
コンテンツ
A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6491704/
要旨
毎年、米国だけで約170万人が外傷性脳損傷(外傷性脳損傷)を受けている。これらの頭部外傷に関連して、イライラ、抑うつ、不安などの精神神経症状が多くみられる。
ミクログリアによる神経炎症は、外傷性脳損傷後に神経精神障害を悪化させたり、原因となったりすることがある。例えば、複数の免疫学的挑戦を受けると、ミクログリアが「プライミングされている」か、または過剰な炎症性の表現型を持った過剰反応性になることが、多くの証拠によって証明されている。ミクログリアのプライミングは実験的な外傷性脳損傷後に起こり、認知機能障害だけでなく、抑うつ症状に似た行動の出現と相関している。
臨界的には、免疫課題は様々であり、病気、加齢、ストレスなどが含まれる。これらの免疫課題のあらゆる組み合わせの影響が、神経免疫環境と外傷性脳損傷への反応を形成している。例えば、ストレスは確実に炎症を誘発するため、外傷性脳損傷後の神経病理学的変化や行動の低下への入り口となる可能性がある。外傷性脳損傷後のストレス関連精神疾患の発生率が増加していることを考えると、ストレスが転帰にどの程度影響を与えるかは特に関心が高い。
このレビューは、外傷性脳損傷後のストレス-免疫経路のコミュニケーションの重要なメディエーターとしての視床下部-下垂体-副腎(HPA)軸の役割を強調することを目的としている。我々はまず、外傷性脳損傷後の不適応な神経炎症について説明し、ストレスが抗炎症メカニズムとプロ炎症メカニズムの両方を介してどのように炎症に寄与しているかを説明する。外傷性脳損傷後のHPA軸の機能不全とストレス反応の変化の結果を記述した臨床的・実験的データについて議論する。
最後に、外傷性脳損傷後に使用される一般的なストレスモデルをレビューし、HPA軸機能障害と外傷性脳損傷後の不適応性炎症との関係をよりよく解明することができるだろう。これらの研究を合わせると、脳損傷後のHPA軸機能障害が一般的であり、脳損傷に対する神経炎症反応のダイナミックな性質に寄与していることが示唆される。
HPA軸に直接関与する実験的ストレス因子は、外傷性脳損傷後の転帰を媒介するストレス-免疫経路の役割をより明確にするために、今後の研究にとって重要な領域である。
キーワード
外傷性脳損傷、精神疾患、神経炎症、ミクログリア、ストレス、HPA軸、グルココルチコイド
序論
米国だけでも530万人以上の人が外傷性脳損傷(外傷性脳損傷)に関連した障害に苦しんでいる(1)。外傷性脳損傷後の障害で最も多く報告されているのは、うつ病、不安、気分障害などの精神疾患の発症または悪化であり、その結果、生活の質が全体的に低下し、長期的な死亡率が増加する可能性がある(2, 3)。例えば、ある研究では、精神疾患を患っている患者のほぼ半数が外傷性脳損傷後に症状を経験するようになったと報告されている(4,5)。外傷性脳損傷後の精神疾患の発症や悪化の主な原因は、不適応な炎症とミクログリアのプライミングである(6,7)。ストレスに対する反応の変化は、心的外傷後の回復を著しく阻害し、脳の炎症状態を悪化させる可能性があることを示唆する証拠が増えてきており、これは外傷性脳損傷後の長期的な悪質な結果を永続させる可能性がある。
大まかに言えば、ストレスとは、積極的に恒常性を維持するために環境の変化に体がどのように反応するかということである(8)。アロスタシスとは、変化によってホメオスタシスが維持されるプロセスである。アロスタシスは、活性化されるとストレスホルモンを放出する視床下部-下垂体-副腎(HPA)軸の協調的な活性化と調節によって達成される。主なストレスホルモンは、ヒトではコルチゾールであり、げっ歯類ではコルチコステロンである。コルチゾール(コルチコステロン) は、体内のストレス応答を調節し、恒常性バランスを維持する他のプロセス、特に炎症と睡眠を制御するように作用する。ストレス性刺激および非ストレス性刺激に対するコルチゾール(コルチコステロン)反応の調節障害は、健康全般および外傷性外傷からの回復に広範な影響を及ぼす可能性がある。
臨床データによると、外傷性脳損傷後のHPA軸の活性化抑制による副腎不全は、外傷性脳損傷症例の4分の1で発生していることが示されている(9)。これらの臨床研究は、外傷性脳損傷が神経内分泌機能のベースラインの変化を誘発することを示しているが、これらの変化がストレス刺激に対する傷害後の反応やストレス刺激からの回復、すなわちアロスタシスに影響を与えるかどうかについての洞察は得られていない。外傷性脳損傷後のHPA軸の機能不全は、ストレスに対する不適切な反応を引き起こし、炎症を調節することができなくなる。どちらのプロセスも外傷性脳損傷後の精神疾患の発症に関与しているが、脳損傷後のストレス-免疫経路間のクロストークについては、前臨床研究と臨床研究の両方でまだ十分に解明されていない。
本レビューでは、外傷性脳損傷後の神経炎症の概要を報告するとともに、脳損傷後のストレス-免疫経路間の相互関係について議論する。次に、外傷性脳損傷後のHPA軸機能障害の臨床的発生率と、実験的外傷性脳損傷モデルでも同様にストレス反応の変化が報告されていることを論じる。最後に、外傷性脳損傷後のHPA軸機能障害を特徴づけるための実験的アプローチについて述べる。神経内分泌機能障害と免疫異常を結びつける分子メカニズムを解明するストレスと外傷性脳損傷の組み合わせモデルの価値に重点を置く。
外傷性脳損傷後の神経炎症
外傷性脳損傷は一次損傷と二次損傷の2つの段階で発生する。
一次損傷
一次損傷は、損傷自体の機械的な力によって引き起こされ、軸索の剪断、出血、挫傷などがある(10)。一次損傷は、軽度、中等度、重度と呼ばれる重症度が様々である。外傷性脳損傷には多くのタイプがあり、大まかにはびまん性外傷性脳損傷と限局性外傷性脳損傷に分類される。
びまん性外傷性脳損傷の例としては、広範囲に損傷はあるが明確な病変を形成しない爆風傷害や低酸素性虚血性傷害があり、焦点性外傷性脳損傷には頭蓋骨骨折や銃弾や榴散弾などの異物による病変が含まれる。びまん性外傷性脳損傷と限局性外傷性脳損傷の両方を表現するために、多くの異なる実験的外傷性脳損傷動物モデルが存在する(11)。びまん性外傷性脳損傷の一般的なモデルは流体打撲損傷(外側液打損傷(FPI))であり、これは脳の無傷の硬膜に流体パルスを印加することで達成される(12)。制御皮質衝撃(Controlled cortical impact:制御皮質衝撃)は、脳に直接ピストンを当てて挫傷を生じさせる局所性外傷性脳損傷モデルである(13)。予防策を開発し、さまざまな形態の一次損傷の影響を軽減するために、このようなさまざまなタイプの損傷モデルを使用することは、外傷性脳損傷研究にとって極めて重要である。
二次損傷
二次損傷は外傷性脳損傷によって間接的に引き起こされ、浮腫や血流の変化など、外傷によって開始されたプロセスが長期化することで生じる。さらに、二次損傷は、ミトコンドリア機能障害(14)酸化的損傷(15)神経炎症(16)などの分子過程を介した神経細胞の損傷と変性を構成しており、外傷性脳損傷後の薬理学的介入の対象となる可能性がある。神経炎症は、傷害後の神経変性疾患の発症に関与していることから、二次傷害では特に注目されている(17, 18)。以前に検討したように、多くの細胞型が外傷性脳損傷後の神経炎症反応に寄与している(19, 20)が、前臨床研究および臨床研究の多くは、中枢神経系(中枢神経系)の自然免疫細胞であるミクログリアの役割に焦点を当てている。ミクログリアが介在する炎症は、運動障害(21)、気分障害(22)、神経変性(23)など、外傷性脳損傷後の多くの症状と関連している。したがって、ミクログリアは、外傷性脳損傷後の二次障害の一部として神経炎症の悪影響を改善するための潜在的な治療標的となる可能性がある。
外傷性脳損傷後のミクログリアが介在する神経炎症は動的である
ミクログリアは通常、小さな細胞体と高度に隆起したプロセスを特徴とする静穏な調査状態にある。外傷性脳損傷後の急性期には、多数の損傷シグナルやIFN-γや損傷細胞からの核タンパク質などのサイトカインがミクログリアに関与して炎症反応を誘発し、亜急性期および慢性期の時点まで持続することができる。中枢神経系の自然免疫細胞として、ミクログリアは、外傷性脳損傷などの堅牢な炎症性刺激に対する保存された応答を持っている。例えば、ラットで実験的に正中線外側液打損傷(FPI)を行った後、Iba-1ラベリングを行うと、ミクログリアは皮質と視床で肥大化して脱アミ化し、外傷性脳損傷後7日と28日まで持続することが示されている。抗原提示型MHCIIは、これらの脱アミノ化ミクログリアで高度に発現している。これらの形態的に異なるミクログリアはまた、CD68の免疫蛍光発現も増加しており、これは、損傷した細胞や死細胞から破片を除去し、びまん性および局所性の実験的外傷性脳損傷後の治癒を促進するためのファゴサイトーシス活性が増加していることを示している(25, 26)。前臨床研究と臨床研究の両方で、これらの脱アミノ化したMHCII+/CD68+ミクログリアを「反応性」として特徴づけ、この炎症促進状態が外傷性脳損傷に対する迅速かつ強固な神経炎症反応を媒介することを明らかにしている。反応性ミクログリアは、IL-1,IL-6,TNF-αなどの炎症性サイトカインを放出し、ミクログリア媒介の炎症を永続させ、他の免疫細胞を勧誘する(27, 28)。マウスで実験的に正中線外側液打損傷(FPI)を行った後、MhcIIおよびCd68 mRNAの発現は傷害後3日目に増加し、外傷性脳損傷後1週間目には上昇したままである(29)。反応性ミクログリアは、マウスの正中線外側液打損傷(FPI)後の炎症性遺伝子発現の時間的変化を含む、皮質炎症の明瞭なプロファイルを促進する(29)。例えば、Ccl7およびIl-1βを含む炎症性ケモカインの遺伝子発現は、外傷性脳損傷後8時間で特異的に制御されている一方で、炎症促進シグナル伝達の永続化に関与するtoll-like receptor(TLR)遺伝子は、外傷性脳損傷後7日目に特異的に制御されている(29)。受傷後8時間後の特異的なミクログリアプロファイルは、一次傷害に応答してミクログリア反応性が急速に増加していることを示しており、一方、外傷性脳損傷後7日後のミクログリアプロファイルは、二次傷害の持続性を特徴づけている。
実験的証拠は、この微小グリア反応性が慢性的に持続することを示しており、炎症が外傷性脳損傷の長期的な結果に寄与するメカニズムを示唆している。マウスで中等度の制御皮質衝撃を行った後、反応性のミクログリアは傷害後1週間頃に炎症状態のピークに達するが、外傷性脳損傷後、特に傷害後52週目には、偽のコントロールと比較して、大脳皮質、大脳皮質、視床で反応性が慢性的に持続することが示されている(30)。さらに、この同じ研究では、慢性的なミクログリア反応性は、病変の大きさの増加によって測定される神経細胞の損失と相関していることが示されている。実験的に中等度の制御皮質衝撃を受けた6ヵ月後と1年後には、結果として生じる病変は大脳皮質と海馬を通って拡大し、同側半球の広範な神経変性を引き起こしている。これらのデータをまとめると、外傷性脳損傷に対する反応性ミクログリアの亜急性期および慢性期の反応は、神経炎症を永続させるだけでなく、慢性的な神経変性と損傷病変の拡大にも寄与していることが明らかになった。これらの所見は、傷害性の非貫通性外傷性脳損傷から得られた死後の患者組織サンプルの臨床分析と一致している。さらに、これらの反応性ミクログリアは、1回の外傷性脳損傷後も数十年にわたって慢性的に持続し、認知機能に悪影響を及ぼすことが臨床的に証明されている。ある臨床研究では、中等度から重度の外傷性脳損傷の生存者のPK結合の非侵襲的PET画像化により、年齢をマッチさせた対照者と比較して、脳内、特に視床部に拡散的に反応性の高いミクログリアが有意に高いことが示されている(32)。受傷後のタイムポイントは11ヶ月から17年までと広範囲にわたるが、各患者は慢性炎症が持続しており、それが認知障害と相関している。患者の処理速度と反応潜時は、年齢を一致させた対照群と比較して、外傷性脳損傷後は有意に遅く、これらの認知障害の重症度は視床微小グリア反応性の亢進と直接相関している。注目すべきは、微小グリア反応性の亢進は傷害部位から離れた場所で観察され、外傷性脳損傷の病変部周辺では観察されないことであり、二次傷害を介して広範囲に損傷を受けていることを示している。
反応性のあるミクログリアは、外傷性脳損傷直後に破片を除去し、シナプスを調整するために必要であるが、慢性的に反応性がある場合には神経毒性を示すことがある。これは、誘導性一酸化窒素合成酵素(iNOS)を介して酸化ストレスと一酸化窒素(NO)などの神経毒性サイトカインを永続させることによって起こる(33)。活性酸素種(ROS)であるNOX2は、マウスの制御皮質衝撃実験の1日後と2日後に大脳皮質で高発現し、形態的に反応性のあるミクログリアと共局在化する(34)。別の実験研究では、Nox2-/-マウスの制御皮質衝撃では、CD68標識が有意に減少し、病変部の面積が減少し、Il-1β、Il-6,Tnfαを含む炎症性サイトカインの遺伝子発現が減少することが明らかになった(35)。このことは、NOX2がミクログリア反応性の細胞毒性産物であるだけでなく、他のミクログリアと関与してミクログリア介在性炎症を永続させることができることを示している。致死的な外傷性脳損傷症例のヒト剖検サンプルの臨床分析でも、対照群と比較して大脳皮質全体でNOX2の発現が有意に増加していることが示されている(36)。一過性のミクログリア反応は傷害に対する適切な免疫応答に必要であるが、持続的な炎症は不適応であり、神経細胞の変性や細胞損失につながる可能性がある。
ミクログリアを実験的に操作することで、外傷性脳損傷後の炎症を制御する上で、これらの中枢神経系の自然免疫細胞がダイナミックな役割を果たしていることが明らかになった。例えば、反応性ミクログリアは、外傷性脳損傷後の炎症の治療標的となる可能性があるが、実験的外傷性脳損傷モデルの後の回復には、ミクログリアが介在する短期的な炎症が必要であることが研究で示されている。神経細胞のフラクタルカインを介してフラクタルカイン受容体(CX3CR1)を活性化すると、ミクログリアの反応性がネガティブに制御され、炎症性表現型が低下する。損傷後、損傷を受けたニューロンはフラクタルカインの放出を減少させ、結果としてミクログリアの反応性を増加させる。外傷性脳損傷後のCX3CR1の作用の時間経過をよりよく理解するために、Cx3cr1-/-マウスに実験的な制御皮質衝撃を投与し、傷害後4日目と5週間目に認知能力と感覚運動能力を試験した。Cx3cr1-/-マウスは、野生型コントロールと比較して、実験的制御皮質衝撃後4日目に知覚運動機能が改善され、Cd68発現が減少した(37)。このことは、外傷性脳損傷後の急性期にミクログリアが介在する炎症が、転帰の悪化を防いでいることを示している。しかし、驚くべきことに、外傷性脳損傷後5週間の時点で、Cx3cr1-/-マウスは野生型マウスと比較して、制御皮質衝撃後に感覚運動機能が低下し、神経細胞数の減少とミクログリア細胞の細胞体面積の増加によって示される病理学的変化が増加している。このように、ミクログリア反応性の亢進による短期的な炎症は外傷性脳損傷直後の回復に必要であるが、慢性的な時期には悪化する可能性がある。適切な長期的な回復には、内因性のミクログリアの制御、ここでは抗炎症性の神経細胞作用が必要である。
外傷性脳損傷後のミクログリア反応性の効果を特徴づけるための薬理学的操作のもう一つの方法は、コロニー刺激因子1受容体(脳脊髄液1R)の拮抗によるミクログリアの排除と再増殖である。脳脊髄液1Rアンタゴニストは、7日、14日、21日後にマウスの成体脳のミクログリア個体群を有意に減少させる(38)。脳脊髄液1Rアンタゴニストが停止すると、ミクログリアは治療後3日目に急速に再増殖を開始し、14日目にミクログリア集団の治療前のレベルに戻る。多くの研究では、反応性のあるミクログリアを休止状態のミクログリアに置き換える試みとして、脳脊髄液1R拮抗薬が使用されている。例えば、CaM/TetDTAマウスは、ドキシサイクリンを食事で与えないと、前脳興奮性および海馬CA1ニューロンでジフテリア毒素A鎖(DTA)を発現し、損傷および疾患を模倣したニューロン死の病変をもたらす(39)。この同じモデルでは、以前に、ミクログリアの除去後に回復する頑健なミクログリア反応性が報告されている(40)。CaM/TetDTAマウスが30日以上にわたって病変を発症すると、ドキシサイクリンを投与してDTAの発現を停止させ、7日間の回復後に脳脊髄液1RアンタゴニストPLX5622を14日間経口投与してミクログリアを除去し、その後21日間ミクログリアの再増殖を行った。これらの再増殖したミクログリアは、再増殖していない動物と比較して、より小さなソーマと隆起した突起で示されるように、形態学的に非反応性であるように見える(41)。さらに、再増殖後の病変動物では、再増殖のない病変動物に比べてCd68を含む炎症性マーカーの発現が有意に減少しており、神経細胞死後の反応性ミクログリア集団の救済が示された。RNAトランスクリプトNanoString解析による遺伝子発現は、IL-1およびiNOSシグナル伝達に関与する炎症性遺伝子の発現が減少し、反応性ミクログリアが減少していることをさらに示した。さらに、高濃度プラス迷路では、病変動物におけるミクログリアの再増殖が不安様行動をコントロールレベルまで回復させ、モリス水迷路では病変動物と比較して空間記憶が改善されていることが示された。このように、再増殖したミクログリアは反応性が低いように見え、神経細胞死後の行動の改善に寄与していることから、反応性ミクログリアが慢性炎症の治療標的となる可能性があることが示唆された。注目すべきは、アストロサイトを介した他のグリア媒介性炎症は、ミクログリアの再増殖の影響を受けず、炎症性反応と微小環境を永続させることである。このように、神経細胞の死によって引き起こされる炎症は、完全にミクログリアに依存しているわけではなく、ミクログリアが介在する炎症がない場合でも継続する可能性がある。外傷性脳損傷の文脈では、実験的ミッドライン外側液打損傷(FPI)の14日前にPLX5622を用いたミクログリア排除は、傷害後7日目にIL-1βおよびNfκbシグナリングを含むNanoString RNA分析を介した炎症性遺伝子発現の減少を再現している。興味深いことに、PLX5622を用いた外傷性脳損傷の前処理は、GFAP免疫蛍光発現の減少を通して決定されるように、アストロサイト関連の神経炎症を有意に減衰させる。このようにアストロサイトを介した炎症が減少しても、ミクログリアの除去は、神経細胞の生存を示す指標であるATF3+細胞数やNeuN+核の数を、傷害後7日目には有意に改善しなかった(29)。これらのデータを合わせると、ミクログリアの排除はミクログリアが介在する炎症にはしっかりと効果があるが、他のグリア細胞や神経細胞の死には有意な影響を与えないことが示された。最終的に、ミクログリアの操作は、傷害後の急性期および外傷性脳損傷からの回復期における炎症反応において、ミクログリアがダイナミックな役割を果たしていることを示している。
外傷性脳損傷後の後遺症に寄与するミクログリアのプライミングを誘導する
外傷性脳損傷後、反応性マイクログリア集団の一部は静穏な調査状態に戻るが、別の集団は中間的な表現型をとる。この集団は、MhcIIおよびCd68のベースライン発現が増加し、二次的な障害に反応して過剰反応性になる閾値が低くなることが特徴である(42)。二次的な挑戦に対する過反応性の炎症反応のために、この中間集団は「プライミングされた」ミクログリアと呼ばれている。ミクログリアのプライミングは、エンドトキシンであるリポ多糖類(LPS)の末梢注射後のプリオン病動物モデルME7で初めて報告された(43)。ME7動物における末梢性LPSチャレンジ後、IL-1β、TNF-α、およびIL-6を含む炎症性サイトカインは、LPSで処置された対照動物と比較して有意に上昇した。また、ME7動物におけるLPSは、対照群と比較して海馬におけるアポトーシス細胞の数を有意に増加させる。このことは、ミクログリアのプライミングによる神経炎症の悪化が、炎症の神経毒性効果を有意に増加させることを示している。特筆すべきことに、ME7動物の脳室に直接LPSを注入しても、末梢投与後に見られるような過剰反応性および強化されたアポトーシスプロファイルは得られなかった。このことは、プライミングされたミクログリア集団が末梢免疫の課題に敏感であることを示しており、したがって、感染による病気は、脳自体の直接的な感染を伴わずに、神経炎症を有意に増加させることができる。以前に検討したように、末梢免疫刺激に対するこの同じように高まった反応は、通常、加齢とともに見られる(44)。これには、IL-6およびIL-1βのようなプロ炎症性サイトカインのレベルの増加および抗炎症性サイトカインIL-4およびIL-10の減少が含まれる。老化したミクログリアは抗炎症機構による調節に抵抗性であり、抗原提示の有効性は加齢とともに低下し、その結果、MHCIIの発現が増加し、適応免疫から自然免疫へのシフトが生じる。これらのデータを合わせると、ミクログリアのプライミングによって引き起こされる過剰な炎症は、既存の状態に相加的に加わり、転帰を悪化させる可能性があることが示されている。
ミクログリアプライミングの増加は精神疾患と関連しており、外傷性脳損傷が変化した行動反応や外傷性脳損傷後の精神医学的後遺症を誘発するメカニズムを提供する可能性がある。実験的に正中線外側液打損傷(FPI)の後、ミクログリアは、プライミングされたミクログリアの永続的かつ反応性の集団を示す、30日後の外傷性脳損傷でMhcIIの発現が増強されている。Il-1βおよびTnfαの発現は、対照と比較して末梢性LPS注射に応答して有意に増加している(45)。さらに、実験的な正中線外側液打損傷(FPI)の後、末梢LPS注射は、注射後24時間でシャム対照および生理食塩水を受けた外傷性脳損傷動物の両方と比較して社会的相互作用を減少させるが、これは病気または抑うつ的な行動の増加の指標である。LPSを受けた外傷性脳損傷の動物はまた、再び炎症が行動障害に関連付けられていることを臨床データをミラーリング、増加した抑うつ症状のような行動を表している他のすべてのグループと比較して、注射後72時間で尾部懸垂試験中に不動のより多くの時間を過ごす。プライム化されたミクログリアは、年齢をマッチさせた対照群と比較して、静止したramifiedミクログリアよりも脱アミ化された反応性ミクログリアの比率が増加することによって見られるように、自殺後のうつ病患者の臨床研究で見られる(46)。単球の誘導に関与するケモカインであるMCP-1の発現もまた、うつ病患者では有意に高く、ミクログリアのプライミングに関連した炎症の悪化をさらに示している。このことは、プライミングを介したミクログリア炎症の亢進と精神症状との間に強い関係があることを示している。これらのデータを合わせると、ミクログリア・プライミングは、LPSなどの二次的な免疫課題に対する炎症反応を悪化させるだけでなく、外傷性脳損傷後の行動や精神症状の悪化にも寄与している可能性があることが示唆される。
ストレスがグルココルチコイドを介して炎症に影響を与える
ストレスとは、広義には、ストレッサーによる恒常性バランスの乱れを指する。ストレス応答をもたらすストレス因子(例えば、物理的、心理的、免疫学的など)には多くの異なるタイプがある。このストレス応答は、HPA軸の活性化によって特徴づけられる(図1A)。HPA軸の神経および非神経構成要素は、性的経験、食欲、または光さえもなどの非ストレス性刺激だけでなく、ストレス性刺激に対しても反応性が適切であることを保証するために多くの調節制御を有する(47)。ストレス刺激を介したHPAの活性化は、恒常性を乱す可能性のある環境イベントや合図と密接に結合した非自発的な反応であり、ストレス刺激を介したHPAの活性化は、非自発的な反応である。異なるタイプのストレス刺激は、視床下部室傍核(PVN)への入力を活性化する。活性化されると、視床下部室傍核ニューロンはコルチコトロピン放出ホルモン(CRH)(コルチコトロピン放出因子としても知られている)とアルギニンバソプレシン(AVP)を分泌する。これらのホルモンは、その後、「マスター腺」である下垂体下垂体前葉では、CRHはコルチコトロピーと呼ばれる特殊な内分泌細胞を活性化し、翻訳後にプロピオメラノコルチン(POMC)を修飾して副腎皮質刺激ホルモン(ACTH)を産生する。その後、ACTHは血液を介して腎臓の上にある副腎この相互作用により、ステロイド生成とグルココルチコイド(グルココルチコイド)特にコルチゾール(コルチコステロン)の放出が開始される。その後、コルチゾール(コルチコステロン)は血液を介して全身を巡り、さまざまな組織に作用して恒常性を回復させる。
図1外傷性脳損傷前後の視床下部-下垂体-副腎軸
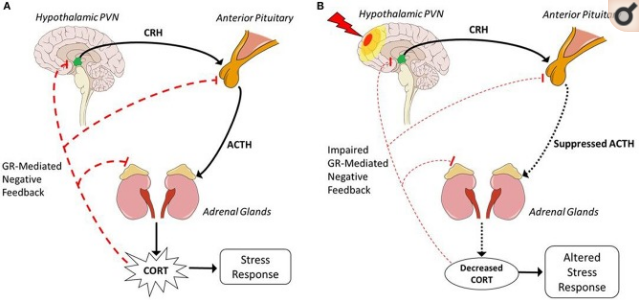
A)ストレス因子に反応して、興奮性ニューロン入力はHPA軸を活性化し、黒の実線で表されるホルモン伝達に変換され、生理学的ストレス応答を生成する。視床下部視床下部室傍核への興奮性ニューロン入力を介した活性化は、CRHとAVPを放出し、正中海綿体を介して視床下部ophyseal下部門脈回路を介して下垂体前部へと導く。CRHは、ACTHの産生を刺激するために下垂体前葉でコルチコトロピーを誘導する。ACTHは血液中に放出され、腎臓よりも上の副腎副腎では、ACTHはコルチゾール(コルチコステロン)の合成を開始する。その後、コルチゾール(コルチコステロン)は血液中に放出され、肺、心臓、筋肉などの複数の組織に作用してストレス反応を誘発する。赤の破線で示されるように、コルチゾール(コルチコステロン)はGC受容体を介したフィードバックを介してあらゆるレベルで作用し、HPAの活性化をネガティブに制御し、コルチゾール(コルチコステロン)の産生を低下させる。
B)稲妻に見立てた外傷性脳損傷は、ストレス因子に反応して下垂体低下症を誘発し、HPA活性化が抑制される(黒点線)。下垂体低下症はACTHの産生が低下しているため、副腎の刺激が低下し、コルチゾール(コルチコステロン)の産生が低下していることを示している。コルチゾール(コルチコステロン)レベルが抑制されていると、赤の破線で示されるように、GC受容体介在性負帰還を介したHPA活性化の継続を抑制することができず、その結果、GC受容体介在性負帰還が障害され、ストレス応答が持続し、ストレス因子に曝露した後の回復時間が長くなる。コルチゾール(コルチコステロン)の減少は炎症の増加と関連しており、これは精神医学的な後遺症の一因となる可能性があるため、傷害によって誘発されるHPA軸の抑制は、外傷性脳損傷後の結果をもたらすメカニズムを示している。矢状脳の模式図。パトリック・J・リンチ、メディカルイラストレーター [CC BY 2.5 (https://creativecommons.org/licenses/by/2.5)]。
HPA軸の活性化に対するグルココルチコイド応答の制御は、低親和性のグルココルチコイド受容体(GC受容体)を介した負のフィードバックに起因するが、基底部のグルココルチコイド分泌は高親和性のミネラルコルチコイド受容体(MR)のフィードバック阻害によって制御される(48)。GC受容体はグルココルチコイドに対する親和性が低いため、HPA活性化がグルココルチコイドレベルの有意な上昇をもたらし、MRが飽和状態になると、抑制的な制御を発揮する。グルココルチコイドは脳、下垂体、副腎でGC受容体に作用し、したがってHPA軸のあらゆるレベルでグルココルチコイド産生を制御することができる。海馬は、海馬の破壊がストレス因子に対するHPA軸の応答を永続させることを示す病変研究を通して見られるように、神経GC受容体に対するグルココルチコイドの作用による主要な調節の領域である(49)。グルココルチコイド はまた、ある研究では帯状皮質への コルチゾール(コルチコステロン) の適用が ACTH と コルチゾール(コルチコステロン) の両方の血漿レベルを低下させることが示されているように、視床下部室傍核 の神経支配を介して HPA 軸の活性化を抑制するために帯状皮質の神経 GC受容体 にも作用する(50)。下垂体前葉では、GC受容体介在性フィードバックがコルチコトロピーに作用してACTH放出を抑制する(51)が、ある臨床研究ではGC受容体アゴニストを静脈内投与すると、外因性CRH刺激に対するACTHおよびコルチゾール(コルチコステロン)応答が急速に抑制されることが示されている(52)。副腎では、グルココルチコイド産生はACTHの適用と直線的に関連しているのではなく、むしろわずかに減少している(53)。このことは、副腎内での動的なグルココルチコイド産生レベルが、GC受容体を介したグルココルチコイドのステロイド産生に対するフィードバック抑制を介して作用していることを示している。視床下部での制御はより複雑である。グルココルチコイドは視床下部室傍核に作用して、ストレス因子に応答してエネルギーと体液の恒常性を調節する(54)。視床下部室傍核におけるGC受容体を遺伝子的にノックアウトすると、ストレスに応答して基底コルチゾール(コルチコステロン)レベルが上昇するが、グルココルチコイドのサーカディアン放出は変化しない(55)ことから、視床下部室傍核におけるGC受容体を介したフィードバックによる抑制性調節は限定的であることが示唆される。ラット脳組織のin situハイブリダイゼーションにより、GC受容体のmRNAは別の視床下部核である弧状核(ARC)で高発現していることが明らかになった(56)。GC受容体 アンタゴニストであるエプレレノン(EPN)を 視床下部室傍核 に投与しても、投与後 80 分まで血漿中 コルチゾール(コルチコステロン) 濃度は有意に上昇しないが、ARC に投与した EPN は投与後 10 分で コルチゾール(コルチコステロン) 濃度を非常に急速に上昇させる(57)。このデータは、GC受容体が介在する多核の負帰還が視床下部のHPA軸反応性を調節していることを示している。注目すべきことに、GC受容体を介したHPA軸の正のフィードバックの例は、脳内にも存在する。例えば、以前に検討したように、恐怖学習に重要な領域では、扁桃体、末梢神経筋床核、内側前頭前皮質へのフィードフォワードグルココルチコイド作用によりHPA軸の活性化がアップレギュレートされ、CRHの放出が増加している(58)。これらのデータから、これらのデータは、適切なストレス応答とホメオスタシスの維持に寄与するGC受容体媒介のグルココルチコイド放出とHPA軸の反応性を敏感かつ厳密に制御していることを示唆している。
HPA 軸の制御に加えて、グルココルチコイドs の GC受容体 介在作用はストレス-免疫軸のコミュニケーションにおいて重要な役割を果たしており、多くの抗炎症・抗炎症作用を有している(図 2)。これまでに検討したように、グルココルチコイド はゲノム経路と非ゲノム経路の両方を介して抗炎症作用を実行している(59, 60)。GC受容体 介在性抗炎症作用は主に、グルココルチコイド応答性エレメント(GC受容体Es)に結合する グルココルチコイド-GC受容体 複合体の核内への転座を介して行われ、例えば、転写因子である核内因子κB(NFκB)介在性のプロ炎症性遺伝子の転写を阻害するが、GC受容体 の非ゲノム作用についてはまだ十分に解明されていない(61, 62)。ミクログリアは、GC受容体 の高発現を介してストレスと免疫の相互作用を統合している(63)ため、グルココルチコイド 免疫抑制の優れたターゲットとなっている。例えば、ラットのミクログリアとニューロンの共培養実験では、IFN-γとLPSを投与すると、誘導性iNOS由来のNOを介して酸化ストレスを介して神経毒性を持つ反応性ミクログリアが誘導された(64)。合成グルココルチコイドであるデキサメタゾン(DEX)の投与は、ミクログリアのNO産生とiNOS mRNA発現を有意に抑制することで神経保護効果を発揮する。このように反応性ミクログリアを介した酸化ストレスの減少は、抗炎症作用を介したグルココルチコイドの高い神経保護効果を示している。さらに、iNOS の発現は NFκB の活性化と関連している。マウス樹状細胞株では、NFκB 活性化を介した iNOS 発現と NO 産生が DEX の前処理により用量依存的に抑制されている(65)。これらのデータは、炎症性刺激がミクログリアの反応性を誘導する前と後の両方で グルココルチコイド が酸化ストレスを軽減することを示しており、グルココルチコイド の抗炎症作用が神経保護作用を持つことを示している。他の免疫細胞は、炎症を減少させるためにグルココルチコイドの作用を媒介するGC受容体を発現している。例えば、以前に検討したように、グルココルチコイド は適応免疫エフェクター細胞である T 細胞の GC受容体 介在性アポトーシスを誘導する(66)。さらに、グルココルチコイドは免疫応答を直接的に永続させないが、血液脳関門(BBB)のような炎症性環境に寄与することができる組織に対して保護効果を有する(67)。DEXで処理した後、試験管内試験ブラスト傷害を受けた不死化マウス脳内皮細胞は、未処理細胞と比較して、経内皮電気抵抗の回復が改善され、タイトジャンクションZO-1免疫染色が増加した(68)。このことは、グルココルチコイドがBBBの回復に重要な役割を果たし、それによって末梢免疫細胞の脳への移行を防ぐことで炎症を減少させることを示している。グルココルチコイドの主な抗炎症的役割は、GC受容体を介したGC受容体Eの活性化であり、その結果、IL-1β、TNF-α、およびIL-6を含むプロ炎症性サイトカインのmRNA発現が抑制される一方で、IL-10およびTGFβなどの抗炎症性サイトカインの発現が増強される(69)。これらのデータを合わせると、免疫細胞の機能を媒介するグルココルチコイドの強力な抗炎症作用が明らかになる。
図2 グルココルチコイドの抗炎症作用と抗炎症作用
グルココルチコイドはストレス因子に対する急性応答において、複数の経路を介して強固な免疫抑制機能を有する。(A)ミクログリアはGC受容体を高発現しており、グルココルチコイドの免疫抑制の主要な標的となっている。GC受容体 を介した グルココルチコイド の作用は、ミクログリアの iNOS 合成を減少させ、NFκB の活性化を防ぐことで神経保護作用を発揮する。B)適応免疫エフェクター細胞である T 細胞は、炎症反応の持続を防ぐ グルココルチコイド 介在性アポトーシスを経ます。C)BBBの内皮細胞はGC受容体を発現している。GC受容体へのグルココルチコイドの結合は、BBBタイトジャンクションの引き締めに寄与し、BBBの完全性を改善して末梢免疫細胞の脳内への侵入を防ぐ。D)グルココルチコイドの主要な抗炎症メカニズムは、IL-1βおよびTNF-αを含む炎症性サイトカインの発現を減少させることによるGC受容体介在性ゲノム免疫抑制である。急性のストレスに直面した場合、グルココルチコイドは主に免疫抑制作用を示すが、慢性的なストレスに直面した場合、グルココルチコイドの炎症促進作用が明らかになった。(E) GC受容体に富むミクログリアは、CD68およびMHCII発現の増加を通じて示されるストレス誘発性のプライミングに感受性があり、その結果、その後の免疫課題に対する過剰反応性の反応を引き起こす。(F) グルココルチコイドはT細胞のアポトーシスを誘導する一方で、IL-2に対するT細胞の感受性を高め、炎症性環境に寄与することでリンパ球の成熟を促進する。G)繰り返された社会的敗北の慢性的な圧力によるグルココルチコイドの生産は増加した不安のような神経炎症性の結果に寄与する骨髄からの単球を動員する。(H) グルココルチコイドはまた、ストレス因子の複数のタイプによるHPA軸の活性化を通じて、外傷や病気に関連する炎症性サイトカインであるIL-6の循環レベルを増加させる。さらに、慢性ストレスは、興奮性細胞死後のIL-1βおよびTNF-αの発現を増加させることにより、炎症プロファイルを悪化させる。これらの抗炎症作用と抗炎症作用は、ストレスに対する炎症反応を媒介するグルココルチコイドの二面性を例証している。
グルココルチコイドの本質的な抗炎症作用により、コルチコステロイドは、傷害後の炎症に対する薬理学的治療法として試験された最初の抗炎症薬の一つであった(70)。外傷性脳損傷後のグルココルチコイドの有効性は、投与時期によって炎症に対する効果が異なるため、議論の余地がある。実験的に体重を落として外傷性脳損傷を行った後、DEXを直ちに投与すると、受傷後24時間と48時間目にミクログリア/マクロファージ炎症のマーカー(EMAP-II、P2X4R、AIF-1)に陽性の細胞数が減少する(71)。しかし、外傷性脳損傷後4日目と6日目までには、生理食塩水対照と比較して炎症マーカーの定量化に有意な差は認められなかった。グルココルチコイドは外傷性脳損傷直後に炎症を減少させるが、これらの効果は一過性であり、臨床試験では死亡率を増加させる可能性がある。外傷性脳損傷の入院・治療後14日以内の死亡率を定量化した大規模臨床試験では、プラセボ群と比較してコルチコステロイドを静脈内投与した場合の死亡率が有意に増加することが示されている(72)。さらに、以前のレビューでは、グルココルチコイド治療の大きな障壁であるグルココルチコイド耐性や不感症を引き起こし、治療効果をさらに低下させることが議論されている(73)。これらの研究は、内因性グルココルチコイドには強力な抗炎症効果があるにもかかわらず、外因性グルココルチコイドによる治療は外傷性脳損傷後の不適応な炎症を抑制するのに有効ではないことを示している。
変化したグルココルチコイド放出は炎症に影響を与える
このようなストレス-免疫軸の緊密な関係は、機能不全の影響を強く受けやすい。具体的には、過剰なHPA軸の活性化による不適切なストレス応答が炎症を増加させることが示されている。グルココルチコイド放出は、グルココルチコイドカスケード仮説(74)として知られるフィードフォワード機構で作用し、その結果、抑制されていないストレス応答とそれに続く炎症反応が生じる。例えば、外傷性脳損傷後の過剰なHPA軸の活性化は、ストレス誘発性のミクログリアのプライミングを通じて神経炎症に影響を与える。以前に議論したように、ミクログリアはGC受容体を高度に発現しているため、過剰なグルココルチコイドレベルの存在下ではストレス誘発性プライミングの影響を受けやすい。ラットに外因性コルチゾール(コルチコステロン)を単独で投与しても、海馬ミクログリアにおける炎症性サイトカインIl-1β、Tnfα、またはIl-6の遺伝子発現は変化しない。しかし、コルチゾール(コルチコステロン)投与後24時間後に免疫チャレンジとしてLPSを投与した場合、海馬ミクログリアは、ビヒクルコントロールと比較して、LPS用量依存的に炎症性サイトカインの発現を有意に増加させた(75)。これは、外因性グルココルチコイドを介したストレスは、その後のLPS免疫チャレンジに反応して、ミクログリアが過剰反応性になるようにプライミングされることを示している。これらの同じ結果は、ストレス因子への曝露による内因性コルチゾール(コルチコステロン)の増加後に見られる。脱出不可能な尾部衝撃の24時間後にLPSを注射すると、海馬のコルチゾール(コルチコステロン)とIL-1βが車のコントロールと比較して有意に増加する(76)。この同じ実験はさらに、脱出不能尾部衝撃の前にGC受容体アンタゴニストであるミフェプリストン(MIF)で前処理を行うと、LPS注射後の海馬のコルチゾール(コルチコステロン)およびIL-1βの増悪した反応が救われることを示している。このことは、グルココルチコイドがストレス誘発プライミングを引き起こすためにミクログリアGC受容体に直接作用することを示している。脳損傷との関連では、実験的な制御皮質衝撃の48時間前にMIFで前処理を行うと、外傷性脳損傷後24時間後のCA1海馬ニューロンの喪失が有意に抑制される(77)。外傷性脳損傷後のニューロン喪失がGC受容体アンタゴニストを介して救われたことは、グルココルチコイドシグナル伝達が初期または二次的な挑戦として、傷害後の不適応な炎症に寄与していることを示唆している。さらに、海馬における神経細胞の生存率が向上すれば、学習、記憶、認知、感情の調節が改善され、外傷性脳損傷後に一般的に見られる精神症状が改善される可能性がある。前臨床の系統的レビューによると、心理社会的ストレスを含むストレス因子は、実験的げっ歯類モデルの海馬と前頭前野におけるミクログリア反応性を有意に増加させ、精神疾患のような行動反応の変化に寄与することが示されている(78)。これらのデータを合わせると、グルココルチコイドはミクログリアGC受容体を介してミクログリアを活性化するように作用し、炎症反応を悪化させることが示されている。ストレスに対するこの炎症反応の増加は、その後、外傷性脳損傷後の神経細胞死や精神合併症の増加につながり、受傷後の回復を阻害し、生活の質を低下させる可能性がある。
ミクログリアと同様に、グルココルチコイドは他の免疫細胞に対しても炎症を促進する作用を持つ。グルココルチコイドを介したアポトーシスは、胸腺におけるT細胞の適切な発達に必要である。さらに、ラットの T 細胞培養物を刺激した場合、コルチゾール(コルチコステロン) の投与は、IL-2 に対する T 細胞の感受性を高めることでリンパ球の増殖を有意に促進し、親炎症性環境に寄与する(79)。慢性グルココルチコイドはまた、末梢組織、特に骨髄に対して顕著な炎症促進作用を有する。副腎摘出術を受けたマウスでは、反復社会的敗北(RSD)ストレッサー後の循環コルチゾール(コルチコステロン)およびIL-6が対照群と比較して有意に減少しており、正常なストレス応答の廃止を示唆している(80)。ストレス後の骨髄中の単球および顆粒球の割合には、副腎摘出マウスと対照群との間に差はないが、血液中の単球の循環レベルは副腎摘出マウスで有意に減少している。このことは、副腎摘出により適切なストレス反応が停止しても、骨髄での単球産生には影響を与えず、血液中への単球の放出が抑制されていることを示している。骨髄における単球の保持に重要なケモカインであるCXCL12の免疫蛍光発現は、ストレス後に副腎が無傷の動物では有意に減少したが、副腎摘出を行った動物では、ストレスを受けていない偽マウスと比較してCXCL12の発現に差は見られなかった。このことは、ストレス因子を介したコルチゾール(コルチコステロン)産生が骨髄における単球の保持に直接影響を与え、結果としてストレスに応答して単球の動員が増加していることを示している。最も注目すべきは、RSD後に副腎が無傷の動物では脳内のマクロファージ集団の割合が有意に増加するのに対し、副腎摘出術はRSD後のマクロファージ集団を減衰させることである。このことは、RSDなどのストレス因子に反応してコルチゾール(コルチコステロン)が放出されると、単球の血中への動員が増加し、脳内のマクロファージ集団が増加し、その結果、不安感の増加などのプロ炎症性環境および神経炎症性の結果をもたらすことを示している(81)。ストレスはまた、慢性的なストレスレベルのコルチゾール(コルチコステロン)の前処理を行うと、カイニン酸誘発性興奮性細胞死後のIL-1βおよびTnfαの遺伝子発現が悪化することから、神経細胞死にも寄与する(82)。これは、以前に議論されたグルココルチコイドの抗炎症作用とは相反するように思われるが、グルココルチコイドの抗炎症作用は、グルココルチコイドの高用量が炎症を永続化させる一方で、グルココルチコイドの低用量から中用量の前処理で見られる。心理的ストレスの免疫学的影響の臨床的システマティックレビューでは、ストレス因子に曝露されてから2時間後までの間に、IL-1β、IL-6,およびTNF-αを含む循環炎症性サイトカインの有意な増加が示されているように、これらと同様の炎症性ストレス効果は臨床集団に見られる(83)。これらのデータは、HPA軸の強力な活性化が複数の組織に作用して炎症を増加させることを示している。グルココルチコイドは強力な抗炎症作用を有するが、HPA軸の活性化に反応してグルココルチコイドが過剰に放出されると、炎症の増加に寄与する可能性がある。
逆に、ストレスに反応してグルココルチコイドの産生が不十分な場合は、通常グルココルチコイドの免疫抑制作用によって打ち消される炎症性サイトカインが増加し、炎症の増加にも寄与する。グルココルチコイドはストレスに反応して放出されて恒常性を回復するため、基底的またはストレスに反応してグルココルチコイドのレベルを抑制すると、炎症を著しく増加させる。例えば、副腎摘出術後にマウスがマウス軟体菌コンタギオーサムウイルスに感染した場合、感染後36時間から72時間の間に80%の死亡率を示したのに対し、非感染の副腎摘出マウスや無傷の副腎を持つ感染マウスでは長期生存率が高い(84)。臨床的には、この抑制されていない炎症は、グルココルチコイドの放出が抑制され、しばしば基底部の炎症が増加していることを示すストレス障害で見られる。例えば、心的外傷後ストレス障害(PTSD)と診断された患者では、基底部コルチゾール(コルチコステロン)レベルは低いが、IL-1β、TNF-α、IL-6などの循環性炎症性サイトカインのレベルが高い(85)。同様に、慢性疲労症候群と診断された患者は尿中遊離コルチゾール(コルチコステロン)レベルが低く(86症状の重症度は活性酸素およびIFN-γレベルの上昇と高い相関がある(87)。これらの研究を合わせると、ストレスが免疫プロセスに影響を与え、HPA軸の調節異常が不適応な炎症につながることが示されている。
外傷性脳損傷後のHPA軸の機能不全
外傷性脳損傷直後には、傷害のストレスによりHPA軸の急性活性化がみられる。マウスの軽度のブラスト外傷性脳損傷では、受傷直後に循環コルチゾール(コルチコステロン)が増加し、対照群と比較して外傷性脳損傷後3時間でピークを迎え、外傷性脳損傷後5時間までにベースラインに戻る(88)。HPA軸の活性化における同様の増加は臨床集団においても見られる。ある研究では、軽度および中等度の外傷性脳損傷後の最初の1~2日に、年齢をマッチさせた対照群と比較して循環コルチゾール(コルチコステロン)レベルが有意に増加することが示されている(89)。この同じ臨床研究では、重度の外傷性脳損傷後、ベースラインの血清コルチゾール(コルチコステロン)レベルは外傷性脳損傷後1~3日で低下することが示されている。このようなストレスの減少は、受傷後の長期的な炎症反応を低下させる可能性があるが、HPA軸の活性化が抑制されると、より重度の外傷性脳損傷後の転帰が悪化する可能性がある。
軽度から中等度の外傷性脳損傷では、重度の外傷性脳損傷に比べてストレス反応が遅れている。実験的に側方外側液打損傷(FPI)を受けた4週間後には、拘束ストレスに対するコルチゾール(コルチコステロン)反応は偽の対照群と比較して有意に鈍化している(90)。HPA軸機能障害に対する傷害の重症度の違いは、外傷性脳損傷の実験モデルでも見られる。例えば、実験的に軽度の制御皮質衝撃では、受傷後7日目と21日目ではシャム対照と比較してストレス因子に対するコルチゾール(コルチコステロン)応答が減衰するが、外傷性脳損傷後34日目と70日目ではストレス因子に対するコルチゾール(コルチコステロン)応答が有意に増加する(91)。中等度の制御皮質衝撃では、受傷後7日目のコルチゾール(コルチコステロン)応答は対照と比較して差がないが、21日目のストレッサーに対するコルチゾール(コルチコステロン)応答は有意に減少し、それは外傷性脳損傷後70日目まで持続する。抑制された内分泌機能については、外傷性脳損傷の臨床症状後にも同様のばらつきがみられる。ある臨床研究では、重度の外傷性脳損傷後、受傷後10日以内に薬理学的に誘発されたストレスに反応して、25~100%の患者がグルココルチコイドの循環レベルが不十分であることが示されている(92)。軽度から中等度の外傷性脳損傷では、受傷後1年以内に16~45%の患者でグルココルチコイドの循環レベルが不十分であることが報告されている(93,94)。外傷性脳損傷後のHPA軸活性化抑制の実際の発生率は、多くの要因により不明である。多くの人は軽度の外傷性脳損傷後に治療を受けようとしないため、軽度の外傷誘発性内分泌機能障害の発生頻度がはるかに高いことが示唆されている。また、内分泌異常のスクリーニングは外傷性脳損傷治療の標準的な治療ではないため、診断されないことが多い。HPA軸の活性化抑制が疑われる場合でも、診断ツールや診断可能な機能障害の閾値は標準化されておらず、代わりに個々の病院や臨床医によって設定されているため、発生報告にばらつきがある(95)。すべての重症度の外傷性脳損傷後のコルチゾール(コルチコステロン)反応の動的で診断が不十分な状態は、外傷性脳損傷後の回復を阻害し、受傷後の長期的な後遺症の発生に寄与している。
外傷性脳損傷後のHPA軸抑制の原因
外傷性脳損傷後のHPA軸機能障害は、HPA軸のどのレベルでも起こりうる。例えば、視床下部が障害され、視床下部室傍核によるCRHの放出を減衰させると、抑制されたグルココルチコイド合成と放出が起こる可能性がある。ラットで実験的に正中線外側液打損傷(FPI)を受けた後、ニューロンのゴルジ体染色によって測定した損傷後1,7,28日目に視床下部のニューロンプロセスの複雑性が増加している(96)。これらの同じ損傷を受けた動物は、外側液打損傷(FPI)後にHPA軸の機能不全を示した。GC受容体介在性フィードバックを介したHPA軸のDEX抑制は、損傷動物の血漿中コルチゾール(コルチコステロン)レベルに有意な差を示さなかったが、偽コントロールではコルチゾール(コルチコステロン)が有意に抑制されていた。このコルチゾール(コルチコステロン)反応の鈍化は、異常なDEX抑制の結果からわかるように、フィードバック抑制の乱れによるものである可能性が高い。GC受容体を介したネガティブフィードバックの障害と併せて視床下部の神経細胞の複雑性の変化は、HPA軸機能障害とHPA軸回路の変化との因果関係を示唆している。これらのデータは、視床下部が傷害によって直接影響を受け、グルココルチコイド放出の抑制と負帰還の障害を誘発するメカニズムを説明している。前述のように、視床下部の弧状核はHPA軸の視床下部室傍核への急速な負帰還を媒介する上で重要な役割を果たしている。マウスの制御皮質衝撃は弧状核の肥大したアストロサイトを誘導し、炎症の増加を示している(97)。臨床画像研究では、8~18歳の小児の受傷後に見られる視床下部の機能障害の可能性も反映している。受傷部位から分離されたこれらの炎症性および形態学的差異は、視床下部の迅速な調節に参加するための視床弓の能力に悪影響を与え、結果としてストレス応答の不適切な活性化と制御をもたらす可能性がある。
外傷性脳損傷後のHPA異常の発生率が最も高いのは、下垂体前葉の機能障害によるものであり、その結果、ACTH放出が減少し、したがってコルチゾール(コルチコステロン)が減少する(図1B)。下垂体は、外傷性脳損傷や外傷による圧力変化または出血に対して特に感受性が高い。長期臨床研究では、受傷後10日目の朝のコルチゾール(コルチコステロン)レベルが異常に高い外傷性脳損傷患者が41%であったことが報告されている(99)。3ヵ月までには32%が朝のコルチゾール(コルチコステロン)値が異常に低かった。この発生率は6ヵ月後と12ヵ月後にも比較的安定しており、コルチゾール(コルチコステロン)値が異常に低い患者はそれぞれ37%と35%で報告されている。下垂体の機能障害はACTHの放出を減少させ、その結果、副腎はグルココルチコイドの合成および産生量を減少させる。この減少に対応して、負のフィードバックの感度は補償するために増加する。このように、抑制の増加は、さらにグルココルチコイド放出を減少させ、ストレス因子に対する応答の変化を引き起こす可能性がある。HPAのすべてのレベルでの直接的、間接的、およびフィードバック調節の複雑さのために、ACTH産生が減少する単一のメカニズムはまだ不明である。
外傷性脳損傷後の副腎の合併症は比較的まれであり、最初の臨床例研究は1997年に報告されている(100)。この患者は、持続的な脱力感、疲労感、吐き気のために外傷性脳損傷後1ヵ月後にリハビリテーションのために入院したが、リハビリテーションで有意な改善が見られなかったため、内分泌検査を受けたところ、外因性ACTHの適用後にグルココルチコイドの放出が抑制されていることが明らかになった。合成グルココルチコイド、プレドニゾン、フルドロコルチゾンによる治療を開始して1週間以内に、めまいや嗜眠の症状が有意に改善した。まれではあるが、副腎の損傷を介した副腎不全の症例は、外傷性脳損傷だけでは症状が著しく重なるため、診断されずに放置されることがある。この合併症は、副腎に直接損傷を与える可能性のある複数の外傷を受けた場合に多く見られ、ほとんどの場合、外傷性脳損傷に直接起因するものではないため、一般的には外傷性脳損傷の直接の症状ではなく、併存疾患と呼ばれている。
外傷性脳損傷後のHPA軸機能障害の結果
外傷性脳損傷後のHPA軸の過剰な活性化と、それが外傷性脳損傷後の回復に及ぼす影響はよく特徴づけられている(7, 101)。例えば、コルチゾール(コルチコステロン)レベルはマウスの正中線外側液打損傷(FPI)後、受傷後30分でピークに達し、受傷後2時間でCRH mRNA発現が偽コントロールと比較して40%増加するため、HPA軸の活性化は持続する(102)。このHPA軸の活性化亢進をCRHアンタゴニストを介して15分後、2,4,6,8時間後に阻害すると、病変体積が45%減少することが明らかになった(103)。このことは、外傷性脳損傷後のHPA軸の過剰な活性化が神経細胞死を増加させ、その結果、神経炎症に影響を及ぼす可能性を示している。前述のように、外傷性脳損傷による過剰なグルココルチコイドもまた、ストレスに反応してミクログリアのプライミングを誘導し、炎症性サイトカインを増加させ、不適応な慢性炎症を引き起こす。
外傷性脳損傷後のHPA軸抑制による炎症性効果は、グルココルチコイド放出の減少が死亡率の増加や外傷性脳損傷後の回復不良の危険因子として同定されているにもかかわらず、よく特徴づけられていない。軽度の外傷性脳損傷に比べて中等度の外傷性脳損傷ではHPA軸の活性化が持続的に減衰していることから、より重度の外傷性脳損傷では長期予後の悪化や障害の発現と関連している可能性が示唆されている。外傷性脳損傷後のコルチゾール(コルチコステロン)レベルの臨床比較研究では、外傷性脳損傷後10日以内に血漿中コルチゾール(コルチコステロン)レベルが平均値の最低四分位にある患者は、他のすべての四分位と比較して有意に高い死亡率を示している(104)。さらに、外傷性脳損傷後10日を超えて持続する持続性コルチゾール(コルチコステロン)欠乏症を呈した患者も、一過性コルチゾール(コルチコステロン)欠乏症を呈した患者と比較して有意に高い死亡率を示した。このように、HPA軸機能障害による血漿中コルチゾール(コルチコステロン)レベルの低下は、外傷性脳損傷後の死亡率の増加と有意な相関がある。コルチゾール(コルチコステロン)レベルの抑制に寄与すると考えられるACTH産生の不足は、ベースラインの血清ACTHが異常に低く、ACTH刺激に対する反応が低下していることから明らかなように、外傷性脳損傷後1年および5年まで持続するという臨床的証拠がある(105)。異常な内分泌反応の持続は、グルココルチコイドの抗炎症作用の低下を介して神経炎症の亢進に寄与している可能性がある。
成人における受傷時の年齢は、ほとんどの研究で年齢との相関がないと報告されているため、外傷性脳損傷後のHPA軸機能障害に対する脆弱性には影響しないようである(93)。しかし、小児の外傷性脳損傷では、12歳未満の小児は、年長の青年に比べて受傷後のHPA軸機能障害の影響を受けやすい(106)。実際、ある研究では、軽度から重度の外傷性脳損傷の調査対象となったすべての小児が、インスリン検査によりHPA活性化に反応してコルチゾール(コルチコステロン)の放出が有意に抑制されていることが明らかになっている(107)。傷害は小児外傷性脳損傷における初期のストレス曝露として作用し、これは不適応なHPA軸の成熟を誘発し、その結果、後の人生においてストレスに対する感受性を恒久的に高める可能性がある(108)。HPA軸機能障害によるストレス反応の変化と組み合わせることで、小児外傷性脳損傷は正常な老化を著しく変化させ、精神医学的な後遺症の可能性を高め、生活の質の低下に寄与する可能性がある。これらのデータは、加齢に伴って受傷し、加齢とストレス反応の変化が持続することにより、外傷性脳損傷に関連した障害を発症するリスクが高くなる可能性のある人が増えていることを示している。
HPA軸の機能不全は、外傷性脳損傷後の精神症状の発症や悪化に大きく関与している。グルココルチコイドは大脳辺縁回路への作用を媒介することで、意欲や感情を調節する(109)。大うつ病性障害は、軽度の外傷性脳損傷後の急性期に見られるように、HPA軸の過活動とフィードバック抑制の異常によって特徴づけられる(110, 111)。外傷性脳損傷後のイメージング研究のレビューでは、前頭前野、腹側前頭葉、前側頭葉などの多くの皮質領域の拡散テンソルイメージングに大きな違いがあることが示されている(112)。以前に検討したように、これらの領域はすべてHPA軸にフィードし、感情的ストレス因子に対する適切なストレス反応の媒介に大きく関与しているため、外傷性脳損傷後の不安や抑うつ症状の増加と関連している(113)。これらの部位の回路における外傷性脳損傷誘発性の変化は、情動調節の障害につながり、過敏性やイライラなどの外傷性脳損傷後の多くの一般的な症状の一因となる。さらに、コルチゾール(コルチコステロン)産生の低下は、疲労、脱力感、体重減少などの症状を引き起こす。これらの症状は外傷性脳損傷後に一般的にみられるもので、広くはポストコンカッション症候群(PCS)と呼ばれている。PCSの他の症状としては、学習・記憶障害、集中力低下、過敏性などがあり、これもHPA軸機能障害の症状を反映している。外傷性脳損傷後にコルチゾール(コルチコステロン)が抑制された症例研究では、このような症状が重なっているため、PCSと関連していると考えられる心理的後遺症を持つ患者がしばしば報告されている(100, 114)。受傷後のHPA軸機能障害に対する認識が高まるにつれ、受傷直後に出現した症状が3ヵ月間持続する場合や、遅発性の症状が3年以内に出現する場合には、外傷性脳損傷後の回復期を通じて内分泌検査を行うことが推奨されるようになってきている(115)。神経内分泌反応の異常と精神行動の悪化の相互作用は、HPA軸機能障害と長期的な外傷性脳損傷回復の重要性を示している。
実験的ストレスモデルは、外傷性脳損傷後のHPA軸機能障害、炎症、および転帰の関係についての洞察を提供する
外傷性脳損傷後のストレス反応の変化が炎症に及ぼす慢性的な影響については十分に理解されていない。ストレス反応の変化は外傷性脳損傷の重要な臨床的帰結として認識されるようになってきているが、HPA軸の機能不全が傷害後の症状に寄与するメカニズムはまだ明らかにされていない。外傷性脳損傷後の長期的な転帰のための最良の情報源の一つは臨床調査であるが、特定の病歴や経験がないために制限されることがある。前述したように、臨床調査は報告数の少なさや内分泌検査の違いによっても影響を受けることがあり、結果として報告数や診断ツールにばらつきが生じる。ストレスの実験モデルは、外傷性脳損傷後のストレスの慢性的な影響とその結果としての炎症をよりよく特徴づけるために必要である。現在、多くの研究で実験的外傷性脳損傷後のストレス反応の変化が示されているが、実験的ストレスが外傷性脳損傷後の炎症に及ぼす影響を調べた研究はほとんどない。ここでは、一般的なストレスモデルを説明し、ストレスモデルが実験的外傷性脳損傷後のHPA軸の機能不全を特徴づける組み合わせ研究をさらに強調する。さらに、これらのストレスモデルが炎症に影響を与え、それによって外傷性脳損傷後の炎症に対するストレスの影響を特徴づけることができるという証拠についても議論する。「ストレス」は様々な刺激によって誘発される可能性があるが、これらの確立されたストレスモデルは、HPA軸の応答を最もよく説明する上で有効であるため、繰り返し使用されている。
拘束ストレス
外傷性脳損傷後のストレス効果を特徴づけるための最も一般的なモデルは拘束誘発ストレスである。拘束ストレスは、固定化という避けられないストレスを利用して、堅牢なHPA活性化とグルココルチコイド放出を誘導する。毎日6時間の拘束を28日間行った後、ストレスを受けたマウスの血漿中コルチゾール(コルチコステロン)レベルは、ストレスを受けていないコントロールと比較して、ストレスを受けている間に有意に上昇したが、これらの効果は、毎日のストレスが終了してから2週間または4週間後には持続しなかった(116)。しかし、注目すべきことに、長時間の拘束ストレスは、28日間の毎日のストレス暴露を通して、強制水泳課題中の動けなくなることで見られるように、抑うつ的な行動を誘発し、拘束ストレスが停止してから1週間後にも持続する。このことから、拘束ストレスは、強い長時間のストレスによってコルチゾール(コルチコステロン)レベルの持続的な変化を引き起こさないかもしれないが、ストレスの行動効果は、ストレス因子が停止した後も持続することが示されている。さらに、マウスで毎日2時間拘束ストレスを10日間受けると、ストレスを受けていないコントロールと比較して、IL1A、IL-6,およびTNF-αを含む炎症性遺伝子の発現が有意に増加することが示された(117)。拘束ストレスのこれらの炎症作用は、ストレス後の行動障害の一因となる可能性があり、傷害によってさらに悪化する可能性がある。実験的外傷性脳損傷後の拘束ストレスは、外傷性脳損傷後の臨床集団で見られるようなストレスに対するコルチゾール(コルチコステロン)反応を抑制する。中間外側液打損傷(FPI)は、外傷性脳損傷後54日目のベースラインコルチゾール(コルチコステロン)レベルを、偽コントロール動物と比較して慢性的に低下させ、外傷性脳損傷を受ける前のベースラインコルチゾール(コルチコステロン)レベルを低下させる(96)。30分間の拘束ストレスに反応して、損傷を受けた動物は、拘束ストレスが終了した1時間後の血漿コルチゾール(コルチコステロン)レベルがシャム対照動物と比較して低下している。これらのデータは、拘束ストレスがHPA軸を特異的かつ強固に活性化し、炎症に影響を与え、外傷性脳損傷後の内分泌機能障害を特徴づける効率的なストレスモデルであることを示している。
強制運動
外傷性脳損傷後の慢性ストレスのもう一つのモデルは強制運動である。強制運動は、密閉されたトレッドミルや車輪を用いて、動物がペースを維持できるように、突っ張ったり、小さな足の衝撃を加えたりして、特定の速度に設定するものである。車輪運動を1時間行うと、自発的な車輪運動と比較して、視床下部室傍核におけるCRH mRNAの発現が有意に増加する(118)。CRHの増加は、強制運動と自発運動を比較して、HPA軸の実質的な活性化を示唆している。別の実験研究では、週 3 回 30 分間のトレッドミル強制運動を 4.5 週間実施すると、運動をしていない対照群と比較して、オープンフィールドタスク中の血漿 コルチゾール(コルチコステロン) 濃度が有意に上昇することが示された(119)。さらに、強制運動にさらされた動物の血漿中コルチゾール(コルチコステロン)レベルの上昇は、脳内のIL-1βレベルの上昇および海馬ニューロンの減少と相関している。このことは、強制運動がHPA活性化を介してコルチゾール(コルチコステロン)を増加させ、炎症および細胞生存に影響を与えうることを示している。別の実験では、軽度/中等度の外側液打損傷(FPI)で傷害を受けた動物に、傷害後1-4日目と7-11日目に20分間の強制的なランニングを毎日2回行ってもらい、対照動物は車輪を使用しないか、または任意の運動のために車輪を使用した(120)。強制運動後、ACTHおよびコルチゾール(コルチコステロン)レベルは、自発的運動および鎮静型コントロールと比較して、偽物および負傷した強制運動群の両方で有意に上昇している。しかし、注目すべきことに、ACTH反応は外側液打損傷(FPI)群では鈍化している。さらに、強制運動は、自発的運動群と鎮静的運動群と比較して、シャム傷害動物の海馬GC受容体の低下を引き起こすが、外側液打損傷(FPI)後の海馬GC受容体にはどの群間でも差は見られなかった。外側液打損傷(FPI)後のコルチゾール(コルチコステロン)レベルの上昇に反応してGC受容体が調節されないことは、外傷性脳損傷後にGC受容体を介した負のフィードバックが損なわれるメカニズムを示していると考えられる。これらのデータは、強制運動が炎症を増加させ、HPA軸の傷害誘発性抑制を示す効果的なストレス因子であることを示している。
強制水泳
強制水泳は一般的な行動テストであるが、堅牢な コルチゾール(コルチコステロン) 反応を誘導するためのストレッサーとしても頻繁に使用される (121-123)。強制水泳では、あらかじめ決められた時間の間、動物が底に触れることができない程度の深さの水の容器に動物を入れる。これは、うつ状態に似た行動の指標として動かない時間を測定することで、行動テストとして機能する。強制水泳の使用は行動テストからストレッサーへと変化しているが、これは強制水泳中の対処プロセスが抑うつ的な行動よりもストレスによって誘発される動けない状態を示すものであるという証拠があるためです(124)。1日30分間の強制水泳ストレスに1日、5日、10日間曝露したマウスでは、ストレス60分前およびストレス直前のベースラインコルチゾール(コルチコステロン)レベルと比較してコルチゾール(コルチコステロン)レベルが有意に上昇していることからもわかるように、頑健なストレス反応が見られる(125)。さらに、毎日 30 分間の強制水泳ストレスを 5 日間受けると、視床下部室傍核 での IL-1β 発現と海馬での IL-6 発現が増加し、強制水泳のストレスに対する炎症反応が増加していることが示された。ラットの軽度および中等度の 制御皮質衝撃 後、傷害後 21 日目と 54 日目に 15 分間の強制水泳を行うと、軽度の傷害を受けた動物では コルチゾール(コルチコステロン) 応答が有意に増加するが、中等度の傷害を受けた動物では コルチゾール(コルチコステロン) 応答が鈍化している(91)。強制水泳は、HPA軸の活性化を介した効果的なストレッサーであり、プロ炎症性プロファイルを増加させ、コルチゾール(コルチコステロン)応答に対する傷害の重症度に差があることが明らかになった。
フットショック
フットショック恐怖条件付けパラダイムは、伝統的に不安に似た行動と条件付き恐怖記憶をテストするものである。動物は、トーンなどの条件付き刺激を足の衝撃と関連付けるように訓練される。訓練後、動物の凍結行動は条件付けされた刺激に反応して測定される。HPA軸はコルチゾール(コルチコステロン)放出を誘導することで恐怖条件付けを調節し、それが海馬に作用して文脈的な恐怖条件付け記憶の統合を増加させる。フットショックの大きさの増加はコルチゾール(コルチコステロン)放出の増加と相関しており、その結果、行動障害が悪化する(126)。また、フットショックはHPA軸に特化した炎症性反応を増加させ、2時間の間に80エピソードの脱出不能なフットショックを受けると、視床下部のIL-1遺伝子発現が増加し、下垂体のIL-6遺伝子発現が増加する(127)。また、足底部の凍結行動は、弱い衝撃とは対照的に強い衝撃のペア刺激に反応して、シャムコントロールと比較して、訓練室のコンテキストだけでなく、条件付きのトーンにも反応して増加する(128)。足部衝撃訓練後の凍結の増加は、不安に似た行動を示しており、外傷性脳損傷による行動障害を示している。さらに、ウエスタンブロット分析では、興奮性NMDA受容体のマーカーであるNR1が基底側扁桃体(BLA)で有意に増加しており、外側液打損傷(FPI)後のBLAと海馬ではGABA作動性抑制性ニューロンのマーカーであるGアルツハイマー病が偽薬と比較して減少する傾向があることが示されている。感情調節の重要な領域への興奮性と抑制性の入力のこれらの変化は、さらに、外傷性脳損傷は精神医学的健康と行動障害に影響を与える可能性がある方法を示している。これらのデータは、フットショックが外傷性脳損傷後の影響を効果的に測定するだけでなく、HPA軸を活性化し、ストレス関連の炎症を増加させることを示している。
ストレスモデルは結果的に睡眠に影響を与える
ストレスの増加は、ストレス要因への曝露が一旦停止しても、睡眠障害を誘発することが 臨床的にも実験的にも証明されている(129)。HPA軸の活性化と環境的な合図を介したグルココルチコイド放出の調整は、正常な睡眠の質と寿命を維持するために不可欠である。HPA の活動の違いは、睡眠の段階を仲介する。ヒトの平均的な睡眠時間は7~8時間であり、ノンレム睡眠(すなわち遅波睡眠)とレム睡眠(すなわち逆説睡眠)の約6サイクルを経験している(130)。一方、マウスは光相の間、12~14時間睡眠し、睡眠時間はわずか2~4分である。さらに、マウスは多相性睡眠を持っており、ノンレム睡眠とレム睡眠のサイクルを周期的に経験し、その間に覚醒期間があることを意味する。このような睡眠構造の違いにもかかわらず、概日睡眠や睡眠の恒常性調節など、げっ歯類の睡眠モデルの特徴はヒトと類似しており(131)、トランスレーショナルリサーチによく用いられている。
睡眠の開始時には、覚醒を最小限に抑えるためにHPA軸が抑制される。HPA軸の活動は、レム睡眠中に交感神経と脳の活動とともに増加し、レム睡眠が終了すると再び抑制される。覚醒が近づくと、HPA軸の活性化によってACTHが放出され、覚醒度が上昇して睡眠が終了する(129)。HPA軸と睡眠は、興奮作用と抑制作用の両方が高度に統合された関係にあり、睡眠の調節や位相に関与するHPA軸のレベルに依存している。睡眠のHPA軸制御は、CRH、ACTH、コルチゾール(コルチコステロン)の間で異なり、時に矛盾しているように思われる。CRHの連続投与はノンレム睡眠とレム睡眠に有意な変化を与えないが、ACTHの連続投与はレム睡眠時間を減少させる。一方、コルチゾール(コルチコステロン)はレム睡眠時間を減少させ、ノンレム睡眠を増加させる(132)。他の研究では、CRHとコルチゾール(コルチコステロン)がレム睡眠を促進することが明らかになっている(133)。この矛盾した関係は、CRHが交叉上核の「ペースメーカー」活動によって直接制御されているため、CRHが覚醒と覚醒の直接的な調節因子であることを示している。グルココルチコイドはCRH産生に対する抑制性フィードバック作用を通じて間接的に覚醒に影響を与えるだけである(134)。レム睡眠では、CRH はレム睡眠に影響を与えない、またはレム睡眠を増加させないという矛盾した結果が報告されているのに対し、コルチゾール(コルチコステロン) はレム睡眠時間を増加させたり減少させたりすることが報告されているため、CRH と コルチゾール(コルチコステロン) の正確な役割は不明のままである(135)。HPA軸活動が睡眠に及ぼす具体的な影響についてはまだ議論の余地があるが、睡眠とストレスの間には高度に統合された動的な関係があることから、ストレスのモデルが実験研究において睡眠に影響を与える道筋があることが示されている。拘束ストレス、強制運動、強制水泳、フットショックはすべて、コルチゾール(コルチコステロン)放出の強力なスパイクを引き起こす急速なHPA軸反応を誘発する能力を介して、ストレスモデルとして機能する。興味深いことに、これらのモデルは睡眠障害との関連性も高い。例えば、暗相の初めに2時間の拘束ストレスに曝露すると、暗相の残りの10時間はレム睡眠が増加することで示される睡眠のリバウンドが誘導される(136)。対照的に、穏やかな操作で睡眠障害を誘発しても睡眠リバウンドは生じず、拘束ストレスによって誘発された睡眠障害と睡眠リバウンドは2時間の間の睡眠喪失によるものではなく、代わりにストレス要因そのものの結果であることを示している。睡眠に対するストレスの具体的な影響は、時間帯、ストレス因子の強さ、代謝状態、睡眠の必要性など、多くの変数に依存して変化する。これにより、異なる量や種類のストレス要因がノンレム睡眠とレム睡眠に相反する形で影響を与えることがある。しかし全体的には、ストレスの結果としての睡眠/覚醒サイクルの変化は、行動や認知に長期的な影響を及ぼす可能性がある。
HPA軸の活性化が睡眠に影響を与えるのと同様に、HPA軸機能に対する睡眠の複雑な調節作用がストレス応答に影響を与える。睡眠は伝統的に、HPA軸の活性化が覚醒行動を誘発する前に、健康的で一貫した睡眠行動を維持するために、HPA軸を抑制する効果を持っている(137)。睡眠自体が変化すると、睡眠中のHPA軸の抑制的な制御がストレス反応の多動化につながる可能性がある。実際、臨床的な睡眠研究では、コルチゾール(コルチコステロン)の分泌プロファイルがヒトでは誘発覚醒と自然覚醒の両方に影響を受けることが示されている(138)。自然覚醒に対するコルチゾール(コルチコステロン)の反応は、ストレスと睡眠の経路が交差し、睡眠障害がストレス反応にどのように影響するかを示している。睡眠による抑制性制御の低下に起因するストレス反応の多動性は、グルココルチコイド による負のフィードバック作用を増加させ、ストレス反応にさらに影響を及ぼす可能性がある。このように、睡眠とストレスは密接に結合した関係にあり、同時に持続的に調節障害が起こるという悪循環が生まれているのである。実験的な強制運動ストレスモデルでは、動物を強制的に起きて活動することで睡眠障害を引き起こし、その結果、睡眠が奪われる。トレッドミルや車輪を使った24時間の強制運動は、ノンレム睡眠とレム睡眠の両方を廃止し、心拍数と体温を有意に上昇させ、HPA軸の活性化の増加を示す指標となる(139)。実験的な強制水泳ストレスモデルにおけるストレス応答とパフォーマンスは、概日リズムのタイミングに影響される。例えば、夜行性条件のラットは、強制水泳の 5 分間の経過において、昼行性条件のラットに比べて脱出行動が少ないことが示されている(140)。これらの行動の違いは血清コルチゾール(コルチコステロン)の変化を反映しており、強制水泳に夜行性で曝露されている間は昼間に比べて増加する。フットショックストレスモデルは、主に条件付きの文脈によって睡眠に影響を与える。例えば、フットショック恐怖条件付けに曝露してから24時間後に、条件付き文脈環境で眠るラットは、中立的な文脈環境で眠る動物と比較して、レム睡眠が選択的に減少する(141)。睡眠軸とストレス軸の相互作用、特にコルチゾール(コルチコステロン)の概日性放出に関しては、ストレス因子への曝露による睡眠の変化を、外傷性脳損傷後の転帰に対するストレスの影響において考慮すべきである。
多くの睡眠障害モデルまたは睡眠遮断モデルは、睡眠に対するストレスの影響を利用してレム睡眠を廃止している。高架プラットフォームモデル(通称「フラワーポット」モデルとして知られている)は、逆さにしたフラワーポットのような単一の小さなプラットフォームを水のプールに入れて、レム睡眠を選択的に阻害するものである(142)。動物はノンレム睡眠を維持することができるが、レム睡眠中の筋緊張の喪失により、動物は水の中に落ちてしまう。この方法は、このようにして、レム睡眠中に複数の覚醒を引き起こし、レム睡眠を完全に廃止するために恐怖条件付けを採用している。高架プラットフォームモデルに72時間曝露した後、ラットの循環コルチゾール(コルチコステロン)レベルと体温は上昇し、レム睡眠遮断後7日間持続する(143)。この制限的プラットフォームモデルの注意点は、単一プラットフォームでは、レム睡眠の廃止だけでなく、運動の制限と社会的孤立によるストレス反応が誘発される可能性があるということである。そこで、レム睡眠のための十分な一貫した空間を確保しつつも、動きの制限を少なくし、複数の動物を一度に試験できるように、水中に多数のプラットフォームを配置したマルチプラットフォームモデルが開発された(142)。ある研究では、複数のプラットフォ ーム法の間に複数のラットを収容すると、単一プラットフォ ーム法と比較して、社会的相互作用による強制的な覚醒を引き起こし、 睡眠発作の回数が減少することが示されているため、複数プラットフォ ーム法の有効性は議論の的となっている(144)。ストレスフリー睡眠モデルでは、ストレスによって誘発される睡眠喪失ではなく、睡眠の操作だけで睡眠が乱されることが保証されている。手動操作は一般的なストレスフリー睡眠モデルで、実験者が動物を優しく操作して睡眠を妨げるものである(145)。睡眠の防止には効率的であるが、手動操作は実験者間で一貫性がない場合があり、マウスは、穏やかな操作中に雌の実験者と比較して、雄の実験者に反応して嗅覚誘発性HPA活性化を有することを示唆する証拠がある(146)。自動睡眠破壊モデルは、実験者の介入なしに動物を目覚めさせるために、エアパフやスイーパーバーなどの機械的破壊物質を使用する。エアパフ法では、動物の睡眠状態を脳波と筋電図でモニタリングすることで、選択的にレム睡眠を阻害する。動物がレム睡眠に入ると、システムは自動的に空気のパフを放出して動物を妨害し、レム睡眠を中断させる(147)。代替的に、空気パフ手段は、予め定められた間隔で睡眠を中断させて、ノンレム睡眠とレム睡眠の両方をグローバルに中断させることができる。自動スイーパーバーも同様に、動物を強制的に覚醒させ、ケージの長さを移動する機械的なバーの上を移動させることによって睡眠を混乱させる(148)。エアパフ法と同様に、これは、脳波および筋電図信号に応答して選択的にレム睡眠を排除するか、または予め設定された間隔を介して全体的に睡眠を中断するために行うことができる。自動睡眠障害システムは、睡眠障害が過剰なストレス因子の存在なしにHPA軸反応性および炎症性プロファイルを調節していることを確実にする。
睡眠障害は外傷性脳損傷後のアウトカムを悪化させる
不眠症、過眠症、睡眠時無呼吸などの睡眠障害は、重症度にかかわらず、外傷性脳損傷を受けた人の30~70%に発生する(149)。これは、HPA軸機能障害と同様に、睡眠障害や障害が外傷性脳損傷後の患者にどの程度影響を及ぼすかが過小報告されていることが多いためである。日中の眠気や過眠症は受傷後の急性期に最もよく報告される睡眠障害の一つであるが、不眠症などの対極にある障害は、外傷性脳損傷後の人の5~25%で報告されている(149)。睡眠における傷害誘発性の変化は、外傷性脳損傷の実験モデルでも認められている。正中線外側液打損傷(FPI)は偽対照群と比較して受傷後最初の6時間の間に全体的な睡眠を増加させる(150)。この増加した睡眠必要性は、受傷後3時間のこの期間に中間線外側液打損傷(FPI)で再び受傷したマウスは、受傷後9時間の時点で2回目の受傷を与えられたマウスと比較して、運動障害、神経学的重症度スコア、不安に似た行動、神経病理が増加しているため、2回目の外傷性脳損傷に対する脆弱性の期間を定義している。この睡眠必要性の程度は限られており、睡眠必要性の増大はマウスの正中線外側液打損傷(FPI)後7日まで持続するが、受傷後2~5週間では外傷性脳損傷の結果として有意な睡眠障害を伴わずに速やかに回復する(151)。外傷性脳損傷の実験モデルでは、睡眠/覚醒行動の慢性的な欠損を再現することはできないが、ヒトへの翻訳のための候補メカニズムの同定には貢献している。例えば、オレキシンとしても知られるヒポクレチンは、主に覚醒行動を促進し、報酬、感情、ストレス回路の調節に関与している(152)。ある研究では、ヒポクレチンノックアウト(HCRT KO)マウスモデルを用いて、ヒポクレチンが外傷性脳損傷後の睡眠/覚醒を媒介する可能性があることを発見した(153)。中等度の制御皮質衝撃の後、HCRT KOマウスは、同じ損傷にさらされた野生型マウスは、より多くの非レム睡眠と少ない覚醒を持っていたが、睡眠/覚醒行動の変化を示さなかった。例えば、ブラストウェーブ外傷性脳損傷に曝露されたマウスは、TGFβシグナル、iNOS、IL-1βなどの神経細胞の生存と炎症を制御する重要な遺伝子において、傷害後8ヶ月の時点でDNAメチル化の長期的な違いを示している(154)。特筆すべきは、概日リズムの調節因子である概日時計3(Per3)や、セロトニンからメラトニンへの変換に関与する酵素をコードするAanatを含む重要な概日リズム遺伝子でメチル化の違いが見られたことである。これらの動物では睡眠障害は測定されなかったが、サーカディアン遺伝子のDNAメチル化の長期的な違いは、外傷性脳損傷による睡眠のエピジェネティックな変調の可能性を示唆している。
睡眠障害モデル、特に睡眠障害のみを介してHPA軸の活性化を調節するモデルは、HPA軸に関与する特異的なストレス因子としての外傷性脳損傷後の睡眠/覚醒障害の影響を探るために必要である。睡眠障害は炎症にも影響を与えるため、外傷性脳損傷後のストレスおよび免疫軸への影響をさらに特徴づけるための生理学的に関連するストレスモデルである。グルココルチコイドの概日性放出は、睡眠の開始と完了に影響を与える。その結果、睡眠はストレス因子に反応してHPA軸の活性化と反応性を低下させたり、増加させたりする。臨床的には、睡眠の喪失は、IL-1,IL-6,およびTNF-αなどの神経炎症反応にも関与する主要な炎症性サイトカインの発現を増加させる(155)。外傷性脳損傷後の睡眠障害に関する臨床調査は数多くあるが、睡眠の変化がHPA軸や炎症などのプロセスにどのように影響を与え、外傷性脳損傷後の転帰に影響を与えるかを調査した実験的研究はほとんどなく、代わりに受傷後の睡眠障害の広範な転帰は示されていない。例えば、ある実験研究では、ラットの強制運動を用いた48時間の完全な睡眠遮断は、軽度の外傷性脳損傷に対する神経細胞の感受性を増加させないことが報告されている(156)。別の研究では、マウスの正中線外側液打損傷(FPI)直後に6時間のジェントルハンドリング睡眠遮断を行ったところ、受傷後1,3,5,7日目の循環コルチゾール(コルチコステロン)レベル、神経学的重症度スコア、または新規物体探索において、睡眠遮断を行っていないマウスと比較して有意な差は認められなかった(157)。急性睡眠障害が実験モデルでは外傷性脳損傷後の回復に影響を与えないことを示す報告は限られているが、慢性的な睡眠障害の影響は依然として不明である。受傷後の持続的な睡眠障害は、外傷性脳損傷を受けた人がストレスや炎症の悪化に対してより脆弱になるメカニズムを示している可能性がある。
睡眠障害は、外傷性脳損傷の結果として精神症状や気分障害に影響を与える可能性がある。爆風に関連した外傷性脳損傷が陽性と判定された約5万人の軍人のうち、PTSDに対する外傷性脳損傷の影響の26%、うつ病に対する外傷性脳損傷の影響の41%を睡眠が媒介していることがわかった(158)。臨床的には、睡眠障害(特に不眠症)の存在は、外傷性脳損傷後の地域社会の統合を低下させ、生活の質を低下させることでうつ病の媒介者または加担者として作用する(159)。また、睡眠障害はPTSD(160)、疼痛(161)、精神疾患(162)などの外傷性脳損傷後の後遺症を媒介したり、寄与したりすることが強く示唆されている。例えば、うつ病や睡眠の質の低下などの自己申告は、外側眼窩前頭前野、背外側前頭前野、帯状皮質のfMRI変化と有意に関連している(163)。さらに、急性期後の外傷性脳損傷患者の大脳皮質MRI解析では、年齢と性別をマッチさせた対照群と比較して、前頭前野と帯状体の連結性と体積変化が一貫して確認されている(164, 165)。これらのデータをまとめると、これらの皮質領域の損傷誘発性変化が睡眠を媒介とする気分障害に寄与している可能性があることが示される。HPA軸機能障害と同様に、小児は外傷性脳損傷後の睡眠障害の影響を強く受けやすい。小児の外傷性脳損傷の実験モデルでも、慢性的な睡眠の変化が見られる。3週齢のマウスを用いて頭部閉鎖型外傷性脳損傷を繰り返し実験した後、11週齢までの環境濃縮は対照マウスのレム睡眠を増加させたが、外傷性脳損傷後の環境濃縮は効果がなかった(166)。睡眠は、外傷性脳損傷が恒常性を維持するための重要なプロセスに悪影響を与える高度に翻訳可能なメカニズムを表している。11~17歳の小児の受傷後、受傷直後の67%が睡眠導入、疲労、眠気に問題があると報告し、38%が受傷後2~3週間まで睡眠の問題が続くと報告している(167)。いくつかの研究では、子どもの睡眠障害は受傷の重症度とは無関係に 慢性的に持続し、受傷後2年まで睡眠障害が続くことが報告されている(168)。HPA軸機能障害や睡眠障害などの受傷後の合併症の影響を受けやすいため、小児外傷性脳損傷後に精神疾患を発症または悪化させる可能性が高くなる(169)。外傷性脳損傷後の精神障害は、精神機能障害、睡眠の変化、傷害の相互作用による症状や結果であることから、ホメオスタシス系とアロスタシス系に対する外傷性脳損傷の影響を理解することの重要性が示されている。
臨床前および臨床外傷研究の将来のステップ
本レビューでは、今後の前臨床外傷研究において引き続き検討すべき事項を明らかにした。
第一に、外傷性脳損傷に対する中枢および末梢免疫応答の時間的な進行は動的であり、回復は以前に受けた、同時に受けた、およびその後に受けた免疫課題の影響を受ける。この実験的軌跡は、外傷性脳損傷とストレスシステムが交差していることを明らかにしたデータによって強調されている。
第二に、脳損傷の長期的な影響を定義するためには、損傷後の慢性的なタイムポイントが必要である。前述したように、HPA軸機能障害の存在は外傷性脳損傷の実験モデルで確認されており、傷害の重症度に対応している。したがって、ストレス-免疫経路と交差する治療介入には、急性期および慢性期の傷害後の時点での特異的な投与戦略が必要となる可能性がある。
第三に、前臨床外傷性脳損傷モデルの開発が劇的に急増している。これは主に、爆発的な爆風や反復性脳震盪に曝露された後の脳損傷の評価が高まったことによるものである。これらのモデルは翻訳を向上させるために必要であるが、モデル特有のストレス-免疫反応を特定するためには多くの研究が必要である。げっ歯類モデルと並んで、他の動物傷害モデルの類似点や注意点をよりよく特定するために多くの研究が行われてきた。特に、ブタなどの大型動物モデルは、脳の大きさ、組織、発達がヒトと似ているため、さらなる洞察を提供する可能性がある(170)。最後に、種特異的な違いを理解しつつ、前臨床から臨床への移植を継続的に 行うことに重点を置かなければならない。
ある臨床調査では、参加者の4分の1近くが生涯にわたる外傷性脳損傷の既往歴を報告しており、これはQOLの低下と関連していた(171)。この生活の質の低下の一因として、障害や収入の低下、慢性的な健康不良のリスクの増加、活動が制限される日数の増加などが挙げられている。特筆すべきは、参加者はまた、一晩の睡眠時間が平均7時間未満であると報告していることである。仕事と健康状態は、外傷性脳損傷の回復と生活をする上で重要なストレス要因である。これらの環境ストレス因子は、受傷後の回復をさらに悪化させる可能性がある。外傷性脳損傷治療をさらに進歩させるために、脳損傷の臨床研究において考慮すべきことは数多くある。
第一に、臨床研究の多くは、診断の改善、多くの場合はバイオマーカー(172)そして外傷性脳損傷の即時治療(173)に焦点を当ててきた。しかし、前臨床研究と同様に、外傷性脳損傷の慢性的な影響を理解することに重点を置けば、回復に影響を及ぼす免疫応答やストレス応答についての必要な洞察を得ることができるだろう。
第二に、外傷性脳損傷患者の適切な慢性治療のためには、ホメオスタティックおよびアロスタティックシステムの長期的な臨床観察が必要である。これは、急性期と慢性期の両方の時点での外傷性脳損傷後のHPA機能障害の評価を標準化することで対応できる。(174). 最後に、急性期と慢性期の治療介入の必要性を認識することで、外傷性脳損傷後のストレス-免疫回復の時間的変化を管理する能力を広げることができるかもしれない。
炎症とストレスのプロセスは、外傷性脳損傷後に急性的にも慢性的にも変化する。ミクログリアのプライミングは、外傷性脳損傷後の神経炎症、神経細胞の変性、行動障害の悪化に寄与している。ここで検討したように、外傷性脳損傷後のHPA軸の機能不全は、ホメオスタシスを維持し、神経炎症性環境を調節する身体の能力に劇的な影響を及ぼす可能性があり、十分に評価されていないが、重大な傷害後の合併症であることが示されている。グルココルチコイドの産生と放出を介した環境の合図への適切な反応は、基底性の炎症性およびストレス反応に必要である。外傷性脳損傷によって誘発されたHPA軸機能障害が炎症に及ぼす長期的な影響は現在のところ不明であるが、受傷後のストレス-免疫軸の破壊は慢性的な炎症、精神疾患の発症や悪化、行動反応の変化に寄与している可能性がある。実験的外傷性脳損傷後のストレスモデルを応用すれば、傷害後の炎症がHPA軸の機能不全によって変調されたり、変調されたりするメカニズムをよりよく解明できる可能性がある。特に睡眠/覚醒障害の変化に対応して、外傷性脳損傷後のHPA軸機能障害は、傷害後の回復を著しく阻害し、生活の質を低下させる可能性がある。免疫軸とストレス軸の間のコミュニケーションの破綻に関与する分子機構の同定は、脳損傷後の治療を改善するための新たな治療標的を明らかにする可能性がある。