Contents
pubmed.ncbi.nlm.nih.gov/35491799
はじめに
多系統萎縮症(MSA)は致死的な成人発症の急速進行性の神経変性疾患であり、自律神経失調症、運動失調症、パーキンソニズムをあらゆる組み合わせで特徴とする[1, 2]。MSAには2つの運動表現型が認められる:パーキンソン病型(MSA-P)はレボドパ反応性に乏しい運動硬直症候群を特徴とし、小脳型(MSA-C)は広範な歩行、四肢運動失調、走査性構音障害、小脳性眼球運動機能障害を呈する。MSAは、これらの運動機能障害以外にも、自律神経障害(泌尿生殖器系、心臓血管系、またはその両方)や、ジストニア、錐体徴候、レム睡眠行動障害、喘鳴などの運動・非運動機能障害を伴う[3] 。
神経病理学的には、ミスフォールディングしたα-シヌクレインは、グリア細胞質封入体(GCI)と呼ばれる不溶性の凝集体を形成し、ミクログリアの活性化と炎症性サイトカインおよび酸素活性種の放出を伴う[4, 5]。α-シヌクレインは主に細胞内タンパク質であるが、α-シヌクレインパチー患者の脳脊髄液(CSF)中には様々なα-シヌクレインが存在する[6] 。MSA患者の脳ホモジネートを接種してα-シヌクレイン封入体病態を発症したマウスを用いた前臨床研究では、異常に折り畳まれたα-シヌクレインが、プリオンのような様式で細胞から細胞へとMSA関連病態を拡大させる可能性が示唆されている[7] 。
これまでのところ、潜在的な疾患修飾療法(DMT)は臨床試験で失敗しているが、現在、MSAを対象とした数多くのDMTが臨床開発中であり、MSAにおける神経変性カスケードのさまざまな主要異常を標的としている(図1に図示)。α-シヌクレインは最も明白な治療標的であり、治療戦略は(ミスフォールドした)α-シヌクレインの凝集、拡散、クリアランスに焦点を当てている。その他の治療戦略は、神経炎症、神経栄養支持、ミトコンドリア機能障害、興奮毒性を標的としている。本論文では、現在進行中のDMTの開発について概説する。PubMedを用い、「MSA」、「多系統萎縮症」、「治療」、「療法」、「疾患修飾」という検索語を用いて、非系統的な文献レビューを行った。MSA患者を対象とした疾患修飾試験の結果を報告する出版物を選択し、これらの報告を批判的に評価・検討した。対症療法に関する研究は除外した。
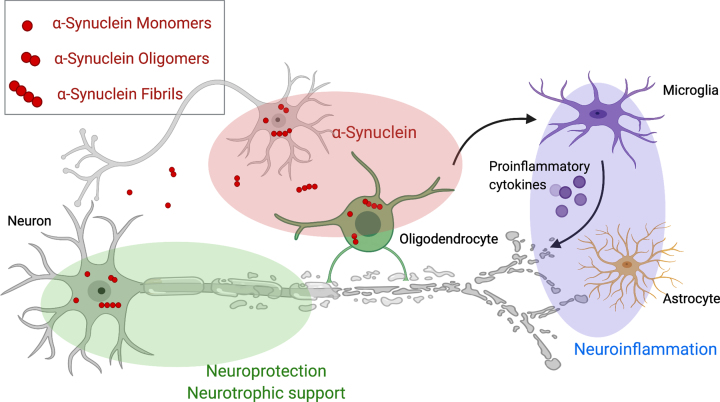
図1 多系統萎縮症における疾患修飾療法の治療標的。この図は、多系統萎縮症の根底にある病理学的メカニズムと、α-シヌクレインの凝集、拡散、クリアランス(赤)、神経栄養支持(緑)、神経炎症のカスケード(紫)を含む潜在的な疾患修飾標的を示している。BioRender.comで作成。
臨床開発
MSAにおけるα-シヌクレインを標的とする治療法を図2に示す。 α-シヌクレインを標的とする治療戦略の概要を表1に示す。

図2 α-シヌクレイン集合体を異なる段階で標的とする疾患修飾療法。疾患修飾療法は、α-シヌクレイン凝集カスケードに沿って異なるレベルを標的とする。ASO、アンチセンスオリゴヌクレオチド。
積極的予防接種と受動的予防接種
α-シヌクレインが神経変性や臨床的パーキンソニズムの発症に絶対不可欠であり、唯一の関連病原体であるという明確な証拠はないが[31] 、α-シヌクレインの寄与の可能性は、上述の前臨床研究やSNCAの増殖が身近なPDを引き起こすという臨床観察によって証明されている[32] 。さらに、α-シヌクレインが誘導する細胞内経路(オートファジーとリソソーム経路)の障害[33] や、エクソソーム放出による分泌とエンドサイトーシスによる再取り込みを通じて起こる可能性が高い細胞間伝達は、α-シヌクレイン指向性治療がさらなる(前)臨床評価を正当化する明確な根拠となる[34] 。しかしながら、免疫療法構築物の大部分は細胞外の病態を緩和することを目的としており、細胞内のタンパク異常はこれらの免疫療法の試みによって直接取り組むことはできないことを認めなければならない。後者の場合、細胞内のα-シヌクレインに干渉するイントラボディーを開発する必要がある。
オリゴデンドロサイトにα-シヌクレインが発現しているMSAのモデルであるMBP-α-シヌクレイントランスジェニックマウスを用いた能動免疫により、MSAの前臨床モデルにおける神経変性病態の改善が示唆された。この研究では、血液脳関門を通過する特異的な抗α-シヌクレイン抗体のワクチンによる産生が観察された。新皮質、線条体、脳梁における神経変性と脱髄は、in vivoのシヌクレイン病モデルで顕著に抑制された。ヒトを対象としたその後の第Ⅰ相試験では、AFFITOPE PD01Aがα-シヌクレインのC末端領域に特異的な抗体反応を惹起し、初期PD患者において安全で忍容性が高いことが報告された[37]。MSA患者を対象とした最近の第I相試験では、PD01AとPD03Aの両方がα-シヌクレインに対する有意かつ持続的な免疫反応を誘導することができ、PD01A群では応答率が高いことが明らかになった[38]。PD01AまたはPD03Aによって誘導された抗体が、実際にMSAの自然経過を変化させることができるかどうかについては、今後の研究が必要である。
受動免疫もまた有望な免疫療法アプローチである。α-シヌクレインに高親和性で結合するいくつかの新規モノクローナル抗体(mAb)が治療パイプラインにある。
Lu AF82422は、健常人とPD患者を対象とした第I相試験で安全性と忍容性が証明されたmAbである)。MSA患者における安全性と有効性を調査する第Ⅱ相試験が2021年末に計画されている[39]。
受動的免疫療法を研究している他の臨床試験は、現在MSAでは研究されていないが、PDのαシヌクレインに対しては有望であり、したがってMSA治療にも有益である可能性がある。プラシネズマブ(PRX002としても知られる)は、PDを対象に開発が進められているmAb治療薬である。第I相試験では、この抗体は安全で忍容性が高いことが報告された[40] 。より大規模な第Ⅱ相試験では、PD患者の運動症状の悪化を遅らせることはできなかったが、MDS-UPDRSサブスコア、MoCA、線条体ドーパミン受容体結合などの副次的・探索的アウトカムで有効性のシグナルが示された, “attol”, “atomic”):“clinical-trial”,”attrs”:{“text”:”NCT03100149″,”term_id”:”NCT03100149″}}NCT03100149and)[41].
もう一つのα-シヌクレイン指向性mAbはMEDI1341である。この抗体は、in vitroではあらかじめ形成されたα-シヌクレイン線維の細胞間伝達をブロックし、in vivoでは細胞外のα-シヌクレインを封鎖できることが示された。健康なボランティアを対象としたMEDI1341の単回上昇用量の安全性と忍容性を評価する第I相試験は最近終了したが、結果はまだ保留中である )。PD患者を対象とした複数回上行投与を評価する第I相試験が進行中である)。
mAbであるBIIB054は、PDとDLBの動物モデルにおいて、α-シヌクレイン負荷量の減少と行動障害の改善に有効であることが証明された[42-44]。健常成人およびPD患者を対象とした無作為化第I相試験では、良好な安全性、忍容性、良好な薬物動態プロファイルが示された[45] 。しかし、PDを対象としたランダム化比較第II相試験では主要評価項目を達成できず、薬剤開発は中止された)。
全体として、能動的および受動的免疫療法はMSAの疾患修飾に役割を果たす可能性があるが、免疫戦略の神経保護効果を確認するためには、さらなる大規模な研究が必要である。しかし、免疫戦略の神経保護効果を確認するためには、さらに大規模な研究が必要である。免疫で標的とされない神経細胞内のα-シヌクレイン凝集体へのアクセス性の限界を克服する必要がある。さらに、アルツハイマー病における過去の免疫研究では、神経変性疾患における免疫化は、ワクチン接種後の髄膜脳炎がかなりの数の治療患者で発生し、有害な結果をもたらす可能性があるため、両極に分かれる可能性があることが明確に示されている[46] 。注目すべきことに、この試験では、免疫によってアミロイド斑が消失し、長期追跡調査によって、アミロイドβに対して積極的に免疫されたアルツハイマー病患者は、14年間実質的にアミロイド斑のない状態を維持したことが示された[47] 。しかしながら、この試験のほとんどの患者は、それでも重度の痴呆に進行していた[48]。最後に、神経炎症が神経変性に関与しているのか、あるいはMSAにおいて有害なαシヌクレイン種からニューロンを保護しているのかについては、まだ研究が必要である。
アンチセンス・オリゴヌクレオチド
α-シヌクレインの細胞内凝集がMSA病態の中心的役割を担っていることを念頭に置くと、α-シヌクレインの産生そのものを減少させることが、疾患修飾の根拠となる。アンチセンスオリゴヌクレオチド(ASO)療法は、SNCA遺伝子のプレmRNAを標的とすることで、α-シヌクレインの細胞内産生を阻害することができる[49] 。動物モデルでは、神経保護効果と髄液中および脳組織中のα-シヌクレインの顕著な減少が示された[49, 50]。しかしながら、α-シヌクレインの完全ノックアウトは神経炎症を悪化させ、有害な影響を及ぼす可能性があることを示唆する前臨床研究によって懸念される[51-53] 。したがって、α-シヌクレインの翻訳を阻害する適切な程度を選択することが重要である。
MSA患者を対象としたASO BIIB101の髄腔内適用による第I相ランダム化比較試験が進行中である)。ロイシンリッチリピートキナーゼ2(LRRK2)を標的としたASOの別の臨床試験が現在進行中である)。
αシヌクレインのミスフォールディングの抑制
緑茶抽出物である低分子エピガロカテキンガレート(EGCG)は、フォールディングされていないα-シヌクレインポリペプチド鎖に結合し、βシート形成を阻害するため、凝集やプリオンのような拡散を防ぐことができる[54-56]。これまでの研究では、EGCGは神経毒性からの保護をもたらす鉄キレーターであることが示唆されている[57]。前臨床モデルでは有効性のエビデンスが示されたものの[58, 59]、MSA患者を対象とした多施設ランダム化比較第III相試験では、52週間の治療後に疾患修飾効果を示すことはできず、一部の患者では肝毒性が観察された[60]。しかし、MRIサブスタディの探索的解析では、EGCGを投与したMSA患者では線条体と前中心回の年間体積減少が少ないことが示された[61]。
MSA治療の可能性があるもう一つの低分子は、α-シヌクレインのミスフォールディング阻害剤NPT200-11Aである。PDモデルマウスを用いた前臨床研究では、大脳皮質におけるα-シヌクレイン病態とアストログリオーシスを軽減する有益な効果が示された。線条体のドーパミントランスポーターレベルの正常化と運動機能の改善が観察された[62, 63]。NPT200-11Aの安全性、忍容性、血中濃度を調べるための健常人を対象とした第I相試験が終了しており、これまでのところ結果は公表されていない)。
αシヌクレインの凝集抑制
Anle138bはα-シヌクレインの細胞内オリゴマーを標的とする低分子化合物である。前臨床試験では、高い経口バイオアベイラビリティと血液脳関門透過性が報告されている。Anle138bは、α-シヌクレインのモノマーに影響を与えることなくオリゴマー形成を阻害し、その生理的機能を維持する。MSAマウスモデルでは、黒質と線条体におけるα-シヌクレイン蓄積の30%減少と相関する行動学的改善と、ミクログリアの活性化の有意な減少が観察された[65, 66]。経口投与されたanle138bの安全性、忍容性、血中濃度を決定するための健康なボランティアを対象とした第I相試験は成功裏に終了し、結果は保留中である[67]。軽度から中等度のPD患者を対象としたanle138bの第1b相試験は、現在患者を募集中である)。
SNpcにおける鉄代謝の調節障害は、α-シヌクレインの凝集を促進し、神経細胞死を引き起こす細胞の活性酸素種の産生を促進する[68] 。新規のキナゾリノン系阻害剤ATH434(以前はPBT434として知られていた)を用いた最初の実験では、PD動物モデルにおいて、α-シヌクレインと酸化ストレスマーカーのレベルが低下し、それに伴って運動が改善することが明らかになった[69]。同様の結果は、トランスジェニックMSAマウスでも再現された[70, 71]。健康なボランティアを対象とした第Ⅰ相試験において、ATH434は安全で忍容性が高く(U1111-1211-0052)、動物モデルにおける有効性と同程度の髄液濃度を達成した[72, 73]。そのため、MSA患者を対象とした第II相試験が現在検討されている[74]。
αシヌクレインの分解促進
MSAでは、オートファジー-リソソーム経路が影響を受けるという証拠が増えつつある[75]。そのような経路の一つが、哺乳類ラパマイシン標的複合体(mTOR)経路である。シロリムスとしても知られるラパマイシンは、mTORの触媒部位へのアクセスをアロステリックに調節することにより、mTORの作用を特異的に阻害する免疫抑制剤である[76]。最近の概念実証研究では、PLP-α-シヌクレイントランスジェニックマウスにラパマイシンを投与したところ、部分的な神経保護とα-シヌクレイン凝集体の減少が証明された。さらなる前臨床研究では、A53Tα-シヌクレイントランスジェニックマウスにおいて、運動機能の改善、4-ヒドロキシノネナール-タンパク質付加物の減少、シナプス傷害の減弱の証拠が示された。しかしながら、単離されたマクロオートファジーは、in vitroでのα-シヌクレインの異常蓄積のクリアランスを改善しなかった。MSA患者を対象とした第II相ランダム化比較試験では、シロリムスの経口投与による疾患進行抑制効果を評価したが、無益性の基準を満たしたため、最近早期に中止された)。
抗生物質リファンピシンは、MSAマウスモデルにおいてα-シヌクレイン線維の形成を阻害し、すでに形成された線維を分解する[80, 81]。大規模な第III相プラセボ対照試験)は、主要評価項目(UMSARS Iスコアの平均変化率)の事前に計画された中間解析の結果、無益性の基準を満たしたことが判明し、早期に中止された[82]。
ニロチニブは、慢性骨髄性白血病の治療薬として承認されているチロシンキナーゼ・アベルソン(Abl)阻害剤として作用する化合物である。前臨床試験では、この薬剤がオートファジー-リソソーム経路を促進することによってミスフォールドしたα-シヌクレインを分解し[83, 84]、酸化ストレスを軽減することが示唆された[85, 86]。残念ながら、ニロチニブはMSAのマウスモデルでは疾患修飾効果を示すことができなかった[87]。しかし、PD患者における所見は、臨床試験において議論のあるものであった。PD認知症およびDLB患者を対象とした小規模の非盲検第I相臨床試験では、良好な安全性と忍容性プロファイルが示され[88] [88]、第II相ランダム化比較試験では、妥当な薬物安全性が確認され、PD患者におけるドパミン代謝物、α-シヌクレインオリゴマー、タウの髄液レベルに対する効果が実証された[89] 。しかし、最近の別の第II相試験では、CSFへの曝露量が低く、6ヵ月間の治療後に有効性は認められなかった。これらの知見は、PDとMSA患者を対象とした試験開発の指針となるだろう。
リチウムは、in vivoおよびin vitroの前臨床モデルにおいて、α-シヌクレインの凝集を抑制し、オートファジーを刺激して神経保護作用を示す[91-93] 。MSA患者を対象としたリチウムの第II相試験は、重篤な有害事象のために中止され、MSAにおけるこの薬物の再利用のさらなる試みは断念された。
前臨床開発
分子ピンセットは、非共有結合効果や静電効果でゲスト分子と結合できる、開いた空洞を持つナノシャペロンである[95]。分子ピンセットCLR01で処理すると、MSAのトランスジェニック・マウス・モデルにおいて、α-シヌクレインの負荷が減少し、GCI密度が用量依存的に減少することが報告された[96, 97]。これらの有望な知見は、MSAや他のシヌクレイン病における疾患修飾の可能性を示唆している。しかし、血液脳関門を通過する浸透性が低いことが懸念され、今後の前臨床研究で対処する必要がある。
ヒト免疫グロブリン骨格と一般的なアミロイド相互作用モチーフを組み合わせた融合タンパク質であるNPT088は、現在アルツハイマー病に対して積極的な臨床開発が行われている)。このタンパク質のモチーフは、アミロイドβやリン酸化タウだけでなく、ミスフォールドしたα-シヌクレインも認識し、PDマウスモデルにおいて凝集したα-シヌクレインの量を著しく減少させた[98]。
低分子化合物SynuClean-Dは、ハイスループットスクリーニングアッセイによって同定された。試験管内およびPDモデルでの最初のテストでは、α-シヌクレイン線維への結合によるα-シヌクレイン凝集の阻害、アミロイド線維の破壊、ドーパミン作動性ニューロンの変性の防止が示された[99]。
c-Ablキナーゼ阻害剤IkT-148009は現在、PD動物モデルで研究されている。また、MSA動物モデルでの研究も計画されている[100]。健康なボランティアとPD患者を対象としたIkT-148009の第I相臨床試験が現在進行中である)。
ニューロシンKallikrein-6は、中枢神経系(CNS)においてα-シヌクレインを切断する能力を持つセリンプロテアーゼである。レンチウイルスベクターを通して導入すると、DLB/PDトランスジェニックマウスモデルにおいてα-シヌクレインの蓄積の減少が示された[101] 。Spencerらによる研究では、カリクレイン-6はR80Q変異により半減期が長くなり、血液脳関門を効果的に通過するためにアポBタンパク質と融合された。このニューロシン(NR)-R80Q-アポB濃縮により、オリゴデンドロサイトとアストロサイトにおけるα-シヌクレインの蓄積が減少し、MBP-α-シヌクレイントランスジェニックマウスの脳梁における髄鞘形成が改善した。さらに、認知と運動活性の行動学的改善も示された。しかしながら、最近の研究では、カリクレイン-6活性の低下は、MSAにおけるα-シヌクレイン蓄積の原因というよりも、代償反応である可能性が高いことが示された[103]。
神経炎症を標的とする
広範な神経炎症とそれに伴うミクログリアの活性化は、MSAにおける主要な病理組織学的所見であり、疾患の病理学的影響を受けた脳領域における神経変性と類似している[104] 。ミスフォールドしたα-シヌクレインが、MSAおよび関連するα-シヌクレイン病変におけるミクログリアの活性化とアストログリオーシスを誘発することを示唆する証拠が増えている[4, 105-107]。神経画像研究では、MSA患者における重篤な神経炎症が、MSA神経病理の根底にある局所パターンと一致していることが示されている[108] 。さらに、炎症性サイトカインレベルの上昇が以前に報告されている[109] 。しかし、神経炎症が神経変性の二次的な結果なのか、MSAの病態生理学的カスケードの独立した一因なのかは、まだ確立されていない。神経炎症の調節因子の疾患修飾能を評価した研究を表2にまとめた。
表2 神経炎症を標的とした臨床試験
| ターゲット | 物質 | フェーズ | デザイン | 主要評価項目 | 結果 | コメント |
| 神経炎症の抑制 | IVIG | フェーズII | OL | AE数 | ポジティブ | 運動機能の改善(サンプル数が少ない、治療期間が短い) |
| ミクログリア活性の抑制 | ミノサイクリン | フェーズII | RCT | UMSARSパートIIスコア | ネガティブ | モーターの改善なし |
| 酸化ストレス軽減 | バーディパースタット | フェーズIII | RCT | 修正UMSARSトータルスコア | ネガティブ | 主要評価項目および主要副次評価項目で失敗 |
IVIG、静注免疫グロブリン、RCT、ランダム化比較試験、OL、オープンラベル試験、UMSARS、統一多系統萎縮症評価尺度。
臨床開発
テトラサイクリン系抗生物質であるミノサイクリンは、ミクログリアの活性化と、炎症性サイトカインの分泌などのその下流事象を抑制することが示された[110, 111]。ミノサイクリンをMSA患者に48週間投与した第II相ランダム化比較試験では、運動機能の改善や神経保護効果は示されなかった[111]。しかし、in vivoで神経炎症を評価するための[11C](R)-PK11195 PETは、MSA患者のサブグループにおいて、皮質下ミクログリア活性化の減少を伴う標的関与が証明された[111]。
MSAにおける神経炎症と毒性サイトカインの産生は、免疫グロブリン(IVIG)静注療法を支持する証拠となる[112-114] 。IVIGは自己反応性T細胞を抑制し、自己抗体を抑制し、サイトカインの産生を阻害する[114] 。Novakらは、9人のMSA患者を対象とした非盲検パイロット試験でIVIG注入の効果を検討し、大多数の患者でUMSARSスコアの低下を示した[114] 。脳MRIに変化はなく、重篤な有害事象も認められなかった。これらのポジティブなシグナルにもかかわらず、MSAにおけるIVIG療法の有効性を確立するには、より大規模な確認試験が必要である。
ミエロペルオキシダーゼ(MPO)は、貪食細胞による活性酸素種の産生に重要な役割を果たしている[115-117]。ベルジパスタットはMPOの強力な阻害剤であり、トランスジェニックMSAマウスモデルにおいてミクログリアの活性化を抑制し、運動機能を改善した[106]。しかし、これらの効果は、進行したMSAのマウスモデルにおける運動障害には影響を与えなかった[118]。健常人を対象としたベルジパスタットを評価したいくつかの第I相試験では、安全性に関する懸念は報告されていない。第2相試験では、PD患者におけるミクログリア活性化の改善が報告された[119] 。MSA患者では、第Ⅱ相試験で臨床的有効性の傾向が示された)。第III相ランダム化比較試験は最近終了し、修正UMSARSスコア、Clinical Global Impression of Improvement(CGI-I)スコア、MSA QOL質問票を含む主要評価項目および主要副次評価項目を達成できなかった)[120]。
前臨床開発
炎症性プロテアーゼであるカスパーゼ-1はα-シヌクレインの凝集を促進する[121] 。概念実証研究では、カスパーゼ-1阻害剤のプロドラッグVX-765がトランスジェニックMSAマウスにおけるα-シヌクレインの凝集毒性を改善した。
最近、トランスジェニックMSAマウスモデルにおいて、α-シヌクレイン指向性抗体と抗炎症治療の併用が評価された[123]。CD5-D5は、α-シヌクレインを標的とするCNS透過性の一本鎖抗体である。レナリドマイドは小さなサリドマイド誘導体で、多発性骨髄腫の抗がん剤として販売されている。この併用療法は、トランスジェニックMSAマウスにおいて、アストログリオーシス、ミクログリオーシス、可溶性および凝集性αシヌクレインレベルの減少を示した。
現在、多発性硬化症の治療薬として販売されている免疫調節薬フィンゴリモドは、脳由来神経栄養因子を増加させることにより、様々な動物モデルにおいて神経保護作用を示す。修飾誘導体であるFTY720-Mitoxyは、脳由来神経栄養因子(BDNF)、グリア細胞系列由来神経栄養因子(GDNF)、神経成長因子の発現を増加させることが知られている[124, 125]。Vidal-Martinezらは、CNP-α-シヌクレイントランスジェニックMSAマウスにおいて、運動障害と神経炎症の軽減、ミトコンドリア機能の回復、GDNF発現の増加によるFTY720-ミトキシの強力な保護効果を報告した[126]。
合成マイクロニューロトロフィンBNN-20は、ミクログリアの活性化を抑え、BDNFを増加させ、神経変性の進行した段階でもドーパミン作動性ニューロンを回復させた[127]。
その他の神経保護戦略
可能性のある神経保護疾患修飾療法を表3にまとめた。
表3 神経保護と神経栄養支持を対象とした臨床試験
| ターゲット | 物質 | フェーズ | デザイン | 主要評価項目 | 結果 | コメント |
| FAF-1 | KM-819 | 第1段階 | RCT | 安全性と忍容性 | 安全で忍容性が高い | – |
| リピドミクス神経毒性 | YTX-7739 | フェーズIb | – | 安全性と忍容性 | – | 継続中 |
| IGF1経路 | 経鼻インスリン | フェーズII | RCT | 言葉の流暢さの合計スコア | – | 運動機能の改善(MSA患者1名のみ) |
| IGF1経路 | エクセンディン-4 | フェーズII | OL | UMSARSパートI&IIスコア | – | 継続中 |
| ミトコンドリア機能障害 | コエンザイムQ10 | フェーズII | RCT | UMSARSパートIIスコア | – | 継続中 |
| 神経細胞/グリア細胞増殖 | 成長ホルモン | フェーズII | RCT | 安全性と忍容性 | 安全で忍容性が高い | UMSARSの悪化が少ない傾向[152]。 |
| 免疫調節、神経保護 | MSCsについて | フェーズII | RCT | UMSARSパートIIスコア | ポジティブ | MSA-Cのみ、画像診断は全例で行われなかった[156]。 |
| ミトコンドリア機能障害 | ラサギリン | フェーズII | RCT | UMSARSパートI&IIスコア | ネガティブ | 運動機能改善なし[160] |
| 神経栄養サポート | フルオキセチン | フェーズII | RCT | UMSARSパートI&IIスコア | ネガティブ | 運動機能改善なし[163] |
| 興奮毒性の低減 | リルゾール | フェーズIII | RCT | UPDRSパートII&III | ネガティブ | 運動機能の改善も生存率もない[137]。 |
| NMDA調節薬 | Tllsh2910 | フェーズIII | RCT | SARAスコア | – | 継続中 |
IVIG、免疫グロブリン静注、RCT、ランダム化比較試験、OL、オープンラベル試験、UPDRS、統一パーキンソン病評価尺度、SARA、運動失調評価尺度、UMSARS、統一多系統萎縮症評価尺度、MSA、多系統萎縮症。
臨床開発
KM-819
FAS関連因子1(FAF1)というタンパク質は、アポトーシスを誘導するために細胞内で発現している。PDではこれらのタンパク質が過剰発現しているため、神経細胞死が増加していることが研究で示されている[128, 129]。KM-819は、FAF1の阻害剤として開発された経口活性の低分子薬である。健康なボランティアを対象とした最初の試験では、安全性と忍容性において有望な結果が得られた[130] 。PDとMSA患者を対象とした第II相試験が検討されている[131]。
YTX-7739
α-シヌクレインに関連する標的をスクリーニングするために、その神経毒性に関するリピドミクス解析が行われた。その結果、ステアロイル-CoAデサチュラーゼ(SCD)によって産生されるオレイン酸が神経細胞に対して神経毒性を持つことが明らかになった[132, 133]。そのため、SCD阻害剤YTX-7739は現在、第Ib相概念実証試験で検討されている[134]。
リルゾール
リルゾールはグルタミン酸拮抗薬であり、筋萎縮性側索硬化症に対して唯一承認されているDMTである[135] 。ナトリウムチャネルとカリウムチャネルを遮断することで、グルタミン酸受容体の刺激を減少させ、興奮毒性による神経細胞死を防ぐことができる。MSAのラットモデルを用いた前臨床研究では、運動障害と線条体病変の体積が有意に減少したことから、神経保護作用の可能性が示唆された[136] 。Seppiらは、10人のMSA患者の少人数グループにおいてランダム化比較試験を行ったが、運動機能の改善は認められなかった。その後、MSAとPSP患者を対象とした大規模なプラセボ対照試験で、リルゾールは疾患の進行と生存に影響を及ぼさなかったと報告された[138] 。
Tllsh2910
小脳のN-メチル-D-アスパラギン酸(NMDA)受容体は、運動学習と協調運動において役割を果たしている[139] 。NMDAモジュレーターであるTllsh2910は、MSAマウスモデルにおいて運動失調性歩行を抑制することが判明している。現在、第III相ランダム化比較単施設試験がMSA-C患者を募集している)。
インスリン様成長因子経路
インスリンは、その神経調節作用、神経栄養作用、神経保護作用により、多くの神経変性疾患において重要な役割を果たしている[140] 。PDやアルツハイマー病では、インスリン様成長因子-1(IGF-1)シグナル伝達が障害されているという証拠がある[141] 。臨床研究では、MSA患者では血漿中インスリン濃度とIGF-1濃度の上昇が観察された一方で[142] 、MSAのトランスジェニックマウスモデルではIGF-1脳内濃度の低下が観察された[143, 144]。14人のPD患者と1人のMSA患者を対象とした経鼻インスリンによる試験的無作為プラセボ対照試験では、治療を受けた患者において重篤な有害事象を伴わずに、Hoehn & Yahr病期分類、UMSARS運動スコア、言語流暢性の改善が認められた[145] 。MSAに対して試験されているもう一つの有望な抗糖尿病薬は、グルカゴン様ペプチドアゴニストであるエキセンディン-4である[144] 。Bassilらは、トランスジェニックMSAマウスにおけるエキセンディン-4治療を評価し、最も重篤な脳領域におけるインスリン受容体密度の増加、線条体における単量体α-シヌクレイン負荷の減少、および黒質ドーパミンニューロンの生存に対する保護効果を観察した[144]。しかしながら、運動徴候はトランスジェニックマウスでは改善されなかった。現在、MSA患者を対象としたエキセンディン-4の第II相オープンラベル試験が進行中である)。
コエンザイムQ10
MSAの大部分は散発性疾患であるにもかかわらず、COQ2変異と小脳型MSAとの因果関係が日本人患者において確立された[146]。これらの変異は、ミトコンドリア呼吸鎖の電子伝達物質であり、強力な抗酸化物質でもあるコエンザイムQ10(CoQ10)の産生低下につながる。MSA患者から採取した人工多能性幹細胞由来のドーパミン作動性ニューロンを用いたin vitro研究では、健常対照群と比較して、MSA患者ではCoQ10レベルが低下し、いくつかのCoQ10生合成酵素がアップレギュレーションしていることが報告されている[147, 148]。これらの変化は、CoQ10の補充によって部分的に回復した[148]。COQ2変異を有するMSA-C患者におけるユビキノール大量投与の症例報告では、3年間の治療後、臨床的または画像的な有益性のエビデンスはなかったと報告されている[149]。とはいえ、すでに募集を終了した第II相ランダム化比較試験が現在日本で進行中である(UMIN000031771)。
成長ホルモン
成長ホルモンは神経細胞とグリアの増殖を刺激し、髄鞘形成と脳の大きさを増大させることが示された[150] 。対照的に、成長ホルモンの欠乏は、新しいニューロンの生存障害、脳の発達と機能の障害と関連している[151, 152]。Holmbergらは、MSA患者を対象に遺伝子組換えヒト成長ホルモン(r-hGH)のランダム化比較試験を行った。12ヵ月間の皮下投与後、r-hGH投与群とプラセボ投与群のMSA患者間で治療効果に差は認められなかったが、サンプル数が少ないことがこの試験の大きな限界であった[152] 。
間葉系幹細胞
間葉系幹細胞(MSC)は、免疫調節作用と神経保護作用があるため、10年以上前からMSA治療の可能性が注目されてきた。最初の臨床試験は、動脈内MSCs治療の実行可能性と安全性を評価する非盲検単施設試験であり、有望な結果を示した[153, 154]。2011年、Stembergerらは、MSAのトランスジェニックマウスモデルにおいて、MSCの潜在的な神経保護効果を確認した[155]。第Ⅱ相無作為化プラセボ対照試験では、MSA-C患者において、自家骨髄由来MSCを動脈内または静脈内投与した場合、プラセボを投与した場合と比較して、UMSARSパートⅡスコアの進行が抑制されたことが報告された[156]。しかし、手技に関連した有害事象(動脈内注入時の小さな虚血性脳病変)が、安全性への懸念を引き起こした。このため、MSA-C患者における自家骨髄由来間葉系幹細胞の動脈内(頸動脈)注入の安全性と忍容性を再検討する別の第I相試験が最近韓国で実施され、その結果は今のところ公表されていない)。
2019年のSingerらは、自己脂肪組織由来MSCの髄腔内注射により、過去のコホートと比較して運動機能の進行が遅くなることを実証した[157]。有害事象の発生率は高用量になるほど増加し、腰痛や後下肢痛を発症する患者がみられ、腰部神経根の肥厚/MRIによる増強が関連した。それ以外の点では、この治療法は安全で忍容性が高いことが証明され、今後の臨床開発が期待される。
ラサギリン
非可逆的なモノアミン酸化酵素B(MAO-B)阻害薬であるラサギリンは、ミトコンドリア代謝の調節により、PD患者において症状の改善と疾患修飾効果の可能性を示している[158] 。MSAのトランスジェニックマウスモデルを用いた前臨床研究では、運動の改善、GCI負荷の軽減、神経細胞の保護が明らかにされた。しかし、MSA-P患者を対象としたラサギリン1mg/日の多施設共同第Ⅱ相ランダム化プラセボ対照臨床試験では、臨床的利益は示されなかった。
選択的セロトニン再取り込み阻害薬
神経栄養因子GDNFとBDNFは神経保護に重要な役割を果たしている。現在抗うつ薬として使用されている選択的セロトニン再取り込み阻害薬(SSRI)は、神経栄養因子の発現に良い影響を与えることが報告されている。トランスジェニックMSAマウスモデルにおいて、フルオキセチンはGDNFとBDNFレベルを増加させ、炎症性サイトカインを抑制することが示されている[161, 162]。MSA患者を対象としたフルオキセチンの第Ⅱ相ランダム化プラセボ対照試験では、UMSARSの総スコアにおいてプラセボに対するフルオキセチンの優越性は示されなかったが、運動と情動の副次的/探索的アウトカムの傾向は、さらなる調査に値する[163] 。しかし、MSA患者600人以上を対象としたレトロスペクティブな長期解析によると、SSRIのいずれかを投与された患者は、SSRIを投与されなかった患者と生存率に差はなかったが、パーキンソニズムや転倒の頻度が高かった[164] 。
GDNFをめぐるもう一つの研究は、無作為化プラセボ対照第I相試験において、MSA患者の被殻へのAAV2-GDNFの両側画像誘導注入の安全性と潜在的臨床効果を評価するため、現在募集中である)。
前臨床開発
ベンズトロピン
ミエリンの形成と修復はオリゴデンドロサイトの主な仕事である[165] 。オリゴデンドロサイトにおけるα-シヌクレインの蓄積は、軸索機能障害と神経細胞喪失をもたらす脱髄を引き起こす。Ettleらは、ムスカリン性アセチルコリン受容体拮抗薬であるベンズトロピンの脱髄促進作用を利用して、MSAのさまざまな前臨床モデルにおける脱髄欠損を回復させようと試みた。この実験では、幹細胞由来のオリゴデンドロサイトのα-シヌクレイン誘導性髄鞘形成が回復し、トランスジェニックMSAマウスにおける神経細胞喪失が予防された[166]。ベンズトロピンは臨床神経学で何十年も使用されているが、脱髄がMSAの主要な標的であるかどうかは議論の余地がある。
モノホスホリル脂質A
Toll様受容体(TLR)は自然免疫反応に重要な役割を果たしている。最近、TLR4はミクログリアによる内因性α-シヌクレインクリアランスの重要なメディエーターとして同定された[167] 。TLR4の機能欠損は、二重MSAトランスジェニックマウスにおいて、α-シヌクレイン蓄積の増加、運動障害の悪化、黒質変性という結果をもたらした[168] 。TLR4アゴニストであるモノホスホリルリピドAで処置したトランスジェニックMSAマウスの前臨床研究では、顕著な全身性炎症反応がないにもかかわらず、ミクログリアのα-シヌクレイン取り込みの増加、著しい運動改善、黒質のドーパミン作動性ニューロンと線条体ニューロンの救出、およびGCI密度の領域特異的減少が明らかになった[169]。このアプローチは、α-シヌクレインクリアランスの内因性メカニズムを強化する興味深い選択肢を提供する。
フェニル酪酸ナトリウム
Sturmらは、エピジェネティックな根源からMSAをターゲットとして、α-シヌクレインがグリア細胞や神経細胞でヒストンのアセチル化に干渉し、アセチル化を阻害して神経毒性を引き起こす可能性があるという仮説を説明した[170, 171]。汎ヒストン脱アセチル化酵素阻害剤であるフェニル酪酸ナトリウムは、PLP-α-シヌクレインマウスにおいて、運動行動の有意な改善と黒質ニューロンの生存を示した[171]。
試験デザインとアウトカム評価
MSAにおける疾患修飾試験の実施には多大な努力が払われたが、試験方法の改善は依然として必要である。レビュー全体を通して強調されたように、MSAにおける過去の臨床試験の失敗の理由については推測するしかない。試験化合物の有効性が不十分であったために失敗した可能性が高いが、追跡期間が短すぎる、サンプルサイズが不十分である、脱落率が高いなど、試験デザインに関連する他の問題も、試験結果に影響を与えた可能性がある。したがって、試験計画においてサンプルサイズを慎重に見積もり、患者の維持と試験のアドヒアランスを向上させる方策を導入することが重要である。さらに、MSA診断のための新しい国際パーキンソン病・運動障害学会(MDS)基準は、確立された疾患だけでなく、早期疾患における診断精度を向上させ、研究者がより均質な患者コホートと早期MSA患者をリクルートできるようにする。
さらに、臨床的尺度を超えて、MSAの疾患進行を定義する信頼性の高い代用バイオマーカーがまだ満たされていない。ここでは多くの努力がなされているが、我々はこの疾患の自然史をより明確にし、マルチモーダルMRIや生体流体マーカーを含む疾患進行の可能性のある代替バイオマーカーについてより多くの研究を行う必要がある。先行研究では矛盾した結果が得られているが[172] 、髄液中のα-シヌクレイン、神経変性マーカー(例、ニューロフィラメント)、グリア機能障害マーカー(例、GFAP)が有用であろう。最近、新しいPETトレーサーが、MSAを他のシヌクレイン病変と区別できることが証明され、今後の研究の画像アウトカムパラメータとして有望な可能性が示された[173] 。最後に、患者ごとに設定されたマイルストーンを把握する臨床転帰評価指標が不可欠であり、最近、UMSARSの現行版の欠点を改善し、対処しようとする取り組みが強化され、データシート内にUMSARSの改訂に取り組む専門家タスクフォースが設置された。
結論と展望
MSA患者の治療管理は、疾患修飾薬の不足と、一過性で部分的な効果しか患者サブグループにもたらさない対症療法により、ほとんどフラストレーションが残っている。それゆえ、MSAの疾患修飾療法に対するアンメットニーズが急務となっている。過去20年間にわたり、MSAにおける分子変化とその根底にある神経病理生理学的事象を徹底的に特徴付けるために、前臨床MSAモデルが開発されてきた。神経毒を用いた動物モデル[174]、トランスジェニック改変によるα-シヌクレインの標的過剰発現[175]、ウイルスベクター[176]などである。より最近の研究では、MSA特異的α-シヌクレイン株の播種特性を研究する試みとして、トランスジェニックマウスにMSA脳抽出物を脳内接種した。ヒトMSA病態の再現は不完全であるにもかかわらず[175, 177]、前臨床研究は、MSAの病態生理学的カスケードや、異常なαシヌクレイン凝集によって引き起こされる二次的変化について、重要な教訓を与えてくれた[178] 。これらのモデルは、疾患修飾作用を有する新規薬剤の開発、および前臨床的な標的関与の確認のための道を開いた。しかし、上述のように、いくつかの治療薬候補は前臨床試験で神経保護効果を示したが、大規模な介入試験では臨床的な効果には結びつかず、前臨床試験での標的関与がどの程度臨床的な効果を予測できるのか疑問が残る。
α-シヌクレインを標的としたこれまでの治療法では、疾患の進行を遅らせることはできなかったが[82, 179]、α-シヌクレインが疾患拡大の重要な一因であり、その毒性作用がMSAにおける細胞死を促進するという、説得力のある前臨床エビデンスが集約されている[8, 22]。したがって、免疫療法、遺伝子組み換え、α-シヌクレインのクリアランスの増強(分解やオートファジー-リソソーム経路の増強による)など、非常に効果的な新規治療戦略を利用したCNSのα-シヌクレイン負荷の低減は、疾患改善への有望なアプローチであり続けている。
酸化ストレスを含む神経炎症とその二次的結果は、MSAにおける神経変性の他の重要な要因である。ミノサイクリン(抗炎症作用のある抗生物質)は、PET画像で標的が関与している証拠があるにもかかわらず、小規模の研究では運動機能を改善することができなかった[111]。最近では、主要な酵素に作用する特異性の高い薬剤が前臨床的に研究されている。その中で、ミエロペルオキシダーゼの不可逆的阻害剤が臨床開発段階まで発展した。MPO阻害剤に関する初期段階の臨床試験で得られた有望な結果に基づいて、大規模な第III相試験が最近終了したが、残念ながらこの試験は主要評価項目と主要副次的評価項目で失敗した。
神経細胞やグリア細胞増殖の刺激、栄養補助による髄鞘形成の促進、細胞補充療法などが、現在追求されている分野である。遺伝子組換え成長ホルモンを用いた初期の研究は否定的であった。しかし、成長ホルモン投与群では運動機能が改善する傾向が見られた。この研究はサンプル数が少なかったため、最終的な結論には至っていない。有害事象の可能性が懸念されたが、間葉系幹細胞はMSA-C患者を対象とした小規模研究で神経保護効果を示した。
まとめると、現在進行中の前臨床試験や臨床試験には有望な治療法が数多くあり、疾患修飾薬の発見に期待が持てる。
謝辞
また、現在進行中のMSAの前臨床および臨床研究に携わっているすべての臨床試験参加者および研究者に感謝したい。
利益相反
WPは、アフィリス社、アルテリティ社、バイオジェン社、ルンドベック社、武田薬品工業社のMSAにおける医薬品開発プログラムに関する有料コンサルタントを務めている。
KSは、Teva、UCB、Lundbeck、AOP Orphan Pharmaceuticals AG、Roche、Gruenenthal、Stada、Lucher Pharma、Biogen、BIALおよびAbbvieからの報酬、国際パーキンソン病・運動障害学会からの謝礼、FWF Austrian science fund、Michael J Fox FoundationおよびAOP Orphan Pharmaceuticals AGからの研究助成金を報告した。
GWは、Biogen社、Biohaven社、Inhibikase社、Lundbeck社、小野薬品工業社、武田薬品工業社、Theravance社からのコンサルタント料および講演料、FWFオーストリア科学基金、オーストリア国立銀行、米国MSA連合、Parkinson Fonds Austria、および国際パーキンソン病・運動障害学会からの研究助成金を報告。
FKは、Institut de Recherches Internationales Servier、Clarion Healthcare、Austrian Society of Neurologyから個人的な報酬を受けている。
他のすべての著者は、利益相反がないことを宣言している。
本研究の全部または一部は、オーストリア科学基金(FWF)[助成金I 4795-B]の助成を受けた。オープンアクセスのため、著者は本投稿から生じたすべてのAuthor Accepted ManuscriptバージョンにCC BYパブリック著作権ライセンスを適用している。
この研究および業績に対する金銭的および物質的な支援はすべて、投稿状に明記された支援内容のリストも含め、原稿に明記する。
