
Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial
https://www.thelancet.com/journals/langlo/article/PIIS2214-109X(21)00448-4/fulltext
Gilmar Reis, PhD
Eduardo Augusto dos Santos Moreira-Silva, PhD(エドワルド・アウグスト・ドス・サントス・モレイラ=シルバ博士
Daniela Carla Medeiros Silva(ダニエラ・カーラ・メデイロス・シルバ)博士
Lehana Thabane教授(PhD
アライン・クルス・ミラグレス(RN
ティアゴ・サンティアゴ・フェレイラ(MD
概要
背景
近年、COVID-19に対するフルボキサミンの治療効果が期待されている。COVID-19の急性症状患者を対象としたTOGETHER試験では、COVID-19による救急搬送、またはCOVID-19による三次病院への転院と定義される入院を予防する上で、フルボキサミンのプラセボに対する有効性を評価することを目的とした。
方法
SARS-CoV-2陽性が確認された高リスクの症状を持つブラジル人成人を対象としたプラセボ対照無作為化適応プラットフォーム試験で、ブラジルの11の臨床施設から、重症化のリスク因子が確認されている適格患者を集めた。患者は、フルボキサミン(100mg、1日2回、10日間)またはプラセボ(またはここでは報告されていない他の治療群)のいずれかに、1対1で無作為に割り付けられた。試験チーム、治験施設のスタッフ、および患者は、治療の割り振りについてマスキングされた。主要評価項目は、COVID-19による救急搬送、またはCOVID-19による三次病院への転院のいずれかとして定義される入院の複合エンドポイントとし、治療意図に基づいて無作為割り付け後28日までとした。修正intention to treatでは、主要アウトカムイベントの発生前に少なくとも24時間の治療を受けた患者を対象とし、per-protocol解析では、高いアドヒアランス(80%以上)を示した患者を対象とした。ベイズ分析の枠組みを用いて、プラセボと比較した介入の効果と成功確率を確立した。この試験はClinicalTrials.govに登録されており(NCT04727424)現在進行中である。
調査結果
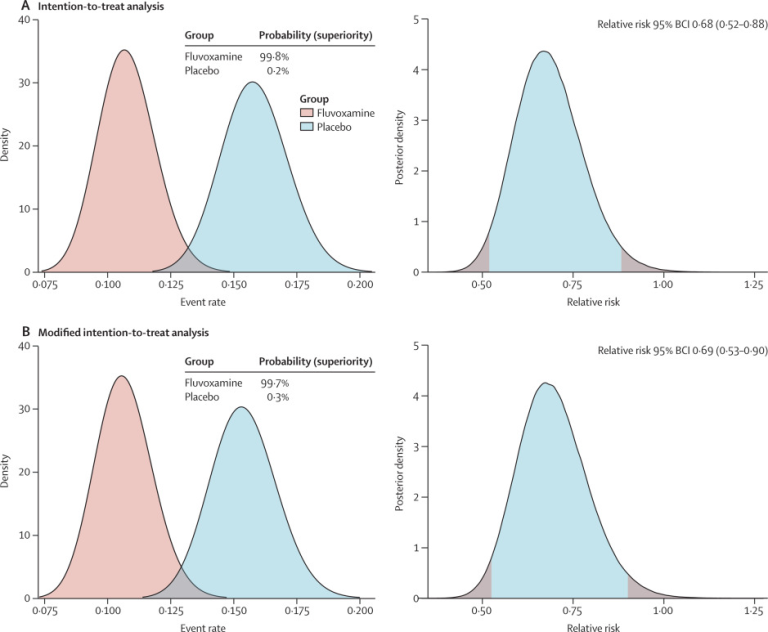
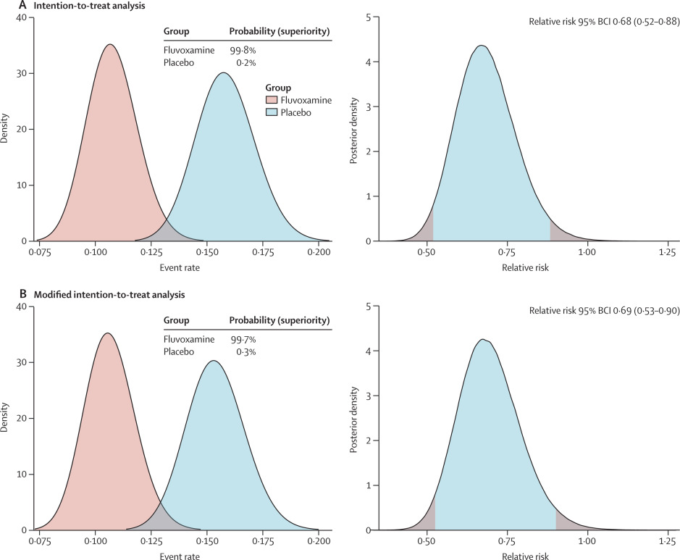
本試験では、9803名の被験者候補者がスクリーニングされた。本試験は2020年6月2日に開始され、現在のプロトコルでは、優越性のために試験アームを停止する20年1月20日から 20年8月5日まで、フルボキサミンへの無作為化が報告されている。741名の患者がフルボキサミンに、756名の患者がプラセボに割り振られた。参加者の平均年齢は50歳(範囲18~102歳)で、58%が女性であった。COVID-19による緊急事態で6時間以上観察された患者、またはCOVID-19により三次病院に転院した患者の割合は、プラセボと比較してフルボキサミン群で低く(741例中79例[11%] vs 756例中119例[16%];相対リスク[RR]0-68;95%ベイズ信頼区間[95%BCI]:0-52-0-88)事前に規定した優越性の閾値97-6%を上回る99-8%の確率で優越性が認められた(リスク差5-0%)。複合的な主要アウトカムイベントのうち、87%が入院であった。主要評価項目の結果は、修正intention-to-treat解析では同様の結果(RR 0-69,95%BCI 0-53-0-90)per-protocol解析ではより大きな結果(RR 0-34,95%BCI 0-21-0-54)が得られた。主要なintention-to-treat解析では、フルボキサミン群で17名、プラセボ群で25名の死亡が認められた(オッズ比[OR]0-68,95%CI:0-36-1-27)。また、per-protocol集団では、フルボキサミン群で1名、プラセボ群で12名の死亡が認められた(OR 0-09,95%CI 0-01-0-47)。また、フルボキサミン群とプラセボ群の患者間で、治療上の緊急性が高い有害事象の発生数に有意な差は認められなかった。
解釈について
早期にCOVID-19と診断された高リスクの外来患者にフルボキサミン(100mg、1日2回、10日間)を投与したところ、COVID-19の救急施設に留まるか、3次病院に転院するかと定義される入院の必要性が減少した。
資金調達
FastGrantsおよびThe Rainwater Charitable Foundationによる。
翻訳
要約のポルトガル語翻訳については、のセクションを参照してほしい。
はじめに
COVID-19に対する安全で効果的なワクチンが開発・配布されているが、特に低資源環境では、その製造、配分、価格に大きな課題が残っている1。そのため、COVID-19に対する安価で広く入手可能な有効な治療法を見出すことは非常に重要であり、特に、広く入手可能で安全性が十分に理解されている既存の医薬品を再利用することは魅力的です2。
研究の背景
本研究以前のエビデンス
2021年9月10日にPubMedを,以下の検索語「(randomized OR trial) AND (fluvoxamine OR antidepressants OR selective serotonin reuptake inhibitors OR SSRIs) AND (COVID* OR SARS-CoV-2 OR SARS-CoV) 」を用いて,日付や言語の制限なく検索したところ,抗うつ薬の使用と挿管または死亡のリスク低下との間に有意な関連性があると報告した観察研究が1件確認された(ハザード比0-56。95%CI 0-43-0-73,p<0-001)症状のあるCOVID-19の成人外来患者にフルボキサミンを投与した場合、プラセボと比較して15日間の臨床悪化の可能性が低かったと報告した無作為化臨床試験が1件あった。この予備的無作為化試験では、152名の参加者が、フルボキサミン100mg(n=80)またはプラセボ(n=72)を1日3回、15日間投与するよう無作為に割り付けられた。主要評価項目は、無作為化後15日以内の臨床的悪化で、息切れ、息切れによる入院、肺炎、酸素飽和度92%未満の基準を満たすこと、または酸素飽和度92%以上を達成するために補助酸素が必要であること、と定義された。
本試験の付加価値
TOGETHER試験は、地域社会におけるCOVID-19患者に対するfluvoxamineの有効性を評価した最大の無作為化試験である。プラセボと比較して、フルボキサミンに無作為に割り付けられた患者は、COVID-19による救急施設への留置またはCOVID-19による三次病院への転院と定義される入院のリスクが低かった。
入手可能なすべてのエビデンスの意味するところ
地域社会におけるCOVID-19患者に対する有効な治療法はほとんどない。今回の結果は、COVID-19疾患による急性罹患率の低下にフルボキサミンが有効であることを示す説得力のある証拠となる。
フルボキサミンは、選択的セロトニン再取り込み阻害剤(SSRI)およびσ-1受容体(S1R)作動薬です3。フルボキサミンがCOVID-19疾患の治療に用いられるメカニズムには、抗炎症作用や抗ウイルス作用の可能性など、いくつかの可能性がある4。小規模なプラセボ対照無作為化試験により、フルボキサミンがCOVID-19外来患者の臨床悪化リスクを低減する可能性が提起されており、より大規模な無作為化プラセボ対照試験の必要性が示唆されている5, 6。
実験室で確認されたSARS-CoV-2の外来患者におけるCOVID-19の進行と入院を予防するためのフルボキサミンの有効性を評価するため、ブラジルのミナス・ジェライス州で無作為化プラセボ対照適応プラットフォーム試験を実施した。このプラットフォーム試験では,8種類の治療法が評価されたが,ここでは,プラセボ対照群を併用したfluvoxamineの臨床評価について報告する。
試験方法
試験デザイン
TOGETHER試験は、ハイリスクの成人外来患者を対象に、COVID-19疾患に対する再製品化された治療法の有効性を検討する無作為化適応プラットフォーム試験である9。本試験は、ブラジルの参加11都市の地域保健当局と共同で設計・実施され、マスタープロトコルによって初期疾患に対する潜在的な治療法を同時に検証したものである。マスタープロトコルでは、無益性による介入の中止、プラセボに対する優位性による中止、新たな介入の追加などの前向きな判断基準が定められている。TOGETHER試験では、これまでに、ヒドロキシクロロキン(プロトコル1)ロピナビル・リトナビル(プロトコル1)10 メトホルミン、イベルメクチン、フルボキサミン、ドキサゾシン、およびペグインターフェロン・ラムダとプラセボの併用(プロトコル2)などが評価されている。TOGETHER試験は、Platform Life Sciences社(カナダ・バンクーバー)が中心となり、Cardresearch社(ブラジル・ベロオリゾンテ)が現地で実施している。統計解析はCytel社(Waltham, MA, USA)が行った。
本試験は、International Conference of Harmonization-Good Clinical Practicesに加え、現地の規制要件にも準拠している。本試験は、ブラジルの地域および国の倫理委員会(CONEP CAAE:41174620.0.1001.5120,承認書5.501.284)およびカナダのハミルトン統合研究倫理委員会(承認書13390)により、研究倫理が承認された。プロトコルと統計解析計画の全文はすでに発表されており9,詳細は付録2(p2)に記載されている。10, 11 独立したデータ安全性モニタリング委員会(DSMC)が本試験を監督した。
参加者
本試験に参加したブラジルの11の臨床施設の都市および治験責任医師は、付録2(p3)に記載されている。各地の治験責任医師は、地域の公衆衛生当局と協力して、地域の医療施設(救急施設、インフルエンザ症状紹介センター、プライマリーケアコミュニティセンター)で参加者を募集した。試験の認知度を高めるために、地域の公衆衛生当局の指示に従い、物理的なメディアやソーシャル・メディアを含むいくつかのコミュニティ・アウトリーチ戦略を用った。
外来診療所に来院すると、地元の医師が参加者をスクリーニングし、参加資格基準を満たしている人を特定した。主な組み入れ基準は、18歳以上で、COVID-19に合致する急性の臨床症状を呈し、スクリーニング日から7日以内に症状が始まった外来患者、またはスクリーニング時に実施したSARS-CoV-2抗原の迅速検査が陽性である患者、または症状が現れてから7日以内にSARS-CoV-2診断検査が陽性である患者であった。対象となる患者は、さらに少なくとも1つの高リスク基準を満たしていた。糖尿病、治療のために少なくとも1種類の経口薬を必要とする全身性動脈性高血圧症、既知の心血管疾患(心不全、先天性心疾患、弁膜症、冠動脈疾患、治療中の心筋症、臨床的に顕在化した心疾患とその臨床的反響)症候性肺疾患またはその治療(肺気腫、線維性疾患)症状を抑えるために薬剤の慢性使用を必要とする症候性喘息。喫煙、肥満(体格指数が30kg/m2以上と定義)移植を受けたことがある、ステージIVの慢性腎臓病または透析を受けている、免疫抑制状態または副腎皮質ホルモン療法(プレドニゾン10mg/日以上に相当)もしくは免疫抑制療法の使用、過去0~5年の間にがんの既往歴がある、または現在がん治療中である、50歳以上である、ワクチン未接種の状態。
以下の重要な基準のいずれかに該当する患者は、本試験から除外された。急性インフルエンザ様症状を伴うSARS-CoV-2診断テストが陰性(インフルエンザ様症状の発症から7日以内であれば、初期に受けたテストが陰性で、数日後に陽性となった患者も対象となる)COVID-19に適合する急性呼吸器疾患がプライマリケアで治療され、過去に入院を必要としたことがある、他の原因による急性呼吸器疾患、SARS-CoV-2のワクチン接種を受けたことがある、など。他の急性・慢性呼吸器疾患や感染症による二次的な呼吸困難(例:非代償性慢性閉塞性肺疾患、急性気管支炎、肺炎、原発性肺動脈性肺高血圧症)現在のSSRIの使用(他のセロトニン再取り込み阻害剤の使用は除外されていない)コントロールされていない精神疾患や自殺念慮、研究のガイドラインや手順に従うことができない、または従う意思がないこと。除外基準の完全なリストは、試験計画書に記載されている。
患者が前述の適格性基準を満たしている場合、試験担当者は書面によるインフォームド・コンセントを得た。インフォームドコンセントを得た後、COVID-19の迅速抗原検査(Panbio, Abbott Laboratories Jena, Jena, Germany)と、妊娠可能な女性のための妊娠検査を行った。COVID-19検査が陰性の場合、または妊娠検査が陽性の場合、その参加者は試験に参加しなかった。インフォームドコンセントの後、試験担当者は無作為化の前に以下のデータを収集した:人口統計、病歴、併用薬、併存疾患、指標となる症例情報への曝露、WHOの臨床的悪化スケール、患者報告式アウトカム測定情報(PROMIS)のグローバルヘルススケール。
無作為化とマスキング
参加者の無作為化は、プロトコルに関連した手順を知らず、このプロセスのために特別に契約した独立したマスクされていない薬剤師が担当し、集中的なコア・ランダム化プロセスによって行われた。各施設は、調整センターの薬剤師にテキストメッセージで無作為化を依頼した。これにより、割付の秘匿性が保たれた。患者は、各参加施設において、年齢(50歳未満または50歳以上)で層別されたブロック無作為化手順により、1対1で無作為に割り付けられた。試験チーム、治験施設のスタッフ、および患者は、治療の割付についてマスキングされた。有効成分とプラセボ錠は、同じ形状のボトルに包装され、有効成分群またはプラセボ群に対応するアルファベットのラベルが貼られた。無作為化の解除を担当した第三者機関の薬剤師のみが、どの文字がどの薬剤またはプラセボに対応しているかを知っていた。本試験はマルチアーム試験であり、すべての有効な介入には対応する不活性プラセボがあるため、対応するプラセボは、その時点での試験のアーム数に対する対照群の割合を表している。
試験手順
すべての参加者は、公衆衛生施設の医療従事者によるCOVID-19の通常の標準治療を受けた。患者は、フルボキサミン(Luvox、アボット社)を1回100mg、1日2回、10日間投与する群と、プラセボを投与する群に無作為に割り付けられ、無作為化後(1日目)に開始された。研究担当者は参加者にウェルカムビデオを提供し、試験、試験薬、有害事象、フォローアップ手順などの情報を提供した。公衆衛生施設で通常の治療を行う臨床医は、通常、症状の管理に重点を置き、細菌性肺炎が疑われる場合にのみ解熱剤を投与したり、抗生物質を勧めたりする。
試験担当者は、1日目、2日目、3日目、4日目、5日目、7日目、10日目、14日目、28日目に、直接、または電話連絡やビデオ会議を利用したソーシャルメディア・アプリケーションを介して、転帰データを収集した。参加者が治験薬を服用しているかどうかにかかわらず、結果データを収集した。有害事象が発生した場合には、臨床治療以外の予定外の訪問(治療期間中)がいつでも発生する可能性があった。
SARS-CoV-2の伝染性の特徴と、陽性者の隔離が推奨されていることを考慮し、バイタルサインデータはほとんど収集しなかった。心臓の安全性は、ベースライン時に6誘導心電図(Kardiamobile, Mountain View, CA, USA)を用いて評価した。記録されたデジタルデータは、識別不可能な状態で中央施設(Cardresearch, Belo Horizonte, Brazil)に転送され、読影された。酸素状態は、非侵襲的に動脈酸素飽和度と脈拍を測定するパルスオキシメーター(Jumper Medical Equipment, Shenzhen, China)を用いて評価し、体温は、研究員が管理する標準的なデジタル口腔体温計を用いて評価した。鼻咽頭スワブまたは喀痰唾液を採取するために、中途半端な大きさの鼻咽頭スワブキットと無菌の受取人保管庫が提供された。PCR検査のための鼻腔スワブは、試験に登録した参加者の第1/4が3日目と7日目に完了した。ウイルスクリアランスを評価し、有効成分による抗ウイルス効果の有無を確認した。
すべての重篤な有害事象および非重篤な有害事象は、現地の規制要件に従って試験担当者に報告された。報告可能な有害事象には、重篤な有害事象、治験薬の投与中止に至った有害事象、および治験薬に関連する可能性があると評価された有害事象が含まれた。
試験結果
主要評価項目は、無作為化後28日以内にCOVID-19関連疾患による医療機関への入院(COVID-19による救急搬送で参加者が6時間以上観察された場合、またはCOVID-19の進行によりさらなる入院を余儀なくされた場合)という複合エンドポイントであった。通常であれば入院するはずの患者の多くが、ピーク時には病院のキャパシティオーバーのために入院できなかったため、複合エンドポイントでは、入院と、入院の代理であるCOVID-19救急病院での待機の両方を取り上げた。ブラジルのこの地域では、50~80床のユニットが、数日間の滞在、酸素吸入、人工呼吸などのサービスを提供し、救急現場で病院のようなサービスを実施していた。6時間という基準値は、臨床医が観察を推奨する時間のみを指し、待ち時間は含まれていない。主要な副次評価項目は、ウイルスクリアランス、臨床的改善までの時間、呼吸器症状のある日数、何らかの原因による入院またはCOVID-19の進行による入院までの時間、全死亡および何らかの原因による死亡までの時間、WHOの臨床的悪化スケールスコア、入院および人工呼吸器の使用日数、有害事象、試験薬による副作用、試験薬の非アドヒアランスの参加者の割合などである。すべての副次的成果は、無作為化後28日までに評価された。
統計解析
Adaptive Design Protocol」および「Master Statistical Analysis Plan」には、サンプルサイズの計算および統計解析の詳細が記載されている9。本試験は適応性があり、サンプルサイズの再評価手法を適用する。各群の計画を立てるために、対照イベント率を15%と仮定し、プラセボとのペアワイズ比較で0-05の両側タイプ1エラーで80%の検出力を達成するために、臨床的有用性を37-5%(相対的リスク減少)以上と仮定した。この結果、当初の計画では、各群681名の被験者を募集することになった。統計チームは、計画された中間解析を行った。中間解析時に優越性の事後確率が40%未満であった場合、無益性の停止閾値を設定した。優越性の事後確率が97-6%の閾値を満たした場合、そのアームは優越性のために停止することができた。
ベースラインの特徴は、連続変数の場合、数(%)または中央値とIQRで報告されている。主要評価項目の解析にはベイジアンフレームワークを適用し、すべての感度解析および副次評価項目にはフリークエンティストアプローチを適用した。ベイズ解析では、試験終了時の治療効果の事後確率を、試験中の意思決定とは無関係に報告することができる。主要アウトカムに対するフルボキサミンの事後有効性は、統計解析計画の付録に詳細が記載されているように、イベント発生率のベータ二項モデルを用いて算出される12。プラセボとフルボキサミンの両方の観察データに基づいて、インフォームド・プライヤーを仮定し、intention-to-treatおよびper-protocol分析(可能な投与量の80%以上を服用すると定義)を行った。修正intention to treat(mITT)とは、主要な結果が出る前に少なくとも24時間治療を受けていることと定義した。イベント発生率の時間的変化については、同時無作為化集団のみを用いて説明した。事前に計画した統計解析計画に従って、サブグループ効果を評価した。治療に必要な数を算出した。
副次評価項目は,事前に設定した頻度論的アプローチで評価した。ウイルスクリアランスについては、無作為化から3日目と7日目に報告された患者の2つの結果(COVID-19陽性-陰性)について、治療法と時間の交互作用項を設定し、被験者のランダム効果を加えた縦断的な混合効果ロジスティック回帰モデルを適用した。時間-イベント間の転帰は Cox 比例ハザードモデルを用いて評価し,二値転帰はロジスティック回帰を用いて評価した。モデルの仮定は、比例性の検定により評価した。サブグループ解析を行い、相互作用のp値を報告した。Per-protocol解析は、結果の頑健性を評価するための感度解析とした。統計解析計画に従い、査読者から要求があった場合にはポストホック解析を行った。すべての解析は,Rバージョン4.0.3を用いて行った。統計解析計画の詳細は,Open Science FrameworkのData Sectionに掲載されている。
本試験では、データおよび安全性モニタリング委員会が独立した監督を行った。我々は 2021年8月2日までのデータに基づいて、fluvoxamine群の4回目の最終中間解析を計画した。ここでは 2021年9月9日までの全患者の追跡調査を紹介する。本試験は、ClinicalTrials.govに登録されている(NCT04727424)。
資金提供者の役割
本試験の資金提供者は,試験デザイン,データ収集,データ解析,データ解釈,報告書の執筆,および出版物への投稿の決定に一切関与していない。実行委員会は、データの完全性およびデータ解析の正確性に責任を負う。試験執行委員会は、試験の実施、完全性、データの正確性および試験実施のプロトコルへの準拠のすべての側面を監督し、委員会はデータの正確性と完全性およびプロトコルへの忠実性を保証した。
結果
現在までに9803人の潜在的な参加者をスクリーニングし、本試験に参加させた。TOGETHER試験では 2020年6月2日に最初の被験者が登録され 2021年1月20日にFluvoxamine群への登録が開始された。本試験は進行中であるため、ここでは、フルボキサミンおよび同時に投与される対照薬に無作為に割り付けられた被験者のみの記述的要約を提供する。2021年8月5日までに、募集した1497人の参加者がフルボキサミン(n=741)またはプラセボ(n=756)に無作為に割り付けられ、1826人がその他の治療群に無作為に割り付けられた(図1)。ここでは 2021年9月9日時点で28日間の追跡調査を終えた全患者のデータを紹介する。年齢の中央値は50歳(範囲18~102歳)で、862人(58%)が女性であった(表1)。ほとんどの参加者は、混合人種と自認しており、1428人(95%)白人が12人(1%)黒人またはアフリカ系と自認している人が10人(1%)残りは不明47人(3%)であった。年齢、肥満度、併存疾患などの共変量に関しては、各グループは概ねバランスが取れていた(表1)。無作為化前の症状の平均日数は3~8日(SD 1~87)であった。
図1試験の概要
表1TOGETHER試験の治療割り付けによる患者の特徴
| Fluvoxamine (n=741) | プラセボ(n = 756) | |
|---|---|---|
| セックス | ||
| 女性 | 409 (55%) | 453 (60%) |
| 男 | 332 (45%) | 303 (40%) |
| 人種 | ||
| 混血 | 709 (96%) | 719 (95%) |
| 白い | 6 (1%) | 6 (1%) |
| 黒人またはアフリカ系アメリカ人 | 5 (1%) | 5 (1%) |
| わからない | 21 (3%) | 26 (3%) |
| 年齢、年 | ||
| <50 | 379 (51%) | 368 (49%) |
| ≥50 | 327 (44%) | 328 (43%) |
| 詳細不明 | 46 (6%) | 49 (6%) |
| 年齢記述統計 | ||
| 中央値(IQR) | 50 (39–56) | 49 (38–56) |
| ボディ・マス・インデックス | ||
| <30 kg / m2 | 355 (48%) | 373 (49%) |
| ≥30kg/ m2 | 376 (51%) | 375 (50%) |
| 詳細不明 | 10 (1%) | 8 (1%) |
| 症状の発現からの時間、日数 | ||
| 0–3 | 328 (44%) | 310 (41%) |
| 4–7 | 239 (32%) | 267 (35%) |
| 詳細不明 | 174 (23%) | 179 (24%) |
| 危険因子 | ||
| 慢性心臓病 | 9 (1%) | 7 (1%) |
| 制御されていない高血圧 | 106 (14%) | 88 (12%) |
| 慢性肺疾患 | 6 (1%) | 3(<1%) |
| 喘息 | 12 (2%) | 16 (2%) |
| 慢性腎臓病 | 2(<1%) | 2(<1%) |
| リウマチ性障害 | 1(<1%) | 0 |
| 慢性神経障害 | 8 (1%) | 6 (1%) |
| 1型糖尿病 | 25 (3%) | 22 (3%) |
| 2型糖尿病 | 104 (14%) | 92 (12%) |
| 自己免疫疾患 | 0 | 2(<1%) |
| その他の危険因子または併存疾患 | 25 (3%) | 24 (3%) |
データはn(%)である。
* 祖先が混血であると自認している人。
すべての患者がCOVID-19の救急医療機関を経由して治療を受けた。COVID-19救急医療機関とのやり取りがあった患者は、フルボキサミン群で180名、プラセボ群で251名であった。COVID-19の救急施設を訪れたことがあるかどうかの相対リスク(RR)は,0-73(95%CI 0-62-0-88)であった。
フルボキサミン群では79人(11%)が主要評価項目のイベントを経験したのに対し、プラセボ群では119人(16%)が経験した(表2)。ほとんどのイベント(87%)は入院であった。ベイズのβ二項モデルに基づき、ITT集団ではRR 0-68,95%ベイズ信頼区間(BCI)0-52-0-88,修正ITT集団ではRR 0-69,95%BCI 0-53-0-90と定義された入院という複合主要評価項目を減少させるフルボキサミンの有益性が示された(図2A)。治療に必要な数は20人だった。Per-protocol解析では、より大きな治療効果(0-34,95%BCI 0-21-0-54)が示された。イベント発生率がプラセボと比較してフルボキサミン群で低くなる確率は、ITT集団では99-8%、mITT集団では99-7%であった(図2A、B)。2021年8月5日に開催されたDSMCでは、この比較が主要評価項目について事前に規定された優越性基準を満たしていたため(事前に規定された優越性基準値97-6%)TOGETHER試験で患者をフルボキサミン群に無作為に割り付けることを中止するよう勧告した。
表2フルボキサミン群とプラセボ群に割り付けられた患者の主要評価項目の割合と、COVID-19による救急搬送またはCOVID-19による三次病院への転院と定義される入院の相対リスク
| 治療意図分析 | 修正されたITT解析 | |||||
|---|---|---|---|---|---|---|
| N | n (%) | 相対リスク(95%BCI) | N | n (%) | 相対リスク(95%BCI) | |
| フルボキサミン | 741 | 79 (11%) | 0·68 (0·52–0·88) | 740 | 78 (11%) | 0·69 (0·53–0·90) |
| プラセボ | 756 | 119 (16%) | 1(参照) | 752 | 115 (15%) | 1(参照) |
BCI=Bayesian credible interval.
図2フルボキサミンとプラセボを比較した場合の有効性の確率とCOVID-19による救急施設への留置またはCOVID-19による三次病院への転院と定義した入院のベイズ相対リスク
副次評価項目の分析結果を表3に示す。7日目のウイルスクリアランス(p=0-090)COVIDによる入院(p=0-10)全原因による入院(p=0-09)入院までの期間(p=0-11)については、フルボキサミンとプラセボの間に有意差はなかった。入院日数(p=0-06)、死亡率(p=0-24)、死亡までの期間(p=0-49)、人工呼吸日数(p=0-90)、回復までの期間(p=0-79)、またはPROMIS Global Physical Scale(p=0-55)やMental Scale(p=0-32。付録2 p8)。)
表3TOGETHER試験におけるフルボキサミンとプラセボの二次的転帰
| フルボキサミン | プラセボ | 推定治療効果(95%CI) | p値 | ||
|---|---|---|---|---|---|
| ウイルスクリアランス(7日目) | 40/207 (19%) | 58/221 (26%) | 0·67 (0·42–1·06) | 0·090 | |
| COVIDで入院 | 75/741 (10%) | 97/756 (13%) | 0·77 (0·55–1·05) | 0·10 | |
| すべての原因による入院 | 76/741 (10%) | 99/756 (13%) | 0·76 (0·58–1·04) | 0·088 | |
| 入院までの時間、日数 | 5 (3–7) | 5 (3–7·5) | 0·79 (0·58–1·06) | 0·11 | |
| 入院期間、日数 | 8 (5–13) | 6 (3–10·75) | 1·23 (0·99–1·53) | 0·059 | |
| 少なくとも6時間の緊急設定訪問 | 7/741 (1%) | 36/756 (5%) | 0·19 (0·08–0·41) | 0·0001 | |
| 少なくとも6時間の緊急訪問までの時間 | 4 (3–7) | 5 (3–8·25) | 0·20 (0·09–0·44) | 0·002 | |
| 死、治療意図 | 17/741 (2%) | 25/756 (3%) | 0·69 (0·36–1·27) | 0·24 | |
| 死ぬまでの時間、日 | 17 (9–21) | 14 (8–20) | 0·80 (0·43–1·51) | 0·49 | |
| 機械的換気 | 26 | 34 | 0·77 (0·45–1·30) | 0·33 | |
| 人工呼吸器の使用時間、日数 | 5·5 (3–12·75) | 6·5 (2·25–12) | 1·03 (0·64–1·67) | 0·90 | |
| アドヒアランス | 548/741 (74%) | 618/738 (82%) | 0·62 (0·48–0·47) | 0·0003 | |
| プロトコルごとの死 | 1/548(<1%) | 12/618 (2%) | 0·09 (0·01–0·47) | 0·022 | |
| 緊急の有害事象の治療 | |||||
| グレード1 | 20/741 (3%) | 11/756 (1%) | 1·88 (0·91–4·09) | 0·096 | |
| グレード2 | 72/741 (10%) | 81/756 (11%) | 0·91 (0·64–1·25) | 0·52 | |
| グレード3 | 38/741 (5%) | 50/756 (7%) | 0·76 (0·49–1·18) | 0·22 | |
| グレード4 | 21/741 (3%) | 20/756 (3%) | 1·07 (0·58–2·01) | 0·82 | |
| グレード5 | 18/741 (2%) | 26/756 (3%) | 0·70 (0·37–1·28) | 0·25 | |
データは特に記載のない限り、n/N(%)または中央値(IQR)。
* 未調整のオッズ比。
† 未調整のハザード比。
‡ 対数変換された線形回帰からの指数化された未調整の推定値。
忍容性の問題により、84名がフルボキサミンを中止し、64名がプラセボを中止した。最適なアドヒアランス(可能な限りの日数で80%以上)を報告した患者を対象としたper-protocol調査の結果、主要アウトカムについてはRR 0-34,95%BCI 0-21-0-54,死亡率についてはオッズ比0-09,95%CI 0-01-0-47で、有意な治療効果が示された。有害事象については、フルボキサミン群とプラセボ群の患者の間で、治療上の緊急性のある有害事象の発生数に有意差はなかった。
事前に規定されたサブグループ解析では、年齢、性別、症状発現からの日数、喫煙状況、併存疾患のサブグループにおいて、フルボキサミンの治療効果がプラセボと比較して軽減されているという証拠は認められなかった(図3,付録2 p9)。
図3TOGETHER試験におけるフルボキサミン対プラセボのサブグループ解析結果
考察
本試験は、我々の知る限り、COVID-19の急性期治療に対するフルボキサミンの有効性を検証した初めての大規模無作為化比較試験である。フルボキサミンを10日間投与した結果、COVID-19による救急搬送、またはCOVID-19による三次病院への転院と定義される入院という主要アウトカムにおいて、臨床的に重要な絶対リスクが5-0%減少し、RRが32%減少したことがわかった。今回の結果は、DSMCがフルボキサミンの投与中止を勧告した後、無作為に割り付けられた患者の28日間の追跡調査を行って得られた試験の完全な解析結果である。フルボキサミンの安全性、忍容性、使いやすさ、低価格、広く普及していることを考えると、今回の結果は、COVID-19の臨床管理に関する国内および国際的なガイドラインに影響を与える可能性がある。
この試験では、高用量のフルボキサミン(100mgを1日3回、15日間)を使用し、主要評価項目のリスクが低いグループを対象としたが、フルボキサミンを投与された80名の患者では臨床的悪化が認められなかったのに対し、プラセボを投与された72名の患者では6例が認められた。フランスで行われた大規模な観察研究では、COVID-19の入院患者7230人という異なる集団を対象とし、SSRIの使用により挿管の使用や死亡が減少したことが報告されている5。
COVID-19感染症に対するフルボキサミンの根本的なメカニズムはまだ不明である。S1Rは、小胞体シャペロン膜タンパク質であり、小胞体ストレス応答-折りたたまれていないタンパク質応答の制御や、炎症誘発物質に反応してサイトカイン産生を制御するなど、多くの細胞機能15に関与している14。フルボキサミンの存在下で、S1Rは、ERストレスセンサーであるイノシトール要求性酵素1αが、インターロイキンIL-6,IL-8,IL-1β、IL-12などのサイトカイン産生の重要な調節因子であるX-boxタンパク質1のmRNAをスプライシングして活性化するのを阻止する可能性があるという。Rosenらによる2019年の研究では、前臨床モデルの炎症や敗血症に対して、このメカニズムでfluvoxamineが有効であることが示された16。
2つ目のメカニズムは、フルボキサミンの抗血小板作用かもしれない17。SSRIは、血小板へのセロトニンの負荷を防ぎ、血小板の活性化を抑制することで、血栓症のリスクを低減する可能性があり、これらの抗血小板作用は心保護作用を示す可能性がある。最後に、もう一つの作用機序として、フルボキサミンが血漿中のメラトニン濃度を上昇させることと関連している可能性がある16。最も可能性の高い機序を明らかにするためには、試験管内試験および動物実験が必要である。今後、無作為化比較試験の一環としてバイオマーカー試験を実施することも、メカニズムの解明に役立つかもしれない。
COVID-19のパンデミックが始まって以来、ClinicalTrials.govには2800件以上の無作為化比較試験が登録されている。しかし、報告されているのは300件に満たず、ほとんどの臨床試験はサンプルサイズが100未満と小規模で、検出力も不十分であった。多くの場合、これらの臨床試験は、地域のパンデミックが波状的に発生し、スタッフを維持するための持続可能なインフラや、リクルートのための地元の関心が不足しているため、リクルートに失敗している。最も明確な医学的理解を得られる試験は、SOLIDARITY17,RECOVERY18,PRINCIPLE11,REMAP-CAP19などの大規模なプラットフォーム試験である傾向がある。そのため、我々は、介入方法が重複する試験を実施している他の研究者と積極的に協力し、我々の試験の決定を認識して、それぞれの試験に影響を与えるべきかどうかを判断できるようにしている。
我々の試験の強みは、重度のCOVID-19を発症するリスクの高い患者を迅速に募集し、登録したことである。本試験の募集戦略では、地域の公衆衛生システムと連携しているため、1日あたり20人以上の患者を頻繁に募集することができた。市販のCOVID-19迅速抗原検査(Panbio, Abbott Rapid Diagnostics Jena, Jena, Germany)を用いて、COVID-19と診断され、症状が出てから7日以内の患者のみを登録した。COVID-19陽性検査とRT-PCRとの一致性を、PCR評価を行った参加者群で評価したところ、ベースラインで採取した両検査の一致率は99%以上であった。この試験では、COVID-19検査が陽性でない被験者や、無症候性SARS-CoV-2陽性の被験者は登録しなかった。主要評価項目は「入院」で、COVID-19による救急搬送を6時間以上継続した場合と、COVID-19が原因で三次病院に転院した場合とで定義した。イベント裁定委員会は、患者の待ち時間を主要評価項目に寄与するものとみなした。ブラジルでのパンデミックに対応するために、専門的な救急医療施設が開発されたが、通常であれば入院するはずの患者の多くが病院のキャパシティオーバーのために入院できなかったため、これらの施設での長期にわたる観察と治療は入院と同等の重要性を持つと考えた。本試験では、主要評価項目の87%が最終的に三次病院への転院となった。また、救急現場と病院の両方で観察された患者は1回のみカウントされた。サブグループ解析では、あらかじめ決められた人口グループを調べ、相互作用の検定では、どのサブグループでも異なる効果は検出されなかった。また、女性はフルボキサミンを、男性はフルボキサミンを好む有意なサブグループとして同定されたが、両グループ間で効果の違いは検出されなかった。
COVID-19の疫学、疾患の進行および転帰に関する我々の理解は 2020年6月にこのプラットフォーム試験を開始して以来、発展してきた。初期の試験では、ウイルス量とクリアランスに対する介入の効果を評価していたが、後期の試験では、より臨床的なアウトカムも評価している。我々は、事前に規定されたルールに従い、適切な倫理審査委員会とコミュニケーションを取りながら試験の調整を行ったことで、高い採用率を維持しながらパンデミックの波に対応することができた。多くの外来患者を対象とした臨床試験とは異なり、本試験では、医学生、看護師、医師による訪問診療や通信によるフォローアップなど、患者との直接的な接触が行われた。また、COVID-19による救急外来や入院の発生率が高いことに加え、患者の募集が迅速に行われたため、予定していた集団の一部が集まった時点で介入の効果を評価することができた。フルボキサミンを投与した患者が最初にリクルートされてから、本試験の最終データカットまでの期間は219日であった。
我々の試験の主な限界は、十分に特徴づけられていない疾患で試験を行うことの難しさに関連している。COVID-19の早期治療のための標準治療は存在せず、様々な支持団体が、本試験や我々の過去の試験で評価されたものを含め、様々な介入を推進している20。さらに、多くの危険因子を持つ患者が早期に回復するのに対し、確立された危険因子を持たない患者は回復しないことから、この疾患による疾患進行の最大のリスクが誰にあるのか、ほとんど理解されていない。我々の集団は、ほとんどの臨床試験で観察されたものよりも入院イベントの割合が高く20,そのため、このような高リスクの集団における治療効果を推論することができる。per-protocol解析よりもintention-to-treat解析の方がより現実的なエビデンスが得られるが、最適なアドヒアランスを報告した患者(可能な日数で80%以上-per-protocol解析)の方が治療効果が高いことがわかった。これは、治療へのアドヒアランスを強化することで、かなりの臨床効果が得られることを示唆している。しかし、アドヒアランスは忍容性と関係があるかもしれない。この理由で、フルボキサミン群では84人、プラセボ群では64人が中止した。最後に、試験開始時にはブラジルではワクチンが入手できなかったが、試験が進むにつれて広く入手できるようになった。試験中に対象基準を変更してワクチン接種を許可したが、試験終了時にCOVID-19ワクチンを1回以上接種したと報告したのは1497人中86人(6%)のみであったため、主要評価項目への影響は最小限であったと考えている。
本試験では、安価な既存薬であるフルボキサミンが、この高リスク集団における進行性疾患治療の必要性を低減することがわかった。フルボキサミンの10日間の投与は、資源の豊富な環境であっても約4米ドルである。本試験は、外来治療のためのモノクローナル抗体を含む、より高価な治療法の治療効果と比較して良好な結果が得られた20, 22, 23。フルボキサミンに関連する重篤な有害事象の絶対数はプラセボよりも少なく、これはフルボキサミンがこれらの参加者の全身の炎症を調節する効果を反映していると考えられる。下気道感染症は、プラセボ群に比べてフルボキサミン群の方が報告頻度が低かった。これは、フルボキサミンを投与されたCOVID-19が確認された患者の入院数が減少したことや、人工呼吸を必要とする患者の数が減少したことと一致している。
フルボキサミンは広く市販されているが、WHOの必須医薬品リストには掲載されていない24が、近縁のSSRIであるフルオキセチンはリストに掲載されている。現在、クラス効果が存在するかどうか、また、COVID-19に対してこれらの薬剤を互換的に使用できるかどうかを確立することが重要だ。今回の試験と類似した集団において、吸入ブデソニドが回復までの時間を短縮し11,入院を減少させる傾向があるという重要な知見が得られたことから、外来診療における代替または追加の介入として評価されるべきであると考えられる。PRINCIPLE試験では、ブデソニドに無作為に割り付けた後、28日目までの自己申告による回復までの期間を評価している11。我々の試験では、WHOによる疾患障害の分類の改善を14日目までと28日目までに評価した点が異なる(付録2 p7)。また、本試験では、主にワクチン接種を受けていない患者を対象としている。ワクチン接種者におけるフルボキサミンの効果を確立するためには、治療効果に関するさらなるエビデンスが必要である。
病気の進行や入院を防ぐために、フルボキサミンを含む介入策を使用することは、高リスクの人を特定することに決定的に依存している。選別されていない集団はリスクが低くなる。患者が治療を選択する動機となる臨床的悪化のリスクの絶対的な減少はどの程度なのか(おそらく我々が観察した約5%ではなく、もっと低い可能性もある)はまだ不明である。以上のことから、COVID-19感染の初期段階にある患者の悪化を予測する有効なルールを開発することの重要性が示唆された。