
コンテンツ
https://ijvtpr.com/index.php/IJVTPR/article/view/86/224

Corinne A. Michels, PhD. niel Perrier, BSEE; Jeyanthi Kunadhasan, MD; Ed Clark, MSE; Joseph Gehrett, MD; Barbara Gehrett, MD; Kim Kwiatek, MD; Sarah Adams, RN; Robert Chandler, MD; Leah A.
Chandler, MD; Leah A. Stagno, BS, AAS; Tony Damian CMT, CST, RMT; Erika Delph, RPh; and Chris Flowers, MD
著者全員 DailyClout Pfizer/BioNTech Documents Investigations Team 3; PO Box 24; Millerton, NY 12546 https://dailyclout.io/; 対応著者:Eメール corinne.michels@icloud.com
要旨
ここで報告する分析はユニークである。それは、ファイザー/バイオエンテックのBNT162b2 mRNAワクチン臨床試験(C4591001)のオリジナルデータを、臨床試験のスポンサーとは無関係のグループが初めて調査したものである。我々の研究は、臨床試験のフェーズ2/3が開始された2020年7月27日から、公式の6カ月中間報告の終了日である2021年3月13日の間に死亡した38人の臨床試験被験者の法医学的分析である。臨床試験フェーズ2/3には44,060人の被験者が参加し、2群に均等に振り分けられ、BNT162b2 mRNAワクチンまたは0.9%生理食塩水からなるプラセボのいずれかを投与された。BNT162b2 mRNAワクチンが米国FDAから緊急使用許可を得た20週目に、プラセボ群の被験者にはBNT162b2ワクチンの接種を受け、ワクチン接種群に切り替える選択肢が与えられた。報告された20,794人の非盲検プラセボ群被験者のうち、19,685人が少なくとも1回のBNT162b2ワクチンの接種を受けた。
驚くべきことに、この試験の33週間における1週間当たりの死亡者数を比較したところ、最初の20週間(プラセボ対照部分)においては、ワクチン接種群とプラセボ群の死亡者数に有意差は認められなかった。第20週以降、プラセボ群の被験者の盲検化が解除され、その大多数がBNT162b2の注射を受けた後、プラセボに固執する被験者の死亡数は減少し、最終的にはプラトーとなった。BNT162b2ワクチン接種群の死亡率は同じであった。
われわれの分析では、6カ月中間報告書に記載された被験者データとファイザー社/バイオエヌテック社の試験実施施設管理者が執筆した出版物との間に矛盾があることが明らかになった。
最も重要なことは、BNT162b2ワクチン接種群では、プラセボ投与群に比べ、心イベントによる死亡数が3.7倍以上増加している証拠を発見したことである。症例報告書への被験者の死亡の報告が遅れたために、心臓の有害事象のシグナルが不明瞭になり、ファイザー社/バイオエヌテック社の緊急使用許可が異議を唱えられることなく進行することができた。
キーワード
BNT162b2ワクチン、心臓イベント、プラセボ対照臨床試験、COVID-19、ファイザー/バイオエヌテック
はじめに
COVID-19と呼ばれる。「新規」コロナウイルス呼吸器疾患のヒト症例が、2019年12月に中華人民共和国武漢市で報告された。しかし、現在では、このウイルスが2019年秋には早くも米国で循環していたことを示唆する重要な証拠がある(Basavaraju et al., u21年;Huang et al., g20年;Wu et al., u20)。2020年1月30日、世界保健機関(WHO)はCOVID-19の流行を国際的に懸念される公衆衛生上の緊急事態と宣言し、2020年3月11日には世界的な「パンデミック」と宣言した。米国では、アレクサンダー・アザール保健福祉長官(当時)が2020年3月10日、COVID-19に対する医療対策のため、公衆準備・緊急事態準備法(PREP法)に基づく公衆衛生緊急事態宣言を発令した。この法律は米国議会で可決され、15年前の2005年12月にジョージ・W・ブッシュ大統領によって署名された。この法律は、保健福祉長官によって宣言された「公衆衛生上の緊急事態」に対応して製造される可能性のある「医療対抗措置」がどのようなものであれ、ワクチン製造業者が責任を問われないよう、事実上不可侵の盾を提供するものであった(Office of the Secretary of Preparedness and Response, April 13, 2021; Martinez, 2021)。こうして、COVID-19に対するワクチン開発競争(Fleming, 2021; Altman et al., 2022)は世間一般に語られるようになり、通常のワクチン開発で必要とされる面倒で時間のかかるプロセス(基礎となる動物実験室での研究、規制当局の審査を経た製造・流通計画の確立)は回避されることになった。
複数の製薬企業が、開発、製造、動物実験、そして大規模な人体実験の実施という、「ワープ・スピード作戦」と呼ばれる挑戦に飛びついた(トランプ大統領、2020年11月13日;米国政府会計検査院、2021年2月11日)。2020年12月10日、HHSのアザール長官の宣言からわずか9カ月後、人体実験開始から6カ月も経たないうちに、米食品医薬品局(FDA)はファイザー/バイオエヌテック社に実験的なBNT162b2 mRNAワクチンの緊急使用許可を与えるという非常に物議を醸す決定を下した。残念ながら、この実験的製品が「安全かつ効果的」であり、「感染や重篤な疾病を予防した」という証拠は、2022年6月まで公開されなかった。
BioNTech社はドイツのバイオテクノロジー企業で、がんや希少疾患、いわゆる。「希少疾病」(他のメーカーが治療対象としていない疾患)の患者特異的治療のための活性免疫療法や、標的タンパク質の置換技術を開発・製造している。2020年初頭、バイオエヌテックはファイザー社と提携し、バイオエヌテックの新規BNT162b2 mRNA SARS2-CoVワクチンの有効性と安全性を判定する臨床試験を実施した。ファイザー社/バイオエヌテック社は、「A Phase 1/2/3 Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals(健康な個人におけるCOVID-19に対するRNAワクチン候補の安全性、忍容性、免疫原性、有効性を評価するための第1/2/3相試験)」と題する多国籍共同臨床試験を米国FDAに申請した(https://Cdn.Pfizer.Com/Pfizercom/2020-11/C4591001_Clinical_Protocol_Nov2020.Pdf)。本申請は承認され、2020年4月に3相試験の第1相試験の被験者登録が開始された。フェーズ1の目的は、ワクチンの至適投与量を決定することである。フェーズ2/3試験は、43,548人以上の被験者を含む「安全性と有効性」フェーズで、2020年7月27日に開始された。
2020年11月20日、Pfizer/BioNTech社は米国FDAに未承認製品審査メモの緊急使用許可申請書(EUA)を提出した(https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)。申請書には、2020年11月14日のデータカットオフまでの臨床試験結果が記載されていた。FDAは2020年12月11日にEUA申請書のコピーをウェブサイトで公開した。これは、同社のBNT162b2 mRNAワクチンの安全性と有効性を裏付けると報告された臨床試験データを、一般市民や医療関係者が評価する最初の機会となった。Polackら(2020)は2020年12月10日、”Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine “と題する学術論文を発表した。Polackら(2020)の著者は、6カ国以上、153の臨床試験施設の施設管理者で構成されている。Fernando P. Polack医学博士はアルゼンチンの臨床試験施設の治験責任医師兼施設管理者であり、筆頭共著者であるStephen J. Thomas医学博士は臨床試験C4591001の主任治験責任医師であった。
従って、これらの出版物の著者は臨床試験の結果を熟知している必要があり、また熟知しているべきだった。2021年9月15日、サイト管理者の同じグループは、「BNT162b2 mRNACOVID-19ワクチンの6カ月までの安全性と有効性」(Thomas et al.) 米国FDAの知識と承認により、オリジナルの臨床試験データは75年間、世界の医学研究コミュニティが研究できるようになることはなかった。
80人以上の公衆衛生担当官と医学研究者からなる非営利連合であるPHMPT(Public Health and Medical Professionals for Transparency)は、2021年9月、テキサス州フォートワースの連邦地方裁判所に、FDAがファイザー(Comirnaty)のCOVID-19 mRNAワクチンを認可した際に依拠したオリジナルの臨床試験データを入手し、普及させるために情報公開訴訟を起こした。この裁判を主導した米国人医師の一人であるアーロン・ケリアティ医学博士の言葉を引用しよう: 「私たちのグループは、臨床試験のデザイン、臨床試験期間の短さ、有害事象の市販後調査のためのパッチワークシステムに懸念を抱いていた。PHMPT事件は承認された。FDAの反対を押し切り、連邦裁判所の判事はファイザーの臨床試験データと文書を月5万5000件のペースで迅速に公開するよう命じた。データの公開は2022年6月初旬に「透明性文書のための公衆衛生および医療専門家」のサイトで開始され、完了までに8カ月かかると予測されていた。残念なことに、見積もりよりもはるかに時間がかかっており、このサイトには文書がダウンロードされ続けている。これらの文書の圧倒的な大きさと複雑さに刺激され、医療専門家、科学者、データアナリスト、統計学者、弁護士などからなるDailyCloutファイザー/バイオエヌテック文書分析ボランティアが結成され、ファイザー/バイオエヌテック臨床試験文書の分析に時間とスキルを提供している。
チーム3は、データ調査に専念するこれらのボランティアのサブセットである。
本報告書は、ファイザー社/バイオエヌテック社の6カ月中間報告書(6カ月有害事象中間報告書C4591001)に記載されている、2020年7月27日の試験開始から6カ月中間報告書のデータ終了日である2021年3月13日までの間に死亡した38名の被験者に焦点を当てたものである。我々の分析により、6カ月中間報告書に記載された被験者データとファイザー/バイオインテックがFDAに提出したこのデータに関する資料との間に重要な矛盾があることが明らかになった: ファイザー/バイオエンテックのFDA緊急使用許可申請書(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)、Polackら(Polack 2020)、Thomasら(2021)である。最も憂慮すべきことは、ファイザー社/バイオエヌテック社が報告していない、BNT162b2ワクチンを接種した被験者の心臓イベントによる死亡数が3.7倍以上増加したという証拠を発見したことである。もしこの情報が重要な時点で判明していれば、BNT162b2 mRNAワクチンの安全性に疑問を呈し、EUAの承認を遅らせ、世界的な展開の中で一般への推奨を変更するのに十分であったかもしれない。
方法
ファイザー/バイオエヌテック社のオリジナル文書は、PHMPT(Public Health and Medical Professionals for Transparency)のウェブサイト(https://phmpt.org/pfizers-documents/)で入手できる。このサイトから以下の文書をダウンロードし、分析の主要なデータ源とした。
- 6カ月中間報告書(16.2.7.4.1有害事象のリスト-16歳以上の全被験者)(6-Month Interim Report (16.2.7.4.1 Listing of Adverse Events – All Subjects≥16 有害事象の6カ月中間報告 C4591001)
- 無作為化スキームと実際に受領したワクチン(16.1.7.1 無作為化スキームと実際に受領したワクチンのリスト-16 歳以上の全被験者)(Listing of Randomization Scheme and Actual Vaccine Received) (無作為化スキームと実際に受領したワクチンのリスト
- 中止された被験者のリスト(16.2.1.1 ワクチン接種および/または試験から中止された被験者のリスト-16 歳以上の全被験者)(中止された被験者のリスト)
- 臨床安全性の6カ月要約(2.7.4 臨床安全性の要約(要約臨床安全性6カ月報告書)
- 被験者の死亡に関する。6 ヵ月中間報告からのナラティブ報告(125742_S1_M5_5351_c4591001-interim-mth6-narrative-sensitive) )
文書 16.2.7.4.1 Listing of Adverse Events (6-Month Interim Report of Adverse Events C4591001)と 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received (Listing of Randomization Scheme and Actual Vaccine Received)を PDF から Excel ファイルに変換し、1 つの Excel Pivot Table ファイルに統合した。これにより、特定の被験者 IDの重複エントリーが削除され、有害事象の特定の優先用語を検索できるようになった。従って、有害事象を示した全ての被験者をリストアップした単一の検索可能なファイルにおいて、投与された用量の種類(BNT162b2 mRNAワクチンまたはプラセボ)、各用量が投与された日付、有害事象の発症日、有害事象のPreferred Term、有害事象に関する試験施設の医師の診断、および事象が試験に関連しているかどうかのファイザーの安全性担当医師の判断を決定することができた。
追加情報は以下から得た
- Pfizer/BioNTech Clinical Trial C4591001 – A Phase 1/2/3, Placebo-Controlled, Randomized, Observer-blind, Dose-finding Study to Evaluate the Safety, Tolerability, Immunogenicity and Efficacy of SARS-COV-2 RNA Vaccine Candidates against COVID- 19 in Healthy Individuals (https://classic.clinicaltrials.gov/ct2/show/NCT04368728).
- 未承認製品に対する緊急使用許可(EUA)(Emergency Use Authorization for an Unapproved Product Review Memorandum Product-Review-Memorandum)
- Analysis Data Reviewer Guide – BLA Analysis for Participants≥16 Years of Age, BioNTech SE and PFIZER INC, Study C4591001 (Analysis Data Reviewer Guide BLA Analysis for Participants≥16 Years of Age BioNTech SE and PFIZER INC. Study C4591001 Content/Uploads/2022/03/125742_S1_M5_c4591001-A-Adrg.Pdf#page=85)
また、DailyCloutウェブサイト(https://vaccines.shinyapps.io/abstractor/)で利用可能なAbstractor検索ツールを使用して、特定の被験者ID、Preferred Terms、または臨床試験データに固有の症例報告書、ナラティブ、およびその他の文書を検索した。
予想死亡数は以下のように推定された。Pfizer/BioNTech 6-Month Interim Report (6-Month Interim Report of Adverse Events C4591001, n.d.)および無作為化スキーム(Listing of Randomization Scheme and Actual Vaccine Received, n.d.)を用いて、153の各臨床試験実施施設で登録された各年齢層の被験者数を割り出した:15-24 歳、25-34 歳、35-44 歳、45-54 歳、55-64 歳、65-74 歳、75-84 歳、85 歳以上。2020年の10万人当たりの年齢調整死亡率は、National Center for Health Statistics Data Brief 427 (Murphy et al., 2021)から得た。各カテゴリーの被験者数に死亡率を掛け合わせ、各年齢層における各臨床試験実施施設で予想される死亡数を推定した。これらの推定値を合計し、33/52.2を乗じて33週間の試験期間を調整した。
結果
2022年7月1日、ファイザー社/バイオエヌテック社は、「16.2.7.4.1 Listing of Adverse Events – All Subjects ≥16 Years of Age (6-Month Interim Report of Adverse Events C4591001)」と題する臨床試験の最初の33週間(2021年7月27日~3月13日)に発生した有害事象に関する報告書を発表した。この文書の3640~3642ページにある16.2.7.7項は「死亡例のリスト-16歳以上の全被験者」である。この最初の期間に38人の被験者が死亡したと報告されている。この文書には、被験者ID、死亡時の性別、年齢、死亡日、および死亡した38人全員の主死因、8人の副死因が記載されている。
文書 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received – All Subjects≥16 Years of Age (Listing of Randomization Scheme and Actual Vaccine Received)を用いて、死亡した各被験者のワクチン接種状況(BNT1626b2 mRNAワクチンまたはプラセボ)と最初の注射を受けた日(投与 1日)を確認した。この2つのPDFファイルを使いやすくするため、Excelファイルに変換し、検索可能なピボットテーブル形式に統合した。
試験開始33週間の概要
Pfizer/BioNTech BNT162b2 mRNAワクチンの臨床試験フェーズ2/3は2020年7月27日に開始された。この日から、スクリーニングにより適格と判断された被験者は、臨床試験のワクチン接種群または対照群に均等に無作為に割り付けられ、それぞれBNT162b2 mRNAワクチンまたは0.9%通常生理食塩水のプラセボのいずれか1用量が投与された。無作為化された被験者のほぼ全員が、2020年11月14日(第16週)までに投与量2を受けた。この16週目までのワクチン接種期間および20週目までの追跡調査期間の初期週において、被験者は有害事象(AE)の発生について追跡調査され、定期検診のために試験実施施設に戻った。Pfizer/BioNTech社では、この期間を「盲検化プラセボ対照期間」と呼び、2020年7月27日から12月10日までのイベントを含む。「盲検化」とは、被験者がBNT162b2ワクチンと生理食塩水プラセボのどちらを接種したかを知らないことを意味する。被験者は無作為に試験群に振り分けられたため、試験群間の違いは、被験者が治療を受けたかプラセボを受けたかだけである。したがって、プラセボは「対照」の状況を表している。プラセボ対照臨床試験では、各群の結果を比較することができ、結果の違いはすべて治療(この場合はBNT162b2ワクチン)に直接帰することができる。
2020年7月27日の他に、4つの重要なランドマーク日がある。
- 2020年11月14日(第16週終了)は、ファイザー/バイオエンテックが米国食品医薬品局(FDA)に申請したBNT162b2 mRNAワクチンの緊急使用承認(EUA)のデータカットオフ日であった(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)。
- この申請は2020年11月20日にFDAに提出され、2020年11月14日までに153の臨床試験施設からファイザー/バイオエヌテック社に提出されたすべてのデータが含まれている。データは治験施設から毎週数回収集された。2020年11月14日は土曜日であったため、1週間後の11月20日の申請で報告されたデータは完全に最新のものであったと推測できる。
- 2020年12月10日(第20週終了)ファイザー/バイオエンテックはFDAのワクチンおよび関連生物学的製剤諮問委員会(VRBPAC)に結果を報告した。ブリーフィング文書(Pfizer-BioNTech COVID-19 Vaccine FDA Briefing Document VRBPAC December 10, 2020 Meeting https://Archive.Org/Details/Vrbpac-12.17.20-Meeting-Briefing-Document-Fda-0)とこの会議のビデオが見られる
- 2020年12月11日、ファイザー/バイオエヌテックが「オープンラベル」または「非盲検」期間と呼ぶ期間が始まった。彼らのEUA申請は2020年12月11日にFDAによって承認された。FDAはまた、臨床試験におけるすべての被験者の盲検化を解除するという彼らの要求を承認した。非盲検化とは、すべての被験者が投与1回目と2回目にBNT162b2 mRNAワクチンを接種したか、プラセボを接種したかを知らされることを意味する。盲検化されていないプラセボ被験者には、BNT162b2 mRNAワクチン、用量3と4が提供された。「オープンラベル」という用語は、バイアル瓶のラベルを被験者に見せることで、今ワクチンを接種していることを保証できることを示すために使用された。
2020年12月11日から2021年1月24日までを「非盲検フォローアップ期間」と呼ぶ。すべてのワクチン接種状況の被験者は、一般的な健康状態およびCOVID-19の感染状態に関して24カ月間追跡され続けたため、「フォローアップ」と呼ばれるようになった。必要なフォローアップの予約が予定され、必要であれば、被験者は緊急医療のために受診した。入院と死亡については治験実施医療機関に報告された。死亡は電子報告システムを通じてファイザー/バイオエンテック社に直ちに報告された。2021年1月24日を選択したことについての説明はないが、この日付はThomasら(2021)で報告されたデータの分析から明らかになった。
- -2021年1月25日は、ファイザー社が「非盲検観察期間」と呼ぶ期間を開始する日であり、6カ月中間報告(有害事象6カ月中間報告C4591001)の2021年3月13日のデータカットオフ日に終了した。全被験者の一般的な健康状態に関する観察は、投与1回目の投与から24カ月後まで継続されることになっていた。
試験の各段階における被験者数を示すフローチャートは、Polackら(2020)およびThomasら(2021)に示されている。報告された人数は、発表された論文内でも、また中止された被験者のリスト(Listing of Discontinued Subjects)に基づいて我々が決定した人数とも、しばしば内部的に一致していないことがわかった。とはいえ、無作為に割り付けられ、用量1を受けたフェーズ2/3の被験者数は、BNT162b2ワクチン接種群22,030人、プラセボ群22,030人、合計44,060人であることを念頭に置くことは重要である。この投与回数は、すべての参加者に同じ日に投与することはできず、再診も、予定されているか否かにかかわらず、同じ日に行うことはできなかった。その代わり、すべての面会は上記の期間中に数週間にわたって行われた。さらに、当初プラセボを投与され、2020年12月11日時点でまだ治験被験者であった20,794人のうち、盲検化解除後にワクチン接種を受けたのは19,685人だけであった。これらの人々へのBNT162b2ワクチンの投与は、第20週から第33週に及んだ。
6カ月の安全期間中の死亡
図1は、Pfizer/BioNTech社の6カ月間中間報告(6-Month Interim Report)に記載された期間中の週ごとの被験者死亡数をプロットしたものである。
6-Month Interim Report(6-Month Interim Report of Adverse Events C4591001)で報告された期間中の週当たりの被験者死亡数をプロットしたものである。
16.2.7.7. この文書は 2021年 4月 1日に作成されたので、死亡日の正確な記載があるはずである。上述した重要な試験日を図1に示す。
第1週は2020年7月27日(月)に開始され、被験者が投与量1の投与を開始した日である。EUA申請データのカットオフ日である2020年11月14日は第16週の終わりである。Pfizer/BioNTech社のEUAが承認された2020年12月11日は、第20週の土曜日であった。6カ月中間報告(6カ月有害事象中間報告C4591001)のデータカットオフ日である2021年3月13日は33週目の土曜日であった。この33週間の期間は、上述し図1に示したように、3つのブロックに分けられた:盲検プラセボ対照期間(2020年7月27日~12月10日)、非盲検フォローアップ期間(2020年12月11日~2021年1月24日)、非盲検観察期間(2021年1月25日~3月13日)。これらの期間の重要性は本報告書の後半で明らかになる。
本試験のBNT162b2ワクチン群とプラセボ群における被験者の死亡数を図1に分けてプロットした。また、各週末に決定された各群の累積死亡数のプロットも示している。最初のプラセボ群死亡は5週目、最初のBNT162b2ワクチン群死亡は7週目であった。試験開始後12週間の死亡例は3例のみで、残りの21週間では35例であった。この対照的な結果は、おそらく試験が進むにつれてワクチン接種を受けた人の数が増加したためであろう。最終的には、当初は生理食塩水のプラセボのみを受けたが、後にBNT162b2注射を受けた人に加わることを選択した約5%を除くすべての参加者が含まれる。図1の結果から2つのことがわかる。
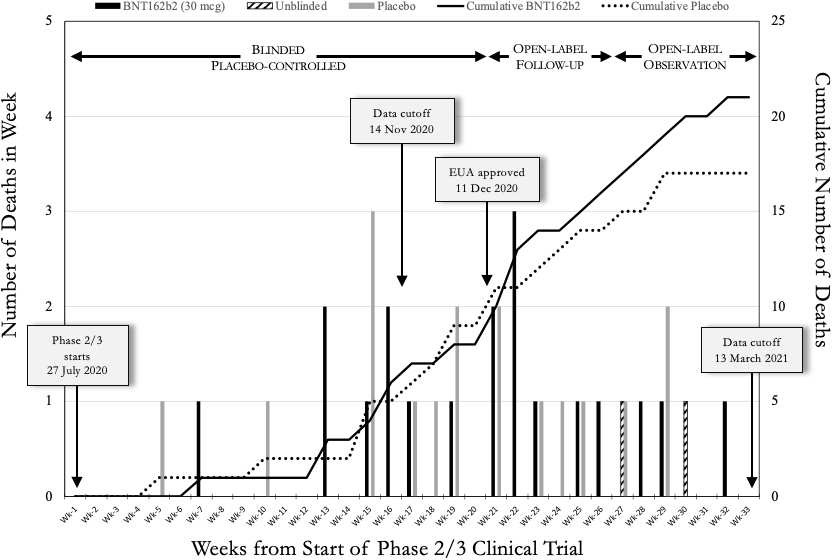
図1:Pfizer/BioNTech臨床試験C4591001の最初の33週間における週ごとの被験者の死亡。死亡した38人の被験者を、2020年7月27日(月)から2021年3月13日(土)までの33週間における死亡日順に示した。グラフの横線間の各棒は、1回の死亡を表している(同じ日に3人以上が死亡したことはない)。黒い実線の棒は死亡したBNT162b2ワクチン接種被験者、灰色の実線の棒はプラセボ被験者、ハッチングを施した棒は2020年12月11日以降にBNT162b2注射を受け入れた盲検化されていないプラセボ被験者である。BNT162b2接種者の累積死亡数を左から右へ上昇する実線で示し、点線はプラセボのみの接種者の累積死亡数を示す。BNT162b2注射を選択して死亡したプラセボ投与者は、BNT162b2投与者としてカウントされている。左から右に3つの試験期間を示す: 盲検プラセボ対照期間、2020年7月27日~12月10日、非盲検フォローアップ期間、2020年12月11日~2021年1月24日、非盲検観察期間、2021年1月25日~3月13日である。
38人の死亡は驚くほど少ない
44,060人の被験者が1回目の投与を受けたという臨床試験参加者の多さを考えると、6カ月中間報告(有害事象に関する6カ月中間報告 C4591001)で報告された38人の死亡例は、特にCOVID-19のパンデミックのさなかでは、予想外に少ないように思われた。このことを検証するために、方法の項で述べたように、2020年の米国の年齢調整死亡率(Murphy et al., y21)に基づいて死亡者数を推定した。我々の推定では、年齢調整死亡率は臨床試験C4591001に参加している他の国の施設における米国の死亡率と同様であると仮定している。153の臨床試験施設のうち、132の施設が米国にあり、被験者の約80%が米国人であった。この注意点を念頭に置いて、我々は2020年7月27日から2021年3月13日までの試験期間中に222人の被験者の死亡が発生するはずであったと推定した。実際の死亡数(38例)は予想数の約18%であった。小規模施設を除き、どの施設でも死亡数は予想より少なかった。
被験者の死亡数が少ないことの説明として考えられるのは、C4591001における「中止された被験者」の多さであり、無作為化された被験者の4.2%である。被験者中止の理由がいくつか挙げられている。その中で最も憂慮すべきは”Lost to Follow-up “である。予定された面会や他の必要なプロトコル作業に現れなかった被験者はLost to Follow-upとみなされた。プロトコルの手順によれば、治験施設スタッフは電話や配達証明郵便、あるいは緊急連絡先を通じてこれらの被験者への接触を試みたが、何度も試みた後、結局その努力は放棄された。6カ月間の中間報告期間中に”Lost to Follow-up “とされた被験者は395名であった: BNT162b2ワクチン群で178人、プラセボ群で217人であった。このうち、203名(BNT162b2ワクチン群99名、プラセボ群104名)は、ファイザー/バイオエヌテックEUA申請のデータカットオフ日である2020年11月14日以前に喪失し、192名(BNT162b2ワクチン群79名、プラセボ群113名)は、それ以降2021年3月13日までに喪失した。
各臨床試験実施施設において、登録被験者数に対する追跡調査対象者数を比較した。各施設の平均被験者数は約300人で、1,200人から4,500人の間に4施設が登録された。153施設のうち96施設では、追跡不能となった被験者は1名以下であった。別の34施設では2-5人の被験者が追跡不能になったと報告した。20人以上の被験者の追跡調査不能を報告した施設は4施設あり、治験施設の被験者の4-5%であった:611人中27人、611人中32人、572人中24人、412人中22人。これらの数字は取るに足らないものではなく、この安全性試験期間中に報告された死亡例の少なさを容易に説明できるものである。各被験者の状態を把握することの重要性を考慮すれば、これらの被験者の所在を突き止めるための努力がもっと必要であったはずである。さらに、Pfizer/BioNTech社は治験実施施設の監督責任を負っていた。過剰な数の追跡不能者を出した施設は、その実績について評価されるべきだった。
全死因死亡率はBNT162B2ワクチン接種によって減少していない
図1は、本試験のBNT162b2ワクチン接種群とプラセボ群における累積死亡数のプロットが、最初の約20週間(2020年7月27日から2020年12月11日)において互いに重なっていることを明確に示している。これは全く予期せぬ所見である。2020年の秋、COVID-19の広がりはピークに達していた。BNT162b2ワクチンが命を救ったというのであれば、ファイザー社/バイオエヌテック社は、ワクチン接種群におけるCOVID-19死亡率の減少による全死因死亡率の減少を示すべきだった。図1は、第20週以降のプラセボ累積プロットが、BNT162b2ワクチン接種群のそれを明らかに下回っている点で、この結論を否定している。第20週は、盲検化が解除された時点、すなわち、被験者にワクチンを接種したかプラセボを接種したかが知らされた時点である。2020年12月11日頃から2021年2月にかけて、この段階に入った20,794人のプラセボ参加者のうち19,685人がBNT162b2ワクチン接種を選択した(Thomas et al., s21)。プラセボによる死亡者数の増加速度が遅くなり、30週目でプラトーになったのは、プラセボ群の規模が徐々に縮小したためと考えられる。もしBNT162b2ワクチンが95%有効であれば、このプロットは逆転していたであろう。
被験者の死亡原因
表1は、図1に示した38人の死亡した被験者の情報を詳細に示している。表1のデータソースは図1と同じで、有害事象の6カ月中間報告書C4591001と無作為化スキームと実際に受けたワクチンのリストである。BNT162b2 ワクチン接種被験者とプラセボ被験者を分けて記載した。各グループ内では、被験者は死亡日順に記載されている。少なくとも1回のBNT162b2ワクチン接種後に死亡した非盲検プラセボ被験者2名は、BNT162b2ワクチン接種群とともに記載され、薄い灰色で強調表示されている。表1には、死亡した各試験参加者の無作為化時に割り当てられた被験者ID、ならびに性別、死亡時年齢、および死亡日が記載されている。表1のデータは確認することができ、DailyClout Abstractorを使用して被験者IDで検索することで、症例報告書と被験者の死亡に関する説明書の原本を入手することができる。
表1には、有害事象の6カ月中間報告書C4591001に記載された一次死因と二次死因も記載されている。各臨床試験実施施設の治験責任医師は、被験者の症例報告書に記載するために、すべての被験者の医療情報をファイザー/バイオエヌテック社に報告する責任があった。被験者の死亡に関する報告書(Narrative Reports on Subject Deaths)には、被験者の死亡に至る時系列と死亡にまつわる状況が要約されている。症例報告書とNarrativeを併用することで、有害事象報告書C4591001の6カ月中間報告書の記載だけでは得られない死因に関する新たな知見が得られる。
死亡は直ちに報告された。Pfizer/BioNTechは、安全性モニタリング試験用の医学用語を列挙した標準化リソースであるMedDRAコード辞書に基づくPreferred Termsのリストを使用した。このリストには1,519の異なるPreferred Termsが含まれているが、驚くべきことに「死」はその中には含まれていない。使用されたPreferred Termsは曖昧で重複していることが多く、診断の混乱を招いた。後述するように、用語に特異性がないため、研究者は、特に心イベントの可能性がある症例において、真の死因を明らかにするために剖検を必要とすることを避けることができた。心イベントは心筋および血管系に限定して定義した。これには、心筋梗塞、うっ血性心不全、心停止、心臓突然死などのPreferred Termsが含まれる。心筋梗塞は心筋組織に対する特異的な低酸素性の不可逆的傷害である。この診断は剖検によってなされるのが最良であるが、血液中のトロポニンの存在など、心臓障害の他の指標も利用可能である。トロポニン値が報告されることもある。
これらの38症例の多くでは、症例報告書や説明報告書に記載された文書が死因診断を十分に裏付けていなかったり、剖検によって心筋梗塞の可能性を除外することができなかった。ファイザー/バイオインテックの医師と治験施設の医療スタッフとの頻繁なやり取りは、症例報告書を見れば明らかである。ナラティブレポートはより短いが、どの被験者が自宅または医療施設で突然死(SAD)または死体で発見(FD)されたのか、また死因を確認するために剖検が行われたかどうかを表1で報告するための詳細を提供している。事故死を除けば、予期せぬ死亡は心筋梗塞や脳卒中であることが多い。残念ながら、ほとんどの場合、剖検は行われなかったか、剖検結果を確認することができなかった。
表1. Pfizer/BioNTech臨床試験被験者*の診断された死因
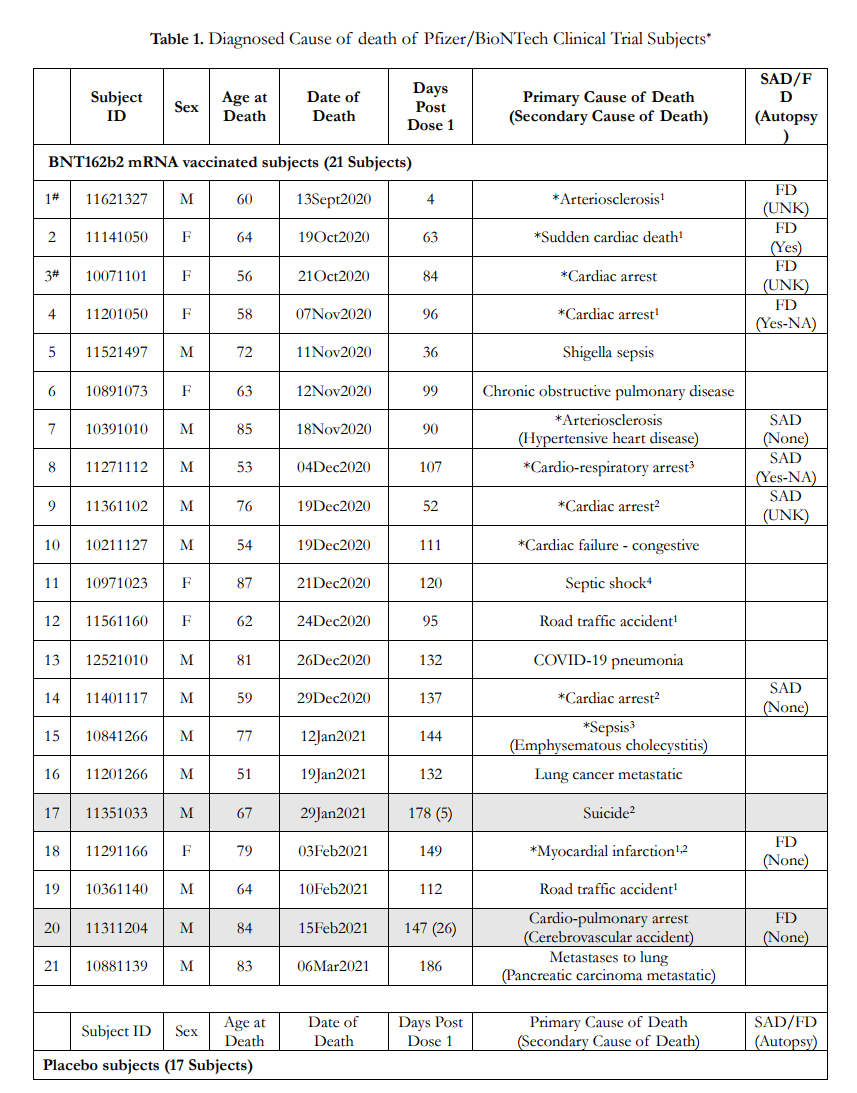
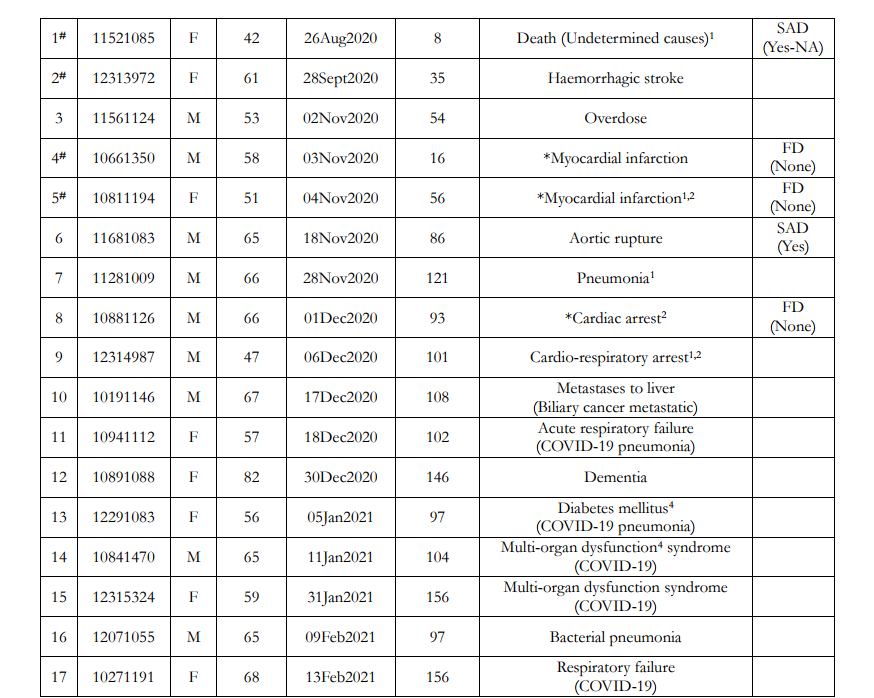
*2020年7月27日から2021年3月13日の期間に死亡した38名の被験者について、臨床試験群、BNT162b2ワクチン接種群、プラセボ群に分けて記載し、初回投与(投与1回目)後の死亡日順に番号を付した。被験者11351033および11311204の行は、これらの被験者が非盲検プラセボ被験者であり、当初のプラセボ群から、非盲検後にBNT162b2ワクチン接種を受けたことを示すため、灰色で影を付けている。括弧内は、これらの被験者がBNT162b2ワクチンの投与量である投与量3を受けた後、死亡するまでの日数である。SADは成人突然死である。FDはFound Deadを示す。剖検は行われなかった(None)、行われたか不明(UNK)、行われたが結果が公表されなかった(Yes-NA)、または行われ結果が報告された(Yes)。
#EUA申請およびPolackら(2020)に含まれる被験者を示す。
*死因診断が心臓イベントとみなされたことを示す。1症例報告書には診断を裏付ける十分な臨床データが記載されていない。2症例報告書に不備があり、診断を確定するには剖検結果が必要である。
4被験者が無作為化の基準を満たさないか、プロトコルから逸脱している。
死亡した被験者に関する38件の症例報告書および報告書はすべてファイザー社/バイオエヌテック社から入手可能であった。一般に、これらの報告書を検討したところ、詳細な情報が不足しており、報告された死因を解釈し確認することは極めて困難であった。多くの場合、被験者の臨床試験前の病歴は欠落していた。また、公判前スクリーニングやその他の定期的な診察で実施された広範な医学的検査の結果も欠落していた。これらの検査結果には、全血球計算、代謝検査、妊娠検査、COVID-19検査、有効薬剤の包括的リストなどが含まれ、被験者の全体的な医学的状態を明らかにすることができたであろう。被験者に関するより詳細な臨床データは存在するが、まだ開示されていない。提供された情報の限界を考慮すると、我々は症例報告書と報告書に記載された情報は、死因に関する治験責任医師の結論を裏付けるには不十分であることが多いと判断した。より顕著なケースについては、表1に上付き文字1および2で示した。興味深いことに、このような懸念の多くは、ファイザー/バイオインテックの担当医師が治験責任医師との対話を担当した際にも指摘された。
DailyCloutのAbstractorを使用し、各症例報告書とナラティブレポートを評価した。死因の診断に関する全体的なコメントと懸念は表1に示した。この評価から得られた知見は特に明白であったので、以下にいくつかの対象について簡単に報告する。特に重要なのは、#1 1271112と#1 0841266の2名の被験者であり、彼らの症例報告書とNarrative Reportには、ファイザー/バイオインテックの死因リストには記載されていない心臓の事象が死因の一因である可能性が高いことが示されていたからである。被験者#1 2291083と#1 0971023は無作為化時に適格要件を満たしていなかったので、この38名の死亡被験者リストから除外されるべきである。被験者#1 0841470は無作為化後に重篤なプロトコル逸脱があった(下記参照)。これら3人の被験者はファイザー/バイオエンテックの38人の死亡リストに含まれているため、解析では除外しなかった。被験者#1 1311204と#1 2314987は、それぞれ心肺停止と心肺停止で死亡したと記載されていた。症例報告書と報告書を分析した結果、彼らの病歴と死亡時の状況から、死因として心臓疾患が考えられた。それにもかかわらず、剖検が行われなかったため、我々は保守的なアプローチをとり、彼らを「心臓イベントシグナル」として含めないことにした。
ナラティブレポートに基づき、表1には被験者の死亡状況も記載されている。6人の被験者(プラセボ2人、ワクチン接種4人)が突然死した(SAD:Sudden Adult Death)。SADは、目撃者がいる中で予期せず急速に起こった死亡と定義した。プラセボ3人、ワクチン接種者6人の計9人が死亡が確認された(FD)。これらは目撃者のいない睡眠中または自宅で一人で死亡した被験者であった。突然死(SAD)または死体で発見(FD)された15人の被験者のうち12人が心臓の有害事象で死亡したことは興味深い。
被験者番号10841266は77歳の男性で、重度の血管疾患、壊疽、糖尿病や他の併存疾患に関連していると思われる複数の足指切断の既往歴があった。彼はBNT162b2ワクチンを単回接種された後、胆嚢炎を発症し、手術を受け、敗血症となり、多臓器不全で死亡した。症例報告書には剖検報告が記載されていないが、これは症例報告書に主な死因について混乱があったことが記載されているため、残念なことである。肺気腫性胆嚢炎は致命的な細菌性の胆嚢感染症であり、この感染症は重度の糖尿病に関連するリスクをさらに高めた。この感染症が死に至る一連の出来事を引き起こした。11月23日に初めて報告されたNSTEMI(非ST上昇型心筋梗塞)は、臓器不全のカスケードの一部であった可能性が高い。11月23日、対象者はトロポニン値の上昇とNSTEMIの疑いで入院した。トロポニン値の上昇は12月1日に確認されたが、12月2日の症例報告書には、NSTEMIはSAE(重篤な有害事象)とはみなされないと記載されていた。この症例では、他の報告事項に加えて、死亡の主原因と被験者が病院でNSTEMIを発症したかどうかに関して、治験実施施設とファイザー/バイオエヌテック社との間で大きな行き違いがあった。「敗血症」が彼の直接的な死因であったようであるが、NSTEMIは少なくとも二次的な死因として寄与因子として記載されるべきである。
被験者番号10841470は肥満した65歳のヒスパニック系男性で、肺線維症と高血圧を含む病歴がある。彼は試験のプラセボ群に属しており、2020年9月30日と10月21日にそれぞれ1回目と2回目の投与を受けた。2020年12月23日、被験者はモデルナmRNAワクチンの投与1回目を受けた。このプロトコルの逸脱は、被験者が2020年12月28日にCOVID-19の症状を報告し、2020年12月31日に入院した後、症例報告書に報告された。入院中に低酸素状態となり、2021年1月2日に挿管された。入院中、治療の一環としてモノクローナル抗体の投与を受けた。このような努力にもかかわらず、対象者は悪化し続け、多臓器不全に陥り、最終的に2021年1月11日に死亡した。被験者番号10841470は「死亡」として中止被験者リスト(Listing of Discontinued Subjects)に記載され、COVID-19を二次的死因とするプラセボ死亡として6カ月中間報告(6-Month Interim Report of Adverse Events C4591001)に記載された。これは被験者の臨床情報の虚偽表示である。この被験者はファイザー社/バイオエヌテック社の臨床試験から中止されるべきであった。なぜなら「被験者は試験以外のCOVID-19ワクチンを接種した」からである。
被験者#1 1271112は53歳のネイティブ・アメリカンの男性で、COPDと「ストレス関連心筋梗塞」の既往歴があった。ナラティブ・レポートによると、2020年12月4日、患者は自宅で階段を昇り降りしていたところ、「足を組み、前傾姿勢で、顔が真っ青」になっているところを母親に発見された。この突然死はBNT162b2ワクチンの2回目の接種から2カ月も経たないうちに起こった。剖検が行われたが、その結果は確認できなかった。12月18日、被験者の死亡後、治験施設の医療モニターは死因を「心筋梗塞に関連した心肺停止」と記載した。12月19日、Pfizer/BioNTech社は、複数の死因を症例報告書に記入することはできないと治験実施医療機関に通知し、「心筋梗塞に関連する」の削除を求めた。医療モニターは記入文言の変更を拒否した。2021年1月5日、ファイザー社/バイオエヌテック社は治験実施医療機関の判断を覆し、死因を「心肺停止」に変更し、副次的死因として「心筋梗塞」を記載しないことを選択した。AESIという具体的な診断が、なぜ後に未確定なものに変更されたのかは不明である。現場の医療モニターの診断を肯定も否定もする重要な剖検報告書がなければ、この対象者を心臓信号事象のグループに含めることが最も適切であると考えた。
被験者番号11621327は、9月10日にBNT162b2ワクチンの1回目を接種した直後に死亡しているのが発見された。彼の遺体は9月13日に警察が福祉チェックを行った際に自宅で発見された(顔面蒼白)。「検死官によれば、死因は動脈硬化性疾患の進行であった。有害事象6カ月中間報告C4591001に記載されている死因は「動脈硬化症」である。しかし、症例報告書には、死因が動脈硬化症であることについて複数の問い合わせがあった。症例報告書には患者の併存疾患として動脈硬化症は記載されていなかった。被験者の症例報告書は127ページしかなく、症例報告書の併存疾患の事前スクリーニング部分、つまり被験者に動脈硬化の既往があるかどうかの証拠となる部分が含まれていない。さらに、剖検が行われていれば、動脈硬化の進行が記録されたはずだが、剖検結果は提供されておらず、入手できなかった。症例報告書に記載された医学的資料だけでは、被験者の死亡を進行した動脈硬化と断定する根拠も、死亡がワクチンと無関係であると結論づける根拠もない。該当被験者の中間報告文書から引用された以下の記述は、根拠がないと我々は考えている。「治験責任医師の見解では、動脈硬化が試験介入、併用薬、または臨床試験手順と関連している合理的な可能性はなく、むしろ疑われる基礎疾患と関連していた。ファイザー/バイオエヌテックは治験責任医師の因果関係評価に同意した。この被験者はワクチン接種後1日か2日以内に死亡した可能性が高い。これは、彼の死亡がBNT162b2ワクチンと関連している可能性を示す明らかな兆候であり、より厳密な調査なしにこれを除外すべきではなかった。我々の意見では、この診断は時期尚早であり、手元にある証拠の判断を著しく誤っていた。
被験者#1 2291083はプラセボを投与され、投与1日目から76日後に死亡した。主な死因は病歴から糖尿病と診断された。血糖値が非常に高かったにもかかわらず、この診断は何度か修正され、最終的にCOVID-19肺炎が二次的な死因とされた。この被験者はHIV陽性で、HIV RNA量が50コピー/mlであり、この試験に組み入れられる許容限度をわずかに超えていた。この被験者は無作為に割り付けられ、試験参加者として承認されるべきではなかった。
被験者ID#1 2314987は47歳の男性で、高血圧、肥満の既往があり、27年間喫煙者であった。彼はプラセボを投与され、投与1日目の82日後に死亡した。予定外の来院で、2020年12月5日午後9時に腹痛、嘔吐、背部痛を呈し、翌6日午前7時に病院で死亡した。剖検の記録はなく、家族の問い合わせにも応じなかった。死因は「非外傷性心肺停止」とされたが、対象者の病歴を考慮すれば、より確実な診断を積極的に追求すべきだった。
被験者ID#1 2315324はプラセボを投与され、投与1日目から136日後に死亡した。主な死因は「多臓器不全症候群」と記載されていたが、症状からCOVID-19と診断された。それ以外は健康な被験者がCOVID-19の症状で入院したようである。患者はICUで肺葉性肺炎のため人工呼吸を必要とし、透析を必要とする急性腎不全であったと記録されている。血管圧迫薬以外に、患者が病院での治療の一環として受けた投薬の記録はない。
結論として、我々は、被験者#1 1271112と#1 0841266を除き、6カ月中間報告書(有害事象に関する6カ月中間報告書C4591001)に記載された死因診断を正確なものとして受け入れるしかなかった。我々の医学的専門知識に基づき、またこれら38名の死亡した被験者の潜在的な安全性シグナルの検索を簡略化する目的で、心筋梗塞、心停止、心臓突然死、うっ血性心不全、動脈硬化という用語を「心臓イベント」という包括的な用語でグループ化した。表1では、心イベントにより死亡したと診断された被験者には*印をつけた。例外的なケースである被験者#1 1271112と#1 0841266の2例では、表1にはファイザー/バイオエンテックが決定した死因が記載されているが、我々の見解では心筋梗塞を死因として除外することはできなかった。従って、#1 1271112と#1 0841266の被験者は、死因の診断の横に*印で示されるように、「心イベント」グループに含まれた。
被験者の死亡に関する報告の不一致
図1にプロットされたデータとファイザー/バイオエヌテックEUA申請書(未承認製品の緊急使用承認審査覚書 https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)、Polackら(2020)、Thomasら(2021)で報告された結果を比較したところ、いくつかの矛盾が明らかになった。
これらの様々なデータソース間の不一致は、特に不愉快である。我々が使用しているデータは、ファイザー社/バイオエヌテック社の臨床試験C4591001の6カ月中間報告書(6-Month Interim Report of Adverse Events C4591001)の「死亡例のリスト-16歳以上の全被験者」というセクションに直接記載されているものである。そのため、他のファイザー/バイオエンテックの文書および公表された報告書に示されたデータと完全に一致しているはずである。これらの不一致は表2に示されている。
表2は、Pfizer/BioNTech社、Polackら(2020年)、Thomasら(2021年)が報告した結果(左欄)と、当社が解析した6ヵ月中間報告(6-Month Interim Report of Adverse Events C4591001)のデータ(右欄)を比較したものである。図1に示すように、盲検プラセボ対照期間~EUA申請データ収集締切日(2020年7月27日~11月14日)、盲検プラセボ対照・非盲検フォローアップ期間(7月27日~2021年1月24日)、非盲検観察期間~6ヵ月中間報告データ収集締切日(2021年1月25日~3月13日)の期間ごとにデータを報告している。2021年1月24日を非盲検観察期間の終了とした根拠は不明であり、Thomas et al.(2021)には説明がない。表2の最後のセクションは、全6ヵ月間(2020年7月27日から2021年3月13日)の要約である。Polackら(2020年)とThomasら(2021年)の両者には、フローチャートで報告された死亡数と原稿で報告された死亡数との間に内部矛盾があることに注意すべきである。これらの矛盾は、どちらの原稿の査読者にも指摘されていないようである。
表2:fizer/BioNTech BNT162b2 mRNAワクチン臨床試験C4591001*の期間中に報告された被験者の死亡の比較
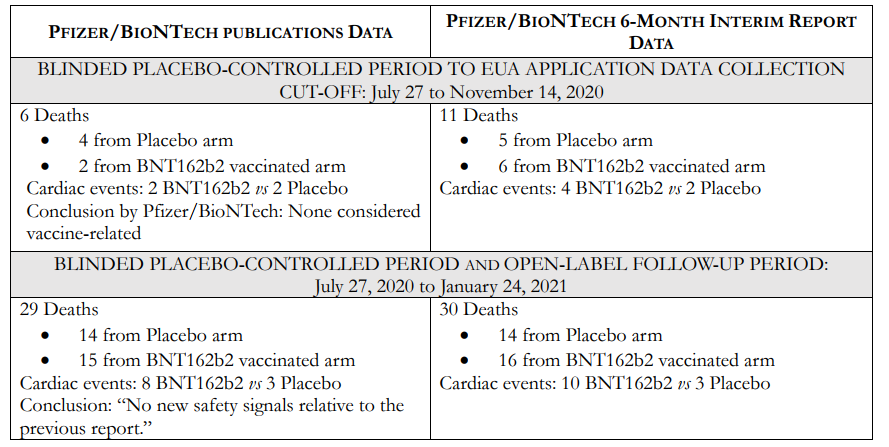

*Pfizer/BioNTech BNT162b2 mRNAワクチン臨床試験C4591001の期間中に報告された被験者死亡の比較。左側の列は、以下の出版物から得られたデータを示している: Pfizer/BioNTechのEUA申請(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)、Polackら(2020)、Thomasら(2021)。右側の列は、Pfizer/BioNTech 6-Month Interim Report on Adverse Events(6-Month Interim Report of Adverse Events C4591001)のデータである。心イベント数は、表1に示された被験者の症例報告書の解析に基づく。結論はPfizer/BioNTechのEUA申請書(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)、Polackら(2020)、Thomasら(2021)の文章から引用した。表は6ヵ月中間報告の4つの期間に分かれている: 盲検下プラセボ対照期間~EUA申請データ収集締切日(2020年7月27日~11月14日)、盲検下プラセボ対照期間および非盲検下追跡期間(7月27日~2021年1月24日)、非盲検下観察期間~6ヵ月中間報告データ収集締切日(2021年1月25日~3月13日)、6ヵ月中間報告サマリー(2020年7月27日~2021年3月13日)である。
EUA申請データ(2020年7月27日~11月14日)
表2の最初のセクションは、ファイザー/バイオインテックの公表データと今回報告された図1および表1のデータを比較したものである。BNT162b2ワクチンを承認するかどうかの決定は、すべてこの結果にかかっていたため、最初の16週間は臨床試験の最も重要な期間である。Pfizer/BioNTechのEUA申請(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)および2020年12月10日に発表され、12月16日に更新されたPolackら(2020)は、2020年11月14日以前に死亡した臨床試験参加者は6名のみであったと報告している:試験のワクチン接種群で2名、プラセボ群で4名であった。Polackら(2020)の死因に関するコメントに基づいて、これら6名の被験者IDを決定した。表1では、これらの被験者に上付き#をつけている。一方、図1および表1に示すように、11月14日(第16週)以前の死亡例は、ワクチン群で6例、プラセボ群で5例、合計11例であった。Polackら(2020)が報告した死亡した6人の死亡日を注意深く検討したところ、死亡日が11月14日以前であったワクチン接種群6人のうち2人、プラセボ群5人のうち4人しか含まれていなかった。これはわれわれの分析で指摘された最初の不一致である。
Pfizer/BioNTechが報告した6人の死亡被験者のうち、表1は、BNT162b2ワクチン接種被験者のうち2人とプラセボ被験者のうち2人が心臓イベントで死亡したことを示している。Polackら(2020)およびPfizer/BioNTechのEUA申請書(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)には、治験責任医師はこれらの死亡がワクチンに関連するとは考えなかったと記載されている。これと比較すると、盲検プラセボ対照期間中に観察された11例の死亡を分析したところ、約半数が心臓イベントによるものであった: BNT162b2ワクチン接種群で4例、プラセボ群で2例であった。この数字は小さいが、BNT162b2ワクチン接種群では心臓イベントが2倍増加したことになる。このことはファイザー社/バイオエヌテック社に、心イベントがワクチンに関連したシグナルイベントである可能性を警告するものであった。しかし、11月14日以前に死亡した5人の被験者の情報がEUA申請時にFDAに報告されていなかったため、そうならなかった。実際の死亡日と被験者の症例報告書に正式に記録された日付との間には長いずれがある(表3)。これらのギャップの原因については後述する。
2020年12月10日、FDAへのプレゼンテーション
12月10日、ファイザー/バイオエヌテックは、FDAのワクチンおよび関連生物学的製剤諮問委員会(VRBPAC)に対し、BNT162b2 mRNAワクチンの緊急使用承認(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-foran-Unapproved-Product-Review-Memorandum)の要求を裏付ける証拠を提示した。このプレゼンテーションは、11月14日のEUA申請データ締切日から25日後に行われた。ファイザー社/バイオエヌテック社にとっては、12月10日に結果を更新する機会であった。その代わり、ファイザー/バイオインテックの代表者は、25日前のEUA申請と全く同じ結果を報告した。C4591001は進行中の臨床試験であった。この期間中に死亡例が追加されることは珍しいことではなかった。実際、6カ月中間報告(6-Month Interim Report of Adverse Events C4591001)を分析したところ、2020年11月14日から12月10日の間にさらに多くの被験者が死亡しており、実際の死亡者数は7月27日から12月10日の間に17人となった。この17人の被験者のうち16人(BNT162b2ワクチン接種8人、プラセボ8人)は、12月10日までにファイザー社/バイオエヌテック社が把握していた。これは、我々の分析で指摘された2番目の不一致である。
死亡した38人の被験者の症例報告書を注意深く調べたところ、6カ月中間報告書に記録された死亡日が、被験者の症例報告書には数日間、時には数週間、正式に記録されていないことが判明した。私たちは、この遅れにパターンがある可能性を探ることにした。表3は、死亡した38人の被験者を、プラセボあるいはBNT162b2ワクチンのいずれを接種したかによってグループ分けしたものである。Pfizer/BioNTechのEUA申請(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)およびPolackら(2020)で報告された6人の被験者は、上付き#で示されている。リストが灰色で強調表示されている被験者は、FDAのVRBPAC会議では死亡が議論されなかったが、発表日である12月10日にファイザー/バイオエヌテックが死亡を把握していた被験者である。表3の情報に基づいて、ファイザー社/バイオエヌテック社は、11月14日から12月10日の間に死亡したさらに10人の被験者を知っており、公式に記録された死亡者数は16人となった(表3の灰色斜線または#印の行を参照)。 被験者#1 0881126の12月1日の死亡は、72日後の2021年2月11日まで症例報告書に公式に記録されなかったため、含まれていない)。Pfizer/BioNTech社が把握しているこれらの16人の死亡は、BNT162b2ワクチン接種群で8人、プラセボ群で8人と、試験の両群に均等に分布していた。死因は不明である。表1に報告された心臓イベントの数の決定に基づくと、心臓イベントに関連した死亡数は、BNT162b2ワクチン接種群で6人、プラセボ群で3人であり、BNT162b2ワクチン接種被験者における心臓シグナルの2倍の増加である。その代わりに、ファイザー社/バイオエヌテック社がこの日までに報告した唯一の心イベントである「心停止」と「心筋梗塞」を用いると、その数はBNT162b2ワクチン接種群で2、プラセボ群で2であり、つまり試験群間でバランスがとれている。
ファイザー/バイオエンテックは、FDAの決定に寄与しうる新情報を自主的に公表すべきだった。そうしなかったのは事実誤認である。一方、VRBPAC会議の出席者全員が、11月14日のデータが古いことに気づくべきだった。驚くべきことに、VRBPACのメンバーは誰も、EUAデータのカットオフ日(11月14日)から今回の会議の日付(12月10日)までの間に発生した有害事象に関する最新情報を要求しなかった
また、死亡原因に関する詳細な情報を求めたり、死亡した被験者の症例報告書を独自に評価したりすることもなかった。死亡した被験者16人は管理可能な数であり、これは承認プロセスにおける重要なポイントであった。FDAがファイザー/バイオインテックのEUAを承認したのは、16週間のデータ、つまり批判的な目で評価されなかった全容の誤った説明のみに基づいていたようである。
盲検プラセボ対照および非盲検追跡調査期間
表2のこのセクションでは、試験の最初の26週間について報告する。ここでは、Pfizer/BioNTech 6-Month Summary Clinical Safety(Summary Clinical Safety 6-Month Report)およびThomas et al.(2021)で報告された被験者の死亡数と原因を、Pfizer/BioNTech 6-Month Interim Reportで報告されたものと比較している。要約臨床安全性(Summary Clinical Safety 6- Month Report)は2021年5月5日にFDAに提出され、2021年3月13日までのデータが含まれている。
この報告書の表7には、投与1回目の受領から盲検化解除日(定義なし)までに各群で死亡した被験者数が記載され、表16には死因が示されている。表16の情報は、Thomasら(2021)が2021年9月15日に出版されたにもかかわらず、更新されることなくThomasら(2021)の表S4に再現されている。両文献で報告された結論は同じ:盲検プラセボ対照期間中の死亡はBNT162b2群で15例、プラセボ群で14例、合計29例であった。
要約臨床安全性(Summary Clinical Safety 6-Month Report)の2.7.4.2.2.1.1項では、200人のHIV陽性フェーズ2/3参加者のサブセット(各試験群から1人ずつ)の中で2人の死亡が発生し、両者とも試験から離脱したと報告している。これらの参加者の被験者IDは#1 1561160と#1 2291083であった。Thomasら(2021)は、試験中の被験者の処分を示すフローチャートの図にこれら2人の被験者を含めているようである。論文本文で報告されている29人の死亡という最終的な数を得るために、これらの被験者のうち1人だけが除外されたようである。被験者#1 2291083の症例報告書を分析したところ、この被験者は無作為化に不適格であったことがわかった(前述)が、被験者#1 2291083は死亡した38人のリストに含まれていたため、表2の計算からこの被験者を除外しなかった。被験者#1 1561160がなぜ除外されたのか、あるいは除外されたのがこれらの被験者であったのかについては情報がない。Thomasら(2021)におけるHIV陽性の被験者の処分と、Thomasら(2021)が29人の被験者の死亡の合計をどのように算出したかは、明確に示されていない。この混乱は、彼らのデータにおける内部矛盾を表しており、30人の被験者の死亡を示す我々のデータとThomasら(2021)のデータの違いの1つを説明する可能性がある。
本試験の最初の26週間における死亡30例のうち、心臓イベントによる死亡は合計13例で、BNT162b2ワクチン接種群で10例、プラセボ群で3例であった。心イベントは明らかにBNT162b2ワクチンの有害事象安全性シグナルである。驚くべきことに、このシグナルについてファイザー/バイオエンテックは言及していない。Thomasら(2021)は、”前回の報告書のデータカットオフ日以降、BNT162b2に関連すると考えられる新たな重篤な有害事象は認められなかった「、”より長い追跡調査期間中、新たな安全性シグナルは認められなかった」と述べている。ファイザー/バイオエンテックが11月14日以前に死亡したと報告した6人の被験者のうち3人は心イベント(心筋梗塞と動脈硬化)で死亡しているので、Thomasら(2021)はおそらくその日以降の死亡を「新たな」有害事象によるものとは考えなかったと思われる。被験者の総死亡数と死因のアンバランスは、我々の解析で指摘された3番目の矛盾である。
2021年3月13日までの非盲検観察期間。Thomasら(2021)とPfizer/BioNTechのSummary Clinical Safety(Summary Clinical Safety 6-Month Report)では、表2の3番目のセクションに示すように、我々が「Open-label Observational Period(非盲検観察期間)」と題した試験期間において、BNT162b2群で3例、非盲検のBNT162b2ワクチン単独プラセボ群で2例の死亡例が報告されている。この期間に死亡した被験者は合計8人であった: BNT162b2群で3人、非盲検のBNT162b2ワクチン接種のプラセボ群で2人、ワクチン未接種のプラセボ群で3人である。Pfizer/BioNTech社がこの最後のグループを除外した理由は不明である。27~33週目に報告した8例の死亡例のうち、BNT162b2ワクチン接種群では1例、プラセボ群では1例も心イベントがなかった。Thomasら(2021)は、「死因はBNT162b2群とプラセボ群でバランスが取れていた」と述べている。繰り返すが、6カ月中間報告のこの最終期間の総被験者死亡数と心臓死におけるわずかな不均衡は、我々の分析で指摘された4番目の矛盾である。
被験者死亡の要約
表2の最後のセクションは、ファイザー/バイオエンテックC4591001臨床試験の最初の33週間で発生した死亡の全容を示している。ファイザー/バイオエヌテック社は、6カ月の追跡調査期間中に死亡した34人の被験者(BNT162b2ワクチンを投与された20人とプラセボ対照群の14人)について説明している。上述したように、6カ月中間報告書に記載された38人の死亡者のうち4人は、その計算には含まれていない。おそらく、#1 2291083と#1 1561160の2人のHIV陽性被験者と、2021年1月24日以降に死亡した2人のプラセボ被験者であろう。BNT162b2ワクチン接種群で21人、プラセボ群で17人である。38例のうち3例は6カ月中間報告書に記載されるべきではなかった。被験者#1 2291083(プラセボ)と#1 0971023(BNT162b2)は適格要件を満たしておらず、無作為化前に除外されるべきであった。被験者#1 0841470(プラセボ)は非試験のCOVID-19ワクチンを受けた。
興味深いことに、COVID-19はこれらのプラセボ被験者の死因として挙げられている。
我々の法医学的分析の基礎資料である6カ月中間報告書(6-Month Interim Report of Adverse Events C4591001)に報告された38例の死亡例のうち、14例が心イベントで死亡しており、これは全死亡例の3分の1以上(36.8%)であった。この14例のうち、11例がBNT162b2ワクチン群、3例がプラセボ群であった。これは、BNT162b2ワクチンを接種した被験者の心臓イベントが3.7倍増加したことを意味する。Thomasら(2021)およびファイザー/バイオインテックの臨床安全性要約(臨床安全性要約6カ月報告書)は、この明らかな重篤な有害事象のシグナルを特定しておらず、またそれについて言及していない。
データの不一致の原因
上記のデータの不一致は、特にEUA承認前に心臓の有害事象シグナルがFDAに報告されなかった理由を理解する上で極めて重要である。上述した表3のデータを分析したところ、ファイザー/バイオエンテック社は、死亡を報告する期間を決定するために、死亡が症例報告書に正式に記録された日を使用しており、実際の死亡日ではなかった。C4591001プロトコルは、被験者の死亡を直ちにファイザー/バイオエヌテック社に通知することを要求していた。ナラティブレポートは、治験施設が被験者の死亡状況を熱心に報告していたことを確認するものである。死亡の正確な日付が記載された報告書は数件しかない。死亡通知プロセスには他のステップも存在するようである。そのうちのいくつかは、死亡詳細報告書のような症例報告書に記載されている。それにもかかわらず、ファイザー社/バイオエヌテック社が、必要な24時間以内に各被験者の実際の死亡日を把握していたことは明らかであるが、記録管理およびレビューの手順が不明であったために、被験者の死亡日を症例報告用紙に記入するのが遅れたのである。
ファイザー社/バイオエヌテック社は、EUA申請書およびVRBPACプレゼンテーションの作成時に、被験者の死亡日を入手可能であったにもかかわらず、なぜ実際の死亡日ではなく、被験者の死亡日を症例報告書に記入した日付を使用したのであろうか。これに答えるため、この記録の遅れから考えられるパターンを探った。実際の死亡日とケース・レポート・フォームに死亡が正式に記録された日とのずれを、死亡した38人の対象者全員について表3に示す。FDAによってEUAが承認された2020年12月11日以前と、EUA承認後の記録の遅れを比較するため、2つの期間に分けた。EUA承認前の死亡はグレーの網掛けで示した。EUA申請およびPolackら(2020)に含まれる6被験者を上付きハッチマーク(#)で示す。
12月10日にVRBPACに報告されるべきであったBNT162b2ワクチン接種被験者8人のうち、報告の遅れの中央値は18日であった(平均17.5日)。プラセボ被験者8名では、遅延の中央値は5日(平均5.9日)であった。12月11日以降の記録遅延を分析すると、両群とも劇的に減少していた。BNT162b2群では7日(平均9.8日)、プラセボ群では3日(平均15.9日)であった。50や72のような少数の外れ値が平均値を歪めるので、中央値は遅延のより良い尺度である。これらの結果は、ファイザー/バイオエンテックが被験者の死亡を通知された日から症例報告書に記入される日までの遅延は、被験者の試験群、および被験者がEUA承認前または承認後に死亡したかどうかによって異なることを示している。
図2は、当初のBNT162b2ワクチン接種群の19人の被験者の報告遅延と実際の死亡日をプロットしたものである。図2Aでは、被験者の死亡が症例報告書に記入された日付がプロットされている。実際の死亡日がEUAのデータカットオフである11月14日以前であった6人の被験者は、被験者ID#で示されている。被験者#1 0071101と#1 1621327は、EUA申請書(四角マーカー)で死亡が報告された2人のワクチン接種被験者である。被験者#1 1141050、#1 1201050、#1 0891073および#1 1521497はEUA申請で報告されるべきであったが、報告が遅れたため報告されなかった(三角印)。残りの被接種者は11月14日以降に死亡しており、塗りつぶした円で示した。点線はデータに対するベストフィットである。
表3:対象者の死亡記録の遅れ*1
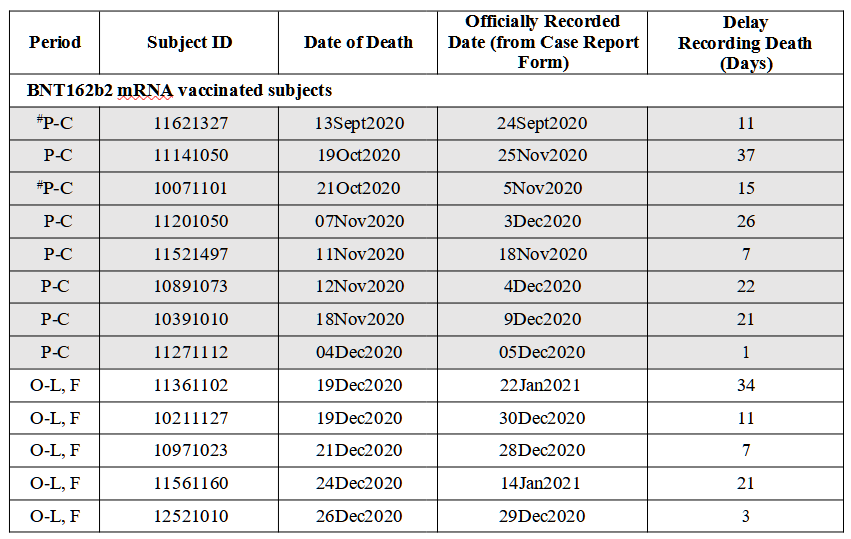
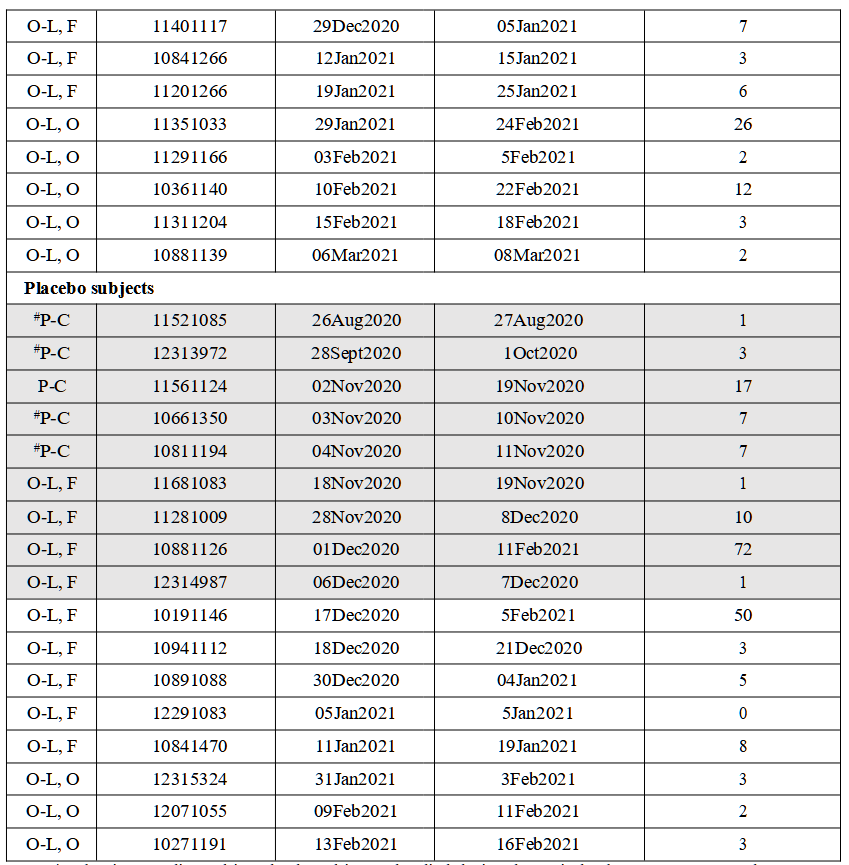
*被験者の死亡記録の遅れ。2020年7月27日から2021年3月13日の期間に死亡した被験者を記載した。BNT162b2ワクチンを投与された被験者は、プラセボを投与された被験者とは別に、真の死亡日順に記載されている。#Pfizer/BioNTechのEUA申請(Emergency Use Authorization for an Unapproved Product Review Memorandum https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum)およびPolackら(2020)で死亡が報告された被験者を含むことを示す。グレーの網掛けをした行は、2020年7月27日から12月10日の間に症例報告書に正式に死亡が記録された個人を強調しており、ファイザー/バイオエヌテックがこの期間に被験者が死亡したことを知っていたことを示している。試験の期間: P-Cはプラセボ対照盲検期間、O-L, Fは非盲検フォローアップ期間(2020年12月11日~2021年1月24日)、O-L, Oは非盲検観察期間(2021年1月25日~3月13日)である。
図2Aから、報告遅延の長さはEUA申請後に有意に減少することがわかる。プラセボ被験者では同様の傾向が観察されないことから、この差について合理的な説明はない(表3)。
11月14日以前に死亡した主要被験者に関する報告書には、ファイザー/バイオエンテックが被験者の死亡日をいつ通知されたかが明記されている。プロトコルC4591001では、死亡や入院のような重篤な有害事象は、ファイザー/バイオエヌテック社に直ちに報告するか、遅くとも24時間以内に報告することが要求されていた。この要件は治験施設スタッフによって遵守されたと思われるが、すべてのNarrative Reportsが正確な日付に言及しているわけではない。図2Bの症例報告ファイルから得た死亡日の代わりに、主要なNarrative Reportで報告された死亡日を使用した。2人の被験者(#1 1141050(37日低下)と#1 1201050(26日低下)のマーカーの位置が劇的に変化し、被験者#1 1621327(11日)の低下は小さく、#1 0891073(3日)の低下はわずかであった。これらの変化を考慮すると、図2Bの最良適合傾向線が示すように、試験期間中の記録遅延は類似している。
ファイザー/バイオエンテックが症例報告書に死亡が記録された日ではなく、実際の死亡日を報告していれば、被験者#1 1141050および#1 1201050はEUA申請に含まれていたであろう。このシナリオを考慮すると、11月14日のデータ締切日前に死亡し、その死亡がEUA申請に含まれるべきであった被験者は、ワクチン接種被験者4名とプラセボ被験者4名であったことになる。このうち、心臓イベントによる死亡は、ワクチン接種群で4例、プラセボ群で2例であった(表1)。したがって、ワクチン接種群の死亡の100%は心臓イベントによるものであった。これらの患者の死亡を症例報告ファイルに記録するのを遅らせ、実際の死亡日を使用しなかったため、EUA承認プロセスの重要な局面で死亡が発見できず、心臓有害事象のシグナルが不明瞭になった。
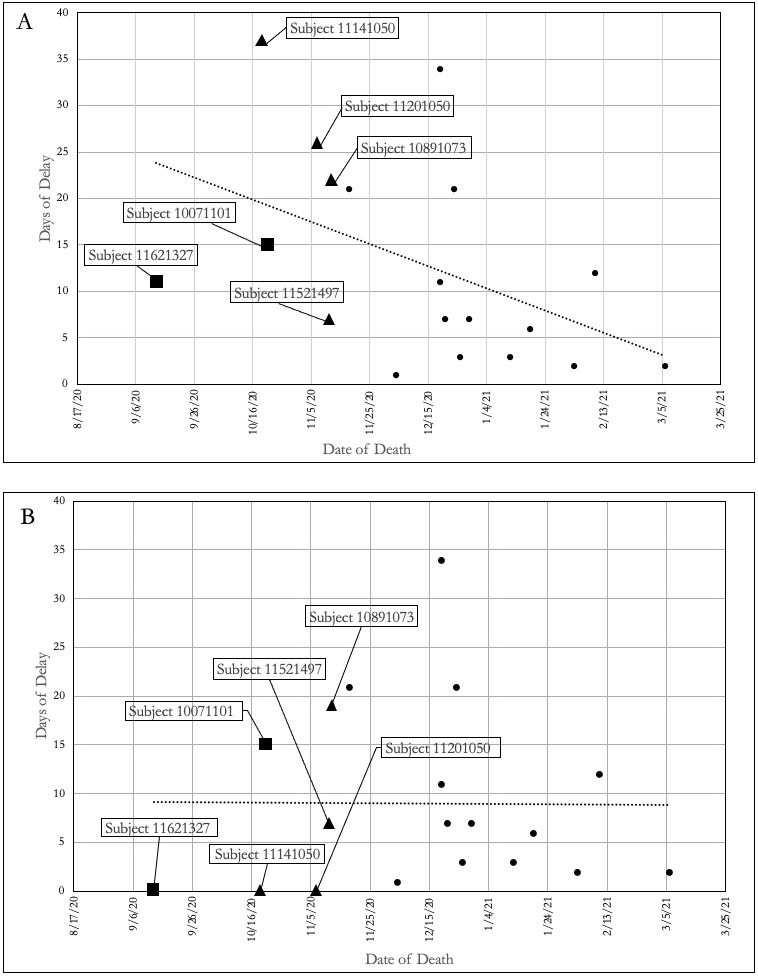
図2:BNT162b2ワクチンを接種した被験者の死亡記録の遅れ。被験者IDは、EUA申請のデータ締切日である11月14日以前に死亡した6人の被験者について記載されている。塗りつぶされた四角( )は、EUA 申請書で死亡が報告された被験者である。塗りつぶされた三角形( )は、EUA で死亡が報告されなかったが、11月 14日より前に死亡した被験者。小さな塗りつぶし円( )は、11月14日以降に死亡した被験者である。図2Aは、被験者の死亡が症例報告ファイルに記録された日をプロットしたものである。図2Bは、被験者の死亡がナラティブレポートに報告された日をプロットしたものである。
要約すると、ファイザー社/バイオエヌテック社がEUA申請書を作成する際に、表2にあるように38名の被験者の実際の死亡日を使用していたならば、FDAがBNT162b2ワクチンを承認したかどうかは疑問である。11月14日以前に死亡した11例のうち、ワクチン接種群6例中4例が心臓の有害事象で死亡しており、プラセボ群5例中2例が心臓の有害事象で死亡していた。もしVRBPACがアップデートを求めていたら、12月10日までにさらに6人の死亡例があったことから、心臓有害事象のシグナルはさらに明白になっていただろう。12月10日までに、ワクチン群で8例、プラセボ群で9例の合計17例が死亡した。ワクチン接種群では8例中6例(75%)が心臓イベントで死亡したが、プラセボ群では9例中3例(33.3%)しか心臓イベントで死亡しなかった。20週間という短期間の臨床試験で、このような明確な心臓有害事象のシグナルが出たことは、FDAの審査担当者が気づいていれば、間違いなく躊躇したはずである。報告の遅れとVRBPACによる好奇心の欠如により、ファイザー社/バイオエヌテック社は、この臨床試験の唯一の真にプラセボ対照無作為化試験の結果の報告を操作することができた。
考察
本研究は、Pfizer/BioNTech社のBNT162b2 mRNAワクチン臨床試験(C4591001)のオリジナル試験データを、試験スポンサーとは無関係のグループが初めて解析したものである。Pfizer/BioNTechの最初の6カ月中間報告(6-Month Interim Report of Adverse Events C4591001)で報告された死亡数が少なかったため、このような大規模なデータセットでは不可能であった詳細なレベルの詳細な調査を実施することができた。そのため、本書はこれら38人の死亡例の法医学的分析として最もよく表現されている。我々は、ファイザー/バイオエンテックとその代理人が公開の場で行ったBNT162b2 mRNAワクチンの臨床試験に関する報告には欠陥があり、実際の試験結果を不明瞭にする重大な報告ミスが含まれていたことを明らかにした。その結果、BNT162b2ワクチンを接種した被験者の心イベントがプラセボ群と比較して3.7倍増加したことが、ワクチン展開時に一般に報告されなかった。
プラセボ対照無作為臨床試験
図1に示した結果は、プラセボ対照臨床試験の価値を証明するものである。薬物であれ治療法であれ、どのような介入であっても、認識されていないリスクや副作用があり、それがせっかくの効果を否定してしまう可能性がある。しかし、介入による効果があることをどうやって知ることができるのだろうか?それにはプラセボ対照無作為化臨床試験が必要であり、これは臨床試験の金字塔と呼ばれてきたものである。被験者集団は特定の基準に基づいてスクリーニングされ、臨床試験の治療群またはプラセボ群のいずれかに無作為に割り付けられる。したがって、被験者集団は類似しており、人口統計学的に既知でなければならない。その結果、両群間の転帰に差があれば、その差は治療に起因することになる。実験科学と同様に、プラセボ群は「対照」として機能し、無作為化された被験者集団のベースライン数を提供する。この対照なしでは、研究中の治療がプラス、マイナス、あるいはまったく効果がないことを確信をもって言うことは不可能である。被験者集団と一般集団を比較することは、まったく不適切であり、科学的にも不正確である。治療を受けることに同意した人は、健康状態、年齢、社会経済的レベルなど、さまざまな点で治療を受けないことにした人とは異なる可能性がある。同様に、VAERS、イエローカード、その他の国の健康監視データベースは非常に貴重ではあるが、プラセボ対照無作為化臨床試験の高い基準を満たしていない。
適切な対照臨床試験の必要性は、2021年1月にWHOアドホック・エキスパート・グループ(WHO Ad Hoc Expert Group on the Next Steps for COVID-19 Vaccine Evaluation)が執筆した論文(WHO Ad Hoc Expert Group on the Next Steps for COVID-19 Vaccine Evaluation, 2021)でも確認されている。この論文では、SARS-CoV-2に対するワクチンのプラセボ対照無作為化臨床試験を、ワクチン導入後も継続することを強く推奨している。”ワクチン配備後に非ランダム化試験から得られた観察データは、信頼性の低い回答をもたらす可能性が懸念された。
観察研究はかなりのバイアスに左右され、明確な解釈が困難である。注意深く分析された観察研究であっても、安全性と有効性について誤解を招くような結果をもたらす可能性がある。委員会の意見では、約22,000人のワクチン被接種者と20,000人のプラセボ被接種者を対象とした対照試験は、比較的一般的な有害事象の検出には十分かもしれないが、まれな有害事象の検出には十分ではない。まれな有害事象を検出するためには、おそらく数十万人というはるかに多数の被験者が必要であり、特に今回分析したファイザー/バイオエヌテック試験のような緊急時に実施される短期試験ではなおさらである。
ファイザー/バイオエヌテック社は、FDAの承認を得て、臨床試験C4591001のプラセボ対照部分を20週目に中止した。被験者には盲検化が解除され(ワクチン接種の状況が知らされ)、プラセボ群の被験者はBNT162b2のワクチン接種を希望することができるようになった。通常の状況では、プラセボの盲検化は、治療/介入によって生命が救われたため、試験を継続することが倫理的に問題があると考えられる場合にのみ行われる。図1のエビデンスは、ワクチン接種が死亡率を減少させないことから、この決定を支持しない。プラセボ被験者の盲検化を解除することで、FDAは2020年12月11日にプラセボ対照臨床試験の体裁をすべて終わらせた。驚くべきことに、生存率の改善はC4591001臨床試験のエンドポイントではなかったため、この情報は無視された。
図1は、このようなBNT162b2 mRNAワクチンのプラセボ対照無作為化臨床試験(少なくとも最初の20週間)の結果である。結果は予想外であった。第一に、被験者の死亡数は予想の5分の1以下である。被験者集団は事前にスクリーニングされたが、除外理由は死亡数の少なさを説明するほど厳密ではなかった。Pfizer/BioNTech、Polackら(2020)、Thomasら(2021)のいずれも、死亡数が少ないことについてコメントしていない。
ワクチン接種群とプラセボ群の死亡率は、試験開始後20週間では同程度であったことから、ワクチンが全死因死亡率を減少させなかったことは明らかである。もしBNT162b2 mRNAワクチン接種がCOVID-19による死亡を減少させているとすれば、この所見はCOVID-19による死亡の減少が他の原因による死亡の増加と釣り合っていることを示唆している。われわれの結果は、このような他の原因の1つが心イベントによる死亡である可能性を示唆している。数が非常に少ないので、これらの他の原因が何であるかについて確固とした結論を出すことは困難である。また、我々の知る限り、ファイザー社/バイオエヌテック社の試験データを評価した国際的な医療規制機関のメンバーや医学文献の査読者が、この所見についてコメントしたり、説明を求めたりしたことはなかった。
ワクチン接種によって死亡が減少しなかったという所見は、南半球における全死因死亡率の研究(Rancourt et al.) 2020年から2022年にかけて行われたこの解析には、4大陸の17カ国が含まれ、BNT162b2を含む異なるCOVID-19ワクチンを使用していた。17カ国すべてにおいて、COVID-19ワクチンが全死因死亡率に有益であるという証拠は見つからなかった。むしろデータは、COVID-19ワクチンのブースター展開と同時かその直前に、全死因死亡率の「前例のない」ピークを示している。
死因は両群間でアンバランスである
表1および表2のデータは、両群の全死因死亡率が同程度であるにもかかわらず、死因のバランスがとれていないことを示している。死亡38例中14例(36.7%)が心イベントの結果であり、臨床試験の治療群では心イベントによる死亡が3.7倍に増加していた。さらに、心イベントによる死亡数の増加は、BNT162b2群における死亡数(21例)とプラセボ群における死亡数(17例)の差を説明する以上のものであった。
心イベントシグナルを発見したことは、ファイザー社/バイオエヌテック社のmRNA抗SARS-CoV-2ワクチンの世界展開後に実施された研究と一致している。本報告書で確認された心イベントシグナルは、ファイザー/バイオエヌテック社のmRNA抗SARS-CoV-2ワクチンの世界的展開後に実施された報告によって確認された。ここで報告された心イベントシグナルのエビデンスは、プラセボ対照無作為化臨床試験から直接得られたものであるため、WHO委員会(WHO Ad Hoc Expert Group on the Next Steps for COVID-19 Vaccine Evaluation, 2021)が指摘したような、ロールアウト後のレトロスペクティブな観察研究に伴う注意点がないことに注意することが重要である。Romeroら(2023)は、VAERS、米国防総省DMED、CDCのV-Safe、イングランドとウェールズの国家統計局、欧州連合のEuroStatなど、米国内外の複数のワクチン安全性データベースのレビューに基づき、急性心筋梗塞の症例数と心臓発作による死亡数が明らかに増加していることを報告している。さらに、30,712,101人の高齢者を対象とした米国メディケア&メディケイドサービスセンター(CMS)のデータを用いて、Wongら(2023)は急性心筋梗塞の統計的に有意な増加(RR = 1.42)を発見した。
Bardaら(2021)は、BNT162b2ワクチンのプラセボ対照臨床試験を模倣するために、イスラエル最大の医療システムによって収集された潜在的な有害事象に関する広範な観察データを使用した。患者はワクチン1回接種後42日間追跡されたが、これは長期的な効果を示すには不十分かもしれないとのことである。この研究では、心筋炎の相対リスク(RR)は3倍に増加したが、心筋梗塞の増加はごくわずか(RR=1.07)であった。同じイスラエルの患者を対象とした別の研究で、Witbergら(2021)は各ワクチンの接種後3-5日目に心筋炎の症例数の増加を認めた。心筋炎全体の推定罹患率は10万人あたり2.13例で、16歳から29歳の若い男性で最も罹患率が高かった。心筋炎は心筋の炎症であり、時間の経過とともに細胞障害や重篤な心不全を引き起こす可能性がある。これらの所見を総合すると、mRNAワクチン接種後に心筋障害が増加することが示唆される。
BNT162b2ワクチンの心臓への副作用の根拠を説明するメカニズムが提案されている。その中心はすべて、ワクチンにコードされたスパイクプロテインの毒性である(Santiago & Oller, 2023);Trougakosら, 2022))。スパイクプロテインによる異常な微小血栓の刺激は、観察された組織損傷の一因として提唱されている(Kell et al., l22);Nyström & Hammarström、2022);De Michele et al., e22)。微小血栓が心筋などの脈管組織への血流を遮断し、酸素欠乏と虚血障害を引き起こす可能性が示唆されている。スパイクプロテイン自体が、心筋ペリサイト上のCD147レセプターに結合することにより、心筋ペリサイトの機能を変化させ、心血管系に障害をもたらすことが示されている(Avolio et al., o21)。彼らはまた、スパイクプロテインがCD147に依存しない機序で周皮細胞に炎症性サイトカインを放出させ、隣接する心筋細胞にダメージを与え、血液凝固を誘発し、血管伝染性を増大させる可能性があることを示している。Krausonら(2023)は、ワクチン接種後の患者20人と非接種対照患者5人を剖検し、様々な組織におけるワクチンmRNAの存在を調べた。人の患者の左心室2検体、右心室2検体からワクチンが検出されたが、これらの患者はすべて死亡後30日以内にBNT162b2を接種されていた。組織領域におけるワクチンmRNAの存在は、心筋傷害の治癒やマクロファージの浸潤と関連していた。また、局所的なスパイクプロテインの発現とスパイク免疫病理による攻撃の可能性を示唆している。Brognaら(2023)は、ワクチン接種後69-187日間、ワクチン接種者だけの血液中にワクチン由来のスパイクプロテイン(PP-Spike)が存在することを証明した。PP-スパイクの持続的発現については説明されていないが、PP-スパイクの毒性作用が長期間持続する可能性が出てきた。
有害事象の方向が透明性を欠く要因
少なくとも5週目から心イベントによる死亡が発生していたことを考えると、なぜ20週目以前に試験のスポンサーから不均衡が報告されなかったのだろうか?C4591001臨床試験における透明性の欠如にはいくつかの要因があるが、その中でも最も顕著なのは、ファイザー/バイオエンテックが被験者のNarrative Reportに記載されている実際の死亡日を報告せず、代わりに死亡が症例報告用紙に記入された日付を使用したことである。
C4591001臨床試験でファイザー/バイオエンテックが使用した症例報告書システムは、一般に認められた業界標準に準拠しておらず、おそらく臨床試験被験者の死因に関する混乱を招いたと思われる(有害事象の6カ月中間報告C4591001)。表1に記載された診断名はエビデンスに基づかないことが多く、症例報告書は透明性に欠け、使い勝手が悪く、ファイザー社/バイオエヌテック社と治験実施医療機関のモニターとの間の回答の完全な「保管の連鎖」を提供していないように思われた。これらの問題は、死因の少なくとも一部が心筋梗塞(MI)または既存の心臓虚血の進行に起因するはずであった被験者11271112および10841266に関して特に関連する。治験コーディネーターは、重篤な有害事象(SAE)の中で最も重篤な死亡38例のみを扱っていた。彼らの最重要課題は真の死因を特定することであったはずである。
プラセボ対照無作為化臨床試験では、すべての参加者の情報を考慮できる試験終了時に、統計学的根拠に基づいて因果関係が決定される。これは、この試験でスポンサーのファイザー/バイオエンテックが行ったように、治療に関して利害の対立する臨床試験の商業スポンサーが行うべき決定ではない。また、今回の臨床試験のように、ケースバイケースで行われるべきでもない。
様々な監視委員会とFDAのVRBPACは、有害事象のシグナルを特定し報告することをファイザー/バイオエヌテックに依存していた。治験依頼者のデータ評価を確認するためのデューデリジェンスは行われなかった。有害事象の6カ月中間報告書C4591001またはファイザー/バイオインテックが公表した報告書のいずれにも、「ワクチン接種に関連する」カテゴリーを含めるべきでなかった。有害事象が調査中の治療に関連するかどうかは、試験を監督する規制当局が判断すべきであった。C4591001は医療上の緊急事態と称される中で進行中の試験であったため、それを行う適切な時期は、ファイザー/バイオエンテックがFDA VRBPACにプレゼンテーションを行った日、2020年12月10日であっただろう。しかし、これは行われず、ファイザー社/バイオエンテック社は12月10日に試験データを更新する必要もなかった。
我々の分析(表2)によると、報告された死亡者数の不一致は、EUA申請のためのデータカットオフ日である11月14日と、ファイザー/バイオエヌテックがFDA VRBACにプレゼンテーションを行った12月10日の2つの重要な時点で観察された。このことは、C4591001プロトコル(https://clinicaltrials.gov/ct2/show/NCT04816643)においてFDAがAdverse Event of Special Interest(AESI)として強調することを望んだ、極めて重要な「安全性」シグナルである心イベントによる死亡数の2倍増を不明瞭にする効果があった。我々は、ファイザー/バイオエンテックが報告書に不必要に混乱させる用語を使用していることを発見した。好ましい用語のリストには、症例報告書の診断に関する具体性の欠如に照らして、正当化されるよりも多くのカテゴリーがあった。さらに、ファイザー社/バイオエヌテック社は、期間の正確な開始日と終了日を示すのではなく、「非盲検観察期間」や「盲検解除後」といった曖昧な表現を用いた。これは特に「非盲検フォローアップ期間」と「非盲検観察期間」の分析に関連しており、Thomasら(2021)によるプラセボ被験者3名の損失につながったと思われる。ファイザー/バイオエンテックの臨床安全性6カ月報告書にある表のタイトルは、そこで報告されたデータを明確にするよりも、読者を混乱させることの方が多い。典型的な例を2つ挙げる: 「表13:投与1回目から投与2回目6カ月後までに少なくとも1件の有害事象を報告した被験者の数(%)、全身臓器クラス別および希望する期間別-投与2回目以降に少なくとも6カ月の追跡期間がある被験者-16歳以上のフェーズ2/3被験者(BNT162b2を最初に投与された被験者)-安全性集団」および「表19: 盲検化解除日からデータカットオフ日(13MAR2021)までの少なくとも1件の重篤な有害事象の発生率、臓器クラス別および希望する期間別、非盲検フォローアップ期間、BNT162b2を最初に投与された被験者、16歳以上のフェーズ2/3被験者、安全性集団」である。
これらのテクニックは、C4591001の臨床試験で明らかにされた真のエビデンスを難解にするものであった。ファイザー社/バイオエヌテック社が使用した症例報告書の書式は、通常期待される水準に達しておらず、透明性が保たれていなかった。被験者の本当の死亡日は被験者のNarrative Reportに記載されるが、治療群に関係なくすべての被験者の症例報告書に適時に記録されなかった。規制当局による臨床試験の商業スポンサーに対する監督もまた欠如していた。さらに、この臨床試験に関する医学文献の出版物は、批判的な目で見直され、編集されたものではなく、おそらく基礎データへの適切なアクセスもなかったであろう。
この報告書は、FDA、CDC、NIHなどの連邦機関が新薬の開発と安全性/有効性評価で使用するプロセスに非常に重大な欠陥があることを明らかにした。1980年の特許商標法改正法バイ・ドール法 2004年のプロジェクト・バイオシールド法 2005年の公共準備・緊急事態準備(PREP)法、そして2016年の21世紀治療法である。これらの法律により、BNT162b2 mRNAワクチンの製造・開発企業であるファイザー/バイオエンテックは、すべての法的責任を回避しながら、臨床試験のオリジナルデータを完全に管理することができた。FDAの承認により、ファイザー/バイオエンテックは、治験ワクチンと利害関係のない医学・科学研究の専門家によるオリジナルデータへのアクセスをブロックすることを許された。44,060人の被験者に関する情報は、ファイザー/バイオインテックの担当者によって収集、監視、評価、保存、分析された。C4591001プロトコル(https://classic.clinicaltrials.gov/ct2/show/NCT04368728)を見直せば、この臨床試験のデータが、数百万件に及ぶ可能性のある医療報告書、臨床試験結果、予定されていた訪問報告書、予定されていなかった訪問報告書などのデータベースを含む膨大なものになることは明らかであろう。FDAやその他の国際的な医療規制機関に提出される報告書も、ファイザー/バイオインテックの担当者が作成し、評価完了まで数日しか与えられなかった。
この特別な臨床試験の審査はユニークなものであった。臨床試験の進行は通常、データ安全性モニタリング委員会(DSMB)によって監視される。DSMBは独立した専門家からなる少人数のグループで、その役割は試験期間中の安全性と有効性のデータを検討し、試験を継続、変更、中止するかどうかについて助言を与えることである。しかし、C4591001試験はそうではなかった。ファイザー社/バイオエヌテック社の臨床試験は米国政府の資金提供を受けていなかったため、ファイザー社/バイオエヌテック社は独自のDSMBを設立することを許された。そのため、ファイザー/バイオエンテックのDSMBメンバーは、利害の対立がない独立したメンバーとは見なされなかった。FDAは11月20日から12月11日まで、この膨大なデータセットをレビューし、安全性に関する決定を下すのに数日しかなかった。おそらくFDAとそのVRBPACは、ファイザー/バイオエヌテックからの膨大なデータバンクに関する要約報告書と、ファイザー/バイオエヌテックのDSMBによる監視の厳密さと徹底性に、あまりにも大きく依存していたのであろう。透明性を求める公衆衛生および医療専門家連盟が起こした裁判が成功していなければ、ファイザー社とバイオエヌテック社以外の誰も、この臨床試験で作成されたオリジナルデータを調査する機会はなかっただろうし、ここで報告されたような矛盾が明らかになることもなかっただろう。米国FDAやその他の国際的な規制機関によるBNT162b2 mRNAワクチンの承認決定は、このワクチンがCOVID-19の「パンデミック」に対応する「安全かつ効果的」な手段であるという基準を満たすことを証明するための、公平で徹底的かつ透明性のある証拠の評価に基づいた情報に基づいた決定ではなかった。
新規ワクチンの安全性を評価するには、20週間では十分ではなかった
RNAの治療利用は大きな可能性を示しているが、まだ発展段階にある(Dolgin, 2021; Sahin et al.) これらに含まれるのが、ワクチンへのmRNAの利用である(Rcheulishvili et al., i22)。当初、mRNAワクチンの使用は、マウスやブタやウシなどの農耕動物を含む動物モデルで検討された(Geall et al., l12;Lutz et al., z17;Schnee et al., e16)。
送達方法は、裸のmRNAからmRNA-LNP粒子まで様々であった。試験された疾患はインフルエンザと狂犬病であった。免疫応答は証明されたが、疾病や疾病伝播に対する免疫学的有効性は決定されなかった。
mRNAワクチン技術の初期のヒト臨床試験は、抗腫瘍免疫療法の開発に焦点を当てていた(Sebastian et al., n19)。CV9201 mRNAワクチンの組成は独自のものであったが、mRNAは5つの腫瘍抗原をコードする混合物であった。CV9201は非常に緩やかな免疫応答を誘導したが、進行期IIIB-IVの非小細胞肺がん標的への影響は観察されなかった。Albererら(2017)は、18~40歳の健康な成人101人を対象に、mRNAベースの狂犬病ワクチン候補CV7201の非盲検非対照前向き第1相臨床試験を実施した。CV7201 mRNAは狂犬病ウイルス糖タンパク質(RABV-G)を遊離型およびカチオン性タンパク質プロタミンとの複合体でコードしていた。この試験では免疫反応の促進が確認された。Aldrichら(2021)は、55人のヒトを対象とした別の非無作為化非盲検対照用量漸増第1相臨床試験において、狂犬病ウイルス糖タンパク質をコードする未修飾のmRNAを用いてmRNA-LNPデリバリーシステムを試験した。その結果、不活化狂犬病ウイルス粒子からなる既存の狂犬病ワクチンRabipurに匹敵する有意な免疫応答が認められた。これらのmRNAワクチンの有効性はヒトではテストされていない。
上記の議論から明らかなように、ファイザー/バイオエヌテックのC4591001臨床試験以前には、mRNA-LNPワクチンの臨床試験はごくわずかしか行われていない。そのどれもが、投与レベルを決定するためのフェーズ1試験以上に進展しておらず、これは、潜在的リスクを十分に認識したごく少数のヒトボランティアを巻き込むものである。過去のワクチン承認では、承認されるまでに5〜10年以上の安全性試験が必要だった。mRNA-LNPデリバリー・プラットフォームの新規性と未試験の性質を考えると、FDAがBNT162b2ワクチンの安全性を宣言するのに、なぜ20週間で十分と考えたのか理解しがたい。このmRNA-LNPデリバリー・プラットフォームの長期安全性は不明である。mRNA-LNPプラットフォームによって刺激される免疫反応の寿命や、SARS-CoV-2ウイルスの伝播が防止されるかどうかも不明である。漏れやすく不完全なワクチンは、より伝播性の高い病原体の進化を促進するという確立された理論(Read et al.)がある。
ワクチンBNT162b2に関して最も懸念されるのは、コードされた抗原であるスパイクプロテインの前臨床試験がファイザー社によって検討されなかったことである。ファイザー社は、スパイクは「内因性タンパク質」であり、細胞内プロセスによって分解されると考えた(オーストラリア保健省への非臨床評価報告書https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf)。Sahinら(2014)は、mRNAワクチンの潜在的危険性をいくつか述べている。一つの懸念は、コードされた抗原としてスパイクプロテインを使用することである。”外来タンパク質の発現と、mRNAバックボーンによって媒介される炎症促進作用が、組織レベルでの免疫病理学を引き起こす可能性も考えられる”。彼らは、コードされるタンパク質の特性や機能に依存する。「内容物」特異的リスクと呼ばれる、さらなる懸念についても論じている。このようなリスクはケースバイケースで評価されなければならない。残念ながら、スパイクプロテインがコードされた抗原として選択されたとき、この点は考慮されなかった。要約すると、BNT162b2ワクチンは、十分な前臨床試験が行われることなく、またこの全く新しいワクチンの安全性を証明するには不十分な期間内に、製造と世界的な流通を急いだのである。
過去50年間、米国はウイルス性伝染病をコントロールするために、何度か集団予防接種プログラムを実施してきた。1976年には、4,500万人に対する豚インフルエンザワクチン接種後の6週間に362例のギラン・バレー症候群が発生したが、これは通常のバックグラウンドの8.8倍の増加であった(Nelson, 2012)。ギラン・バレー症候群はまれな疾患であるため、安全シグナルとして認識されやすい。死亡や心臓発作は、はるかに一般的な有害事象である。そのため、警告シグナルとして認識されにくい。これらを重篤な有害事象の安全性シグナルとして特定するためには、極めて多数の治験被験者とより長い追跡調査期間が必要である。豚インフルエンザワクチンが数千万回接種された後、FDAが豚インフルエンザワクチン接種の中止を決定したのは、9つの州で3人の高齢患者が死亡してからであった(Schmeck, 1976; Schwartz, 1976)。
もしFDAがこの報告書に記載された心臓疾患のシグナルを知っていたら、1976年の豚インフルエンザワクチンの大失敗に見られたように、規制当局はmRNAワクチンの安全性の問題について考え直したかもしれない。mRNAワクチンの市販後に報告された早期警告シグナルやその他の有害事象の妥当性が証明されているにもかかわらず、この新しいタイプのワクチン・プラットフォームは市場から排除されておらず、6カ月の小児にさえ承認されている。なぜなのだろうか?
結論
- 1. 22,030人のワクチン接種被験者と22,030人のプラセボ被験者を対象としたC4591001プラセボ対照無作為化臨床試験は、ファイザー/バイオインテックのBNT162b2ワクチンを公平に評価する世界で唯一の機会であった。
- 2. 20週目からプラセボ被験者の盲検化を解除したことにより、プラセボ対照臨床試験が終了し、有害事象シグナルの可能性に関する偏りのない評価がすべて終了した。
- 3. mRNA-LNPプラットフォームは新規のものであり、ヒトでの第2/3相試験は行われておらず、PP-スパイクプロテインの毒性は未知であった。これらを総合すると、20週間のプラセボ対照臨床試験だけでは、最も一般的な安全性懸念事項を除き、何かを特定するには不十分である。
- 4. 全死因死亡数はBNT162b2ワクチン接種によって減少することはない。
- 5. 6カ月間の有害事象中間報告で報告された38例の死亡例のうち、BNT162b2ワクチン接種群では21例が死亡し、プラセボ群では17例が死亡した。
- 6. 治験実施計画書に違反する症例報告書への被験者の死亡報告の遅れにより、EUAは異議を唱えられずに進行した。
- 7. 被験者の死亡数は、米国の年齢調整死亡率に基づく予想数の17%であった。1つの可能性として、”Lost to Followup “となった395人の被験者がある。
- 8. BNT162b2ワクチンを接種した被験者では、プラセボと比較して心イベントが3.7倍増加した。
- 9. 成人突然死(SAD)または死体発見(FD)となった15人の被験者のうち、12人が心臓イベントで死亡し、うち9人がワクチン接種を受けていた。
- 10. 心臓有害事象のシグナルは、ファイザー/バイオエンテックが把握していた被験者の正確な死亡日を被験者のNarrative Reportに報告するのが遅れたために不明瞭になった。
競合利益
なし。
Forensic Analysis of the 38 Subject Deaths in the 6- Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial