Contents
Endogenous Mechanisms of Neuroprotection: To Boost or Not to Be
www.ncbi.nlm.nih.gov/pmc/articles/PMC7916582/
オンラインで2021年2月10日公開
Sara Marmolejo-Martínez-Artesero,1 Caty Casas,1,† and David Romeo-Guitart1,2,*.
Paola Pizzo(学術編集者)
www.sciencedirect.com/science/article/pii/S0033062019300386
概要
神経細胞のような分裂終了細胞は、一生をかけて生きていかなければならない。そのために、生物・細胞は自己修復機構を備えて進化し、長寿命化を実現してきた。
ここ数年の神経保護剤の発見ワークフローは、神経変性における神経細胞の損失につながる病態生理学的なメカニズムをブロックすることに焦点を当てている。しかし、残念ながら、神経変性の進行を遅らせたり、防いだりすることができたのは、これらの研究から得られたわずかな戦略だけであった。
しかし、生物や細胞が本来持っている自己修復メカニズム(一般的には細胞の回復力と呼ばれる)を支持することで、神経細胞を武装させ、その自己修復を促進することができることを示す説得力のある証拠がある。これらのメカニズムを強化することは、まだ十分に注目されていないが、これらの経路は、神経細胞の死を防ぎ、神経変性を改善するための新たな治療の道を開く。
ここでは、主な内因性の保護機構を取り上げ、神経変性時の神経細胞の生存を促進する上での役割について説明する。
キーワード
オートファジー、細胞の回復力、内因性メカニズム、神経保護、神経細胞の生存、アンフォールドタンパク質反応
1. 神経衰弱のプロセス
先進国の長寿化に伴い、アルツハイマー病(AD)、パーキンソン病(PD)、ハンチントン病(HD)などの神経変性疾患、すなわち加齢に伴う神経系の性能低下の頻度は増加すると考えられている。これらの病態には、神経細胞、アストログリア、ミクログリアなどの要素があることを示す証拠がいくつかあるが、生活機能の低下は、神経細胞の減少が進行することによって引き起こされる。神経細胞は、そのターンオーバーの少なさから、一生をかけて生きていかなければならない分裂終了細胞である。このため、神経細胞は、その終焉の原因となる外的・内的な傷害に対処するための強力な内在的な保護機構を必要とする。これらの外部/内部の危険は、外傷や興奮毒性化合物、活性酸素種(ROS)タンパク質凝集体、およびその他の毒性分子である。幸いなことに、細胞には内在する機械があり、回復機構を活性化したり再生経路を促進したりすることで死を阻止している。若い神経細胞はこれらの自己修復保護機構が適切に機能しているが、加齢によってこれらが乱れ、神経細胞は無防備な状態になる。同様に、神経変性疾患においても、これらの自己修復機構の機能不全が指摘されている。
この数十年間、新規かつ効果的な神経保護治療法を得るために多大な努力が払われてきた。しかし、これらの治療法は病態生理学的なメカニズムを標的としており、結果的には神経細胞の死を加速させることになる。そこで、神経細胞が本来持っているメカニズムを利用して、効果的な神経保護法を実現してはどうだろうか。
この保護ネットワークは、さまざまな細胞プロセス(アンフォールドタンパク質応答(UPR)オートファジーなど)のクロストークによって駆動されるが、これらは、細胞がストレスに適応して生存するという同じプロセスに収束する[1,2,3]。最近,私たちは,神経保護剤を発見するための新たな理論的根拠を検討した。それは,健康と神経変性・加齢に共通する,生存か死かという正反対の表現型をもつ2つの異なる神経損傷後に,神経細胞がどのような分子メカニズムを働かせるかを解読することである。そのために、私たちは、内因性の保護メカニズムの機能性または機能不全を模倣した2つの生体内試験ベースの末梢神経損傷モデルを使用した。これらのモデルでは,体幹-損傷間の距離に応じて、運動ニューロン(MN)の死(根部剥離(RA))または生存(遠位軸索切除(DA))が引き起こされる [2]。これらのモデルを用いて,システムバイオロジーに基づいたアプローチにより,RA後の運動ニューロンの死が,神経変性疾患で観察される神経細胞の減少と類似していることを確認し,神経損傷後に運動ニューロンがどのようなメカニズムで生き延びるかについても説明した[2]。
変性プロセスとは、アポトーシス、ネクローシス、アノイキス(細胞―マトリクス間接着の喪失により誘導されるアポトーシス)、小胞体ストレス、核小胞体ストレス、細胞骨格の再編成、ミトコンドリア機能不全などである。
一方、生存の原動力となるのは、正しいUPR、熱ショック応答、オートファジー経路、ユビキチン・プロテアソームシステム、シャペロンシステム、小胞体関連分解装置、抗酸化防御である(表1)。
興味深いことに、これらのメカニズムはすべて何年も前に別々に記述されており、プレコンディショニング傷害と呼ばれている(以下参照)。
表1 内因性の神経保護メカニズムごとに関与するタンパク質と、その効果が媒介される分子メカニズムの概要
| 関与するタンパク質 | 病理学 | 保護のメカニズム | 参考文献 |
|---|---|---|---|
| オートファジー | |||
| ATG5 | PD 1 | ATG5はオートファジーを誘導し、ATG16L1-ATG12と複合体を形成することによってオートファゴソームの伸長を調整する。ATG5の過剰発現は、オートファジーフラックスを増加させることによって保護する。 | [ 4 ] |
| 軸索切断の運動ニューロン 2 | [ 5 ] | ||
| ATG7 | PD | ATG7は、オートファゴソーム形成を促進する複合体ATG12-ATG5とATG5 -ATG16L1-ATG12の結合を促進することにより、オートファジーを誘導する。ATG7の過剰発現は、オートファジーフラックスを増加させることによって保護する。 | [ 4 ] |
| p62 | タンパク質凝集体を特徴とするハエモデル | p62は、オートファゴソームの電荷負荷を管理し、オートファゴソーム形成の後期段階で重要な役割を果たす。p62は、ポリグルタミンタンパク質オリゴマーのオートファジー分解を介して保護的な役割を果たす。 | [ 6 ] |
| TFEB | PDモデル | TFEBの過剰発現は、リソソームの生合成と機能、およびオートファジーフラックスの維持に不可欠な転写ネットワークを調節する。 | [ 7 ] |
| AD 3のマウスモデル | [ 8 ] | ||
| Beclin1 | 基礎条件 | Beclin1は、オートファジーの開始と核形成に不可欠であり、抗アポトーシス能力を持っている。 | [ 9 ] |
| 小胞体ストレス応答 | |||
| BIP | PDモデル | BIPは、保護的でタンパク質の折り畳みを制御し、UPRコンポーネントを調整するERシャペロンである。BiPの過剰発現は、誤って折りたたまれたタンパク質の折り畳みを促進することによって保護する。 | [ 10 ] |
| PERK | PERKの活性化された細胞質ゾルドメインはeIF2αをリン酸化し、翻訳を阻害し、ATF4 mRNAの優先的な翻訳を誘導する。これにより、必須遺伝子がアップレギュレートされ、細胞の恒常性が回復する。小胞体ストレスが長引くと、適応促進型UPRが失敗すると、PERKは下流のCHOPの活性化を通じてアポトーシス促進性シグナルをトリガーし、アポトーシスを促進する。 | ||
| 脳内出血 | 損傷後のPERKの早期活性化は、タンパク質恒常性を回復することによって神経を保護する。 | [ 11 ] | |
| タウ病理学 | PERKの過剰発現または薬理学的活性化には保護効果がある。 | [ 12 ] | |
| スクランブルマウスモデルとAD | PERKの活性化を回避すると、神経細胞死が減少し、加齢に伴う記憶が改善される。 | [ 13、14 ] | |
| プリオン病のinvivoモデル | アストロサイトにおけるPERKの特異的阻害は、ニューロンの喪失を遅らせる。 | [ 15 ] | |
| CHOP | 視神経挫滅モデル | CHOPは、複数の疾患モデルにおける小胞体ストレス後のアポトーシスに関連している。CHOPを削除すると、網膜神経節細胞の生存が促進される。 | [ 16 ] |
| IRE1a | 肝不全 | IRE1αは、哺乳類と下等真核生物の両方でUPRの開始に主要な役割を果たすER膜貫通タンパク質である。IRE1の活性化された細胞質ゾルドメインは、その基質Xbp1から 26bpイントロンを切断し、その翻訳を促進して、UPR遺伝子の転写を活性化する転写因子Xbp1を形成する。 | [ 17 ] |
| Xbp1 | Xbp1は非常に活性の高い転写因子であり、ERシャペロンとERADに関与するタンパク質の発現を促進する。 | ||
| 心臓の虚血/再灌流 | Xbp1の活性化は心臓の保護を促進する。 | [ 18 ] | |
| AD、PDおよび脳卒中後 | Xbp1の活性化は神経保護を促進する。 | [ 19、20、21 ] | |
| 糖尿病および虚血誘発性網膜症 | XBP1を介したUPR活性化の活性化とNF-κB活性化の阻害による保護効果。 | [ 22 ] | |
| 視神経挫滅モデル | Xbp1の過剰発現は、ニューロンの生存を増加させる。 | [ 16 ] | |
| ATF6 | ATF6は、細胞のトランスクリプトミクスネットワークを変化させることによってERタンパク質恒常性を仲介する。 | ||
| 神経損傷 | ATF6の切断を増加させることは、UPRの神経保護調節である。 | [ 23 ] | |
| 虚血モデル | 薬理学的ATF6活性化はUPRを誘発する。 | [ 24 ] | |
| 脳卒中 | ATF6の強制発現はオートファジーを誘発する。 | [ 25 ] | |
| ATF5 | 人間のてんかん | ATF5は、非神経細胞の小胞体ストレス応答時に選択的に翻訳され、発達中の脳で高度に発現し、神経前駆細胞の増殖を調節する。成体ニューロンは、生存促進メカニズムとして小胞体ストレス下でATF5レベルを増加させる。 | [ 26 ] |
| 抗アポトーシス | |||
| IAP | IAPタンパク質は、カスパーゼの活性化を阻害することでアポトーシスを抑制する。 | ||
| 虚血モデル | IAPの活性化は、標的剥奪後の運動ニューロンに対するGDNFの生存促進効果を仲介する | [ 27 ] | |
| 新生児神経軸索切断 | ターゲット剥奪後の運動ニューロンのアポトーシス死。 | [ 28 ] | |
| 成人期の軸索切断 | IAPの過剰発現は神経細胞死を阻止する。 | [ 29 ] | |
| 虚血 | 虚血性プレコンディショニングにより、IAPはカスパーゼ死のカスケードを停止し、神経細胞死を防ぐことができる。 | [ 30 ] | |
| AKT | 基礎条件 | AKTは、アポトーシスをブロックすることにより、生存促進のプレーヤーである。 | [ 31 ] |
| 基礎条件 | AKTはp53が誘導するアポトーシスを阻害する。 | [ 32、33、34 ] | |
| 基礎条件 | AKTはFOXOをリン酸化し、細胞の生存率を高める。 | [ 35 ] | |
| アンチアノイキス | |||
| PI3K / AKT | 基礎条件 | PI3K / AKTは、分化した細胞の生存を促進する。 | [ 36、37 ] |
| MMP9 | MMPは細胞外マトリックスの主成分を分解する。 | ||
| 脳虚血 | MMP9阻害は、虚血時のラミニン分解を抑制する。 | [ 38 ] | |
| ALS 4マウスモデル | MMP9阻害は運動単位を保護する。 | [ 39、40 ] | |
| ADモデル | MMP9阻害は運動単位を保護する。 | [ 41 ] | |
| 細胞骨格 | |||
| KIF5c | 興奮毒性invitroモデル | KIF5cはミトコンドリア輸送において主要な役割を果たしている。KIF5cは、ミトコンドリア機能を調節し、死の信号の前で良好な細胞の健康を維持することにより、神経保護を仲介する。 | [ 42 ] |
| DCTN1 | 破骨細胞の分化 | DCTN1は、細胞質モータータンパク質ダイニンの成分であり、細胞骨格の組み立てと組織化に関与している。DCTN1の過剰発現は、アポトーシスによる死を防ぐ。 | [ 43 ] |
| RILP | 基礎条件 | ダイニンアダプターRILPは、オートファゴソーム生合成と逆行性輸送のプロセスを統合して、オートファジーフラックスを制御する。 | [ 44 ] |
| ミトコンドリア | |||
| PINK1 | PINK1は、損傷したミトコンドリアの外膜に蓄積してE3ユビキチンリガーゼパーキンを動員し、マイトファジーを開始する分子センサーである。 | ||
| HDのフライモデル5 | PINK1レベルを上げると、ミトコンドリアの完全性が向上し、神経保護が促進される。 | [ 45 ] | |
| Nrf2 | 細胞保護および解毒遺伝子の発現を調節して、酸化ストレスおよび神経炎症と闘い、神経損傷を軽減する。 | ||
| HD | アストロサイトの小分子によるKeap1–Nrf2–ARE経路の活性化は、非興奮毒性のグルタメート毒性に対するニューロンの耐性を強化する。 | [ 46、47、48 ] | |
| PD | Nrf2の薬理学的活性化はPDの進行を防ぐ。 | [ 49、50 ] | |
| SOD1変異モデル | Nrf2の星状細胞特異的過剰発現は、脊髄運動ニューロンの生存を改善し、マウスの寿命を延ばする。 | [ 51、52 ] | |
| 脳虚血-再灌流障害 | p62とKeap1–Nrf2クロストークは、オートファジーによってROSを除去し、酸化的損傷を防ぎ、小胞体ストレスを調節する。 | [ 53 ] | |
1 パーキンソン病(PD)2 運動ニューロン(MN)3 アルツハイマー病(AD)4 筋萎縮性側索硬化症(ALS)5 ハンチントン病(HD)
私たちは、薬理学的治療によってこれらの内因性神経保護メカニズムを後押しすることで、運動ニューロンが、異なる種から異なる発生段階に至るまで、さまざまな死のシナリオの中で生き延びることができることを実証した[23,54,55]。
2. 内因性メカニズムの最初の証拠。プレコンディショニング
内因性の保護メカニズムの表現型の効果は,40年前に記述されている.1986年のことである。Murryらは,プレコンディショニング傷害として知られる亜致死の生理的ストレスが,心臓の組織回復を促進することを報告した[56]。ここから、これらの治癒メカニズムは、脳や脊髄(SC)でも観察されるようになった[57]。例えば,神経損傷後や心臓再生時には,これらの細胞応答が観察され,それぞれ活性酸素や細胞外小胞の産生が機能回復を促している[58,59,60]。驚くべきことに、特定の臓器をプレコンディショニングすることで、他の臓器を損傷から守ることができる[61]。
これらの効果には、いくつかの特定のエフェクターが関与している。プレコンディショニングによる傷害後、さまざまなメディエーター(一酸化窒素や活性酸素)が産生されると、ホスファチジルイノシトール3キナーゼ(PI3K)/プロテインキナーゼB(AKT)プロテインキナーゼC(PKC)などのシグナル伝達経路が活性化され、低酸素誘導因子1α(Hif1-α)やNF-κBなどの転写因子が調節されるようになる。
その結果,一酸化窒素合成酵素(iNOS),熱ショック蛋白質(HSP),シクロオキシゲナーゼ-2(COX-2)などの「エンドエフェクター」と呼ばれる物質が産生され,将来の傷害に対する組織内の保護作用が促進されることになる[61]。
これらの研究を総合すると,生物・細胞には内因性の防御機構があり,それを高めることが効果的な治療戦略となりうることが示唆される。
3. 内因性神経保護メカニズム
3.1. オートファジーの微調整
神経細胞が恒常性を維持するためには、細胞内の物質を継続的にリサイクルする必要がある。マクロオートファジー(以下、オートファジー)は、真核細胞内で高度に調整された分子ネットワークであり、リソソームでの分解を通して細胞質内の物質を再利用しようとするものである。当初、この分解機構は飢餓状態でのみ観察されていたが、近年の研究により、細胞はタンパク質のホメオスタシスを調節するためにオートファジーの基礎レベルを持っていることが明らかになった。これらの基本的なレベルは、正常な状態での神経細胞の軸索維持と生存に不可欠である[62,63]。機能的なオートファジー・フラックスは、さまざまなオートファジー関連(ATG)遺伝子、キナーゼ、その他の制御タンパク質によって高度に調整されたプロセスである。これらはすべて、オートファゴソームの正しい開始、核形成、伸長、閉鎖、および細胞質負荷を分解するためのリソソームとの融合を調和させるために協働する[64]。海馬では、加齢に伴ってオートファジーの流れが悪くなることが観察されているが、そのレベルが再び確立されると、新しい記憶の形成が促進される[65]。神経細胞におけるオートファジーの障害または機能不全は、神経変性と関連しており、一方、オートファジーの活性化は神経保護をもたらす[5,54]。筋萎縮性側索硬化症(ALS)では,初期相と伸長相に関連するタンパク質の変化が観察されており[66,67],ラパマイシンなどのオートファジーの誘導剤は,脳虚血,外傷性脳損傷(TBI),ADの後に神経保護作用を発揮する[68,69,70]。ATG5またはATG7の神経細胞特異的ノックアウト(KO)は,神経変性,細胞質内封入体の蓄積,神経細胞の死を引き起こすが[62,71],これらの過剰発現は,PDのモデルにおいて有益である[4]。最後に,オートファゴソーム内の電荷を管理し,オートファゴソーム形成の後期に重要な役割を果たすp62は,神経変性疾患の特徴であるタンパク質凝集体を特徴とするハエモデルにおいて,神経保護作用を示す[6].
いくつかの研究では,神経変性の過程でオートファゴソームやオートリソソームが蓄積することが示されており,オートファジーが過剰に活性化され,神経細胞死の引き金になっている可能性が示唆されている。細胞質内でのオートファジー過程の異常な蓄積は、過剰に活性化されたオートファジーではなく、リソソームの機能障害が原因である可能性がある[72]。TBI後、オートファジーは適切に開始されるが、リソソームの機能不全によりオートファゴソームが排除されず、未解決のオートファジーが起こり、神経細胞死を促進する[73]。こうしたリソソームの非機能的な経路は、脊髄損傷(SCI)後にも見られ、機能回復を妨げている[74]。オートファゴソームのクリアランスにおける同様の閉塞は,神経変性疾患(すなわち,ADのヒト脳)においても記述されている[75]。これらの証拠を総合すると、オートファジーの解消を強化することで、保護効果が得られることが示唆される。プラットは最近、神経変性を防ぐためにリソソームタンパク質の機能を向上させるという治療の道を強調した[76]。リソソームの生合成と機能に不可欠な転写ネットワークを調節する転写因子EB(TFEB)を過剰発現させると,PDのラットモデル[7]やADマウスモデル[8]で神経保護効果が促進された。
オートファジーの誘導は、思うようにはいかない。カノニカルな保護機構ではあるが、その機械や過剰な活性化は細胞死を促進することがある[77,78]。ヒトのプリオンにさらされた後にオートファジーを阻害すると、神経細胞の損傷が減少するが、これはオートファジーの誘導が死をも促進することを示している[79]。また、オートファジーの開始を減少させると、新生児および成体マウスのSC切断後の機能回復が促進され、アポトーシスが防止され、虚血後の錐体細胞の死が減少する[80,81,82]。軸索切断された神経細胞に注目すると、オートファジーをブロックすることで、脊髄のものは神経保護され[80]、ATG5のレベルを上げることで脊髄の運動ニューロンが保護される[5]。さらに、化学療法を受けた癌細胞は、治療によるアポトーシス死を克服するためにオートファジーを活性化するが、運動ニューロン依存性のオートファジーはアポトーシスを抑制するという論争もある[54]。さらに、ATGは神経細胞死を引き起こすこともある。ATG5は、切断されると自食促進能力を失い、その活性を細胞死の誘導に向かわせる[83,84,85]。ベクリン1は、正常な状態では抗アポトーシス作用を持っているが、C末端で切断されると、アポトーシスシグナルに対して細胞を感作する[9]。したがって、両方の細胞プロセスの間にはクロストークが存在し、細胞はそれらを方向転換して、傷害に対処するための生存の可能性を高めることができる[83]。
では,神経保護のためには何が重要なのであろうか?オートファジーを促進するのか、阻害するのか。ファインチューンがその答えである[86]。微調整されたオートファジーを誘導することで、以下のような有益な効果が得られる。
- 機能していないタンパク質/小器官を除去する、
- 細胞が新しい状況に再適応できるようにする、
- 神経細胞の死を媒介する炎症やアポトーシス誘導物質などの有害な作用を分解する [87,88]。
しかし、このオートファジーは、細胞死を誘発する過剰な分解を避けるために、非常に特定の時間帯に活性化されなければならない。
最後に、オートファジーは、炎症反応の調節、新しい記憶の形成[65]、シナプスのホメオスタシスの維持[89]、細胞内の貨物の輸送[90]など、非正規の/分解的な機能も持っている。そのため、完全にブロックしてしまうと、神経系および/またはニューロンに不可逆的なダメージを与えることになる。
3.2. 小胞体ストレス応答のセクシーな部分への挑戦
神経細胞は、折りたたまれていないタンパク質や凝集体に対して非常に敏感である。小胞体は、細胞のプロテオスタシス(タンパク質の合成、折り畳み、選別)を担っている。小胞体の機能が低下すると、ミスフォールドしたタンパク質が蓄積され、小胞体ストレスが生じ、小胞体過負荷応答(ERO)小胞体関連分解(ERAD)経路、またはUPRが活性化されるが、これらは非常によく保存された細胞応答である。小胞体とUPRの分布と形態の変化は,神経変性疾患[91,92,93]や神経損傷後に神経細胞を分離した際に観察されている[16,94]。Binding immunoglobulin protein (BIP) は、GRP78としても知られる小胞体常駐型のシャペロンであり、UPRの主要なセンサーである。不活性な状態では、BIPは3つの主要なUPRエフェクター、すなわち、C/EBP homologous protein (CHOP)を誘導するRNA活性化プロテインキナーゼ様ERキナーゼ (PERK)、X-box binding protein 1 (Xbp1)のmRNAをスプライシングするinositol-requiring protein-1α (IRE1α)、および活性化転写因子-6α (ATF6)と結合している[95,96]。BIPがミスフォールドしたタンパク質を検出すると,これらのトランスデューサーが活性化され,特定のタンパク質(すなわち,シャペロン,転写因子)の遺伝子発現の変化を促す。その目的は,遺伝子発現の調節,ミスフォールドしたタンパク質のクリアランスの促進,あるいはタンパク質合成の抑制によって,細胞がタンパク質を正しくフォールドする能力を高め,細胞がストレスに適応して生存できるようにすることである[97]。その証拠に,ドーパミンニューロンにBIPを過剰発現させると,その生存率が高まり,逆にダウンレギュレーションすると,ナイグラルドーパミンニューロンの死が誘導される[10]。さらに、BIP +/-マウスでは、プリオンの病因が加速して伝播することが示された[98]。全体的に見て、UPRの調節は、神経変性に対して保護効果を発揮する可能性がある[94]。
UPRの活性化は、神経変性疾患の初期のイベントであり、その正確な調節は、病理の進行に有益な効果がある[100,101]。UPRは、細胞を保護する内因性のメカニズムとして作用するかもしれないが、その(過剰な)活性化は、アポトーシスを促進する[102](すなわち、PERK軸は、プロアポトーシスまたはアンチアポトーシスの能力を持っている[91])。その上、最近の研究では、小胞体の異なる擾乱がUPRの3つの枝を異なる形で活性化することが示唆されており、これらの枝の協調的な活性化が常に存在するわけではないことを示している。したがって、細胞は特定の傷害に対応するための特定のプログラムを持っている。例えば,CHOPの阻害やXbp1の過剰発現は,神経損傷後の神経細胞の生存率を高めるが,これは,各枝が神経細胞死に異なる役割を果たしていることを示している[16]。
脳損傷後にPERKが早期に活性化されると神経保護効果が得られるが,この経路を介したシグナル伝達が持続すると細胞損失が悪化する[11]。PERKの過剰発現や薬理学的な活性化は、タウの病理を軽減し[12]、PERKの持続的な活性化を回避することで、神経細胞死を減少させ[13]、加齢に伴う記憶力の低下を改善する[14]。アストロサイトにおけるPERKの阻害は、プリオン病の生体内モデルにおいて神経細胞の損失を遅らせる。興味深いことに、アストロサイトにおけるPERKの活性化は、セクレトームを乱し、シナプス形成機能を変化させ、シナプスの消失を引き起こす[15]。同じ著者は、PERKのこの有害な作用に関与する主な下流メカニズムは、細胞外マトリックス-細胞接着経路であり、UPRとアノイキスを架橋していると記述している(下記、3.4項参照)。活性化転写因子5(ATF5)のレベルは、PERK/真核生物翻訳開始因子2a(eIF2a)の活性化に直接依存している。ATF5は、ヒトのてんかんにおいて、死に対してより抵抗力のあるニューロンと直接関連している[26]。しかし、これらの効果のその後の影響については、それほど明確ではない。ATF5は、2つの抗アポトーシスエフェクター(後述)であるB細胞リンパ腫2(Bcl-2)と誘導性骨髄性白血病細胞分化タンパク質(Mcl-1)の発現を誘導し、アポトーシスを阻害することになる[103]。また、ATF5は、非神経組織において、オートファジーの主な調節因子であるラパマイシンの機械的標的(mTOR)を調節し、UPRとオートファジーを相互に関連させている。
IRE1αの活性化は肝不全を改善し[17]、その下流のエフェクターであるXpb1は心臓の保護を促進し[18]、ADの神経保護、PDの神経保護、脳卒中後の神経保護を促進する[19,20,21]。驚くべきことに,糖尿病および虚血誘発性網膜症を対象とした研究では,UPRの保護効果がXbp1によって媒介されることが示された[22]。それにもかかわらず、IRE1αブランチの慢性的な活性化は、Tumor necrosis factor-a (TNF-α) receptor-associated factor 2 (TRAF2)のリン酸化を引き起こし、さまざまな方法でアポトーシスによる細胞死を誘発することになる[104,105,106]。Ire1αを異所性に過剰発現させると、PDのショウジョウバエモデルでは、オートファジー依存性の神経細胞死を引き起こす[107]。したがって、特定の時間帯にIRE1α-Xbp1を調節することで、保護効果が得られる可能性がある[108]。
我々は最近,NeuroHeal薬理学的治療またはサーチュイン1(SIRT1)の過剰発現が神経損傷後の運動ニューロンの生存を誘導し,IRE1αのリン酸化を減少させながら切断されたATF6の存在を増加させることを報告した[23].ATF6を薬理学的に活性化すると,プロテオスタシスが活性化され,さまざまな虚血モデルで保護効果が得られるが[24],この転写因子を阻害すると,悪影響を及ぼす.詳細には、ATF6は、抗酸化反応関連タンパク質の発現を調節し、ROSホルミシスを調節する[109]。ATF6を強制的に発現させると、脳卒中後の機能的転帰が改善されるが、著者らはこの効果がオートファジーの誘導を介している可能性を示唆している[25]。
では、治療的に興味深いのは、UPRを活性化したり、弱めたりすることであろうか?UPRの特定の枝を活性化することが重要なポイントである。UPRを正確に活性化すると、細胞がプロテオスタシスを回復するのを助け、保護作用を促進することができる。しかし、ストレスが続いてプロテオスタシスが回復しないと、UPRが神経細胞のアポトーシスを誘発し、これはPERKやIRE1αの枝によって媒介されるので、この概念は注意が必要である[110]。さらに、UPRはオートファジーとも関連しており、その逆もまた然りである。BIPは、オートファジー反応を仲介し、神経細胞の生存を促進する。最後に,UPRの3つのブランチは,ATGの転写を調節しており[112],両細胞のプロセスが複雑に関連していることを示唆している。
3.3. 「今日ではない」なアポトーシス
アポトーシスは、カスパーゼ依存性のプログラムされた細胞死(PCD)で、細胞の細胞膜や細胞小器官の完全性を維持する[113]。アポトーシスの調節不全は、多くの癌、神経変性疾患、炎症性疾患の原因となっている。カスパーゼによって誘導される死は、高度に制御されたプロセスであり、最終的な細胞死を引き起こすためには、複数のプレイヤーが協調して作動する必要がある[114]。アポトーシスのような死の特徴は、筋萎縮性側索硬化症(ALS)のマウスモデルやAD、PDで見られるが、アポトーシスが神経細胞死の最終的な実行者であるかどうかは不明である[115]。進化の過程で、細胞は、必要のないときに死なないように、あるいは、早すぎるPCDを避けるために、いくつかのメカニズムを発達させていた。細胞は、プロアポトーシス機構とアンチアポトーシス機構のバランスが死に向かって突き進んだときにのみ、効率的なアポトーシス死を引き起こす。我々の生体内試験モデルによると、RAはアポトーシス経路を誘導するが、同時に抗アポトーシス経路も誘導し、それらのバランスが古典的なアポトーシスではない代替的な未知の死をもたらすことが観察された[2]。この分野の最近の発表では、カスパーゼは、細胞死を促進することなく神経系を再構築することによっても作用し、その活性は細胞内の位置に依存することが示唆されている[116]。したがって、神経変性組織で見られるカスパーゼの活性型は、死に関係のない役割を果たしている可能性があり、最終的な神経細胞の死は他の致命的なメカニズムによるものである。
アポトーシスは、3つのタンパク質ファミリーによって駆動される抗アポトーシス経路によって阻害されることがある。FLICE-阻害タンパク質、Bcl-2,Inhibitors of Apoptosis Proteins (IAPs)である。IAPは,虚血モデルで神経保護作用を示したり[27],新生児期の神経損傷後に運動ニューロンの死を回避したりしている[28]。IAPは、大人になってからの軸索切断後の神経細胞死を阻止する役割を担っていると提案されている[29]。同じ方向性で,X-linked-IAP (XIAP) の翻訳後修飾は,その抗カスパース3機能を阻害し,PDの病因の一因であるとされている[117].
虚血の有害な影響を部分的に軽減する虚血性プレコンディショニングは,IAPを介して作用し,カスパーゼカスケード活性化後の細胞の生存を可能にする[30]。IAPはまた、新生児軸索切断後の運動ニューロンに対するグリア細胞由来向神経性因子(GDNF)の生存促進効果を媒介する[28]。アポトーシス促進タンパク質を調節することで細胞死を回避する他の分子経路は、細胞外シグナル調節キナーゼ(ERK)とAKTである。この意味で、AKT経路は、アポトーシスをブロックすることで生存を促進するプレーヤーとして記述されている[31]。AKTは、アポトーシス誘導因子であるp53の分解を促進することで、そのアポトーシス促進能力を阻害する[32,33,34]。それ以外にも、カスパーゼがAKTを切断することで阻害することができ、細胞の生存と死を細かく調節していることを指摘している[118]。一方、AKT活性は、Forkhead box protein O (FOXO) 転写因子をリン酸化する。FOXOは、アポトーシスに関連しており [119]、その修飾によって細胞の生存率が上昇する [35]。AKT依存性のFOXOのリン酸化は、その核への侵入を回避し、Bcl-2-interacting mediator of cell death (BIM)やBcl-2 nineteen-kilodalton-interacting protein 3 (Bnip3)などのアポトーシス促進遺伝子の誘導を回避する[119,120,121]。一方、移行後のFOXOの修飾は、細胞内のその転写ネットワークを微調整し、アポトーシスではなくオートファジー誘導の方向へと向かわせる[54,121,122,123]。したがって、FOXOファミリーを特異的に調節することは、アポトーシスを抑制することで神経細胞の生存を促進するための新たな手段となる[54,124]。
最後に、神経細胞の活動は、NMDA依存性の抗アポトーシス遺伝子のアップレギュレーションによる抗アポトーシスの促進因子でもある[125,126]。これらのアップレギュレートされた遺伝子のいくつかは、ミトコンドリアがストレスに対してより耐性を持つようにして[126]、細胞が損傷を生き残るのを助ける。
3.4. アンチアノイキスによる再接着
細胞と細胞外マトリックス(ECM)の間の相互作用は,組織内で正しく機能的に統合されるために不可欠である。この相互作用が阻害されると、細胞はアポトーシスと共通の経路を持つ「アノイキス」と呼ばれるPCDによって死滅する。興味深いことに、内在性のアノイキスプログラムの崩壊は、腫瘍細胞に悪性度を与え、死なずに他の組織に逃れて再付着するのに十分な細胞回復力を与える [127,128]。これらの相互作用の主要なエフェクターはインテグリンタンパク質であり、αサブユニットとβサブユニットの組み合わせによって形成される。この組み合わせにより、リガンドの特異性と細胞内シグナルが決定される。ECMシグナルはインテグリンを介して神経細胞に伝達され、細胞の形状、生存、運動、増殖、発生、神経細胞の結合、シナプス可塑性に不可欠である[129]。インテグリンはまた、成長因子の細胞内シグナル伝達にも重要であり[130]、成長因子は、死に至るメカニズムをブロックすることで神経細胞の生存を調節することでよく知られている。β1インテグリンサブユニットは、細胞とECMの相互作用に不可欠であり、その遮断は、アノイキス[36]や神経細胞のアポトーシスを引き起こすのに十分である[131]。さらに、このサブユニットの細胞内シグナル伝達は、網膜神経節細胞の生存に関係しており[132]、その欠陥は神経変性疾患に見られる[133]。
それにもかかわらず、細胞は、死に対抗するために、抗アノイキスのサブルーチンを発達させていた。これらのサブルーチンは、チロシンキナーゼ、低分子GTPases [128]、NF-κB [134]、PI3K/AKT、癌原遺伝子チロシン-プロテインキナーゼ(Src)またはERK軸、およびオートファジーによって開始される [135,136]。NF-κBは、Bcl-2やIAP-1などの抗アポトーシスタンパク質を誘発することで、抗アノイキスを調節する[135]。一方、PI3K/AKTの細胞生存における役割は広く知られており、分化した細胞の生存に貢献している[36,37]。また、ECMの剥離は、アポトーシスを回避する自己防御機構であるオートファジーを誘導する[135]。これらの証拠は、自己保護メカニズムの複雑なネットワークを示唆している。
アノイキスは、ECMタンパク質を破壊するマトリックスメタロプロテイナーゼ(MMP)の増加により、TBI後の神経細胞死にも見られる[137]。MMPの発現とレベルは神経外傷後に変更され、軸索変性、グリアの瘢痕形成、シナプスのリモデリングにおいて異なる役割を担っている。神経細胞の生存に関しては,MMP9を阻害することで,ラミニンの分解が減少し,脳虚血の保護効果が得られる[38]。また,MMPは神経変性にも関与している[138]。最近の研究では,MMP9を阻害することで,ALSマウスモデルの運動器に保護作用があることが報告されている[39,40]し,ADモデルにおいても同様である[41]。したがって、特定のMMPを阻害する治療法は、間接的に神経細胞内の抗アノイキスプログラムを維持し、神経細胞の生存を促進する。
3.5. 細胞骨格とモータートランスポーター
神経細胞の細胞骨格は、微小管(MT)中間フィラメント(IF)アクチンマイクロフィラメントの3つの異なる構造複合体で構成されている。これらはそれぞれ異なる細胞機能を持つ。微小管は神経突起や樹状突起の動きを制御し[139]、アクチンは細胞の形態を担当し[140]、アクチンマイクロフィラメントは細胞骨格構造に力学的安定性をもたらす[141]。これらの構造複合体の欠損は,神経変性疾患,末梢神経障害,シナプス機能障害などで観察され,成熟した脊柱の消失につながる[141,142,143,144,145,146]。
微小管の動態は高度に制御されたプロセスであり、その不均衡は神経細胞の生存や軸索の性能に壊滅的な結果をもたらす一方で[142]、その安定化は神経細胞の死を阻止し[147]、中枢神経系における軸索の成長を促進する[148]。より詳しく言えば、細胞骨格構造は鉄道であり、キネシンとダイニンのモータータンパク質は、それぞれ前向性または逆向性の輸送によって貨物を運ぶ列車である。したがって、モーター複合体は神経細胞の生存にも不可欠である。キネシンファミリーは,キネシン-1(歴史的にはKIF5cと命名)とキネシン-3(KIF1A,KIF1Bα,KIF1Bβ)のメンバーによって形成されている[149].KIF5cは、運動ニューロンで濃縮されており[150]、その遺伝子の切断は、運動ニューロンの疾患や麻痺と関連している[149,151]。最近では,ALSの発症にも関与していることが示唆されている[152].KIF5cの微小管との相互作用が損なわれると、軸索の変性とそれに続く神経細胞の死を引き起こす [153]。KIF5cの破壊は、ミトコンドリアダイナミクスの障害を引き起こし、その結果、刺激に応じて神経細胞の生存または死をもたらす。さらに、KIF5cはミトコンドリアの機能を微調整し、細胞の健康状態を左右する(後述、セクション3.6.参照)[42]ので、その調節は神経保護を促進する可能性がある。アミロイドβのようなタンパク質の凝集体は,KIF5aの安定性に有害な影響を与え,ミトコンドリアの動きや機能の低下につながる[154].
逆行性タンパク質も神経保護作用を発揮する。それらはダイニンのもので、異なるタンパク質によって形成される多タンパク質複合体であり、p150glue(dynactin1/DCNT1)が最も豊富なサブユニットである。機能不全のダイナクチンサブユニット1(DCTN1)は、ALSマウスモデルとして使用されており、その変異は、軸索輸送の欠陥を引き起こし、マウスにALS様の表現型をもたらす[155,156]。KOマウスでは、加齢に依存した運動ニューロンの死が見られ、これはオートファジーの阻害を伴う[157]。DCTN1は、神経細胞体内の自食胞輸送に明確な役割を持っており、その障害は遠位軸索にアンフィソームの蓄積を引き起こし、AD様の表現型を導く[158]。ダイニンのアダプターであるRab-interacting lysosomal protein (RILP)は,オートファゴソームの形成と輸送に重要な役割を果たしており,その阻害によりオートファゴソームの蓄積が引き起こされる[44]。このように、微小管の機能不全は、キネシンやダイニンの局在異常とともに、リソソームの機能不全を引き起こし、ADにおけるオートファゴソームの蓄積やシナプス前部のジストロフィーを誘発することが観察されている[159]。DCTN1を破骨細胞に過剰発現させると、アポトーシスによる死を防ぐことができることから、モータータンパク質は他の細胞種や組織においても細胞死を回避する役割を持っていることが示唆される[43]。
要約すると、軸索輸送の減少は、多くの神経変性疾患や神経系の損傷後に見られる。その欠陥は、微小管の構造および/または軸索輸送に必要な分子モーターの変化をもたらす[5]。適切な軸索輸送は、神経細胞が正常に機能するために重要であり、このプロセスの障害は、神経細胞の終焉に寄与する。細胞骨格を安定化させたり、モータータンパク質のレベルや活性を高めたりして、細胞の輸送機構を高めることは、神経細胞内の正しいオートファジーの流れを再構築することで、神経保護につながることが実証されている[5]。
3.6. ミトコンドリアの機能不全
神経細胞の機能は、エネルギーとカルシウム(Ca2+)のバランスに依存しており、ミトコンドリアの機能は神経細胞にとって非常に重要である。ミトコンドリアは固定された小器官ではない。ミトコンドリアは、細胞内で形、大きさ、数、局在などを変化させ、細胞の要求に応じて融合や分裂を行う能力を持っている。トリカルボン酸サイクル(TCA)と電子輸送チェーン(ETC)を介した酸化的リン酸化(OXPHOS)によってエネルギーを生産している。OXPHOSが活性化されると活性酸素が発生し、生理レベルでは幅広い機能(分化、オートファジー、免疫反応)を持ち[160]、軸索再生も可能となる[60]。それにもかかわらず、生理的レベルを超えた活性酸素は、脂質、DNA、タンパク質の損傷を引き起こすため有害である。このような変化は、神経変性疾患、脊髄損傷、TBIなどに関連している。また、ミトコンドリアは、神経細胞死を調節する経路に関与することで、神経細胞生存の中核的な調節因子として機能している。
ミトコンドリアは、細胞骨格、モータータンパク質、および適切なアダプターによって細胞内を輸送される。神経細胞では,ミトコンドリアは主に,アダプターであるMiroおよびMilton/trafficking kinesin-binding protein 1 (TRAK)タンパク質によって微小管上を輸送される[161]。神経細胞内でのこれらのミトコンドリアの動きは,シナプス内での最適なフィットネスを維持するために不可欠であり,エネルギーを生産し,Ca2+を緩衝するなどの役割を果たしている。[162]. ミトコンドリアはしばしば小胞体の近くに局在し、ミトコンドリア関連小胞体膜、すなわちミトコンドリア関連膜(MAMs)を形成する。これらの膜マイクロドメインは、脂質の合成/輸送、Ca2+の動態/シグナル伝達、オートファジー、ミトコンドリアの形状とサイズ、アポトーシス、エネルギー代謝など、様々な細胞プロセスを共同で制御し、影響を与える可逆的な境界線である[163]。AD,PD,ALSなどの神経疾患では,MAMが変化している[164]。ミトコンドリアはATGのハブとして機能し,オートファゴソームの形成に必要な膜を供給し,オートファゴソームのフラックスを調節している[165]。また,ミトコンドリアはUPR(mt)を起こし,活性化された経路によっては,ミミズやマウスの寿命延長と関連するが[166],過剰に活性化されると神経変性を引き起こす[167]。
ミトコンドリア機能障害は,ミトコンドリアの数が不足したり,ミトコンドリアに必要な基質が供給されなかったり,あるいはミトコンドリアの電子輸送およびATP合成機構の機能障害によって生じる.高レベルの活性酸素とそれに関連する活性種(RNS)は、ディスムターゼ酵素や抗酸化物質によって中和することができる[168]。ALSやPDなどの神経変性疾患では,これらの酵素や特定のミトコンドリア呼吸複合体の変化が観察されている[169].ミトコンドリアの数や機能の乱れは,細胞のホメオスタシスを著しく低下させ,病気の発症の引き金となる。そのため,細胞は,ミトコンドリアの生合成と消去という相反するプロセスの間で動的なバランスを保つことを追求している。機能不全のミトコンドリアが蓄積したり、ミトコンドリアの生合成が失われたりすると、細胞死が生じる。神経変性を予防するための最近の治療法は,NAD+の調節 [170],エピジェネティック・マークの調節 [171],または脳内のセロトニン軸の調節 [172]によって,ミトコンドリアの生合成を促進することを目的としている.マイトファジーによるミトコンドリアの機能不全のクリアランスは,神経保護をもたらす。マイトファジープロセスの開始に不可欠なPTEN誘導キナーゼ1(PINK1)の過剰発現は,HDのハエモデルにおいて神経細胞の生存率を高めている[45]。また,PINK1変異体を用いたPDモデルでは,NAD+を補充することで神経毒性が軽減される[173].
ミトコンドリアの機能は,活性酸素や細胞の抗酸化反応とクロスリンクしている。そのようにして,転写因子であるNuclear factor erythroid-derived factor 2-related factor 2 (Nrf2)は,細胞保護遺伝子や解毒遺伝子の発現を調節して酸化ストレスや神経炎症に対抗し,神経損傷の軽減を目指している。したがって,神経変性疾患の病気の進行を遅らせるための効果的な操作となりうる[174,175,176]。活性酸素の刺激を受けると,Nrf2はKelch-like ECH-associated protein (Keap1)から解離し,それによって抗酸化酵素の発現が調節される[177]。Keap1がp62のユビキチン化を媒介することが記載されている[178]。Keap1の発現が低下すると、p62が細胞内に蓄積されて細胞障害を引き起こす一方、Keap1を過剰に発現させると、オートファジー経路を介してp62の分解が促進される。一方,p62は,オートファジー経路を介してNrf2を活性化し,p62-Keap1-Nrf2-antioxidant responsive element (ARE) 経路を形成し,活性酸素による酸化的損傷に対抗する[179]。さらに,Nrf2は,ミトコンドリアの生合成の制御に関わる調節ループを形成する。Nrf2は,mtDNA転写の制御に直接関与するペルオキシソーム増殖剤活性化受容体γ共役因子1α(PGC-1α)および核呼吸因子(NRF1)の発現を増加させる。最後に,Nrf2は,マイトファジーの誘導に重要な役割を果たすPINK1の発現を制御していることから[180],細胞の抗酸化能力がミトコンドリアの状態にも影響を与えることが示唆される。
神経変性疾患は,Nrf2経路の阻害とオートファジーの機能不全の両方に関連しており,活性酸素,老化した小器官,ミスフォールドしたタンパク質の蓄積を引き起こす [181,182].神経変性疾患は、多くのタンパク質凝集体と活性酸素に関連しており、神経細胞の保護メカニズムであるp62-Keap1-Nrf2の正帰還軸を誘導する[183,184]。Nrf2の発現は、ADの動物モデルやアルツハイマー病患者の脳では低い[185]。Nrf2のAREへの結合は、疾患の進行中にすぐに起こり、これは活性酸素生成の増加と対応している[186]。Nrf2は、活性酸素の発生とAβを介した活性酸素誘発毒性を減少させることで神経保護を行う[187,188]。HDでは、ミトコンドリア複合体IIの機能障害があり、活性酸素の増加を引き起こしている[48]。HDの初期段階では、Nrf2アゴニストを投与すると、アストロサイトとミクログリアでKeap1-Nrf2-AREを介して重要な細胞保護遺伝子が増加する[189]。アストロサイトの小分子によるKeap1-Nrf2-ARE経路の活性化は、非殺傷性のグルタミン酸毒性に対するニューロンの抵抗性を促進する[46,47,48]。ミトコンドリアの機能、生合成、マイトファジーの変化は、PDの重要な病理学的特徴であり、Nrf2は、ミトコンドリアの品質管理と恒常性を制御する重要な転写因子である[190]。PDでは,Nrf2-AREシステムの活性化が見られ[191,192],その薬理学的活性化はPDの進行を阻止する[49,50].Nrf2の活性化は、スーパーオキシドディスムターゼ1(SOD1)変異タンパク質によって引き起こされる活性酸素や細胞死に対して保護的な役割を果たす。さらに、アストロサイトのNrf2を過剰発現させると、SC 運動ニューロンの生存率が高まり、SOD1トランスジェニックマウスの寿命が延びる[51,52]。また,脳虚血再灌流障害における活性酸素の除去,酸化的損傷の防止,小胞体ストレスの調節には,オートファジーを介したp62とKeap1-Nrf2経路とのクロストークが重要な役割を果たしていると考えられている[53]。
最後に、ミトコンドリアが神経細胞の生存を促進するのは、ミトコンドリアが内外の死のイニシエーターを感知し、ミトコンドリアに収束するシグナルカスケードを誘発し、その後、異なるタイプの細胞死(内在性アポトーシスなど)につながる1つ以上の細胞死経路に再分岐するからである[193]。
4. 全身の変調を狙う
4.1. カロリー制限
カロリー制限(CR)は、さまざまな生物で寿命を延ばし、いくつかの器官に保護作用を及ぼす。CRは、全身環境から様々な細胞内集団まで、生体全体に影響を与える。2010,Kromerと共同研究者は、CRの効果がSIRT1依存性のオートファジーに依存していることを示唆した[194]。一方、PD疾患においてCRはグレリン-AMPK軸による神経保護作用があり、AMPKはオートファジーの重要な誘導因子であることが指摘されている[195]。長期的に CR を維持することは明らかに不可能であることから、治療上の関心は、生体内で CR の 生理学的効果を模倣した新しい CR「模倣薬」(CRM)の発見に向けられた[196]。CRとCR模倣薬の両方とも、ADラットモデルにおいて、オートファジー誘導による認知機能の改善という有効性が確認されており[197]、神経変性を治療するための新たな治療手段となっている。
4.2. 運動
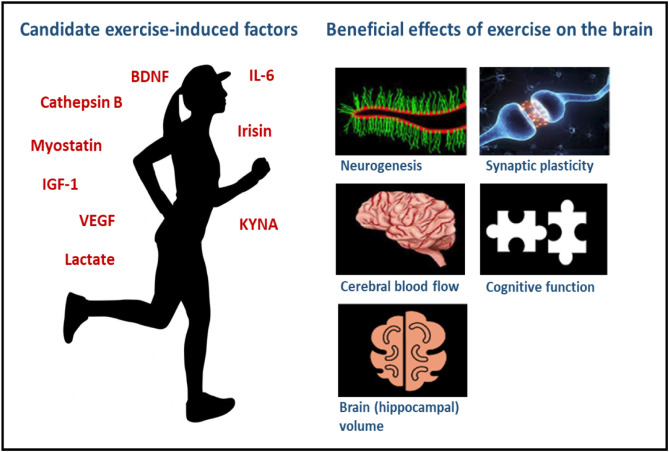
運動は、神経障害性疼痛などの病態を軽減したり、脳卒中モデルの機能的転帰を改善する能力があるため、関心を集めている[198]。また、運動は、炎症反応を抑制し、抗酸化バランスを高めることで、PDの進行を遅らせることができる[199]。運動は、内因性の向神経性因子のレベルを増加させることで作用すると言われている[200,201]。さらに、運動は筋肉ホルモンの分泌を調節し、脳の保護作用、神経新生を促進し、脳の老化を改善する[202]。実は最近、同じホルモンであるイリシンが骨形成に関与していることが記載されており[203]、運動が体全体に影響を与えることがわかる。
5. 効果的な神経保護剤の発見:何があり、どこへ行くのか
神経変性疾患に共通する特徴として、UPRの非正常な活性化、オートファジープロセスの蓄積、ミトコンドリアの機能不全などが挙げられる。これらが重なると、神経細胞を圧倒し、その死を引き起こすことになる。効果的な神経保護剤は、これらのメカニズムを修正し、老化や障害に対する完全な回復力を細胞に与えるものでなければならない。そのためには、細胞内の分子ネットワークを完全に修正し、機能を完全に回復させる必要がある。ALSに対するリルゾール[204]のような承認された薬剤や、ALSに対するラパマイシン[204]、ADに対するスペルミジンやDH[205,206]のような現在進行中の臨床試験は、これらの変性プロセスのうちの1つだけをターゲットにしており、神経細胞は他のプロセスに圧倒されている。これらの治療法は有益な効果をもたらすかもしれないが、私たちは、1つの標的だけでなく、異なる分子経路を支持する遺伝学的または薬理学的なアプローチを見つけることを提案する。
SIRT1,BIP、ATG5などのタンパク質を特異的に過剰発現させると、神経損傷後の神経細胞の生存が促進され、神経変性疾患においても神経保護効果を発揮する。これらは主にUPRやオートファジーのネットワークを微調整するものである。トランスジェニックマウスやウイルスベクターを用いて SIRT1 を活性化すると、ALS、AD、HD などのさまざまな神経変性疾患 [207,208,209] において、また神経損傷後にも神経保護効果があることがわかっている [55]。SIRT1のデアセチラーゼ活性は、オートファジー、PERKの減少によるUPRの調節、ATF6の切断の増加など、さまざまな内因性の保護メカニズムを支持し[23,210]、抗アポトーシス効果を持ち、AKT活性を調節してアノイキスを抑制する[211,212]。したがって,それを正確に調節することで,細胞の回復力を高めることができるかもしれない。私たちの最近の研究から、SIRT1 脱アセチル化酵素の活性を調節することは、細胞の回復力を実現するための分子ネットワークに不可欠なノードであると結論づけている[54,55]。最後に、BIPの過剰発現は、凝集体を防ぎ、オートファジーやマイトファジーを誘導することから[99]、BIPの調節は、異なる神経保護経路をクラスター化する有効なアプローチでもある。
6. おわりに
内在性の神経保護機構を高めることは、神経変性疾患の治療や神経外傷後の組織の恒常性維持のためのエキサイティングな治療の道を開くものである。この分野は今日では未開拓の分野であるが、具体的な病態生理学的特徴を阻害するよりも、より効果的な生物医学的結果をもたらす可能性がある。したがって、遺伝学的、薬理学的、あるいは全身的な調節療法によってそれらを支持することで、病態の進行を遅らせ、機能回復を促進することができる。最適な治療戦略には、内因性の保護メカニズムを具体的に調整して、ネットワーク全体を再構築し、保護を実現することが必要である。
