Contents
www.ncbi.nlm.nih.gov/labs/pmc/articles/PMC8538968/
Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways
オンラインで2021年10月18日公開
Amanda Cano,1,2,3,4,* Elena Fonseca,5,6 Miren Ettcheto,2,7,8 Elena Sánchez-López,2,3,4 Itziar de Rojas,1,2 Silvia Alonso-Lana,1 Xavier Morató,1 Eliana B. Souto,9,10 Manuel Toledo,5,6 Mercè Boada,1,2 Marta Marquié,1,2,† and Agustín Ruíz1,2,†
ジュゼッペ・ビアジーニ(学術編集者) Giuseppe Biagini
要旨
てんかんは、神経細胞の電気的不均衡を特徴とする中枢神経系の慢性疾患である。てんかんは、神経細胞の電気的不均衡を特徴とする中枢神経系の慢性疾患であり、世界で5,000万人が罹患している2番目に多い神経疾患である。また、てんかん患者の30%は利用可能な治療法に反応しない。現在、てんかん発作の引き金となり、細胞死につながる神経毒性作用を促進する分子プロセスに関する主な仮説は、さまざまな要因によるグルタミン酸経路の悪化と神経細胞へのCa2+の大量流入に焦点を当てている。しかし、それ以外のメカニズムも提唱されており、そのほとんどが、アルツハイマー病、パーキンソン病、ハンチントン病、多発性硬化症などの他の神経変性疾患でも報告されている。興味深いことに,このような共通の分子的関連性と,これらの疾患に対する有効な治療法がないことを主な理由として,いくつかの抗てんかん薬がこれらの病態における治療の可能性を評価するために研究されてきた。そこで,この総説では,てんかんと主要な神経変性疾患との間の共通の分子経路を徹底的に調べ,これらの集団におけるてんかんの発生率を検討し,難治性のてんかんやその他の神経変性疾患の治療における現在の,そして革新的な抗けいれん薬の使用を探求する。
キーワード:てんかん、神経変性疾患、アルツハイマー病、パーキンソン病、ハンチントン病、多発性硬化症
ハイライト
- てんかんは2番目に多い神経疾患であり、神経変性疾患の患者にも現れることから、両者の間には分子的なつながりがあると考えられている。
- βアミロイド斑、神経原線維変化、αシヌクレイン、ハンチンチンタンパク質の変異などの出現と、発作に先立つ神経細胞の興奮性亢進とを関連付ける証拠が増えてきている。
- アトルバスタチン、セフトリアクソン、ロサルタン、アナキンラ、ラパマイシン、フィンゴリモドなどの承認されているいくつかの薬剤は、抗てんかん薬として動物モデルで研究されている。
- レベチラセタム、ゾニサミド、バルプロエートなど、一般的に使用されている抗てんかん薬は、他の神経変性疾患でも研究されている。
1. はじめに
てんかんは,神経細胞の電気的活動の不均衡を特徴とする中枢神経系(CNS)の慢性疾患であり,様々な再発性かつ予測不可能な発作を引き起こす[1]。てんかん症候群の中には、皮質の菲薄化や脳容積の減少が進行し、脳の複数の領域で神経細胞が死滅するものがある[2,3]。最新のGlobal Burden of Disease調査によると、てんかんは、障害調整生命年の観点から、世界で2番目に深刻な神経疾患と考えられている[4]。2016年には、全活動期のてんかん患者数は世界で4,590万人、年齢標準化された死亡率は10万人あたり1.74人と推定されている[4]。全世界では、毎年240万人がてんかんと診断されていると推定されている。Brainstorm Consortiumによると、てんかんは最も遺伝性の高い神経疾患であるとされている[5]。先進国では、一般人口10万人あたり、年間30〜50人の新規患者が発生している。一方、発展途上国や低開発国では、この数字は最大で2倍にもなる。これは、風土病や出産関連の傷害のリスクが高まること、医療インフラにばらつきがあること、予防的な健康プログラムの利用が少ないことなどが原因である[6]。Fiestらが行ったメタアナリシスでは、てんかんの生涯有病率は、原因不明のてんかんと、有病率の高い全般発作を伴うてんかんを含めて、全世界で1000人あたり7.60人であると指摘されている[7]。
てんかん発作は、脳内の神経細胞が異常に過剰または同期して活動することで、さまざまな症状を引き起こす。発作には、特定の脳領域またはネットワークが関与するもの(焦点性発作)と、同期的な両半球の放電が関与するもの(全般性発作)がある[8]。てんかんの分類は複雑で、発作のタイプから、発症年齢、特定の病因、併存疾患などのいくつかの臨床的特徴を包含するてんかん症候群まで、さまざまなレベルが含まれている[8]。国際抗てんかん連盟(International League Against Epilepsy: ILAE)の最新の分類によると、てんかんの病因は、構造的病因、遺伝的病因、感染的病因、代謝的病因、免疫的病因、および未知の病因に分類される[8]。
分子レベルでは,これらの疾患は,シナプス前膜の脱分極を促進し,これが神経細胞の過興奮性の主な原因となり,てんかん発作に特徴的な異常な電気活動を引き起こすと言われている(図1)。過剰刺激を受けると、いくつかのイオンチャネルや膜受容体の構造変化が起こり、Ca2+イオンやNa+イオンが神経細胞内に大量に流入し、K+イオンが流出する。そして、このイオンの不均衡が、神経毒性作用や神経可塑性の変化を促進するさまざまなシグナルカスケードの活性化を引き起こし、最終的に細胞死に至る[9]。
図1 てんかんとそれに伴うASDにおける発作活動の発生の一般的な分子メカニズム
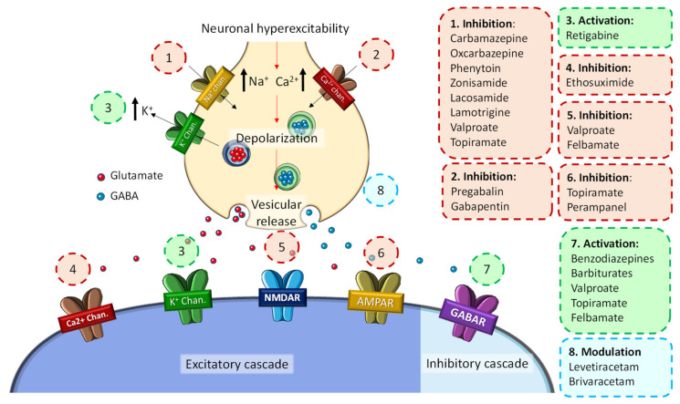
Hughlings Jacksonが発作は局所的な神経細胞の発火によるものであると提唱した19世紀後半以降、大脳皮質が解剖学的に発作の主な原因であると考えられてきた[10,11,12]。近年、病理組織学的、電気生理学的、定量的な神経画像学的研究の結果から、焦点性発作と全般性発作の両方に、皮質と皮質下の構造の神経ネットワーク間の多様な相互作用が関与していることを示す十分な証拠が得られている[13]。同様に、発作は異なる脳構造間の一般的な変化だけでなく、過剰または非同期の発作的な活動に支配された神経ネットワークの機能不全によるものであることが説明されている[13]。局所てんかんは、成人のてんかんの中で最も一般的なタイプであり、発作の主な発生部位は側頭葉であるが、前頭葉、頭頂葉、後頭葉にも発生の病巣が観察されている(頻度の高い順)[14]。同様に、扁桃体-海馬複合体は、てんかん発生プロセスに関与する重要な解剖学的回路の1つである。海馬硬化症は、側頭葉てんかんにおける典型的な組織学的所見であり、神経細胞の損失の代表的な形態である[15]。
てんかん症候群には多くの種類があり、その原因、てんかん原性病巣、症状も異なるため、てんかんの治療法も複雑で、多くの場合、効果がない。20世紀初頭、最初の抗てんかん薬(フェノバルビタール、バルプロエート、ベンゾジアゼピンなど)が登場し、第二世代の薬(ガバペンチン、プレガバリン、ラモトリギン、レベチラセタム、トピラマートなど)が新たな治療法として臨床現場に登場したのは1990年代に入ってからであった[17]。そのため,近年では,第3世代の薬剤(ラコサミド,ルフィナミド,ペランパネルなど)が登場している。これらの物質は、強化された制御中枢活性と、より良好な薬物動態プロファイルを有している(図1)[17]。しかし,これらの薬剤は,ほぼ発作抑制のみに焦点が当てられており,てんかんの発生メカニズムには焦点が当てられていないため,現在,抗てんかん薬(ASD)と呼ばれている[17]。そこで,この総説では,てんかんと主要な神経変性疾患との間の共通の分子経路を徹底的に調べ,これらの集団におけるてんかんの発生率を検討し,難治性てんかんやその他の神経変性疾患の治療における現在の革新的なASDの使用法を探ることにした。
2. 神経変性疾患におけるてんかん
2.1. てんかんとアルツハイマー型認知症
アルツハイマー病(AD)は、世界で5,000万人が罹患している最も一般的な形態の認知症であり、神経変性プロセスに関連した記憶喪失と認知機能の低下を特徴としている[18]。ADにおける神経毒性とシナプス機能障害の主な仮説は、この病気の典型的な病理学的特徴、主にリン酸化タウ(p-tau)の細胞内神経原線維変化(NFT)と細胞外アミロイドβ(Aβ)老人斑に焦点を当てているが、ADの病因に関与する他の多くのメカニズムが説明されている[19]。
アルツハイマー病患者におけるてんかんの頻度については、アルツハイマー病患者は、ADでない人に比べて、てんかん発作を起こすリスクが80倍以上高いことが報告されている[20]。その一方で、てんかん患者は、数年後に認知症を発症するリスクが高いとされている[21]。これらの理由から、ADにおける発作の発生率の増加は、ADの発症が一般的に65歳以降であることに起因しているだけではないかと主張されている[22]。しかし、てんかんとADの関係については、多くの議論がなされている。いくつかの研究では、アルツハイマー病患者の間で発作の発生率が高いことが示されているが、多くの著者は、使用したモニタリングの種類や調査した集団に応じて、ADにおける発作の有病率は3.5%から64%の範囲であることを強調している[23]。
ADの病理学的メカニズムとてんかんとの間の分子的関係は、脳のグルコース代謝低下、空間記憶やナビゲーションの障害、海馬ニューロンの損傷、側頭葉の全般的な神経変性など、共通して広く見られる証拠から、広く研究されている[20]。興味深いことに、老人斑は、ADの症例が初めて報告されるよりも10年以上前に、てんかん患者で初めて報告された[24]。実際、ADとてんかんの関係を評価した最初の臨床研究は、1950年代初頭にさかのぼる[25,26]。どちらの疾患も神経細胞の損傷を伴い、また、双方向の関連性があるようである[27]。
コール博士の研究グループは,ボストンのマサチューセッツ総合病院とハーバード・メディカル・スクールのてんかんサービスで,興味深い研究を行った。その結果、アルツハイマー病患者は、臨床症状を伴わない不顕性発作を睡眠中に起こすことがわかった。この研究は、発作がADの病的なペースを調整、促進、または加速するのではないかという仮説を浮き彫りにした[28]。同様に、10年前に行われた研究では、アルツハイマー病患者の42%が潜在性発作を発症したのに対し、対照群では11%であった。このてんかん活動は、主に睡眠の深い段階で側頭葉に由来するものであった。さらに、5年間で、これらのアルツハイマー病患者は、潜在性発作のないAD対照群と比較して、認知機能の低下が進んでいった[20]。
てんかんとADを結びつけるメカニズムはいくつか報告されている。最近の実験データによると、神経細胞の興奮性亢進自体が、ADの神経病理学的負担や認知機能の低下を促進する上で重要な役割を果たしている可能性が示唆されている[22]。したがって、ADに特徴的なアミロイドβ(Aβ)およびタウペプチドレベルの上昇は、発作を誘発する分子経路と関連していると考えられる(図2)。
図2 アルツハイマー病の主要な病理学的分子経路に由来する発作活性

アルツハイマー病の病理学的特徴は、ACh受容体やNMDA受容体、Na+/Ca2+チャネルを介して、神経炎症や細胞内Ca2+の増加を促進する。これにより、神経炎症や神経細胞の興奮性亢進が促進され、その結果、神経変性プロセスが増加する(逆もまた然り)という悪循環に陥る。NE, norepinephrine(ノルエピネフリン)。
2.1.1. てんかんにおけるAβの役割
アミロイド前駆体タンパク質(APP)、プレセニリン-1(PS1)、プレセニリン-2(PS2)遺伝子の変異が典型的な原因である遺伝性ADに罹患した患者は、特に発作を起こしやすい集団であり、発作発生率は30%以上である[29]。これらの知見は、てんかん感受性におけるAβの重要な役割を支持するものである(図2)。さらに、派生した仮説では、ADの分子変化がてんかん発作を促進し[22]、それがADの病理を悪化させるという悪循環が記述されている[29]。ADでは、Aβプラークではなく、可溶性オリゴマーAβが神経細胞の興奮性亢進の主な原因であると報告されている[22]。そのため、Aβ可溶性ペプチドの中で最も毒性の高いAβ1-42は、K+電流を選択的に阻害することで神経細胞の興奮性を高めることがわかっている[30]。グルタミン酸シグナルもまた、アルツハイマー病患者のAβによって変化することが記載されている。神経細胞やグリアのグルタミン酸再取り込みの障害は、グルタミン酸のスピルオーバーを引き起こし、その結果、興奮毒性を引き起こす可能性がある。同様に、グルタミン酸の興奮毒性は、N-methyl-D-aspartate receptor (NMDA-R)のトラフィッキングに対するAβの影響によっても悪化する[31]。Kamらは、Aβによるコリン受容体やCa2+チャネルの活性化が、臨床的なADに先行する初期の潜在的なてんかん活動を引き起こすのではないかという仮説を立てた[32]。間接的には,Aβの形成に関与する主要なタンパク質の1つであるβセクレターゼ1(BACE1)も,てんかん発生プロセスの促進に関係している(図2)[22]。BACE1は,電位依存性Na+チャネルのβ2およびβ4サブユニットを切断することが,いくつかの研究で報告されている.β2 の切断は,細胞表面上の受容体の転写および発現を変化させ[33],β4 の切断は,細胞内のNa+レベルを著しく上昇させる[34].この2つのプロセスは,全般的な神経細胞の興奮性亢進をもたらし,最終的には発作の発生につながる。Kimらは、前臨床研究において、BACE1欠損マウスのナトリウムチャネル代謝の生理的変化を明らかにした[35]。彼らは、BACE1-nullマウスの脳では、Nav1.1タンパク質レベルおよびNavβ2プロセッシングが野生型マウスに比べて有意に低下していることを発見した。興味深いことに、BACE1-nullマウスでは、海馬表面のNav1.1レベルが有意に低下していたが、Nav1.2レベルは上昇していた。これは、表面のNav1.1レベルの低下に対する代償メカニズムであると考えられる。これらの結果は、BACE1活性を治療的に阻害することで、アルツハイマー病患者のNa+代謝に影響を与え、神経細胞膜の興奮性を変化させる可能性があることを示唆している[35]。同様に、BACE阻害剤が痙攣の発生に関与している可能性があると言われている。この点に関しては、最近、BACE阻害剤は、学習や記憶に影響を与えることなく、発作関連遺伝子ファミリーを持つ人に多動性を誘発することが報告されている[36]。
また、神経炎症の発生、ひいては発作の発生にAβが関与している可能性も指摘されている(図2)。神経炎症は,病的なプロセスに対する反応として,中枢神経系において免疫反応が引き起こされることを特徴とし,てんかんとADの両方で検出されている[37]。中枢神経系における炎症は,主にミクログリア,アストロサイト,およびオリゴデンドロサイトによって媒介される[38]。Aβによるグリアの活性化は,多数の炎症性サイトカイン(すなわち,TNF-α,IL-6,またはIL-1β)の放出を引き起こし,全般的な神経炎症の出現を引き起こす。このプロセスは、神経毒作用を促進し、最終的には神経細胞の過興奮性の出現につながり、さらに神経変性プロセスを増加させるという悪循環に陥いる[22]。同様に,IL-1βなどの炎症性サイトカインは,アストロサイトによるグルタミン酸の放出を促進し,その再取り込みを減少させることによって[39],あるいはNMDA-Rをアップレギュレートして細胞内Ca2+の流入を増加させることによって,神経細胞の興奮性亢進を増大させることが記載されている[40]。さらに、生体内試験および試験管内試験の研究では、炎症の悪化と発作の間に双方向の関係があることを示す証拠が得られており、両方の事象が悪循環の中で互いにフィードバックされている[39]。
2.1.2. てんかんにおけるタウの役割
てんかん発作の発生におけるタウの役割を理解する上で、動物モデルは非常に有用である(図2)。トランスジェニックAPP/ノックアウトタウマウスを用いた前臨床モデルでは、タウタンパク質がAβのてんかん発生作用の必要なメディエーターであることが示唆された[41]。この研究では、トランスジェニックマウスは、野生型マウスよりも発作の頻度が低く、重症度も低かった。また,タウタンパク質は,細胞外グルタミン酸の増加とNMDA-Rの機能不全によって,顕著な神経細胞の興奮毒性を促進することが示されている[42]。同様に、タウは、てんかんの発症に深く関わる海馬での神経細胞の移動異常にも関係している[43]。
2011年に行われた慢性てんかん患者の死後調査では、解析した脳のほぼ70%に軽度または中等度のADのタウ病理が認められた[44]。タウ負荷は、進行性の認知機能低下と有意に関連しており、慢性てんかん患者では、特発性てんかんや遺伝性全般てんかんの患者よりも、焦点性てんかんの方がより高いタウ負荷と関連していることが多かった[44]。同様に、てんかん発生の3つの異なる動物モデルを用いた研究では、細胞内のリン酸化/脱リン酸化を司る酵素であるホスファターゼ2A活性の低下が認められ、これがてんかん発生脳領域でのp-tauの増加につながっていた[45]。
2.1.3. ADとてんかんにおけるアロプレグナノロンの役割
アロプレグナノロンは、プロゲステロンというホルモンに由来する天然の神経ステロイドである。蓄積された証拠は、アロプレグナノロンとAD発症との間の分子的関係を示している[46]。何人かの著者は、アルツハイマー病患者の前頭葉皮質におけるアロプレグナノロンの血漿および脳内レベルの低下を報告している[46]。不思議なことに、Luchettiらは、ADの神経病理学的初期段階の脳において、アロプレグナノロンの合成につながるアルドケトリダクターゼC2という酵素のmRNAレベルの増加を報告している[47]。この増加は、アロプレグナノロンのレベルを上げるための前頭前野の代償メカニズムであるという仮説が立てられているが、この事象を完全に理解するにはさらなる研究が必要である。アロプレグナノロンだけでなく、他の神経ステロイドも減少すると、神経保護作用が低下することが示唆されている。このことは、アポトーシスや神経細胞の減少を引き起こす原因の一つと考えられ、神経変性プロセスや興奮性亢進の原因となり、最終的には発作の発生につながると考えられる。同様に、アロプレグナノロンの減少は、アストロサイトとミクログリアを慢性的に活性化させる可能性があることも報告されている[46]。プラーク周辺で活性化したミクログリアは、神経毒性のあるサイトカイン、ケモカイン、活性酸素や窒素種の産生を促進し、これらはまた、ニューロンの興奮性を高め、最終的には発作の原因となっている。
2.2. てんかんとパーキンソン病
パーキンソン病(PD)は、黒質および線条体におけるドーパミン神経終末の進行性喪失を特徴とする神経変性疾患であり、運動および協調症状を引き起こすだけでなく、認知機能の低下、抑うつ、不安などの症状も引き起こす[48]。PDは、2番目に有病率の高い神経変性疾患であり、最も一般的な運動障害である[49]。PDの起源はまだ明らかになっていないが、特定の遺伝子の変異や環境的な原因が関与しているのではないかという仮説が立てられている[48]。パーキンソン病患者は、ドパミン作動性の低下と、PDの発症に重要な役割を果たしていると思われるシナプス前のタンパク質であるα-シヌクレインの構造の変化を示します[50]。ドパミン神経細胞は,α-シヌクレインのオリゴマーの毒性,小胞体ストレス,オートファジープロセス,カルシウムホメオスタシスの障害,ミトコンドリアの機能と構造の変化などの結果,損傷を受ける可能性がある[51].α-シヌクレインは,PDの認知症と密接に関連するレビー小体の主成分でもあり,パーキンソン病患者の50%以上の青斑核で発見されている[52].α-シヌクレインのミスフォールドや凝集は、散発性PDの発症時によく現れる。これらの凝集体は、腸神経系や嗅球から大脳皮質に至るまで、細胞から細胞へとトランスシナプス的に伝播する可能性があると報告している著者もいるが、これらのタイプの神経細胞においてα-シヌクレインのトランスシナプス的な動きが決定的に証明されているわけではない[53]。
典型的な症状は、振戦、硬直、または徐変であるが、原型PDや他の形態のパーキンソン病では、てんかん発作やてんかん様症状を示すこともある[54]。Brainstorm Consortiumによると、PDとてんかんの間には遺伝的な相関関係はないとされている[5]。パーキンソン病患者のてんかん発作の発生率に関する既存の観察研究は、横断的データ、小規模で不均一な研究集団、または交絡因子を調整していないデータに基づいている。しかし、Feddersenらは、パーキンソン病患者の2.6%がてんかんを発症すると報告している[54]。この値は、20年前にBodenmannらが2.4%の有病率を示して報告した値と非常に似ている[55]。Gruntzらが最近行ったネステッドケースコントロール分析を用いたレトロスペクティブコホート研究では、23,086人の偶発的パーキンソン病患者と92,343人のPDフリーの人のうち、898人の患者が偶発的にてんかん発作を起こしたことが確認された[56]。パーキンソン病患者コホートにおけるてんかん発作に罹患した人の数は、PDフリーコホートの2倍で、それぞれ266.7/100,000人年、112.4/100,000人年であった。また、てんかん発作の調整済みオッズ比(OR)は、パーキンソン病患者ではPD非発症者と比較して1.68であった。同様に、発作を誘発する複数の合併症を有するパーキンソン病患者は、発作を誘発する複数の合併症を有さないPDフリーの人と比較して、てんかん発作のリスクが最も高かった。この研究は、PDの発症がてんかん発作の発症リスクの増加と関連していることを明確に示唆している[56]。しかし、本研究では、これらの知見が分子レベルでの違いによるものなのか、本研究の患者が服用している併用薬によるものなのか、あるいは因果関係の程度は明らかにされていない。したがって、これらの問題を明らかにするためには、さらなる研究が必要である。
利用可能な治療法に関しては、L-DOPAやアポモルフィンなど、多くのPD治療薬が抗てんかん作用を有しており、両疾患間の横断的有病率の実測値を変化させる可能性があることを強調しておく必要がある[57,58]。
2.2.1. てんかんにおけるα-シヌクレインの役割
PDとてんかん発症の引き金となる病態生理学的メカニズムにおけるα-シヌクレインの役割は、ミトコンドリア機能障害と密接に関連している(図3A)[51,59,60]。上述したように,ミスフォールドしたαシヌクレインの蓄積は,主に大脳基底核に位置する感受性の高い神経細胞にレビー小体を形成させる.同様に、異常なαシヌクレインは、さまざまなレベルでミトコンドリアの構造に影響を与えることが報告されている[51]。(小胞体とミトコンドリア間のカルシウム輸送に関与するミトコンドリア膜に存在する電位依存性アニオンチャネルの変化により,Ca2+が大量に流入し,その結果,ミトコンドリア機能不全を誘発する小胞体の興奮性亢進が生じる.(ii) TOM22受容体との結合により、ミトコンドリア外膜からのタンパク質の輸入が阻害され、その結果、複合体Iの活性が低下し、ミトコンドリアが脱分極し、Ca2+のホメオスタシスが障害され、活性酸素種(ROS)が過剰に産生される。(iii) ミトコンドリアの電子伝達系の複合体IおよびVの直接的な阻害 (iv) ミトコンドリアの脱分極、その結果、セリン・スレオニンキナーゼPINK1のミトコンドリア外膜への蓄積が起こり、損傷したミトコンドリアのオートファジーによる除去が開始される。酸化ストレスの防止やミトコンドリアの機能維持に重要な役割を果たしている酵素であるミトコンドリアのサーチュイン3が阻害されると、ミトコンドリアのバイオジェネシスやダイナミクスが損なわれる[51,60]。
図3 パーキンソン病とてんかんの関連分子経路

(A) 異常α-シヌクレインの蓄積に由来するミトコンドリア機能不全を介した神経細胞の興奮状態。異常なα-シヌクレインは、ミトコンドリア機能障害やレビー小体形成の誘導を通じて、膜の脱分極、細胞内Ca2+の大量流入、酸化ストレスを促進する。これにより、神経炎症や神経細胞の興奮性亢進が促進され、その結果、神経変性プロセスが増加する(逆もまた然り)という悪循環に陥る。(B)ドーパミンのD1/D2ファミリー受容体への結合による抗てんかん/プロピルプシーの特性 ドーパミンがD1Rに結合すると、cAMPの増加が促進され、NMDA-Rの活性化とGLUT1の遮断が起こり、細胞内Ca2+の大量流入とグルタミン酸の再取り込みの低下が促進される。これにより、神経炎症や神経細胞の興奮性亢進が生じ、その結果、神経変性プロセスが増加する(逆もまた然り)という悪循環に陥る。ドーパミンがD2Rに結合すると、cAMPの産生が抑制されるため、D1R活性化の場合とは逆の作用が促進される。NEはノルエピネフリン、ROSは活性酸素種を意味する。
ミトコンドリア機能障害とレビー小体はともに、活性酸素レベルの上昇と酸化ストレス、膜脂質の過酸化による膜の破壊の促進、グリアの活性化、炎症性サイトカインの放出などの悪循環の引き金となり、神経炎症の増加、神経変性、そして最終的には神経細胞の興奮性亢進につながります(図3A)[53]。
2.2.2. てんかんにおけるドーパミンとノルエピネフリンの役割
前述のように、ドーパミンは抗てんかん作用を持つと言われている。しかし、この効果は、ドーパミンが結合する受容体のファミリーによって条件付けられている[53]。ドーパミン受容体には,D1とD5からなるD1ファミリーと,D2,D3,D4からなるD2ファミリーの2つのファミリーがある。ドーパミンが両方のサブタイプに結合すると,その効果は逆になる[53]。D1様受容体の活性化は,アデニルシクラーゼの活性化を促進し,それによってcAMPが増加し,その結果,NMDA-Rの活性化とGLUT1の遮断につながる。これらの結果,グルタミン酸,細胞内Ca2+,酸化ストレス,炎症性サイトカインが増加し,神経細胞の興奮性亢進が刺激され,発作が引き起こされる(図3B)[61]。これに関して,90年代に行われた研究では,難治性てんかん患者においてD1受容体を活性化すると,皮質の興奮性が高まり,てんかん活動の発現が促進されるが,D2受容体アゴニストは逆の作用を示すことがすでに示されている[62]。
確立されたパーキンソン病患者の死後の脳分析では,神経伝達物質であるノルエピネフリン,アセチルコリン,セロトニンのレベルが広範囲にわたって低下しており,ノルエピネフリンが最も影響を受けていた[63].ノルエピネフリンの神経回路の大部分がそこにあるため,最も影響を受けたのは,青斑部の神経回路であった。興味深いことに、レビー小体の蓄積のほとんどは、この脳領域にも見られる[53]。この減少は,PD関連のうつ病だけでなく,ノルエピネフリンが神経細胞の興奮性を調節することから,てんかん活動の出現にも関連している可能性がある[64]。前臨床研究では、ノルアドレナリン系を病変させた動物は、海馬のキンドルや発作に対して脆弱である[65]。しかし、これらの記述がヒトにも当てはまるかどうかは完全には明らかになっていないため、この仮説を確認するためにはさらなる研究が必要である。
2.2.3. PDとてんかんにおけるアロプレグナノロンの役割
パーキンソン病患者における神経ステロイド濃度の変化を分析した研究がいくつかある。Bixoらは20年前、対照群の黒質と尾状核でアロプレグナノロンのレベルが上昇していることを発見し、この神経ステロイドの合成がドーパミン系で行われていることを示した[66]。対照的に,パーキンソン病患者では,di Micheleらは,脳脊髄液中のアロプレグナノロンのレベルが低下していることを報告しており,この疾患におけるプロゲステロン代謝物の分子的関連性が示唆されている[67]。さらに,アロプレグナノロンを合成する2つの酵素,5α-リダクターゼ1型(SRD5A1)とアルドケトリダクターゼC3(AKR1C3)のmRNAの発現が,パーキンソン病患者の末梢血単核細胞で有意に低下していることが判明した[46].このことは,プロゲステロンの代謝を制御する酵素機構に全般的な欠陥があることを示唆している.同様に,黒質ではSRD5A1の発現が低下していたが,興味深いことに,尾状核ではAKR1C2の発現が上昇しており,神経変性プロセスにアロプレグナノロンが関与していることが示唆された[46]。これらの事実はすべて、神経保護機能の低下と神経細胞の興奮性の増大に関連しており、最終的には発作の発生につながると考えられる。しかし、これらの知見を検証するためには、大規模な患者コホートを用いたさらなる研究が必要である。
2.3. てんかんとハンチントン病
ハンチントン病(HD)は、常染色体優性の希少な神経変性疾患であり、運動機能障害、協調性の欠如、舞踏病やジストニア、行動障害、認知機能の低下などを伴う[68]。PDと同様、HDでも尾状体と基底核が最も影響を受ける部位である。HDは、ハンチンチン(HTT)遺伝子の変異が引き金となり、ミスフォールドしたハンチンチンタンパク質(mHtt)が過剰に産生されることで発症する[69]。第4染色体のエクソン1では,変異した遺伝子がCAG三塩基反復の病原性ゲノム拡張を示している。一般的に、CAGリピートの数が多いほど、HDの発症が早いとされている[70]。
早期発症のHD(若年性HDとも呼ばれる)は非常に稀で(症例の10%未満)父方からの遺伝が優先的に関連し、重度で急速な病状の進行を示します [53,71]。この患者群では、特に小児期に発症したHDではてんかん現象がよく見られるが、成人期に発症したHDではほとんど見られない[53,71]。記録されているHD患者の最も一般的な発作型は全身性強直間代発作とミオクロニー発作であり、皮質と辺縁系の構造が関与していることが示唆されている[53]。HDにおけるてんかんの発生率に関しては、あまり情報がない。Cloudらが若年性HD患者を対象に行った研究では、被験者の38%にてんかん発作が認められた[72]。全般性強直間代性発作が最も多い発作タイプで、強直発作、ミオクロニー発作、凝視発作がそれに続いた。さらに彼らは、HD発症時の年齢が低いほど、発作のリスクが高まることを発見した。逆に、Spilaらは成人発症のHD患者におけるてんかん発作の頻度を調査し、成人発症のHD患者におけるてんかんの有病率は一般人口と同様であると報告した[73]。しかし,これらの研究はレトロスペクティブなものであったため,決定的な結果を得るには限界があった。したがって、これらの知見をすべて検証するためには、より多くの患者が登録された将来の前向き研究が必要である。
2.3.1. てんかんにおけるmHttの役割
HTT遺伝子の突然変異は1983年にGusellaらによって報告されたが[74]、HDの発症と進行におけるmHttの役割はまだよく知られていない。てんかんでは、mHttは様々なメカニズムで神経細胞の興奮性亢進に寄与していることが報告されている(図4A,B)[53]。一方で,mHttはミクログリアを活性化し,炎症性サイトカインの大量分泌,神経炎症の増加,神経変性,そして最終的には神経細胞の興奮性亢進につながる[75]。一方で,アストロサイトの膜のGLUT1トランスポーターを損傷することで,グルタミン酸の取り込みを阻害する。その結果,シナプス腔内のグルタミン酸が増加し,この神経伝達物質に特有の興奮毒性カスケードが引き起こされる[75]。同様に,mHttは,脳由来向神経性因子(BDNF)の遺伝子などの必須遺伝子の転写異常を促進することが報告されており,これにより,グルタミン酸作動性反応の増強とGABA作動性反応の抑制を通じて,神経細胞の興奮性亢進が引き起こされる[76]。また,mHttがミトコンドリアの機能を変化させ,Ca2+のホメオスタシス障害,異常な活性酸素の産生,ミトコンドリアのタンパク質輸入量の変化,ミトコンドリアの断片化の増加,そして最終的にはATP産生量の減少を引き起こすことが示唆されている[75].PDの場合と同様、これらのミトコンドリアの変化は、てんかんの発作活動を引き起こす興奮毒性分子のいくつかのカスケードを生じさせる。
図4 ハンチントン病とてんかんの関連分子経路

(A) mHttがてんかんの発症につながる一般的なメカニズム。(B) mHttによって促進された損傷に由来するミトコンドリア機能不全を介した神経細胞の興奮性。mHttは、ミトコンドリア機能不全やミクログリアの活性化、アストロサイトのGLUT1RやBDNF、GABA神経細胞の抑制などを誘導することで、膜の脱分極、細胞内Ca2+の大量流入、酸化ストレスを促進する。これらはすべて、神経炎症と神経細胞の興奮性亢進を促進し、その結果、神経変性プロセスを増加させる(その逆もまた然り)という悪循環に陥る。
2.3.2. てんかんにおけるBDNFの役割
HDでは、BDNFレベルの低下と、このタンパク質に親和性の高い受容体(TrkB)の機能低下が報告されている[76,77]。これらの変化は、主にmHttによって引き起こされるBDNFとTrkBの両方の神経細胞の遺伝子転写の低下と関連している[53]。しかしながら、てんかんにおけるBDNFの役割は非常に複雑である。てんかん発作時に生じる興奮毒性に対するBDNFの保護作用について言及している著者もいるが、BDNFの寄与はほとんどが抗てんかん作用であると考えられる[53]。90年代に行われた研究では、BDNFが大幅に増加すると、GABAニューロンの反応が低下し、間質のグルタミン酸レベルが上昇することで、ニューロンの興奮性亢進が直接促進されることがすでに報告されている(図4A)[78,79]。対照的に,他の研究では,明確な抗てんかん活性を有することが示されているNPYペプチドを介して,BDNFの持続的なレベルが抗てんかん効果を促進する可能性が示唆されている[80]。興味深いことに,HD患者ではNPY/ソマトスタチン介在ニューロンが増加していることから,これらの患者では大脳皮質が過興奮状態になる前に代償機構が存在することが示唆されている[53]。さらに,海馬のBDNF発現は,てんかん後遺症モデルラットの認知能力にプラスの効果を与える可能性が示されている[81]。同様に,BDNFは,抗アポトーシス作用や抗酸化作用,オートファジーの抑制を通じて,神経変性を保護する役割を果たすことが報告されている[82]。これらの結果は,てんかん発生の治療のための分子標的の可能性を提起するものであるが,認知効果がBDNFのシグナル伝達に直接由来するのか,あるいは重要な活動の抑制による二次的なものなのかは不明である。一方、BDNF シグナルが検証されているてんかん発症モデルは、ほとんどが構造由来のてんかんに基づいており、これらのシグナル伝達経路が異なる病因で共有されているかどうかは、依然として議論の対象となっている。
2.4. てんかんと多発性硬化症
多発性硬化症(Multiple Sclerosis: MS)は、脱髄過程と軸索の損傷を特徴とする中枢神経系の不均一で複雑な自己免疫疾患である。世界中で200万人以上が罹患しており、中枢神経系の最も一般的な慢性炎症性疾患であると考えられている[83]。MSは純粋な神経変性疾患には分類されていないが、その典型的な病理学的プロセスは、神経組織の長期的かつ不可逆的な破壊をもたらす[84,85,86]。
MSの発症原因は完全には解明されていないが、MSの発症には遺伝的要因と環境的要因が組み合わさっていることが知られている。興味深いことに、遺伝学的データは、MSの病因が様々な非中枢神経系自己免疫疾患と重要な特徴を共有していることを示唆している[83,87]。さらに、MSの潜在的な原因として、腸管伝染性の亢進の存在も強調されている。この変化は、血液中への物質(例えば、ウイルス、細菌、毒素)の制御されない通過を可能にし、免疫系の異常な反応を引き起こす可能性がある[88]。
MSの病変は、中枢神経系全体に現れる可能性があり、脱髄、炎症、グリア反応の焦点領域として白質で最も容易に認識される。MSの組織損傷は、免疫系、グリア(ミエリンを作るオリゴデンドロサイトおよびその前駆体、ミクログリア、アストロサイト)およびニューロンの間の複雑かつダイナミックな相互作用から生じる。MSの自己免疫性炎症障害に関与する細胞は、主にリンパ球(TおよびBリンパ球)マクロファージ、およびミクログリアである。MS患者では、血液脳関門(BBB)が損傷し、自己反応性のTリンパ球が通過する。脳内では、これらのT細胞がミエリン鞘を破壊し、他の免疫細胞やサイトカイン、抗体などの可溶性成分によって周囲の炎症が促進される(図5)[88]。
図5 多発性硬化症の主要な病理学的分子経路に由来する痙攣活性

自己免疫反応は、脱髄と軸索の損傷を促進し、それがミクログリア、オリゴデンドロサイト、マクロファージの活性化を引き起こし、神経炎症と神経変性を開始する。このようにして、神経細胞の過興奮性が高まり、それが神経変性プロセスを増加させるという悪循環に陥る。
MSの臨床症状は非常に異質である。典型的には、感覚障害および/または運動障害、視神経炎、疲労、三叉神経痛、またはめまいを呈します[89]。PDと同様に、Brainstorm Consortiumは、MSとてんかんの間に遺伝的な相関関係はないと報告している[5]。しかし、発作はMS患者に現れることがある[90]。脱髄病変の解剖学的多様性を考慮すると、MS患者では多種多様な発作タイプが観察されている[91]。MS患者310人を対象に実施されたあるレトロスペクティブ研究では、3.2%がてんかんを患っていることが報告されている。これらの患者では、発作が最初のMS症状であり、最も頻度の高い発作タイプは部分的二次性全般化発作であった。さらに、これらの患者は、てんかんを発症していないグループに比べて若く、MS症状の発現も早く、全員に皮質病変が認められた[92]。
てんかんとMSの分子的な関連性は完全には解明されていないが、いくつかの仮説が提唱されている。脱髄過程を引き起こす自己免疫の亢進により、アストロサイトとミクログリアの両方が活性化されるとともに、オリゴデンドロサイトのアポトーシス過程が開始される[93]。このようなメカニズムにより、炎症性サイトカインが大量に放出され、中枢神経系の炎症が全般的に増加する。その結果、神経変性プロセスが促進され、脱髄プロセスが刺激され、神経組織が破壊されるという悪循環に陥ってしまう。これらの病態生理学的変化は、神経細胞の興奮性亢進につながり、発作発生の主な原因となります(図5) [94]。同様に、抗体、Tリンパ球、炎症性サイトカイン、マクロファージなどによる直接的な軸索の損傷も、ニューロンの電気的活動の不均衡に直接寄与する。この変化は、神経細胞の膜電位振動に影響を与え、神経細胞の興奮性亢進、そして最終的には発作につながります(図5)[94]。
MSおよびてんかんにおけるアロプレグナノロンの役割 いくつかの研究では、アロプレグナノロンがMSおよびてんかんの共通の病理学的経路を標的にしている可能性が報告されている[95]。MSに関しては、多発性硬化症で神経ステロイドの合成が損なわれていることが報告されている[96]。この意味で、Noorbakhshらは、自己免疫性脱髄のマウスにアロプレグナノロンを投与すると、神経行動障害が改善され、神経病理と神経炎症が改善されることを示した[97]。同じ著者は、アロプレグナノロンを含むいくつかのニューロステロイドのレベルが、MS患者の白質で抑制されていることを示した[96]。てんかんに関しては、Melettiらは、てんかんにおいて、アロプレグナノロンがGABA-A受容体を介した抑制性電流の正のモジュレーターであることを明らかにした。同様に、Lévesqueらは、アロプレグナノロンが発作の発生と興奮性神経細胞活動の発生を調節する効果があることを示した。さらに、中側頭葉てんかんの動物モデルにおいて、アロプレグナノロンの投与が自発的な発作の発生を遅らせることも明らかにしている[98]。
行ってみよう。
3. てんかん治療の現状と難治性てんかんの基準
最初のASDは、19世紀後半に偶然発見された。その数年後、動物モデルの使用により、さまざまな分子やその誘導体の開発が可能となり、現在では、てんかん患者の発作の発生を予防するために、さまざまな薬剤が利用できるようになっている。
ASDは、その主な作用機序により、大きく4つのカテゴリーに分類される。(ASDは、その主な作用機序により、電位依存性イオンチャネルの調節、GABAを介した抑制性神経伝達の増強、グルタミン酸を介した興奮性神経伝達の減弱、シナプス前作用による神経伝達物質の放出調節の4つに大別される。ASDの中には複合的な作用機序を持つものもあり、場合によっては完全には解明されていないものもある(表1)[99,100]。このように多様な治療法があるにもかかわらず、てんかん患者の3分の1は、治療に抵抗性のあるてんかん発作を抱えている[101]。現在、てんかんの臨床試験は、主に薬剤耐性てんかん患者の発作を予防できる分子の開発に焦点を当てている。規制当局は最近,難治性てんかんに特化した最初の薬剤であるセノバメートを承認した。セノバメートは,無作為化二重盲検臨床試験において,発作の頻度を減少させることが示されている[102]。この薬剤はターニングポイントとなり,難治性てんかん患者の発作の制御に貢献できる新しい分子の開発に光を当てた。
表1 現在使用されているASDの主な作用機序
| 分子標的 | 抗けいれん薬 | 提案された作用機序 | |
|---|---|---|---|
| 電位依存性イオンチャネル | Na +チャネル | フェニトイン、フォスフェニトイン、カルバマゼピン、オクスカルバゼピン、酢酸エスリカルバゼピン、ラモトリジン、ラコサミド、セノバメート*、ルフィナミド、トピラメート、ゾニサミド | Na +チャネルの急速/低速不活性化の強化、活動電位の伝播の抑制 |
| Ca2 +チャネル | エトスクシミド | Ca2 +電流を調節することにより過興奮を抑制します | |
| K +チャネル | レチガビン(エゾガビン) | 膜電位を安定させる閾値以下のK +電流を生成します | |
| GABAを介した阻害 | フェノバルビタール、プリミドン、ベンゾジアゼピン、スチリペントール*、トピラマート、フェルバメート、セノバメート、レチガビン(エゾガビン)、チアガビン、ビガバトリン、アセトアゾラミド、トピラマート、ゾニサミド、ラコサミド* | シナプス抑制の増加とグルタメート活性の低下 | |
| シナプス放出機構 | SV2A | レベチラセタム、ブリバラセタム | 興奮性神経伝達物質放出の抑制 |
| 電位依存性Ca2 +チャネルのα2δサブユニット | ガバペンチン、プレガバリン | 興奮性神経伝達物質放出の抑制 | |
| AMPAレシーバー | ペランパネル | 細胞外Ca2 +濃度とニューロンの興奮性を阻害します | |
| 混合/不明 | バルプロ酸、フェルバメート、セノバメート、トピラマート、ゾニサミド、ルフィナミド、副腎皮質刺激ホルモン、カンナビジオール | ||
Sills and Rogawski (2020)から引用している。*考えられる作用機序で、まだしっかりと確立されていない。
しかし、これらの薬剤はすべて、発作の発生を抑制する有効な薬剤であることが実証されている。発作発生とてんかん発生は重要な違いがあり、これらは異なる生理学的プロセスを表しているため、その治療目標も異なるはずである。発作発生は、発作間欠状態から発作に移行する過程を表し、てんかん発生は、特定のグループや神経回路が過剰興奮状態となり、自発的にてんかん発作を発生させる過程を表する。てんかんに関連するいくつかの特定の疾患の遺伝学および病態生理学に関する知識の進歩により、結節性硬化症複合体におけるエベロリムス[103]や神経性セロイドリポフスチン症におけるリソソーム酵素置換[104]など、いくつかの症候群に対する特異的な治療法が開発されている。しかしながら、特に成人期のてんかんにおいては、特定の病因が不明なてんかんやてんかん症候群が未だに多く存在しており、そのような患者群に対する特異的な治療薬は現在のところ存在しない。興味深いことに、てんかんと神経変性プロセスが双方向に関連している可能性があることから、てんかんの経過を変えることができる可能性のある新たな分子標的の開発への道が開かれている。
ASDの中には、動物モデルにおいて抗てんかん作用を示すものがあるが、より大規模な臨床研究ではその効果は確認されていない[105]。また、アトルバスタチン、セフトリアキソン、ロサルタン、イソフルラン、N-アセチルシステイン、アナキンラ、ラパマイシン、フィンゴリモドなど、いくつかの承認薬の潜在的な抗てんかん作用が動物モデルで報告されている[106,107,108,109,110,111,112]。これらの薬剤の再配置は、いくつかの特定の病因において魅力的な選択肢となりうるが、これらの結果は臨床試験では確認されていない[113]。この効果は、てんかん発生に関するほとんどの実験的研究がキンドリングモデルに強く影響されており、ヒトにおけるキンドリングの存在を支持する証拠には議論があるという事実によって説明できるだろう[114]。これらの研究のほとんどは、外傷後または脳卒中後のてんかんに基づいており、これは特定可能な後天性脳損傷に続発するてんかん発生の典型的な例である。前臨床研究を臨床試験に結びつけるのが難しいのは,病因が多岐にわたっていることと,他のてんかん症候群ではてんかん発生のメカニズムがおそらく異なっていることが原因であると考えられる[113]。
4. 神経変性疾患における抗てんかん薬
てんかんと他の神経変性疾患との間には分子的なつながりがあるため,これらの病態における抗けいれん薬の治療可能性や,併存疾患としてのてんかんに対する治療アプローチを評価するために,さまざまな研究が行われている。抗けいれん薬は、その種類の多さと作用機序の違いから、原因が不明であったり、治療効果が不十分であったりする中枢神経系の病態に対して、非常に興味深い候補として位置付けられている。しかし、これらの病態における抗けいれん薬の潜在的な神経保護作用については、まだ不明である。
4.1. アルツハイマー型認知症におけるASD
アルツハイマー病においても,ASDの薬理作用の可能性を明らかにしようとする研究者がいる。例えば,Mucke博士の研究グループは,ヒトAβが異常に多く,異常な神経細胞ネットワーク活動やてんかん発作を示すhAPPマウスモデルにおいて,レベチラセタム(LEV)の慢性投与の効果を評価した[115]。著者らは,LEVの投与により,異常なスパイク行動やてんかん状の放電を抑制するだけでなく,これらのマウスの神経細胞ネットワークの機能障害を抑制し,シナプス障害や認知障害を回復させることができることを見出した。さらに、LEVの効果をアルツハイマー病患者で評価することを目的とした臨床試験もいくつか行われている。例えば、ジョンズ・ホプキンス大学医学部の研究グループは、認知症の軽度認知障害(MCI)患者の記憶機能に対するLEVの効果を評価する第2相試験を実施した(NCT01044758)。LEVは,海馬歯状回およびCA3領域の異常亢進を抑制し,内嗅皮質の異常低活性化を後押しし,走査型記憶課題のパフォーマンスを改善することが報告された[116]。同様に,LEVをADの興奮性亢進や発作活動の治療に用いることを評価する臨床試験(NCT03875638,NCT03461861,NCT01554683)や,てんかんに関連する神経精神症状に対するLEVの効果を検討する臨床試験(NCT04004702)も実施されている[117]。さらに、LEV、フェノバルビタール、またはラモトリギンを服用しているアルツハイマー病患者を対象とした前向き無作為化3群並行群症例対照研究では、これら3つのASDの間で有効性に有意な差はなかったが、LEVは他のASDよりも有害事象が少なく、認知能力の改善と良性の神経心理学的副作用と関連していた[118]。同様に、ハーバード・メディカル・スクールの研究者らは、軽度のアルツハイマー病患者におけるLEVの急性投与の神経生理学的および認知学的効果を評価するフィージビリティー・スタディを実施した。その結果、LEVは、脳の振動連結性を表す脳波の低周波帯および高周波帯をポジティブに変化させることがわかった。このことは、LEVがアルツハイマー病患者に対して有益な効果を持つことを示唆している[119]。したがって、LEVは、アルツハイマー病患者にとって認知的に安全なASDと考えられる。しかし、LEVの効果が通常の加齢と比較してADに特有のものであるかどうか、また、より長期の投与が有益な臨床効果と関連しているかどうかを明らかにするためには、より大規模な縦断的研究、および年齢をマッチさせた健常対照者を用いた研究が必要である。
4.2. パーキンソン病に対するASD
ドパミンアゴニストとドパミン補充のためのレボドパが現在のPD治療のアプローチである。しかし、これらの物質の効果は徐々に低下し、神経変性の進行を止めることができなくなる。そのため、PDに効果のある新規または既存の化合物を見つけるために多くの努力がなされてきた。この点では、いくつかのASDが研究されており、特にゾニサミド(ZNS)が興味深い結果を示している。
ZNSがその有益な効果を発揮するメカニズムはいくつか提案されている。(i)モノアミン酸化酵素Bを阻害し、MAO-B経路によるドーパミン誘導活性酸素の産生を減少させ、黒質変性に寄与する[120,121]、(ii)T型カルシウムチャネルを遮断し、PD症状を改善する[122,123]、(iii)線条体におけるレボドパ-ドーパミン代謝を調節し、ドーパミン合成を促進し、細胞外ドーパミン濃度を増加させる[124]。(iv)アデノシンA2AおよびエンドカンナビノイドCB1受容体の発現を低下させ、レボドパ誘発性ジスキネジアを改善すること [125]、(v)ドーパミンのターンオーバー、シナプス伝達、遺伝子発現を調節し、向神経性因子を誘導したり、神経炎症、酸化ストレス、アポトーシスを抑制することで、神経保護を行うこと [126]。
異なる病期のPDの治療に対するZNSの有効性を探るため、多くの臨床試験が行われてきた。疾患の初期段階では、非盲検臨床試験により、ZNSの単回投与が運動機能障害と睡眠機能障害を改善することが示唆された[127]。進行期では、いくつかの研究で、運動変動の補助療法としてのZNSの可能性が評価されている。第2相および第3相臨床試験では、進行したパーキンソン病患者において、ジスキネジアを悪化させることなく、ZNSが運動機能およびウェアリングオフ現象を改善することが実証された[128,129]。PDの後期では、非盲検の第2相試験のみが行われた。その結果、ZNS 300mg/日の投与により、PD症状、特にウェアリング・オフ現象に由来する症状の発現が抑制された。著者らは、ZNSによるドーパミン合成の長期的な活性化が、PD症状、特にウェアリング・オフ現象を改善すると推測した[130]。しかし、この研究の参加者数が少なすぎたため(n = 10)明確な結論を出すことができず、これらの知見をすべて検証するにはさらなる研究が必要である。現在、進行性PDにおけるZNSの役割を評価するため(NCT04182399)と、PDのジスキネジアに対するZNSの忍容性と有効性を検討するため(NCT03034538)に、ZNSを用いた2つの臨床試験が展開されている。予備的な結果はまだ知られていない。
4.3. ハンチントン病に対するASD
HDの症状は非常に多様であるため(コリア、ジスキネジア、ミオクローヌス、アカシジア、ブラキシズム、うつ病、認知・コミュニケーション障害、記憶障害など)他の病態で広く使用されている多くの薬剤がHDでも検討されている[131]。例えば,ASDはミオクローヌスエピソードの治療の主要な候補となっている。ミオクローヌスとは,突発的な筋肉の収縮のことで,てんかん発作のスパムやジャークに似た短時間の不随意な収縮であるが,てんかんとは関係がない。HDでは、ミオクローヌスは主に若年性に見られるが、遅発性のものにも見られる。興味深いことに、若年型では、非てんかん性のミオクローヌスがてんかんと共存することがある[131]。これらのHD症例では、バルプロエートの単独またはクロナゼパムとの併用が推奨される[131]。また、同じ適応症でバルプロエートに代わる治療法としてLEVが推奨されている。同様に、バルプロエートとオランザピンの併用は、HDに伴う焦燥感や攻撃性の緩和に役立つことが報告されている[132]。ミオクローヌスがてんかん発作に関連しない皮質由来のものである場合、ピラセタムの処方が認められている[132]。
4.4. 多発性硬化症に対するASD
MS患者は、生活の質に大きな影響を与える神経障害性疼痛に悩まされることが多く、プールされた有病率は63%である[133]。ASDは、これらの患者の神経障害性疼痛の治療に広く使用されている。現在、神経障害性疼痛に使用されている抗てんかん薬は、カルバマゼピン、オクスカルバゼピン、ガバペンチン、ラコサミド、ラモトリギン、クロナゼパム、レベチラセタム、フェニトイン、プレガバリン、トピラマート、バルプロエートである。とはいえ、この適応症の認可状況は国によって異なる可能性がある[134]。一般に、ASDが神経因性疼痛を軽減する作用機序の仮説は、高周波の神経細胞の発火を減少させる能力に基づいている。3つの標準的な説明が記載されている。(i)増強されたγ-アミノ酪酸(GABA)の抑制(例:クロナゼパム、バルプロエート)(ii)イオンチャネルの調節による神経細胞膜の安定化作用(例:ガバペンチン、ラモトリギン)(iii)NMDA受容体部位の抑制[134]。
LEVは、MS患者の神経因性疼痛の軽減だけでなく、相性痙縮の減少にも有効であることが示されている。Hawker 氏らは、テキサス大学の多発性硬化症プログラムに参加している患者を対象に、レトロスペクティブな医療記録レビューを行った。その結果、LEVによる治療後、すべての患者でPenn Spasmスコア(位相性痙縮の指標)が低下し、一部の患者では神経因性疼痛の改善も報告された[135]。これらの有望な結果にもかかわらず、これらの知見を確認するためには、大規模かつ良好な対照試験が必要である。同様に、バルプロエートもMSのマウスモデルで研究され、様々な症状に対するバルプロエートの有効性が評価された。その結果、バルプロ酸はT細胞のホメオスタシスを回復させ、これらのマウスの病態を改善することがわかった。しかし、この結果を確認するためには、さらにヒトでの研究を行う必要がある[136]。
臨床試験に関しては、完了した研究では、MS患者におけるオクスカルバゼピン(NCT02104661)[137]、ラモトリギン(NCT00257855)[138]、LEV(NCT00423527)の保護的役割も評価されている。しかし、これらの研究から一貫した結果はまだ得られていない。これまでに見つかったエビデンスを検証するためには、より多くのサンプルサイズの研究が必要である。
5. おわりに
てんかんは世界中で約5,000万人が罹患している。発展途上国では、出産関連の傷害、医療インフラのばらつき、予防医療プログラムの利用率の低さなどにより、最も影響を受けている。てんかんの発作に先立って起こる神経細胞の興奮性亢進には、神経細胞へのCa2+の大量流入が主なメカニズムとして関与している。しかし、それ以外にも、発作の発生やてんかんの発生に関連するメカニズムが数多く提案されており、その多くは主要な神経変性疾患のメカニズムと関連している。
ADでは、Aβペプチドやp-tauが神経炎症や神経変性の発生、NMDA-Rs、AChRs、イオンチャネルの調節に果たす役割がよく説明されている。これらの変化は、最終的には発作の出現につながる。同様に、PDでは異常なα-シヌクレインが、HDではmHttが出現すると、ミトコンドリアの損傷が起こり、神経細胞膜のイオンバランスに大きな影響を与える。同様に、酸化ストレス、細胞内Ca2+、炎症性サイトカインなどの増加も現れ、神経細胞の異常な興奮性を助長する。PD、MSともに、てんかんとの遺伝的な相関関係は認められていない。しかし、多くの研究では、これらの患者にてんかん発作が出現することが強調されている。PDでは、発作の発生に関係するドーパミンの二重作用が示されている。D2ファミリーの受容体が活性化されると、発作の発生を防ぐ経路が誘発されるのに対し、D1ファミリーはてんかんを誘発する経路を活性化すると考えられている。MSでは、自己免疫反応によって促進される典型的な脱髄と軸索の損傷が、ミクログリアの反応を増大させ、神経変性を促進し、最終的には神経細胞の興奮性を増大させる。
これらの知見は、てんかんと主要な神経変性疾患との間の分子的な相互関係を強調するものである。これらの変化を管理することは、てんかんだけでなく、他の神経疾患に併存するてんかんの治療にも有効であると考えられる。現在、多くのASDが利用可能であるが、かなりの割合の患者が薬剤抵抗性のてんかんを患っている。そのため、アトルバスタチン、セフトリアクソン、ロサルタン、アナキンラ、ラパマイシン、フィンゴリモドなど、いくつかの承認済みの薬剤が抗てんかん薬として動物モデルで研究されている。とはいえ、これらの使用可能性は臨床試験で確認する必要がある。同様に、LEV、ZNS、バルプロエートなど、一般的に使用されているASDも、主にこれまでに報告されている分子的な関連性と、これらの疾患に対する有効な治療法がないことから、他の神経変性疾患での使用が検討されている。この点に関しては、いくつかの臨床試験が行われているが、これらの治療法を臨床に導入するためには、さらなる研究が必要である。
謝辞
A.C.は,助成金Juan de la Cierva (FJC2018-036012-I)によるスペイン科学・イノベーション・大学省の支援に謝意を表する。著者は、Instituto de Salud Carlos III (ISCIII) Acción Estratégica en Salud(スペイン国家R+D+I計画に統合され、ISCIII Subdirección General de EvaluaciónとFondo Europeo de Desarrollo Regional (FEDER “Una manera de hacer Europa”) grant PI17/01474がM. B. Boadaに授与した助成金の支援に謝意を表する。 M.E.は、スペイン経済・競争力省のプロジェクトSAF2017-84283-RおよびCIBERNEDのプロジェクトCB06/05/0024の支援を受けたことを認めている。E.B.S.は,ポルトガル科学技術財団(FCT)の戦略的資金(UIDB/04469/2020)の支援に謝意を表する。A.R.は、CIBERNED (Instituto de Salud Carlos III (ISCIII)), EU/EFPIA Innovative Medicines Initiative Joint Undertaking, ADAPTED Grant Nº 115975, from EXIT project, EU Euronanomed3 Program JCT2017 Grant Nº AC17/00100, from PREADAPT projectの支援を受けている。神経変性疾患共同プログラム(JPND)グラント番号AC19/00097,およびグラントPI13/02434,PI16/01861 BA19/00020,およびPI19/01301から。Acción Estratégica en Saludは、スペインの国家RCDCI計画に組み込まれており、Instituto de Salud Carlos III (ISCIII)-Subdirección General de EvaluaciónおよびFondo Europeo de Desarrollo Regional (FEDER-“Una manera de Hacer Europa”)、Fundación bancaria “La Caixa” and Grífols SA (GR@ACEプロジェクト)による資金提供を受けている。
略語
Aβ:アミロイドβ、AD:アルツハイマー病、APP:アミロイド前駆体タンパク質、ASD:抗てんかん薬、BACE1:βセクレターゼ1,BBB:血液脳関門、BDNF:脳由来向神経性因子、中枢神経系:中枢神経系、GABA:γ-アミノ酪酸。HD(ハンチントン病)LEV(レベチラセタム)mHtt(折りたたまれていないハンチンチンタンパク質)MS(多発性硬化症)NFT(神経原線維変化)NMDA-R(N-メチル-D-アスパラギン酸受容体)PD(パーキンソン病)p-tau(リン酸化されたタウ)WHO(世界保健機関)ZNS(ゾニサミド)。
著者の貢献度
A.C.は概念設計と文献検索を行い、原案を執筆し、図をデザインした。E.F.は、セクションの執筆、表のデザイン、原案の内容修正に貢献した。M.E.とE.S.-L.は、1つのセクションの執筆と原案の内容修正に貢献した。I.d.R.、S.A.-L.、X.M.は、原案の言語および内容の修正に貢献した。E.B.S.、M.T.、M.B.は、原案の監督、執筆/レビュー、編集に貢献した。M.M.とA.R.は、監督、執筆/レビュー、編集、プロジェクト管理、資源や資金の獲得に貢献した。すべての著者が本研究に実質的に貢献した。すべての著者は、本稿の公開版を読み、同意した。
資金調達
本研究は外部資金を受けていない。
