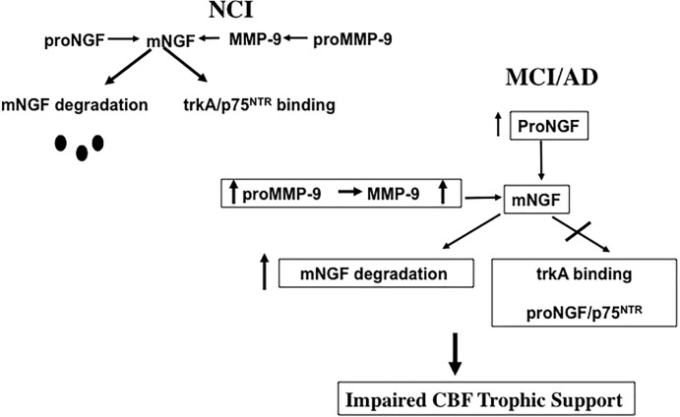
アルツハイマー病の進行中のNGF代謝経路
NGF/proNGF複合体の成熟・分解を制御する代謝経路の欠損は、脳血流機能障害に重要な役割を果たしている可能性がある。最近、プラスミノーゲン、組織プラスミノーゲンアクチベーター(tPA)ニューロセルピン、マトリックスメタロプロテアーゼ9(MMP-9)マトリックスメタロプロテアーゼ1の組織阻害剤(TIMP-1)の協調的な活性によって、proNGFを成熟したmNGFに変換し、細胞外空間でmNGFを分解するプロテアーゼカスケードがアルツハイマー病では欠損していることが示された(Bruno and Cuello, 2006)。これに関連して、MCIおよびアルツハイマー病では前頭前野および頭頂皮質でMMP-9タンパク質レベルおよび活性のアップレギュレーションが報告されており、これは認知パフォーマンスと逆相関しており(Bruno et al 2007)NGF/proNGF活性の変化を駆動している可能性がある(図7)。我々は、proMMP-9およびMMP-9の増加が、NCIからMCIへの移行の間に脳血流ニューロンのNGFサポートを損なうことを示唆している(図7)。興味深いことに、皮質proNGFの同様の増加(Iulita et al 2014)およびTrkA陽性脳血流ニューロンの減少(Sendera et al 2000)は、ダウン症候群(DS)で報告されており、これらの障害におけるNGF神経栄養障害の重複を示唆している。アルツハイマー病とDSの両方の症例は、皮質SPとNFTの病理を示し、中年期までに認知症を発症する(Mann and Esiri, 1989)ことから、これらの神経疾患はさらに関連している。血液、尿、脳脊髄液(脳脊髄液)中のメタロプロテアーゼ(MMPs)のレベルがアルツハイマー病の潜在的なバイオマーカーとして機能する可能性が示唆されている(Zucker et al 1999;Lorenzl et al 2003,2008)。アルツハイマー病の初期に安定した代謝NGF/proNGF複合体の調節を可能にする、MMPが前膜で調節障害されているかどうかを調べることは興味深いことである。注目すべきことに、proNGFレベルは、可溶性アミロイドβ1-42またはフィブリラーアミロイドβ[3H]ピッツバーグ化合物B(PiB)結合の増加とは関連しておらず、代わりに、アルツハイマー病におけるコンパクト/コア6-CN-PiB-陽性プラークと関連していた(Perez et al 2015)その可溶性形態ではなく、アミロイドβのフィブリラー堆積物が、前膜で見出されたproNGFのアップレギュレーションにおいて役割を果たしている可能性があることを示唆している。神経変性は、アミロイドβオリゴマーが野生型マウスではなくp75NTR欠損マウスの脳に送達されたときに、アミロイドβ1-40がp75NTRに結合すること(Knowles et al 2009)および脳血流ペリカリアの結果である(Simmons et al 2014)。興味深いことに、脳血流変性は、アルツハイマー病のマウスモデルにおいて、p75NTRの神経トロフィン結合ドメインの枯渇に続いて停止した(Knowles et al 2009)。これらの知見は、p75NTRシグナルがアミロイドβ誘導性変性に関与していることを示唆しており、アルツハイマー病治療のターゲットとして示唆している。
図7 proMMP-9/MMP-9の増加が、アルツハイマー病の進行中にコリノトロフィー性前脳基底ニューロンをサポートするNGFの能力に与える影響を示す図
アルツハイマー病の進行中に海馬のproNGFと下流経路
海馬は中側頭葉記憶回路の一部である。それは、アルツハイマー病の初期段階では広範なNFTを発症するが、アミロイド病理は少ない(Hyman et al 1984, 1990; Braak and Braak, 1991; Arriagada et al 1992)と、内側中隔および垂直辺縁の対角帯ニューロンから主要なコリン作動性入力を受ける(Mesulam et al 1983)。これらの海馬中隔コリン作動性突起ニューロンもまた、その生存のためにNGFおよびその認知受容体に依存しており、アルツハイマー病では退化するので、海馬NGF/proNGFシステムの変化を決定するために研究が行われた。ウェスタンブロット分析は、大脳新皮質とは対照的に、アルツハイマー病では海馬proNGFレベルの有意な増加を明らかにしたが、MCIではなかった(Mufson et al 2012b)。興味あるのは、NCIおよびアルツハイマー病と比較してMCIの海馬におけるTrkAタンパク質レベルの有意な減少の観察であり、MCIからアルツハイマー病への移行の間にNCIレベルに戻る(Mufson et al 2012b)(図8)。アルツハイマー病初期の安定したproNGFに直面してTrkAの減少は、proNGF/p75NTR/sortilin/NRH2結合を強化し、最終的には海馬におけるプロサバイバルからプロアポトーシスシグナル伝達へのバランスをシフトさせる可能性がある(図8)。海馬のTrkAレベルのアップレギュレーションは、病気の進行を遅らせるためのヒトの脳の回復力(Mufson et al 2016a,b)のもう一つの例である。
図8 アルツハイマー病の進行中の海馬神経栄養タンパク質レベルの変化を示す模式図
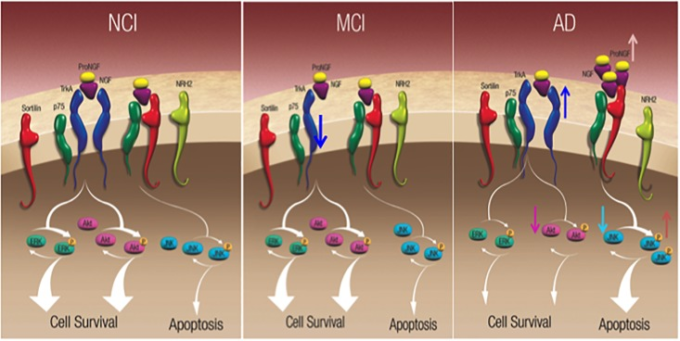
(Mufson et al 2015)から転載。
神経成長因子およびproNGFは、下流の多数の細胞生存およびアポトーシスシグナル伝達経路をそれぞれ活性化する(図8)。TrkAリン酸化によって活性化される細胞生存タンパク質Erkは、遺伝子転写に関与する核内エフェクターを活性化する(Zhu et al 2001)。総Erk、phospho-Erk、およびphospho-Erk/Erk比の前帯(Perez et al 2015)および海馬(Mufson et al 2012b)レベルは、NCI、MCI、およびアルツハイマー病の間で変化していない。対照的に、ストレス活性化キナーゼphospho-JNK、およびphospho-JNKとJNKの比は、アルツハイマー病前膜(図8)(Perez et al 2015)および海馬(Mufson et al 2012b)で有意に増加したが、総JNKレベルはアルツハイマー病海馬と同様に安定していた(Mufson et al 2012b)。アポトーシス酵素の活性化に関与するJNKシグナル伝達経路の構成要素であるBcl2は、アルツハイマー病ではなく、MCIでは前膜でアップレギュレートされた(Perez et al 2015)。特に興味深いのは、ホスホ-JNKおよびAT8タウ陽性NFTおよびニューロピルスレッド(NT)の密度が、アルツハイマー病の発症時に正の関係にあるという知見であり、JNKの活性化がSer202/Thr205(AT8部位)でのタウのリン酸化を媒介するという観察を支持するものであった(Goedert et al 1997;Reynolds et al 1997)。したがって、JNKのプロアポトーシスシグナルの活性化は、アルツハイマー病におけるエピソード性記憶障害に役割を果たしている可能性がある。
アルツハイマー病進行中のコリノ栄養基底前脳ニューロン遺伝子発現
認知症の発症時に、比較的余裕のあるニューロンと比較して「選択的に脆弱な」脳血流ニューロンの遺伝的特徴を明らかにすることは、認知症の発症に介入する治療法をターゲットとした転写支援型のドラッグデザインを開発する上で極めて重要である。転写駆動型の治療アプローチは、認知症の発症と発症に重要な役割を果たすNGF依存性脳血流システムを含む脳のコネク トームを保存する可能性が高く、特に疾患プロセスの初期段階ではその可能性が高いと考えられる(Mufson et al 2012a)。p75NTRによって同定された脳血流ニューロンの遺伝子発現プロファイルを比較した研究(Mufson et al 1989)では、選択されたシナプス関連マーカー(例えば、シナプトフィシンおよびシナプトタグミン1のダウンレギュレーション)、タンパク質ホスファターゼ/キナーゼ(例えば、タンパク質ホスファターゼ/キナーゼのダウンレギュレーション)の調節障害が示されている。プロテインホスファターゼ1および2サブユニットのダウンレギュレーションおよびサイクリン依存性キナーゼ5のアップレギュレーションなど)およびエンドソーム-リソソソームマーカー(例えば、リソソソームマーカーであるカテプシンD、rab4,rab5,およびrab7のアップレギュレーション)が、年齢をマッチさせたNCI被験者と比較してMCIおよびアルツハイマー病において有意なダウンレギュレーションを示した(Ginsberg et al 2006a,b,2010; Counts et al 2011)。さらに、NCIと比較して、MCIおよびアルツハイマー病のnbMからマイクロ吸引した単一脳血流ニューロンでは、TrkA、TrkBおよびTrkCの有意なダウンレギュレーションが認められた(Ginsberg et al 2006b)(図9A)(Ginsberg et al 2000,2010)。これらの知見は、NCIと比較してアルツハイマー病の最大の減少を伴うMCIの中間的な減少を明らかにした。さらに、発現配列タグ付きcDNA(EST)[すなわち、Trk受容体の細胞外ドメイン(ECD)とチロシンキナーゼ(TK)ドメインの両方を標的としたEST]がダウンレギュレーションされていた。Trk発現の「ステップダウン」障害は、一部では、アルツハイマー病の臨床症状に関連した脳血流ニューロンの衰退の原因となっている可能性がある。この概念を支持するのは、TrkAのダウンレギュレーションが、MMSE、複合グローバル認知スコア(GCS)Episodic、Semantic、Working Memory、Perceptual Speed、およびVisuospatialドメイン、ならびにBraak NFTステージおよび前脳基底部および海馬内のニューライトプラーク(NP)負荷を含む認知機能低下のいくつかの尺度と関連していたという知見である(Ginsberg et al 2006b 2019)。したがって、Trk遺伝子の発現欠損は、MCIからフランクアルツハイマー病への移行のための分子マーカーを提供する可能性がある(Ginsberg et al 2019)。対照的に、p75NTR転写物レベルは、臨床診断群にわたって脳血流ニューロンにおいて安定であった(Ginsberg et al 2006b)が、これは、MCIおよびアルツハイマー病におけるp75NTR免疫陽性のnbMペリカリアがNCIと比較して有意に減少したことと比較して、興味をそそられる所見であった(Gilmor et al 1999)。脳血流ニューロンにおけるp75NTRタンパク質と転写産物の発現の不一致は、疾患発症時のmRNA転写とタンパク質翻訳の間の断絶を示唆している。死後のヒトの脳組織における脳血流単一集団の観察は、アルツハイマー病の進行中に認知性NGF受容体ファミリーの変化に相対的な選択性があること、および神経栄養障害が認知機能低下および神経病理学の初期段階に先行または発生することを示唆している。
図9 アルツハイマー病の進行中に単一のコリノ栄養ニューロンのプロファイリングから得られた知見
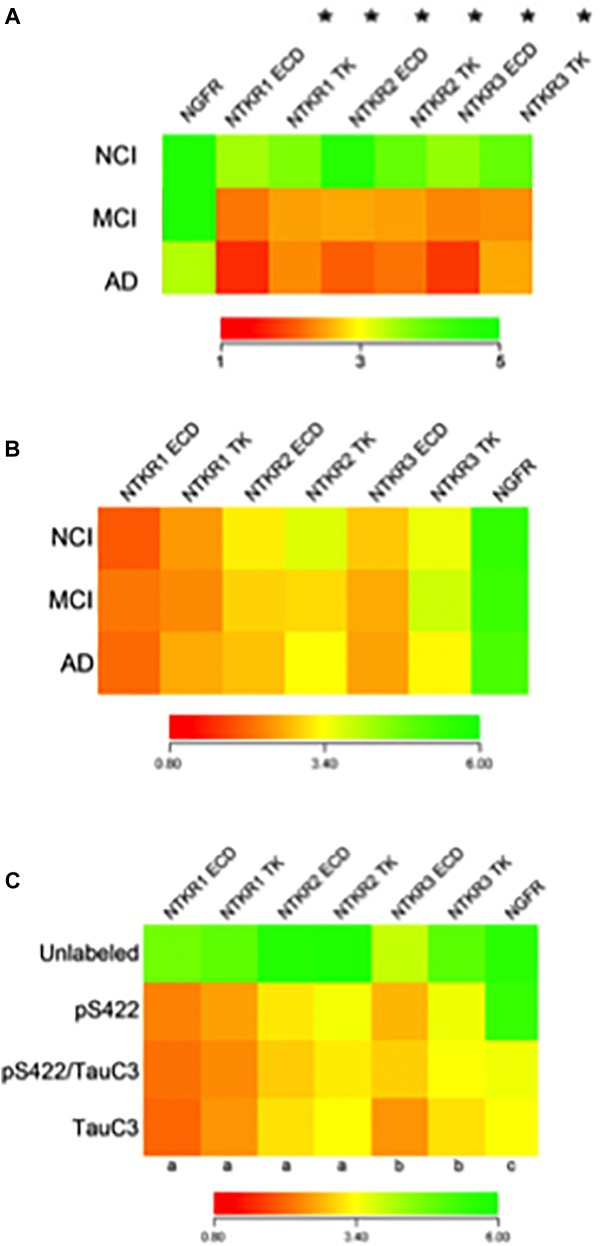
A)Ntrk1(TrkA)Ntrk2(TrkB)およびNtrk3(TrkC){細胞外ドメイン(ECD)とチロシンキナーゼドメイン(TK)の両方}のダウンレギュレーションを示すヒートマップ (アスタリスク)ではなく、認知症の進行中にNgfr(p75NTR)が発現することを明らかにした。MCIにおけるTrk遺伝子発現はアルツハイマー病までの中間的なものであり、NCI→MCI→アルツハイマー病へのステップダウン効果を示唆している。B)pS422+ nbMニューロンにおける選択されたニューロトロフィン転写物の相対的な発現プロファイルのヒートマップ。Trk受容体やNgfrにはNCIと比較してMCIやアルツハイマー病で差は見られなかった。(C) Trk受容体のダウンレギュレーションは、非標識対照ニューロンと比較してpS422+ nbMニューロンで認められた。NgfrのダウンレギュレーションはネオエピトープTauC3と関連していた。キー。(a)非標識> pS422,p < 0.001; (b)非標識> pS422,p < 0.01; (c) pS422 > pS422+/TauC3+、p < 0.01。(A) Ginsberg et al 2006b)の許可を得て翻案。(B,C) Tiernan et al 2018a)からの許可を得て、Adapted。
アルツハイマー病の進行中のコリノトロフィー神経細胞TAUの病理学
疾患進行中の神経栄養因子の機能障害と同時に、脳血流ニューロンは、MCIおよびアルツハイマー病においてNTと同様にグロボースNFTとして現れる細胞内タウ介在物を発現する(Sassin et al 2000; Mesulam et al 2004; Wu et al 2005; Vana et al 2011)。ヒトの脳には、3つのタンデムリピートを持つ3つのタウアイソフォーム(3Rtau; Mapt1,Mapt3,Mapt5)と4つのタンデムリピートを持つ3つのタウアイソフォーム(4Rtau; Mapt2,Mapt4,Mapt6)が存在する。脳血流ニューロンのカスタムデザインされたマイクロアレイ評価では、アルツハイマー病、MCIおよびNCI被験者間で6つのタウ転写物のいずれにも変化は見られなかった(Ginsberg et al 2006a)。しかし、すべてのタウ転写産物について、4Rtauレベルに対する3Rtau発現の減少を伴う3Rtau/4Rtau比の有意なシフトが観察された。タウ転写産物のデータは、MCIおよびアルツハイマー病における脳血流ニューロン内の3Rtauおよび4Rtauの遺伝子ドーズ量の変動を示唆しているが、これは正常な加齢には見られなかった(Ginsberg et al 2006a)。
単一ニューロン発現プロファイリング調査は、RROSによって提供されたNCI、MCIおよびアルツハイマー病症例から得られた無標識ニューロンと比較して、プレタングルマーカーpS422+、後期カスパーゼ切断タウマーカーTauC3+またはpS422/TauC3+で標識された個々のnbMニューロンにおいて、ニューロトロフィン受容体をコードする転写物のレベルがどの程度変化するかに取り組んできた(Tiernan et al 2018a)。定量分析は、臨床段階間またはタウニューロン表現型間のトランスクリプトシグナル強度を比較した。各臨床ステージからマイクロアスピレートしたpS422+ nbMニューロンにおけるトランスクリプト発現を比較したところ、統計的差異は認められなかった(図9B)。しかしながら、臨床診断とは無関係に解析した場合、ニューロトロフィン受容体発現を調節する主要遺伝子の発現レベルは、nbMニューロンにおいて、非標識からpS422+、pS422+/TauC3+からTauC3+への表現型の移行によって分類されるように変化した(図9C)。非標識と比較して、pS422+ nbMニューロンは、ニューロトロフィン受容体TrkAの細胞内TKおよび細胞外ECDドメインをコードする6つのmRNAの有意なダウンレギュレーションを示した(Ntrk1 TK、50%ダウンレギュレーション。Ntrk1 ECD、53%)TrkB(Ntrk2 TK、45%;Ntrk2 ECD、42%)およびTrkC(Ntrk3 TK、38%;Ntrk3 ECD、35%)をコードするmRNAを発見した(図9C)。さらに、これらの同じ転写物が、初期のプレタングルタウ抗体タウオリゴメリックコンプレックス1(TOC1)を含むニューロンにおいて有意にダウンレギュレートされていることを発見した(カウンツ、未発表の観察)これは、神経栄養障害がフランクNFT形成の前に起こるという仮説を支持するものである。
対照的に、パン-ニューロトロフィン受容体p75NTR(Ngfr)をコードするmRNAの転写レベルは、TauC3が出現するまで低下しなかった。この発現データは、TauC3とp75NTRがタンパク質レベルで脳血流内に共局在していないことを実証した我々の立体構造学的知見を補完するものである(Mufson et al 2002b; Vana et al 2011)。先行研究では、MCIおよびアルツハイマー病におけるnbMニューロンにおいて、NCIと比較してTrk受容体の発現レベルがダウンレギュレートされていることが報告されている(Ginsberg et al 2006b)。一方、NcI、MCIおよびアルツハイマー病症例からのpS422+ニューロンではNtrk発現に差が見られなかったのに対し、nbM非標識ニューロンからpS422+ニューロンへの表現型移行により、Ntrk転写物がダウンレギュレートされることが明らかになった(図9C)。
我々は以前に、TOC1(Patterson et al 2011;Ward et al 2013)およびp75NTRに対する抗体イムノ染色を行って、最も神経毒性の高いタウの種である可能性が高いプレタングルタウオリゴマー集合体を定量化した(Berger et al 2007;前田 et al 2007;前田 et al 2007)。2007; Maeda er al 2007; Kopeikina er al 2011; Lasagna-Reeves er al 2012; Sahara er al 2013)は、アルツハイマー病の進行中に脳血流ニューロン内のタウオリゴマー集合体を定量した(Tiernan er al 2018b)。ここで、p75NTR+/TOC1+ nbMニューロンの数は、NCIからMCI、アルツハイマー病まで進行して増加したのに対し、単一のTOC1+ nbMニューロンはNCIとMCIでは低かったが、アルツハイマー病では増加した。Braakスコアが低い(Stages I-II)NCI症例のp75NTR+、p75NTR+/TOC1+、およびTOC1+ nbMニューロンのサブ解析を行ったところ、Braakスコアが高い(Stages III-V)NCI高病理症例と比較して、NCI高病理症例ではp75NTR+/TOC1+デュアル免疫標識ニューロンの数が有意に増加していた。p75NTR+ nbMニューロンの減少は、GCSおよびMMSEパフォーマンステストのスコアの低下と関連していた。TOC1は主にNCIではpS422と共局在していたが、MCIおよびアルツハイマー病への移行は、TOC1+/pS422+からトリプルラベルのTOC1+/pS422+/MN423+ニューロンへのシフトによって特徴づけられ、疾患進行中のnbMのコリノトロフィーニューロンにおけるエピトープ出現の特異的で直線的な順序を示唆していた。この配列は、異常なリン酸化により、タウタンパク質がさらなるリン酸化およびコンフォメーションイベント(Luna-Munoz et al 2007;Bertrand et al 2010)に向けてプライミングされ、オリゴマー化が促進されることを示唆している(Iqbal et al 2013)。プレフィブリル性タウの病理学がnbMニューロン内の分子および細胞の変化と関連しているという知見(Tiernan et al 2016,2018b)を考えると、プレフィブリル性オリゴマー性タウの出現は、おそらく細胞の喪失に先行していると思われる。神経栄養機能障害のドライバーとしてタウ病理を暗示する証拠は、タウトランスジェニックマウスおよびタウトランスフェクトされたニューロン型細胞において見られ、この細胞は、栄養物質である脳由来神経栄養因子(BDNF)のダウンレギュレーションを示す(Rosa et al 2016)。一方、NGFは、タウのターンオーバー(重度アルツハイマー病ot et al 1996)およびリン酸化、開裂およびユビキチン化を含む翻訳後修飾を調節する(Nuydens et al 1997;SheltonおよびJohnson et al 2001;Babu et al 2005;A軽度アルツハイマー病oro et al 2011)ことから、神経栄養異常が脳血流ニューロン内でタウ病理を開始することが示唆される(Canu et al 2017)。さらに、タウの微小管結合能の低下および/またはタウの体液性蓄積は、軸索変性および関連するNGF/TrkAシグナル伝達障害に寄与する可能性がある(Mufson et al 2002a; Schindowski et al 2008)。さらに、proNGFは、GSK3βの増強された活性を介して試験管内試験でタウの高リン酸化を誘導する(Shen et al 2018)。これらの集合的な結果に基づいて、将来的には、異常なタウ代謝とコリノトロフィー性のnbMニューロンにおける神経栄養シグナル伝達の障害との間の潜在的な相互作用の分析が行われる(Capsoni et al 2000)。
アルツハイマー病の進行中のコリノトロピックエピジェネティックな変化
ヒストンアセチル化と脱アセチル化は、ChATの制御を介して脳血流ニューロンの機能にも関与している(相澤・山室 2010;相澤 et al 2012;Bekdash 2016)ことから、アルツハイマー病におけるニューロンの選択的脆弱性におけるエピジェネティクスの潜在的な役割が示唆されている。神経細胞の核および細胞質内に位置する脱アセチラーゼ活性を有するエピジェネティック酵素であるヒストン脱アセチラーゼ(HDACs)が、アルツハイマー病の発症に役割を果たしているという証拠が増えている(Ding et al 2008;Guan et al 2009;Xu et al 2011;Cook et al 2012;Graff et al 2012)。いくつかのHDACは、小胞体ストレス(HDAC4)(Shen er al 2007)ミトコンドリア輸送(HDAC6)(Chen et al 2010)タウ過リン酸化(HDAC6)(Ding et al 2008)およびアミロイドβおよびタウ蓄積(SIRT1)(Julien et al 2009;LallaおよびDonmez 2013)。しかし、エピジェネティックな調節異常がコリノトロフィー性のnbMニューロンで起こるかどうかは、アルツハイマー病ではまだ未調査のままである。
HDACのうち、HDAC2は、クロマチン可塑性調節を介して認知に関与する転写物の調節に役割を果たすことから、広範な研究が行われてきた(Dawson and Kouzarides, 2012; Graff and Tsai, 2013; Volmar and Claes, 2015)。これに関して、臨床病理学的調査により、アルツハイマー病発症時にnbMニューロン内のHDAC2-免疫反応性(ir)核の変化が明らかになった(Mahady et al 2019)。この研究では、通常は丸みを帯びたHDAC2-ir nbM核が、MCI、軽度アルツハイマー病(軽度アルツハイマー病)および重度アルツハイマー病(重度アルツハイマー病)において、ソーマ内に卵形、平坦化、および偏心的に位置するように出現したことが明らかになった(図10A-H)。重度アルツハイマー病では、HDAC2の核強度が他の臨床グループと比較して有意に減少していた(図10D,H)。さらに、HDAC2-irの核内強度は、NCI群およびMCI群と比較して軽度アルツハイマー病で有意に減少した(図10I)。この研究はさらに、HDAC2 nbM核強度がNCIおよびMCIにおいて有意差がないことを示した。重度アルツハイマー病核面積は、NCI、MCIおよび軽度アルツハイマー病で観察されたものよりも有意に小さかった(図10J)。軽度アルツハイマー病では、HDAC2-ir核はNCIと比較して有意に小さい面積を示した。HDAC2-ir核の強度は、ワーキングメモリおよびGCSと相関することが判明した(Mahady et al 2019)。nbM p75NTR免疫反応性ニューロンの数の減少は、病期を越えて減少し、HDAC2核免疫反応性の減少と関連していた(Mahady et al 2019)。同様に、HDAC2核免疫反応性の低下は、AT8プレタングルを有するnbMニューロンの数の増加と逆に関連していた。HDAC2核の強度を定量したところ、軽度アルツハイマー病および重度アルツハイマー病では、NCIおよびMCIと比較して、非タングルベアリングのp75NTR陽性ニューロンが有意に減少していることが明らかになった。p75NTR、プレタングルメーカーAT8,または後期タウエピトープであるTauC3のためにトリプルラベルされたNbMニューロンは、アルツハイマー病において、各病期の非タングルベアリングp75NTRニューロンと比較して、HDAC2免疫反応性のさらに大きな減少を示した(Mahady et al 2019)。グループ内分析では、HDAC2-irは、各臨床グループにおいて非タングルベアリングコリン作動性ペリカリーアにおいて最も高かったことが示された(Mahady et al 2019)。興味深いことに、MCIおよび軽度アルツハイマー病において、HDAC2核免疫反応は、HDAC2/AT8/チオフラビン-SまたはHDAC2/TauC3/チオフラビン-Sニューロンにおいてさらに低下した。これらの知見は、HDAC2発現の低下はフィブリル性タウ病理の発症前に起こり、この変化はアルツハイマー病の進行中にnbMニューロンのリン酸化されたタウエピトープやコンフォーマルなタウエピトープによって悪化していることを示唆している。ChAT mRNA発現およびタンパク質レベルは、ChaT転写物のコアプロモーター領域のハイパーアセチル化によってエピジェネティックに調節されるが(Aizawa er al 2012)HDAC2核レベルの低下はMCIのコリン作動性nbMニューロン内で見られたが、ChAT nbMタンパク質レベルの有意な低下はアルツハイマー病のみで見られた(Mahady er al)。 これらの現象は、HDAC2核タンパク質のダウンレギュレーションが疾患過程の初期にはニューロンのChAT活性を変化させないことを示唆している。大脳皮質コリン作動性nbM投射部位におけるHDACの免疫組織化学的分析は、地域的に異なる所見を実証した。例えば、HDAC1とHDAC2はアルツハイマー病内耳皮質で減少しているが(Mastroeni et al 2010)HDAC2は、HDAC1またはHDAC3ではなく、HDAC2は、コントロール被験者のHDAC2と比較して、アルツハイマー病の海馬と内耳皮質のニューロンで増加している(Graff et al 2012)。前頭皮質のウエスタンブロットでは、HDAC2レベルが安定したままであったのに対し、NCIと比較してMCIおよび軽度アルツハイマー病ではHDAC1,HDAC3,HDAC4,HDAC6が有意に増加していることが明らかになった(Mahady et al 2018)。これらの知見は、アルツハイマー病で影響を受ける脳領域全体で、差動的なエピジェネティック調節が起こることを示唆している。
図10 HDAC2単体(A-D)とHDAC2とクレシルバイオレット二重染色(E-H)された基底核ニューロンの光顕微鏡写真
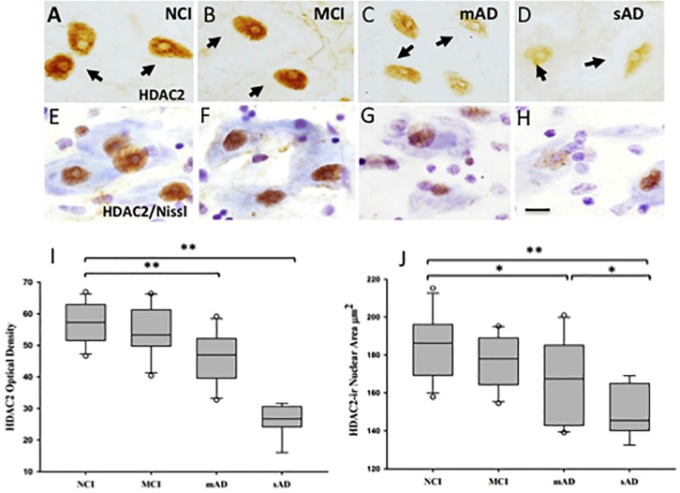
HDAC2陽性の核は丸みを帯びた形状を失い、MCI群、軽度アルツハイマー病群、および重度アルツハイマー病群ではソーマ周辺部に変位していた。I)ボックスプロットは、HDAC2-irがNCIおよびMCI群と比較して軽度アルツハイマー病群で有意に減少したことを示す(p<0.001)およびNCI、MCIおよび軽度アルツハイマー病群と比較して重度アルツハイマー病群で有意に減少したことを示す(p<0.001)。J)HDAC2-ir核の面積がNCI(p = 0.03)群と比較して軽度アルツハイマー病で有意に減少し、NCI(p < 0.001)群、MCI(p < 0.001)群、および軽度アルツハイマー病(p = 0.04)群と比較して重度アルツハイマー病で有意に減少したことを示すボックスプロット。箱グラフ中の丸印は外れ値を示す。∗p < 0.05;****p < 0.001。スケールバー。10μm。Mahady et al 2019)より転載。
アルツハイマー病の進行のためのコリノトロフィーバイオマーカー
上述のように、proNGFのタンパク質レベルは、NCIと比較してMCIまたは軽度アルツハイマー病の臨床診断を受けて死亡した被験者の死後大脳皮質(Peng et al 2004)および海馬(Mufson et al 2012b)においてそれぞれ増加しており、これは死後の認知テストのスコアの低下と相関していた(Peng et al 2004;Mufson et al 2012b)。これらの観察は、proNGFの変化した脳脊髄液レベルがNCIからMCIおよびアルツハイマー病への移行をマークするかどうかの調査を開始した。RROS参加者から心室脳脊髄液を死後に入手し、ワシントン大学ナイトアルツハイマー病研究センターでCDR 0(認知症なし)CDR 0.5(MCIまたは非常に軽度のアルツハイマー病)またはCDR 1(軽度のアルツハイマー病)と臨床的に診断された被験者から死前の腰部脳脊髄液を収集した(Counts et al 2016)。心室脳脊髄液の定量的ウエスタンブロットにより、NCIと比較してaMCIではproNGFレベルが有意に(50%)増加し、NCIと比較してアルツハイマー病では70%増加しており、心室脳脊髄液 proNGFレベルの増加と認知機能の悪化との間に有意な逆関係が示された(Counts et al 2016)。腰部脳脊髄液 proNGFレベルは、CDR 0.5例とCDR 1例ではCDR 0例と比較して有意に(30%)上昇した。アミロイドβ1-42,総タウ、phospho-tau、またはphospho-tau/アミロイドβ1-42のレベルには群間で差は見られなかったが、総タウ/アミロイドβ1-42レベルの比率は、CDR 0例の被験者と比較してCDR 1例では50%高かった。proNGF/アミロイドβ1-42,proNGF/総タウ、およびproNGF/phospho-tauの比率を計算し、脳脊髄液 proNGFレベルを含めることでこれらのバイオマーカーの信頼性が向上したかどうかを判断した。興味深いことに、proNGF/アミロイドβ1-42レベルはCDR 0と比較してCDR 0.5とCDR 1で50%高く、一方、proNGF/total tauとproNGF/phospho-tauはグループ間で変化がなかったことから、proNGFをバイオマーカー候補に含めることで、アルツハイマー病の前臨床段階または前駆段階の人を識別するために必要な診断確率が向上することが示唆された。
アルツハイマー病の治療戦略としてのNGF療法
コリノトロフィック皮質投影系を救済するための治療戦略としてNGFを採用した研究から得られた証拠は、脳血流ニューロンの萎縮の防止、および実験的損傷または正常な加齢に起因する行動障害の修正に関するいくつかの有望な結果を明らかにした(Hefti, 1986; Williams et al 1986; Hartikka and Hefti, 1988; Hatanaka et al 1988; Nabeshima et al 1994; Burgos et al 1995; Charles et al 1996; Humpel and Weis, 2002)。この証拠から、NGFを促進する治療法は、アルツハイマー病のコリノトロフィー機能障害を逆転させるのに有益であるという概念につながった。しかし、初期のNGFの全身投与に関する研究の検討では、標的ニューロンに到達するためのニューロトロフィンのバイオアベイラビリティー、制御されていない神経伝達物質の放出、神経緊張の亢進、ニューロンの芽生え、交感神経刺激、抗体の誘導、悪液質、および痛覚過敏などのいくつかの弱点が示された(Sramek and Cutler, 1999; Jonhagen, 2000; McArthur et al 2000; Apfel, 2001)。 1990, 1991, 1996; Blesch and Tuszynski, 1995)および過去の失敗例(例えば、薬物送達の不良および望ましくない全身的副作用)を考慮して、アルツハイマー病に対するex vivo NGF遺伝子治療の有用性を決定するための第I相臨床試験が実施された(Tuszynski et al 2005)。その目的は、ヒトNGFを頭蓋内に送達することにより、NBM内のコリン栄養ニューロンを変性から保護し、残存するコリン作動性ニューロンの機能を増強することにあった。臨床試験では、アルツハイマー病患者さんを対象に、NVM内のコリン作動性ニューロンを対象に、ex vivoまたは生体内試験遺伝子導入によるNGF転写療法が行われた。退化した nbM ニューロンは NGF に反応し、NGF 源に向かって軸索が芽生えていることが確認された(図 11)。一方的に遺伝子導入を受けた参加者では、NGF処理を受けたコリン作動性のnbMで神経細胞の肥大が見られた。
図11
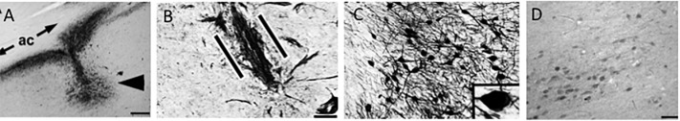
A)神経成長因子(NGF)免疫標識は、前交連(AC)の下のメイナート(矢印)のヒト基底核におけるNGF遺伝子の送達部位を示す。スケールバー=325μm。患者は3年前に注射を受けた。B)p75NTRニューロトロフィン受容体の免疫標識は、直線的な方法で移植片(平行線の間)に浸透基底前脳コリン作動性軸索を示している。スケールバー= 25μm。C)神経成長因子を発現するニューロンは、注入部位(D)から3 mmに位置するメイナートニューロンの基底核のあまり強くないラベリングと比較した。C)のスケールバーは、(C)= 100μmで、(D)= 325μmで。画像はTuszynski et al 2015)より転載。
さらに、アデノ随伴ウイルスベクター(血清型2)媒介のNGF遺伝子導入を持続した患者は、細胞シグナル伝達および機能マーカーの活性化を示した。興味深いことに、病理学的にタウを示したnbMニューロンとタウ免疫陰性であったニューロンの両方がNGFを発現しており、タングルを有するニューロンが細胞シグナル伝達を活性化する治療用遺伝子に感染し得ることが示された。これらの試験では、NGF治療に関連した副作用は認められなかった。以上の結果から、変性ニューロンが NGF に反応して軸索芽生え、細胞肥大、機能マーカーの活性化が可能であることが明らかになった。これらの研究は、NGF誘発芽出しがNGF遺伝子導入後10年間持続したこと、およびこの治療法が長期間にわたって安全であると思われることを実証した(Tuszynski et al 2015)。これらの研究は、二重盲検、プラセボ対照臨床試験ではないことに留意すべきである。遺伝子治療アプローチのさらなる臨床研究が望まれている。
アルツハイマー病治療のための低分子ニューロトロフィン化合物
NGF受容体の低分子部分アゴニストおよびアンタゴニスト活性化剤は、アルツハイマー病治療のために検討されてきた (Skaper, 2008)。例えば、TrkAに対する低分子アゴニストのハイスループットなスクリーニングアッセイにより、伝統的な漢方薬で使用されるアルカロイドであるガンボジックアミド(Gambogic amide)が候補として同定された(Jang er al)。 ガンボジックアミドは TrkA に選択的に結合し(TrkB や TrkC には結合しない)TrkA のチロシン残基をリン酸化し、Akt や Erk TrkA を介した NGF シグナル経路を活性化する。ガンボギンアミドは、PC12細胞における興奮障害を改善し、神経細胞の成長を促進し、マウスにおけるカイニン酸ニューロン誘導細胞死を減少させることが実証されている(Jang et al 2007)。

en.wikipedia.org/wiki/Garcinia_prainiana
また、いくつかの証拠は、p75NTRによって促進される変性シグナル伝達の調節がアルツハイマー病の潜在的な治療標的であることを示唆している。アンリガンド状態でのその構成的活性、またはそのプロニューロトロピン(proNT)リガンドによって刺激された活性を介して、p75NTRは、JNK、カスパーゼおよびRhoAの活性化を含む変性シグナル伝達機構を促進する(Casaccia-Bonnefil et al 1996; Harrington et al 2002; Troy et al 2002; IbanezおよびSimi 2012; CoulsonおよびNykjaer 2013)それぞれがアルツハイマー病関連の変性に寄与する可能性が高い(図12)。様々なADマウスモデルとp75NTRノックアウトマウス構築物を交配させると、神経細胞の変性が減少した(Sotthibundhu et al 2008; Knowles et al 2009; Murphy et al 2015)。複数の研究は、アルツハイマー病リスクの増加と関連するproNT結合を媒介するsortilinおよびSorCS2を含むp75NTR、proNGF、proBDNF、またはp75NTR共受容体をコードする遺伝子における遺伝的多型を同定している(Cozza et al 2008;Di Maria et al 2012;Anastasia et al 2013;Reitz et al 2013;Andersson et al 2016;Matyi et al 2017)。p75NTRと相互作用し、そのシグナル伝達を調節することが発見された最初の低分子リガンドは、NGFのループIドメインをモデル化した合成ペプチドおよびニューロトロフィンリガンドの変異解析に基づくin silicoスクリーニング戦略を用いて同定された(Masa et al 2006)。その結果、ニューロトロフィンの添加が神経細胞の生存に依存している細胞培養条件下では、ニューロートロフィンの添加が神経細胞の死滅を抑制することが判明した。これらの低分子リガンドは、効果的にp75NTRポジティブモジュレーターとして作用し、proNGF誘発細胞死を阻害し(Masa et al 2006)proNGFのp75NTRへの結合をブロックした(Tep et al 2013)。これらの条件下では、低分子リガンドはp75NTRの「アンタゴニスト」として記述され得る。LM11A-31は、インターロイキン-1受容体関連キナーゼ(IRAK)生存アダプターのp75NTRへのリクルートを刺激し、下流のNF-κBおよびAktプロ生存シグナル伝達をアップレギュレートすることが示されている。神経栄養活性およびシグナル伝達は、p75NTR-/-ニューロンを用いた培養物においても、p75NTR-ECDブロッキング抗体の存在下においても、全く認められなかった(Masa et al 2006)。LM11A-31は、脊髄損傷(Tep et al 2013年)神経原性膀胱機能障害(Ryu et al 2018年)および関節炎(Minnone et al 2017)のモデルにおけるproNGF誘発性変性機構をブロックする。
図12 p75NTRとアルツハイマー病関連の変性シグナル伝達ネットワークとの関連を示す図
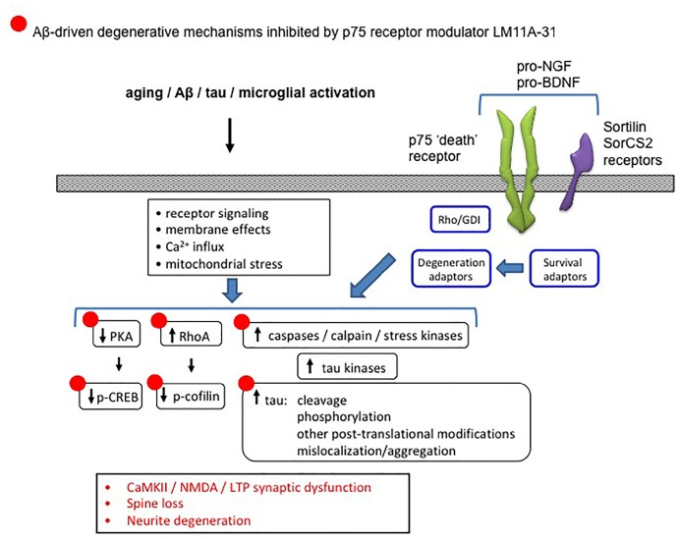
老化やアミロイドβなどにより、神経細胞やシナプスの機能や変性に関わるシグナル伝達経路に多くの変化が生じる。p75NTRは、そのアンリガンド状態で、あるいはプロニューロトロフィンとp75NTRおよびそのソルチリンファミリーの共受容体との結合に応答して、1つ以上の受容体アダプターを介して促進される変性シグナル伝達により、アルツハイマー病に関連する変性プロセスを可能にするか、あるいは促進することが知られている。LM11A-31は、プロニューロトロフィンの変性シグナル伝達を阻害し、p75NTRを介した支持・生存シグナル伝達を促進することで、これらのアルツハイマー病関連作用の多くを逆転させる(-で示されている)。
複数の前臨床研究により、アルツハイマー病関連の変性メカニズムの進行を遅らせるための治療アプローチとして、低分子でのp75NTRのモジュレーションの役割が示されている。p75NTRに連結された細胞内シグナル伝達ネットワークは、アルツハイマー病に関与する変性シグナル伝達ネットワークと実質的に統合されている(Nguyen et al 2014)。培養海馬ニューロンをアミロイドβオリゴマーに曝露したADモデルにおいて、LM11A-31およびLM11A-24は、以下のアミロイドβ誘発性変性メカニズムを阻害した:カルパイン/cdk5,GSK3β、JNK c-Jun、p38キナーゼ、RhoAの活性化;過剰なタウリン酸化;およびAktおよびCREBの不活性化(Yang er al)。 これらのリガンドはまた、アミロイドβ誘導性神経ジストロフィーおよび海馬長期増強(LTP)障害をブロックした。hAPPLond/Swe ADマウスモデルを用いた研究では、LM11A-31を1日1回3ヶ月間投与することで行動障害が修正され、タウリン酸化とミスフォールディング、神経突起ジストロフィー、ミクログリア活性化、アストロサイト活性化を含む神経変性分子および細胞病理学的を阻害した(Knowles et al 2013;Nguyen et al 2014)。しかし、可溶性アミロイドβやアミロイドプラークレベルを低下させる効果はなく、p75NTRの変調がタウ関連分子変性イベントやシナプス障害を含む神経変性を促進するアミロイドβの能力を阻害するメカニズムと一致した。hAPPLond/Swe ADマウスにおけるミクログリア活性化の尺度を減少させるLM11A-31の能力は、ミクログリア活性化の複数のマーカーおよびトランスロケータータンパク質(TSPO)に向けられたPETリガンドを用いたマイクロPETイメージングを用いて確認された(James et al 2017)。後期の治療を採用した hAPPLond/Swe アルツハイマー病 マウスの研究では、LM11A-31 の適用は、おそらく特に堅牢な生物学的効果を示す、神経変性の部分的な反転をもたらした(Simmons et al 2014)。図12は、proNT変性シグナル伝達を阻害し、p75NTRを介して支持/生存シグナル伝達を促進するp75NTRモジュレーターLM11A-31のアルツハイマー病変性メカニズムを阻害/逆転させる能力を要約したものである。
LM11A-31は、生体内試験での前臨床試験に基づき、新規ターゲットを対象としたファーストインクラスの化合物として、LM11A-31を改良した製剤を健常者を対象とした第1相臨床試験を実施し、安全性が確認された。本化合物は現在、軽度から中等度のアルツハイマー病患者を対象とした無作為化二重盲検第2a相探索的エンドポイント試験が実施されている(NCT03069014)。治療は6ヶ月間毎日経口カプセルを介して投与され、ベースラインおよび治療後の測定は以下の通りです:複数のバッテリーを使用した認知テスト、MRIの容積測定とFDG-PETイメージング、および脳脊髄液 アルツハイマー病コアバイオマーカーとターゲットの関与と作用機序に関連するバイオマーカー。
おわりに
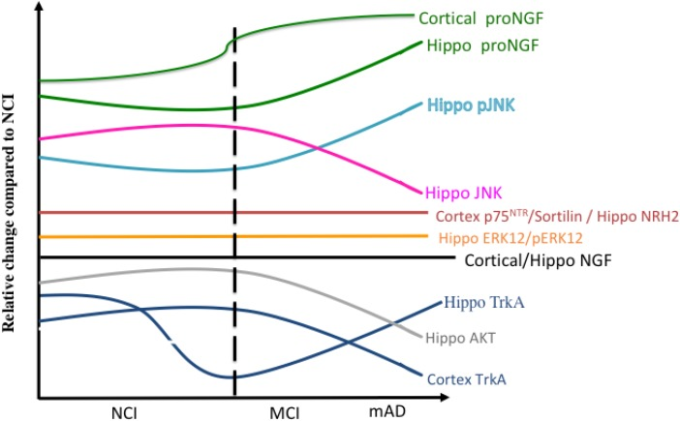
図13は、アルツハイマー病の進行中に大脳皮質と海馬内のNGF上流および下流のシグナル伝達経路の相対的な変化を要約している。データの優勢は、NGFとその認知受容体の正常レベルがコリノトロフィー系の生存と維持に必要であることを示している。MCIおよび軽度アルツハイマー病におけるTrkAおよびp75NTR陽性ニューロン数の減少に直面してコリン作動性のnbMニューロンが保存されていることは、コリン作動性ペリカリア自体の直接的な損失ではなく、疾患プロセスの初期に受容体タンパク質の表現型ダウンレギュレーションが存在することを示している。トランスクリプトとタンパク質の両方のデータは、脳血流ニューロンの機能不全がTrkA媒介の生存シグナルとproNGF/p75NTR媒介のプロアポトーシスシグナルの間の不均衡と関連していることを示している。このような変性事象にもかかわらず、コリノ栄養系は、前駆期(DeKosky et al 2002)および疾患の後期においても、細胞の回復力および/または神経可塑性を発揮することができる(Mufson et al 2012b)。神経栄養機能障害に加えて、核内エピジェネティックタンパク質の変化は、アルツハイマー病の進行中にコリノトロフィー性nbMニューロン内で起こり、特にHDAC2は、転写産物発現の変化に関連したメカニズムを示唆している。死亡後の心室脳脊髄液または死亡前の腰部脳脊髄液で定量されたプロNGFの増加は、MCIおよびアルツハイマー病への移行を示し、このプロニューロトロフィンが疾患進行の有用なバイオマーカーであることを示唆している。臨床試験は、NGF遺伝子治療がアルツハイマー病における脳血流変性の予防のための治療アプローチになる可能性があるという証拠を提供している。おそらく、この治療アプローチと、再生シグナル伝達を促進するためのTrkAに対する低分子アゴニストの開発(Jang er al)。
図13 アルツハイマー病の進行中の大脳皮質と海馬におけるNGF関連タンパク質の相対的なレベルの変化を示す要約図。