
Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets
www.ncbi.nlm.nih.gov/pmc/articles/PMC6890857/
Si Yun Ng1,Alan Yiu Wah Lee1,2,*。
要旨
外傷性脳損傷(TBI)は、世界的に見ても、民間人や軍人の罹患率と死亡率の主要な原因の一つである。TBIの複雑な病態生理に関する知識が進歩しているにもかかわらず、その基礎となるメカニズムはまだ十分に解明されていない。初期の脳損傷は急性かつ不可逆的な一次損傷を伴うが、その後の二次的な脳損傷は数ヶ月から数年かけてゆっくりと進行することが多く、治療的介入のための窓口となっている。現在までのところ、中枢神経系の二次障害の特徴的な事象として、軸索のワレリア変性、ミトコンドリア機能不全、興奮障害、酸化ストレス、ニューロンやグリアのアポトーシス細胞死などが挙げられている。これらのプロセスに関連した薬物投与可能な標的の同定に向けて、広範な研究が行われている。さらに、細胞標的への生理活性物質の効率的、特異的、制御されたデリバリー戦略を考案することにより、中枢神経系への治療薬のバイオアベイラビリティーを向上させるために、多大な努力が払われてきた。ここでは、TBIの病態とその背景にある分子機構について概説した後、新たな治療標的となる薬剤について紹介する。また、最近の中枢神経系への薬物送達に関する様々なアプローチの開発についても議論する。
キーワード 中枢神経系外傷、二次損傷、神経細胞再生、細胞貫通タンパク質、バイオポリマー、薬物放出制御
序論
外傷性脳損傷(TBI)は、あらゆる年齢層において、罹患率、障害、死亡率の主要な原因の一つとなっている(Bruns and Hauser, 2003; Dewan er al)。 世界的には、毎年5,000万人以上の人がTBIに苦しんでいる(Maas et al 2017)。2005年の時点で、約317万人のTBI生存者が神経学的、心理社会的問題から長期的な障害に至るまでの心的外傷後合併症を経験している(Zaloshnja et al 2008; Bazarian et al 2009)。TBI患者の臨床管理への膨大な支出とそれに伴う社会経済的問題は、医療制度と社会に大きな負担を強いている(Finkelstein et al 2006)。TBIの臨床的特徴や複雑な病態生理学的メカニズムの理解が進み、前臨床試験や第I/II相試験で有望な効果を示す新規治療法が開発されてきたが、その多くは第III相臨床試験では不成功に終わっている。実際、過去30年間で30件以上のTBI治療薬の診断・治療目的の臨床試験が失敗に終わっている。本レビューでは、TBIの病態形成における分子・細胞イベントの概要を紹介する。薬物化可能な標的と治療の新たな方向性についての最新情報を提供し、その後、これらの治療薬を制御された方法で送達するための様々なアプローチについての議論を行う。
TBIのカテゴリー
侮辱の固有の物理的メカニズムによると、TBIは3つのカテゴリーに分類される。(i)頭部閉鎖型、(ii)貫通型、(iii)爆発性爆風TBIである。TBIの臨床的特徴には、長時間の昏睡、頭痛、吐き気、失語症、発作、健忘症、および攻撃性や不安などの行動異常が含まれ、これらはTBI後数秒から数分以内に起こるが、これらの症状の中には数ヶ月から数年まで持続するものもある(Bruns and Hauser, 2003; Andriessen et al 2010)。
閉鎖性頭部TBIは一般的に、主に自動車事故、転倒、スポーツ活動による鈍的衝撃によって引き起こされる。この形態のTBIの発生率は、一般市民の間で最も高い。強い鈍的・圧迫的な接触力により、衝撃部位の直下にある脳の正常な機能が破壊され、脳血管系や神経細胞に即時的な損傷を与える。また、衝撃時に発生した振動や衝撃による脳の変位は、脳組織の圧迫や脳血流の低下を引き起こす。どちらのメカニズムも最終的には、局所的な局所挫傷または他の脳領域へのびまん性損傷をもたらす。
貫通型TBIは、異物が頭蓋骨を貫通し、硬膜を通って脳実質に侵入した場合に起こる。閉塞性頭部TBIと同様に、脳組織の裂傷は主に局所的な損傷、頭蓋内出血、脳浮腫および虚血を引き起こす。動きの速い投射物の侵入により、組織の空洞化が起こり、傷害をさらに悪化させることがある。神経学的損傷の種類および重症度は、脳を貫通する外部体の大きさ、速度、経路および強度に依存する。過酷な環境に脳組織がさらされているため、このタイプのTBIでは感染の可能性が比較的高くなる。このタイプの外傷は侵襲性が高いため、貫通型TBIは閉鎖型頭部TBIに比べて呼吸不全、肺炎、低血圧、脳脊髄液漏れなどの急性の合併症を伴う(Black et al 2002)。
20世紀のアフガニスタンやイラクを中心とした戦争関連TBIによる死傷者の有病率が高かったことから、最近では爆発性爆風TBIが新たなカテゴリーとして考えられている(Warden, 2006)。閉頭式TBIや貫通型TBIとは異なり、爆発によって発生した急激な圧力衝撃波によって脳が障害され、頭蓋骨から封じ込められた脳実質に膨大な量のエネルギーが伝達される(Ling and Ecklund, 2011)。爆風傷害の影響は、爆風誘発傷害の異なる段階での傷害結果に応じて、一次(衝撃波による内部損傷二次(貫通三次(爆風波による身体傷害四次(最初の三種類以外)のパターンに分けることができる(Cernak and Noble-Haeusslein, 2009; Risdall and Menon, 2011)。爆風で発生した運動エネルギーは脳の変形を引き起こし、その結果、灰白質と白質の両方に広範なびまん性の損傷を生じ、神経細胞死、軸索損傷、血液-脳-脳バリア(BBB)障害、血管痙攣、偽動脈瘤形成、充血、挫傷、および脳浮腫を引き起こす(Cernak and Noble-Haeusslein, 2009)。上記の臨床的特徴とは別に、外傷後ストレス障害は爆発性爆風TBIとの関連性が高く、TBI生存者の発生率が高いことが研究で示されている(Risdall and Menon, 2011)。
TBIの病態生理
TBIに伴う神経組織の損傷は2つのカテゴリーに分類される。
(i)一次損傷は、最初の脳梗塞の際に機械的な力によって直接引き起こされるものであり、(ii)二次損傷は、一次損傷後にさらに組織や細胞が損傷を受けることを意味する。
一次脳損傷
脳へのさまざまな機械的傷害の即時的な影響は、2つのタイプの一次傷害を引き起こす可能性がある:焦点性脳傷害とびまん性脳傷害である。研究では、中等度から重度のTBIを受けた患者では、両タイプの損傷が共存することが一般的であることが示されている(Skandsen et al 2010);しかしながら、びまん性軸索損傷(DAI)はTBI症例の約70%を占める。裂傷、圧迫、脳震盪の結果として、閉頭部TBIや貫通型TBIでは、頭蓋骨の骨折や損傷部位の中心部に局所的な挫傷を伴う局所的な脳損傷を示す(coup; Schmidt et al 2004)。神経細胞やグリア細胞の壊死領域は、血液供給が低下したクーデターに集中しており、脳の限られた層で血腫、硬膜外、硬膜下、脳内出血が発生する。二次性挫傷は、脳が跳ね返って頭蓋骨に衝突したときの二次的な衝撃により、クーデター(contre-coup)と反対側またはクーデターを取り囲む組織に発生することがある(Schmidt et al 2004)。受傷の重症度に応じて、認知障害、行動変化、片麻痺などを引き起こすことがある。局所損傷とは対照的に、びまん性脳損傷の主なメカニズムは、急激な減速と加速の非接触力であり、それによって脳組織に剪断損傷と伸張損傷が生じる。この強い引張力により、神経軸索、乏突起細胞、血液血管が損傷し、脳浮腫や虚血性脳損傷を引き起こす(Smith er al)。 びまん性TBIの特徴は、主に脳幹や胼胝などの皮質下や深部白質組織での軸索損傷が広範囲に及ぶことであり、軸索輸送の障害や軸索細胞骨格の劣化を伴う。注目すべきことに、これらの軸索損傷はTBI後も数ヶ月間持続することがあり、出血や脳浮腫などの遅発性の二次病理との関連が示唆されている(Saatman et al 2008)。軸索損傷と神経変性の程度は、TBIの重症度を決定する。興味深いことに、爆発性爆風TBIは慣性力ではなく衝撃波の結果であるが、典型的なびまん性脳損傷の特徴を示している。
二次脳損傷
一次損傷時に発生する生化学的、細胞的、生理学的事象は、しばしば数時間から数年に及ぶ二次損傷へと進行する。メカニズム的には、多くの因子が二次損傷に寄与しており、その中には、興奮毒性、ミトコンドリア機能不全、酸化ストレス、脂質過酸化、神経炎症、軸索変性、アポトーシス細胞死などが含まれる(Ray et al 2002; 図1)。
図1 外傷性脳損傷(TBI)の病態生理を模式的に示したもの
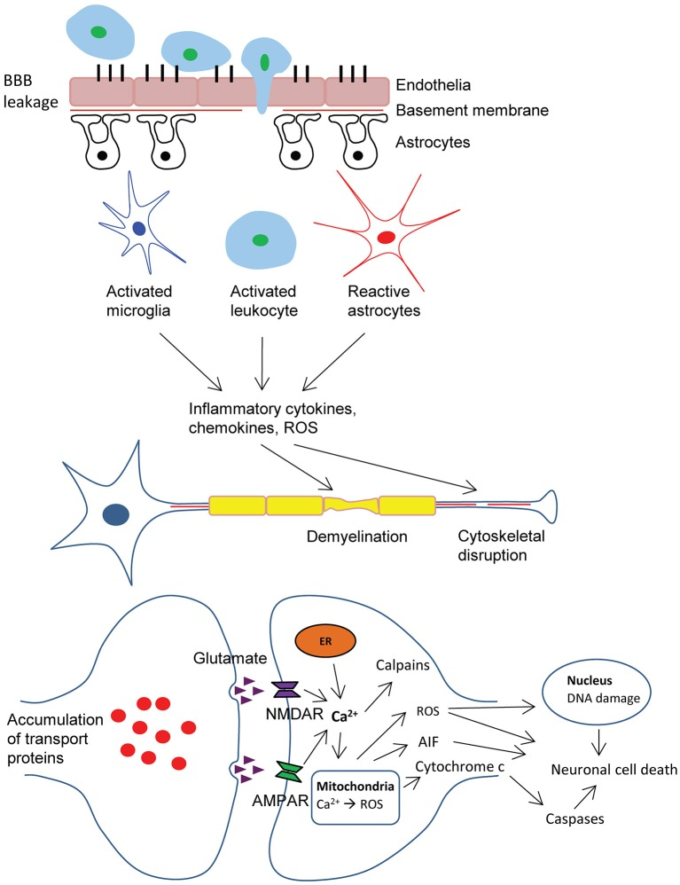
TBI傷害によるBBB機能障害は、活性化した白血球の脳実質への移行を可能にし、細胞接着分子のアップレギュレーションによって促進される。活性化した白血球、ミクログリア、アストロサイトは、サイトカインやケモカインなどの活性酸素や炎症性分子を産生し、軸索の脱髄や細胞骨格の破壊に寄与し、軸索の腫脹や末端への輸送タンパク質の蓄積を引き起こし、神経細胞の活動を低下させる。進行性の軸索損傷は神経変性をもたらす。また、病変部のアストログリア症はグリア瘢痕形成を引き起こし、軸索の再生を阻害する非寛容な環境を作り出する。一方、切断された神経細胞からの流出によるシナプス空間へのグルタミン酸・アスパラギン酸神経伝達物質の過剰蓄積、シナプス前神経末端からのグルタミン酸誘発性放出の悪化、外傷性・虚血性脳での再取り込み機構の障害は、シナプス後膜に位置するNMDA・AMDA受容体を活性化し、カルシウムイオンの流入を可能にする。細胞内貯蔵庫(ER)からのCa2+イオンの放出とともに、これらの事象は活性酸素の産生とカルパインの活性化につながる。ミトコンドリアの機能不全の結果、アポトーシス誘導因子(AIF)やチトクロームCなどの分子がサイトゾルに放出される。Fas-Fasリガンドの相互作用を含むこれらの細胞および分子イベントは、最終的にカスパーゼ依存性および-非依存性の神経細胞死につながる。BBB、血液脳関門、ROS、活性酸素種、AMPA、α-アミノ-3-ヒドロキシ-5-メチル-4-イソオキサゾールプロピオン酸、NMDA、N-メチル-d-アスパラギン酸、ER、小胞体。
興奮毒性
動物とヒトの両方の研究で、TBI時のBBB破壊および一次神経細胞死がシナプス前神経末端からグルタミン酸やアスパラギン酸などの興奮性アミノ酸の過剰放出を誘発することが実証されている(Faden et al 1989; Chamoun et al 2010)。TBI時の過剰なグルタミン酸の存在は、グルタミン酸トランスポーターの機能不全によるグルタミン酸再取り込みの障害によっても寄与している。TBI後24時間以内にアストロサイトナトリウム依存性グルタミン酸トランスポーターGLAST(EAAT1)およびGLT-1(EAAT2)の発現が40%減少し、グルタミン酸の再吸収の有意な減少につながることを示す証拠がある(Rao et al 1998; van Landeghem et al 2006)。これらの興奮性アミノ酸は、イオントロピックグルタミン酸受容体(iGluR)とメタボトロピックグルタミン酸受容体(mGluR)の両方を活性化する。N-メチル-d-アスパラギン酸(NMDA)受容体やα-アミノ-3-ヒドロキシ-5-メチル-4-イソオキサゾールプロピオン酸(AMPA)受容体のようなiGluRのメンバーは、グルタミン酸と結合したときにNa+、K+、Ca2+のイオン流束を可能にし、神経細胞の膜脱分極を引き起こすリガンドゲートイオンチャネルである(Meldrum, 2000)。NMDA受容体は電圧依存性であり、Ca2+イオンにも透過性があるという特殊な性質を持っている。過度のグルタミン酸によるAMPAおよびNMDA受容体の過剰活性化は、細胞外Ca2+およびNa+イオンの流入を可能にし、シナプス後のニューロンにおけるイオンの恒常性を変化させることが示されている(Sun et al 2008; Brustovetsky et al 2010)。NMDAによる細胞内Ca2+の急増は、Ca2+/カルモジュリン依存性プロテインキナーゼII(Folkerts et al 2007マイトジェン活性化プロテインキナーゼ(MAPK;Lu et al 2008)およびプロテインホスファターゼ(Bales et al 2009)を含む様々な下流のシグナル伝達分子の活性化を開始する。プロテインキナーゼCもまた活性化されてNMDA受容体に結合し、それによってシナプス後神経細胞へのCa2+流入を促進する(Luo et al 2011)。同様に、AMPA受容体の活性化もまた、カルシウム依存性のメカニズムを介してMAPK経路を引き金とすることができる(Schenk et al 2005)。グルタミン酸によるNMDA受容体の活性化は、活性酸素種(ROS; Reynolds and Hastings, 1995; Girouard et al 2009)および一酸化窒素(NO; Sattler et al 1999)の産生を促進し、これは二次的な細胞傷害をさらに悪化させる。iGluRとは異なり、mGluRはGTP結合タンパク質を介してCa2+および下流のシグナル伝達を調節する。mGluRのグルタミン酸刺激はホスホリパーゼC/イノシトール-1,4,5-三リン酸の活性化を引き起こし、細胞内貯蔵から細胞質へのCa2+放出を促し、損傷を受けた中枢神経系のシグナル伝達カスケードの引き金となる(Weber, 2012)。また、細胞質内の過剰なCa2+は、カルシニューリン、カルパイン、カスパーゼなどのアポトーシス細胞死を引き起こす多くのタンパク質を活性化する。さらに、Ca2+と活性酸素の蓄積は、ミトコンドリア機能の障害を引き起こし、Ca2+と活性酸素の恒常性の調節障害をさらに悪化させる。要約すると、興奮性神経伝達物質の大量放出によるグルタミン酸受容体の過剰な刺激は、外傷後の酸化ストレスおよび長期間にわたる興奮性細胞死を引き起こし、これは死亡率の増加および6ヶ月間の神経学的転帰の悪化と相関している(Deshpande et al 2008年;Chamoun et al 2010)。
ミトコンドリア機能障害
ミトコンドリアの機能不全は、TBIの特徴的なイベントの一つであり(Xiong et al 1997細胞死を引き起こす代謝および生理学的な調節障害に寄与している。細胞内Ca2+の隔離およびミトコンドリアへの過剰なイオンの流入は、活性酸素の産生、ミトコンドリア膜の脱分極、およびATP合成の阻害をもたらす(Lifshitz et al 2004; Singh et al 2006)。これは、電子輸送鎖の破壊と酸化的リン酸化過程の障害をもたらし、したがって、細胞生存のための代謝反応の回復とカルシウムサイクルの調節を阻害する。また、ミトコンドリア透過性遷移孔(mPTP)もこのような条件下で活性化される。内膜タンパク質であるアデニンヌクレオチドトランスロケーター(ANT)がシクロフィリンDと結合して構造変化を起こすと、mPTPが開き、内膜透過性が上昇する(Susin er al)。1998; Naga er al)。2007; Tsujimoto and Shimizu, 2007)。ミトコンドリアを電子顕微鏡で解析したところ、クリスタエ膜の破壊や膜電位の低下など、ミトコンドリアの著しい膨潤や構造的損傷が明らかになった。さらに、アポトーシス細胞死に重要な役割を果たすチトクロムCやアポトーシス誘導因子(AIF)などのミトコンドリアタンパク質がサイトゾルに放出されている(Sullivan et al 2002;Singh et al 2006)。
活性酸素の放出と脂質過酸化
蓄積されたエビデンスは、酸化ストレスがTBIの発症にかなりの程度まで寄与していることを示唆している。内因性の活性酸素とフリーラジカルは、酵素過程、活性化された好中球、興奮毒性経路、ミトコンドリアの機能不全など、様々な原因からTBI後に絶えず発生している(Xiong et al 1997; Shohami and Kohen, 2011)。一方、TBI後のCa2+の蓄積は、一酸化窒素合成酵素(NOS)の活性を高め、NOの産生を助ける。過剰なNOとフリーラジカルのスーパーオキシドとの反応はペルオキシナイトライト(PN)の形成をもたらし、これは酸化的損傷を誘発し、3-ニトロチロシン(3-NT)や4-ヒドロキシノネナール(4-HNE; Hall et al 2004年)のような酸化的マーカーを検出することによって測定することができる。生体内研究では、TBI後の同側皮質および海馬における3-NTおよび4-HNEのレベルの増加が示されている(Hall et al 2004; Singh et al 2006; Deng et al 2007; Ansari et al 2008a)。酸化ストレスはまた、損傷を受けた大脳皮質と海馬のシナプス可塑性の障害と関連しており、損傷後24時間から48時間の間にシナプシン-1と脳卒中後認知症-95というシナプス蛋白質の損失を伴う(Ansari et al 2008a,b)。これらの活性酸素は、タンパク質やDNAだけでなく、膜リン脂質中の多価不飽和脂肪酸と反応するだけでなく、順番にリポペルオキシラジカルを形成し、さらに細胞膜を損傷する。ミトコンドリア膜の透過性の増加と膜タンパク質の酸化は、イオン輸送の変化につながる。例えば、異常なCa2+蓄積は、長時間の興奮毒性に深く関係している(Praticò er al)。 つまり、TBIにおける高活性酸素フリーラジカルの持続的な放出とそれに伴う活性酸素媒介性脂質過酸化のレベルの上昇は、脳の可塑性、脳血流に悪影響を与え、免疫抑制を促進する(Ansari et al 2008a)。
神経炎症
TBI後の24時間の急性期内では、BBBの機能不全により、循環好中球、単球およびリンパ球の損傷した脳実質への浸潤が可能になる(Lotocki et al 2009)。TBI患者の脳脊髄液(脳脊髄液)および死後組織の分析(Buttram et al 2007年;Frugier et al 2009年;Goodman et al 2009)およびげっ歯類の組織の分析(Ahn et al 2004年;Lotocki et al 2009年;Semple et al 2010)により、これらのポリモンは、脳脊髄液(脳脊髄液)および死後組織に存在することが明らかになった。2010)では、これらの多核白血球が補体因子やIL-1β、IL-6,TNF-αなどの炎症性サイトカインを放出していることが明らかになり、それに対応するmRNAとタンパク質の24時間後の増加によって明らかになった。様々なサイトカインの持続的なアップレギュレーションは、BBB透過性の変化、浮腫の形成、神経学的障害と関連していることがわかった。Fasスーパーファミリーの一員として、TNF-αはFasリガンドと密接に相互作用し、プログラムされた細胞死に不可欠なカスパーゼを活性化する(Morganti-Kossmann et al 2002)。MIP-α、MCP-1およびIL-8(CXCL8)などのケモカインは、外傷後に有意に増加し、これらのケモカインは相乗的に作用し、白血球の傷害部位へのさらなるリクルートに関与している(Kossmann et al 1997;Buttram et al 2007;Bye et al 2007;Semple et al 2010)。さらに、内皮細胞および白血球細胞接着受容体のリガンドであるICAM-1およびVCAM-1の発現のアップレギュレートは、白血球および免疫細胞の内皮との相互作用を促進し、それゆえに、それらの損傷部位へのリクルートを促進する(Calllossman et al 1997;Rancancan et al 2001)。神経炎症の長期化と遅延は、マクロファージのリクルートを促し、常駐するミクログリア細胞を活性化し、アストログリア症を促進する(Morganti-Kossmann et al 2007; Bye et al 2011)。TBI生存者のマクロファージおよび活性化ミクログリアの蓄積は、受傷から数年後のTBI生存者における進行性のファゴサイトーシスおよび持続的な炎症反応によって明らかである(Gentleman et al 2004年;Johnson et al 2013)。
軸索変性
ワレリアン変性は、DAI後数分以内に広く観察される。即時の機械的損傷は、縦方向に配向したニューロフィラメントと微小管からなる軸索細胞骨格ネットワークの混乱をもたらす(Tang-Schomer et al 2010)。急性軸索損傷は、カルシウムを介した恒常的なタンパク質分解とともに進行し、最初の外傷の数日後から数ヶ月後に遅発性および二次性軸索切断に発展し、ミエリン鞘の分解、軸索輸送の障害、および軸索輸送タンパク質の蓄積を特徴とする(Povlishock, 1992; Saatman et al 2003; Büki and Povlishock, 2006)。軸索接続の解離および結節における軸索輸送タンパク質の蓄積に起因する退縮球根の形成は、最終的には、損傷した軸索の長期にわたる腫脹およびニューロンおよびオリゴデンドロサイトのアポトーシス細胞死をもたらし得る(Büki and Povlishock, 2006)。DAIの特徴として、これらの退縮球根は軸索マーカーであるβアミロイド前駆体タンパク質(β-APP)とニューロフィラメント(NF)によって、びまん性TBIの実験モデルでは早ければTBI後1日、最長で2週間で検出される。屈折球は、主に脳幹の冠状皮質と錐体路に存在するが(Pierce et al 1996; Hellewell et al 2010海馬、大脳皮質、帯状体、内被膜、外被膜にも存在することが報告されている(Hellewell et al 2010)。Hellewell er al)。 (2010)は、冠状動脈の軸索損傷と神経炎症性細胞(ミクログリアやマクロファージ)の浸潤との関連性を示しており、これが血流障害、軸索の劣化、オリゴデンドロサイトの損傷、白質の変形につながることを示している。
グリア瘢痕とミエリン関連軸索成長阻害剤
中枢神経系への傷害はしばしばアストロサイトの活性化と増殖を引き起こす。その結果、反応性アストロサイトは病変部位に浸潤し、反応性アストログリア症を発症する。アストロサイトとオリゴデンドロサイト、髄膜細胞、ミクログリア、線維芽細胞が混ざり合うことで、瘢痕のような構造を形成し、これが軸索再生の主要な物理的障害となり、TBIの回復を阻害することが示唆されている(Fawcett and Asher, 1999)。しかし、最近の所見では、中枢神経系の損傷後にアップレギュレートされるグリア瘢痕のニューロカンやバーシカンなどのコンドロイチン硫酸プロテオグリカン(CSPG)が、実際には軸索再生を阻害する分子障壁であることが示唆されている(Asher et al 2000年、2001年、2002)。グリア瘢痕の他の抑制性分子、例えばテナシンやセマフォリン3Aなどとともに、これらの分子は軸索成長のための非許容的な環境を構成している(Zhang et al 1997; Pasterkamp et al 2001; De Winter et al 2002)。興味深いことに、RhoA経路は、RhoA活性またはその下流のエフェクターを遮断することで、これらの基質上での神経軸索の寛容な成長を促進するため、これらの抑制効果を媒介することに関与している(Winton et al 2002; Monnier et al 2003)。例えば、グリア瘢痕におけるセマフォリン3Aによって誘発されるシグナル伝達カスケードは、ニューロピリン-プレキシン受容体複合体およびRhoGTPアーゼの活性化に関与しており、これらは、F-アクチン細胞骨格の調節を介して成長コーンの崩壊を誘導すると考えられている(PasterkampおよびKolodkin、2003)。
さらに、切断された軸索内のミエリンが損傷すると、ミエリン関連糖タンパク質(MAGオリゴデンドロサイトミエリン糖タンパク質(OMgpNogo-A)などの軸索伸長阻害剤の暴露を引き起こす(Chaudhry and Filbin, 2006)。興味深いことに、これらのミエリン関連阻害剤は、神経細胞膜上のNogo受容体(NgR)複合体に特異的に結合し、その複合体は、p75NTR、Troy、およびLINGO-1という共受容体から構成されている(Wang er al)。 これらの阻害剤は、RhoA GTPasesおよびRhoキナーゼの活性化を誘発し、成長円錐体の崩壊および神経突起の後退を誘導することができる(Nash et al 2009)。実際、外傷を受けたヒトの脳組織の死後分析では、反応性グリアと腫脹した神経突起におけるRhoAおよびRhoBタンパク質の発現が増加しており、これはTBI後も数ヶ月間持続することが明らかになっている(Brabeck et al 2004)。実験的に誘発された焦点性脳損傷では、外傷後18時間後に病変部の皮質と海馬に活性なRhoAが蓄積していることが明らかになった(Dubreuil et al 2006; Zhang Z. et al 2008)。TBIにおけるRho GTPase経路の正確な役割については、さらなる研究が必要であるが、脊髄損傷や脳虚血などの中枢神経系損傷への関与は十分に確立されている(Dubreuil et al 2003; Yagita et al 2007)。RhoAは軸索の再生を阻害するだけでなく、軸索や神経細胞の再生を阻害し、アポトーシス反応にも関与していることが示唆されている。
アポトーシス細胞死
ニューロンとオリゴデンドロサイトのアポトーシス細胞死は、二次脳損傷の特徴である(Beer et al 2000; Grady et al 2003)。Smithら(1997)は、ヒト海馬ではTBI後最大1年間、ニューロンの細胞死が明らかであることを報告している。これらのアポトーシス事象は、カスパーゼやカルパインなどのシステインプロテアーゼの活性化を伴い、細胞外シグナル調節キナーゼ(ERKp38MAPK、ジャナスキナーゼ/シグナル変換器および転写活性化因子(JAK/STAT;Kawasi et al 1997;Mori et al 2002;Raghupathi et al 2004;Zhao et al 2011)などの様々な神経化学的、細胞的および分子的経路の相互作用によって誘発され得ることが報告されている。カスパーゼ依存性のメカニズムによって引き起こされるアポトーシス細胞死は、外因性の死の受容体経路または内因性のミトコンドリア経路によって誘導され得る(Stoica and Faden, 2010)。外因性経路は、TNFおよびFasと細胞表面上の特定の受容体との相互作用を伴うのに対し、内因性経路は、ミトコンドリアの脱分極後にチトクロームcが放出されたときに活性化される(Sullivan et al 2002)。シトクロムcはアポトーシス活性化プロテイン-1とATP依存性複合体を形成し、サイトゾル内でATPと反応する。どちらのメカニズムも、最終的にはカスパーゼ3の切断および活性化につながるカスパーゼ8および9のアップレギュレーションおよび活性化を介して、カスパーゼ依存性の下流シグナル伝達を活性化する(Clark et al 1999, 2000; Zhang et al 2003)。一方、TBIにおけるカスパーゼに依存しないアポトーシスは、スペクトルリンおよびNFタンパク質などの細胞骨格タンパク質のタンパク質分解を介したカルパインの活性化(Deng et al 2007)およびAIF(Hong et al 2004Smac/DIABLO、Omi/HtrA2,ポリ(ADP-リボース)ポリメラーゼ-1およびエンドヌクレアーゼGなどのミトコンドリアタンパク質の放出(Mammis et al 2009)によって開始され得る。これらのミトコンドリアタンパク質は、核内に移動し、下流のシグナル分子を活性化し、その結果、神経細胞およびグリア細胞において、DNA損傷およびクロマチンの縮合をもたらす。アポトーシスは、Bcl-2ファミリーのような抗アポトーシスタンパク質およびBaxのような死を誘導する因子によって制御され得る(Wennersten et al 2003)。研究は、Bcl-2タンパク質の発現が、TBI患者の脳組織において有意にアップレギュレートされていることを示している(Clark et al 1999)。同様に、Baxタンパク質の25%の増加が外傷性ラット脳で観察された(Raghupathi et al 2003)。
オートファジーとリソソソーム経路の障害
オートファジーは、リソーム依存性分解経路を介して細胞内小器官やタンパク質のターンオーバーを制御する適応的な恒常性プロセスである(Mizushima er al)。 オートファジーは、細胞が切断されたりストレスを受けたりした際に、細胞内の異常なタンパク質や小器官を排除することで、細胞の安定性を維持し、生存を維持するために重要な役割を果たしているが、アポトーシスによる細胞死、炎症、適応免疫応答の調節にも関与している(Maiuri er al)。 マクロオートファジーは、最もよく知られているオートファジーのサブタイプの一つであり、オートファゴソームとして知られている二重膜構造に損傷を受けた小器官やタンパク質などの細胞質成分を隔離し、その後、タンパク質分解が起こるリソソソームとの融合を伴う多段階のプロセスである(Mizushima, 2007)。このオートファゴシックフラックスは、ATG9,オートファゴソームマーカータンパク質LC3-II、オートファゴソーム形成に必須のベクリン1タンパク質などのオートファジー関連(ATG)タンパク質ファミリーのメンバーによって厳密に制御されている。蓄積されたエビデンスは、TBIやSCIの二次損傷過程におけるオートファジーリソソーム経路の関与を示唆しているが、オートファジーリソソーム経路が有益な役割を果たすか有害な役割を果たすかについては、まだ議論の余地がある。オートファジーマーカーのアップレギュレーションとオートファゴソームの蓄積は、二次損傷の初期段階で観察されており、それは重症度と相関し、数週間から数ヶ月間持続することがある(Diskin et al 2005; Clark et al 2008; Sakai et al 2014; Au et al 2017)。ラパマイシンによって増強され得るオートファジーフラックスの増加は、神経行動機能の改善、神経細胞の生存率の向上、炎症の減少、および損傷を受けた脳におけるグリオシスと関連している(Erlich et al 2007;Zhang Y. B. et al 2008)。実際、多くの神経保護剤は、オートファジーを活性化することにより、TBI誘発二次傷害を緩和する(Ding et al 2015年;Gao et al 2017年;Zhang et al 2017)。それにもかかわらず、リソソソーム機能は、リソソーム膜透過性の増加を伴うTBIにおいてしばしば損なわれることが発見されている。これは、オートファゴフラックスの障害とオートファゴソームおよびその貨物の病理学的蓄積をもたらし、神経細胞死を引き起こし、外傷の重症度を悪化させる(Sarkar et al 2014)。
潜在的な治療法
TBIの一次損傷は通常、急性の物理的損傷と壊死性細胞死を伴うため、治療法は主に損傷部位の安定化と二次損傷の予防を目的としている。上述したように、二次的損傷は、様々な危険因子の配列によって引き起こされ、進行性に発達する。これは、持続的な炎症反応、興奮毒性、酸化ストレス、アポトーシス細胞死を含む、傷害震源地を越えてニューロンおよびグリア細胞のさらなる喪失を誘発しうる事象への治療的介入の窓を提供する(Ray et al 2002)。二次性脳損傷の基礎となるメカニズムをよりよく理解するために、広範囲にわたる研究が行われてきた(表1)。
表1 外傷性脳損傷の病態生理、治療標的、可能性のある治療法のまとめ

興奮毒性に対する神経細胞とグリアの保護
グルタミン酸受容体拮抗薬
非競合的NMDA受容体拮抗薬であるHU-211(デキサナビノール)は、ニューロン培養においてNMDA受容体を介した神経毒性を減衰させることが示されている(Nadler et al 1993)。それは、外傷後3日目に投与されたときにラットの脳におけるNMDA誘発性Ca2+蓄積の有意な減少によって明らかなように、生体内試験でも同様に強力である(Nadler et al 1995)。HU-211の外傷後投与は、グリアおよびニューロン細胞のBBB機能不全、脳浮腫、TNF-α産生、ならびにアポトーシスを減少させる(Eshhar et al 1995; Shohami et al 1997)。同様に、別のNMDA受容体アンタゴニストMK801(ジゾシルピン)は、酸化ストレス、ミクログリア活性化、酸化ストレス、軸索損傷および神経細胞死を減少させることが示されている(Goda et al 2002;Imer et al 2009)。重要なことに、これらの効果は、認知機能および神経学的転帰の改善と関連している(Shohami et al 1995,1997)。同様に、AMPA受容体アンタゴニストNBQXは、神経軸索およびオリゴデンドロサイトにおける損傷を減衰させることが示された(Follett et al 2000;Goda et al 2002)。これらのグルタミン酸受容体拮抗薬は、実験的TBIの様々なモデルで神経保護効果を示すが、臨床試験ではTBI患者の神経学的転帰を改善することはできなかった(Maas et al 2006, 2010; Jain, 2008)。動物を用いた前臨床試験と患者を対象とした臨床試験との間に矛盾が生じたのは、グルタミン酸介在性興奮毒性が一次神経損傷直後の急性現象であることに起因していると考えられる。外傷を受けたヒトの脳におけるグルタミン酸の持続的な上昇は、むしろ、一次海馬ニューロンにおけるNMDA受容体拮抗薬のアポトーシス促進作用を実証した以前の報告(Hardingham et al 2002)に裏付けられているように、免れたニューロンの生存を維持する神経保護機構である可能性がある。実際、NMDARは、神経保護効果と神経毒性効果の両方を媒介することが知られている(Hardingham, 2009)。対照的な機能は、四量体NMDARのGluN2サブユニットの異なる特性および差動分布に起因すると考えられている。GluN2Aを含む受容体は主にシナプスに局在するが、GluN2Bを含む受容体はシナプスおよびシナプス外の両方の場所に存在する。GluN2Aは生存促進作用を有することが知られているのに対し、GluN2Bは興奮性グルタミン酸刺激後の細胞死を促進することが知られている(Liu et al 2007)。したがって、NMDARの機能を無差別にブロックすることは、興奮毒性を低下させず、プロ生存シグナルを抑制する可能性がある。
カルシウムチャネルおよびカルシウム活性化酵素の阻害剤
L-およびN-カルシウムチャネルのような電圧感受性の高いイオンチャネルの過剰活性化は、カルシウムの恒常性の長期的な変化を引き起こし、TBIにおける二次傷害時の興奮毒性を助長するもう一つの重要な因子である。実際、多くのカルシウムチャネル阻害剤が実験的TBIにおいて神経保護効果を示すことが示されている。流体打撲脳損傷ラットモデルでは、カルシウムチャネル遮断薬SNX-111(Ziconotide)は、外傷後6時間の時点で同側領域における外傷誘発性カルシウム蓄積を50-70%減少させることが明らかになっている(Samii et al 1999)。別のカルシウムチャネル阻害剤(S)-エモパミルは、脳浮腫および脳血流を減少させることが示されている(Okiyama et al 1992, 1994)。SNX-111および(S)-エモパミルの両方とも、脳損傷に関連する運動障害および認知障害を改善することができる(Okiyama et al 1992; Berman et al 2000; Verweij et al 2000)。45%のアミノ酸類似性を有するSNX-185は、SNX-111と同様のメカニズムで作用するが、バイオアベイラビリティが改善され、脳内での持続性が延長されている(Newcomb et al 2000年;Lee et al 2004)。L型電圧感受性カルシウムチャネル拮抗薬ニモジピンもまた、ラットの記憶障害に有益な効果があることが判明したが、血圧降下作用とTBI生存者で観察された頭蓋内圧の改善が見られなかったため、臨床試験は中止された(Bailey et al 1991; Veng et al 2003; Maas et al 2010)。さらに、カルシウムチャネル遮断薬であるニカルジピンの試験でも、臨床的な効果は控えめです(Compton et al 1990)。最近の研究では、カルパイン阻害剤であるMDL-28170は、TBIと低酸素性虚血性障害の両方において、神経細胞の損傷部位に局在する細胞骨格タンパク質α-spectrinの分解を抑制し、カルパインとカスパーゼ-3の阻害を通じて壊死とアポトーシスの減少と関連していることが示唆されている(Kawamura et al 2005年;Thompson et al 2010)。TBI動物をMD-28170で前処理すると、損傷した軸索のAPP(軸索輸送障害のマーカー)およびRMO-14(神経フィラメント圧縮のマーカー)の免疫標識が減少することで示されるように、軸索構造の保存および軸索膜漏出の減少を介して神経保護効果も発揮する(Buki et al 2003; Ai et al 2007; Czeiter et al 2009)。同様に、カスパーゼ-3阻害剤Z-DEVD-fmkは、神経細胞-グリア共培養において神経細胞死を減少させ、マウスおよびラット脳の誘導損傷における神経機能の改善および病変体積の減少に十分である(Clark et al 2000年;Knoblach et al 2004)。
ニューロンとグリアへの化学的ストレスとの闘い
酸化防止剤
mPTPの強力な調節因子である免疫抑制剤シクロスポリンAは、TBIの実験モデルにおいて神経保護効果を有することが実証されている(Kulbe et al 2018)。シクロスポリンAの正確な作用機序はまだ十分に理解されていないが、TBI後のその投与は、細胞質ホファスターゼであるカルシニューリンとmPTPでのCyp-Dとの結合を介したCa2+の蓄積の減少と関連している。シクロスポリン治療はまた、チトクロムcのミトコンドリアへの放出およびCa2+のミトコンドリアへの流入を阻害する(Sullivan et al 2005)。さらに、シクロスポリンAは、脂質過酸化を抑制することで抗酸化作用を示す(Turkoglu et al 2010)。これらの作用は、軸索損傷およびミトコンドリア機能不全の改善につながり、その結果、皮質損傷の減少および神経学的転帰の改善をもたらす(Okonkwo and Povlishock, 1999; Okonkwo et al 1999; Scheff and Sullivan, 1999; Sullivan et al 1999, 2000, 2010; Alessandri et al 2002; Mbye et al 2008)。それにもかかわらず、TBIにおけるシクロスポリンAの小規模無作為化臨床試験では、驚くべきことに、健常者と比較して患者の神経学的転帰および生化学的パラメータの改善が認められなかったことに留意すべきである(Mazzeo et al 2009)。しかし、シクロスポリンAを有効成分とするNeuroViVe社が開発した薬剤NeuroSTATの欧州多施設共同第II/III相臨床試験が最近開始され、TBI患者を対象とした結果はまだ評価されていない。
メチルプレドニゾロンは合成グルココルチコイドであり、主に抗炎症作用や中枢神経系の浮腫を抑制する作用があることから、急性期の中枢神経系損傷の臨床治療に広く用いられている。興味深いことに、メチルプレドニゾロンの高用量投与は、その抗酸化作用により神経保護効果を発揮し、特に外傷後の脂質過酸化を抑制する。メチルプレドニゾロンの抗酸化作用のメカニズムについてはほとんど知られていないが、メチルプレドニゾロンは脂質二重層の構造に溶け込み、細胞膜をより強固にし、それによって脂質過酸化ラジカルの移動性を制限すると考えられている(Hall, 1992)。メチルプレドニゾロンは、最大の抗炎症効果と神経保護効果を確保するために、中枢神経系損傷の初期段階で最適な濃度で投与されなければならない。メチルプレドニゾロンは、2004年にCRASHとして知られる無作為化プラセボ対照試験に組み込まれたことがある。この試験では、10,000人以上のTBI患者を対象に、2週間の追跡調査期間を設けて大規模なサンプルが募集された。それにもかかわらず、結果は死亡率の増加という望ましくないものであった(Thompson and Bakshi, 2005)。実際、メチルプレドニゾロンで処置されたラットはまた、視床下部、下垂体および海馬における神経細胞のアポトーシスの有意な増加を示した(Chen et al 2011;Zhang et al 2011これらは記憶および学習障害と関連している(Chen et al 2009)。
抗炎症剤および抗アポトーシス剤
半合成テトラサイクリン誘導体であるミノサイクリンは、BBBを介して移行・拡散する能力を有しており、脳卒中、SCI、アルツハイマー病、TBIなどの神経疾患の様々な実験モデルにおいて、抗炎症作用および抗アポトーシス作用を示すことが明らかにされている。数多くの研究により、ミノサイクリンの神経保護効果は、ミクログリアの活性化、増殖およびIL-1β、IL-6およびTNF-αなどの炎症性サイトカインの産生を阻害することに起因することが実証されている(Sanchez Mejia et al 2001年;Bye et al 2007年;Choi et al 2007年;Parachikova et al 2010年;Garrido-Mesa et al 2013)。例えば、閉鎖性頭部損傷のマウス実験モデルでは、ミノサイクリン投与により大脳皮質のIL-1βレベルが50%も著しく低下し、同時にミクログリアの活性化が抑制され、神経学的転帰が改善された(Bye et al 2007年;Ng et al 2012)。興味深いことに、ミノサイクリン治療は、マトリックスメタロプロテアーゼを阻害し、BBBの完全性を維持し、脳浮腫の軽減につながることがわかっている(Homsi et al 2009)。ミノサイクリンはまた、カスパーゼ活性を阻害することにより抗アポトーシス特性を示すことが示されている(Sanchez Mejia et al 2001)。さらに、Siopiら(2011)は、ミノサイクリンの投与により、神経保護性可溶性APPαのレベルがトラウマの24時間後に有意に回復し、それゆえに損傷した軸索の保護に寄与することを報告している。最近の研究では、ミノサイクリンの早期投与により、MCP-1やS100βなどの様々な炎症性およびグリアタンパク質マーカーが外傷後51日目に減少し、それに伴って軽度の爆風TBIのラット実験モデルにおいて運動、不安、空間記憶が有意に改善することが報告されている。このことは、ミノサイクリンが長期的な神経保護効果を有する可能性を示唆している(Kovesdi et al 2012)。
エリスロポエチン(EPO)は、1型サイトカインスーパーファミリーに属している。EPOとEPO受容体の両方の発現は、正確なメカニズムはまだ解明されていないが、神経保護に重要な役割を果たしているTBIで有意にアップレギュレートされている(Brines et al 2000)。EPO/EPORの相互作用により、受容体関連のJak-2がリン酸化され、その結果、カスパーゼ、Ras/MAPK、核内因子Kappa B、Stat-5などの様々なシグナル伝達経路が活性化されることが明らかになっている(Fujitani et al 1997; Mammis et al 2009)。さらに興味深いことに、EPOはEPO受容体が存在しない場合にも神経保護効果を発揮することが示されている。これらのEPOを介したメカニズムは、炎症反応やアポトーシス細胞死に顕著な役割を果たすことが明らかになっている(Yatsiv et al 2005; Xiong et al 2010)。ラットを用いた研究では、EPO治療が神経炎症を抑制し、接着分子、NF-kB、およびIL-6,IL-1βおよびTNF-αなどのプロ炎症性サイトカインの有意なダウンレギュレーションを示す証拠が示されており(Chen et al 2007アストロサイト反応およびミクログリアの活性化の減少も示されている(Yatsiv et al 2005)。また、EPOは、抗アポトーシスタンパク質phospho-AktおよびBcl-XLのアップレギュレーションにより、抗アポトーシス効果を有することが示されている(Yatsiv et al 2005年;Liao et al 2008)。さらに、Bcl-2遺伝子の発現が増加し、それに伴ってBaxレベルが低下する(Liao et al 2009)。他の有益な効果としては、神経新生の亢進、NO産生の減少、および脳の腫脹、皮質組織および軸索損傷の改善が挙げられる(Lu et al 2005年;Yatsiv et al 2005年;Cherian et al 2007)。EPOのこれらの効果は、認知機能および運動機能の改善と関連している(Lu et al 2005年;Yatsiv et al 2005年;Xiong et al 2010)。2010年には、実験的なTBIにおけるEPOの神経保護効果が、オーストラリアとニュージーランドの共同研究で中等度から重度のTBI患者を対象とした臨床試験で成功を収めた。それにもかかわらず、EPOは重度の神経機能障害患者を減少させないことが示された(Nichol et al 2015)。
神経細胞の再生促進
神経栄養因子
血管内皮成長因子(VEGF脳由来神経栄養因子(BDNF神経成長因子(NGF基本線維芽細胞成長因子(bFGF)および上皮成長因子(EGF)を含む神経栄養因子は、神経細胞およびグリア細胞の外傷後の運命を決定することができる。TBI後にこれらの成長因子を投与すると、神経学的転帰を改善することができる(Wu et al 2008; Sun et al 2009)。例えば、外因性VEGFは、Akt経路およびRaf/MEK/ERKカスケードの活性化を介して、TBIの実験モデルにおいて、アストロサイト反応を増加させ、血管新生を促進し、神経新生を促進する(Wu et al 2008; Thau-Zuchman et al 2010; Lu et al 2011)。また、VEGFはアポトーシス細胞死を減少させ、Rho依存性経路を介して神経細胞の成長を促進する(Jin et al 2006)。
損傷を受けた成体ラット脳の側脳室または傍脳実質へのNGFの投与は、コリン作動性中隔ニューロンの生存を促進し、ニューロン細胞死を減少させることが示されており、これは記憶保持および認知障害の改善と一致している(Kromer, 1987; Dixon et al 1997; Sinson et al 1997)。同様に、BDNFの外因性注入は、興奮毒性、脳虚血およびSCIの実験モデルにおいて、組織学的欠損および神経学的機能の改善、および軸索再生の促進に寄与する(Burke et al 1994; Schäbitz et al 1997; Namiki et al 2000)。しかし、Blahaら(2000)は、BDNFの外傷後の髄質内注入後に、損傷を受けた大脳皮質と海馬の両方において、記憶喪失の改善とアポトーシス細胞死の変化が認められなかったことに留意すべきである。ラット海馬スライス培養を用いた試験管内試験の局所外傷モデルでは、bFGFとEGFの投与は、NeuN免疫染色とBrdU陽性新生前駆細胞の有意な増加によって明らかなように、歯状回における既存のニューロンの生存と新生ニューロンの形成を促進した(Laskowski et al 2005)。TBIラットの脳室にbFGFを投与すると、同様の有益な効果が観察され、その結果、TBIによって誘発された神経学的欠損が有意に回復した(Sun et al 2009)。
ラット脳へのbFGFの注入は、側方流体打撲によって誘発された損傷の3時間後にも、神経細胞の損傷および病変体積を有意に減少させることができる(Dietrich et al 1996)。実際、切断された中枢神経系は、傷害後に様々な成長因子を産生することが判明している。Chiarettiら(2008, 2009)は、重度のTBIを受けた子供の脳脊髄液におけるNGFの有意な上昇を示しており、これはグラスゴー回復スコアの改善と相関している。また、霊長類以外の動物においても、皮質病変部位におけるBDNFとその受容体のアップレギュレーションが観察されている(Nagamoto-Combs et al 2007)。これらの研究から、神経栄養因子がTBI後の神経保護に有効であることが示唆されている。
RhoA GTPaseの抑制
損傷を受けた中枢神経系では、中枢神経細胞は再生の可能性を持っているが、その過程は非寛容な環境によって大きく抑制されていることが、蓄積された証拠によって証明されている。最近では、小さなGTPaseであるRhoAが、グリア瘢痕や損傷を受けたミエリンの軸索再生を阻害する分子の効果を媒介する重要な役割を果たしていることが明らかになっていた。外酵素C3トランスフェラーゼは、Clostridium botulinumに見られる酵素であり、NADからのADP-リボース部位をRhoタンパク質のアクセプターアミノ酸残基アスパラギン-41に移動させることでRhoタンパク質をADP-リボシル化し、それによって成長円錐崩壊と軸索再生の阻害を引き起こす下流のシグナル伝達を遮断する(Aktories et al 2005)。軸索再生促進におけるC3トランスフェラーゼの効果は、試験管内試験および生体内試験のSCIおよび末梢神経損傷の動物モデルで広範囲に研究されている(Tan et al 2007; Höltje et al 2009; Boato et al 2010; Huelsenbeck et al 2012)。実験的脊髄損傷を受けたラットは、C3ペプチドで治療すると神経学的転帰の改善を示した(Boato et al 2010)。オリジナルのC3細菌毒素外酵素と同じ酵素活性を有するC3誘導体BA-210は、脊椎損傷の動物モデルにおいて機能的な再生を強化することが実証されている(Lord-Fontaine et al 2008)。重要なことに、それは低温で18ヶ月間保存した後も安定性を維持することができる(Lord-Fontaine et al 2008)。医薬品セトリン/VX-210(BA-210が有効成分である)は、脊髄損傷患者における安全性、忍容性および治療効果を評価する第I/IIa相オープンラベル臨床試験に合格しており(Fehlings et al 2011年;McKerracher and Anderson、2013年現在、急性外傷性頸部脊柱管損傷患者における有効性および安全性を評価する第IIb/III相試験を実施中である。軸索および神経突起の再生を促進する重要な役割に加えて、C3はまた、p53NTRとの相互作用を通じてアポトーシスを制御する(Dubreuil et al 2003)。中枢神経系の再生促進におけるC3トランスフェラーゼの広範な細胞機能を考えると、C3トランスフェラーゼと他の神経保護剤との組み合わせ療法は、相加的な効果をもたらす可能性がある(McKerracher and Guertin, 2013)。TBIの実験モデルにおけるC3トランスフェラーゼの意義はまだ明らかにされていないが、脊髄損傷で観察された有益な効果は、これら2つの形態の中枢神経系外傷の類似性を考えると、TBIにも適用可能であると考えるべきであろう。
ミエリン由来軸索成長阻害剤に対するDNAワクチン
切断された軸索に暴露されたミエリン関連の軸索成長阻害剤は、成長円錐崩壊を引き起こし、軸索の再生を阻害することが知られている。最近の研究では、ミエリン由来の阻害剤Nogo、MAGおよびOMgpに対するDNAワクチンが、ラットのTBIおよび脳卒中の実験モデルにおいて、皮質小脳突起の軸索修復を促進し、神経学的転帰を改善することが報告されている(Zhu et al 2007年;Zhang et al 2009)。脊椎損傷後のNogo受容体(NgR)に対するラットの免疫化もまた、軸索再生および機能回復を促進する(Yu et al 2007,2008)。DNA ワクチン化は、抗原をコードする遺伝子操作された DNA を体内に注入して免疫応答を誘導し、宿主の免疫系を活性化させる新しい比較的簡単な手法である。これらの研究により、ミエリン由来の阻害剤に対するDNAワクチンが、損傷を受けた中枢神経系の回復を促進する有望なアプローチであることが明らかになった。これらのDNAワクチンの効果を検証し、その作用機序や副作用の可能性をよりよく理解するためには、より詳細な調査が必要である。
グリア瘢痕の克服
最近の知見によると、グリア瘢痕は軸索再生を阻害する物理的な障壁として機能するだけでなく、そこに存在するCSPG、テナシン、セマフォリンなどの阻害性分子の複雑なカクテルも軸索成長のための非許容的な環境を形成していることが示唆されている(Fawcett, 2006)。グリア瘢痕におけるニューロカン、ホスファカン、バーシカン、NG2などのCSPGの大幅なアップレギュレーションは、中枢神経系損傷後の軸索再生の失敗に寄与している。CSPG分解酵素であるコンドロチナーゼABCの投与は、24時間以内にCSPGのレベルを低下させ、病変部のキャビテーションを減少させる(Lin et al 2008)。脊髄損傷の生体内試験研究では、損傷を受けた軸索の芽生えと成長の促進、およびそれに伴う再保存におけるコンドロチナーゼABCの効果が確認されている(Bradbury et al 2002; Yick et al 2003; Chau et al 2004; Barritt et al 2006)。重要なことに、軸索病理の改善は、神経学的欠損の改善と関連している(Bradbury et al 2002;Barritt et al 2006)。トランスジェニックマウスにおけるコンドロチナーゼABCの過剰発現はまた、アストロサイトスカーを介した軸索の再生を示している(Cafferty et al 2007)。したがって、グリア瘢痕に存在する阻害分子は、TBIにおける再生を促進する有望なターゲットであると考えられる。
幹細胞治療
ニューロンとグリアの喪失は、切断された中枢神経系の主要な特徴である。したがって、これらの細胞の置換は有効な治療法である。骨髄間質細胞は、試験管内試験でも生体内試験でもグリアやニューロンを含む複数の細胞株に分化することができる(Sanchez-Ramos et al 2000; Lu et al 2001)。TBI後にラットまたはヒト骨髄の間質細胞をラットに静脈内投与したところ、病変した大脳皮質に移行し、グリア(GFAP)およびニューロン(NeuN)マーカーによってそれぞれ識別されるように、アストロサイトおよびニューロンの表現型を示すことが判明した(Lu et al 2001; Mahmood et al 2004b)。骨髄間質細胞もまた、打撲傷、脳室下帯、海馬における新たなBrdU+増殖細胞の存在によって示されるように、TBI後の神経新生の誘導に重要な役割を果たしている(Mahmood et al 2004b)。これらの組織学的所見は、神経学的および運動機能の持続的な改善と相関していた (Lu et al 2001; Mahmood et al 2004b)。同様に、間葉系幹細胞もまた、試験管内試験および生体内試験でのTBI研究において有益な効果を示している。マウスから分離した間葉系幹細胞は、神経幹細胞培養において増殖を促進し、GFAPの発現を誘導する。間葉系幹細胞を急性TBIモデルに注入すると、IL-1β、IL-6,TNF-α、CCL2,CCL11,CXCLなどの様々な炎症性プロサイトカインやケモカインの発現が低下する(Galindo et al 2011)。間葉系幹細胞は、抗炎症作用に加えて、カスパーゼ-3の活性化を抑制し、AKT活性を増加させることで、海馬や大脳皮質の神経細胞の損失を減少させる(Kim et al 2009)。ヒト間葉系幹細胞は、移植後2週間後にTBIラットの神経機能を改善することも示されている(Kim et al 2009)。
ヒトの幹細胞は、神経保護効果が知られているNGFやBDNFなどの神経栄養因子を放出する能力があるため、多くの研究で使用されている。例えば、ヒト胎児幹細胞の移植は、運動機能や記憶力の持続的な改善をもたらし、それは病変部の体積の減少や病変部の神経細胞の損失と関連している(Riess et al 2002; Skardelly et al 2011)。これらはまた、血管新生の促進と、損傷後の活性化ミクログリアの抑制に起因することができる(Skardelly et al 2011)。重要なことに、胎児幹細胞は、グリア由来の神経栄養因子を放出することで、損傷を受けた海馬や大脳皮質でニューロンやアストロサイトに分化することが明らかになった(Riess et al 2002; Gao et al 2006)。若年のTBI患者を対象とした自家骨髄間質細胞移植の小規模第I相臨床試験では、神経学的には中程度の改善しか認められなかったが、副作用は認められなかった(Cox et al 2011)。Tianら(2013)は亜急性期のTBI患者を対象に、自己骨髄由来の単核球を髄腔内投与した第I/II相試験を行った。主要な合併症は認められなかったが、機能の改善は植物状態と運動障害が持続する患者の半数以下にとどまった(Tian et al 2013)。より多くの被験者を募集して本研究を拡大することで、このアプローチの実現可能性についての洞察が得られるであろう。
細胞外ベシクルとmiRNA
幹細胞治療はTBIの再生を促進する有望な効果を示しているが、これらの治療法は様々な合併症を伴う。胎児胚性幹細胞の使用には、間違いなく法的・倫理的な問題がある。幹細胞の多能性は、制御されない増殖や腫瘍形成のリスクをもたらす(Jeong et al 2011)。幹細胞を体内に投与すると、微小血管が閉塞し、免疫反応を引き起こす可能性がある (Furlani et al 2009)。また、幹細胞を単離し、調製し、生存可能な状態を維持することは困難である。上述のように、間葉系幹細胞は近年、TBI治療の有望な候補として浮上してきている。体内に投与された間葉系幹細胞は、損傷した組織部位に優先的に移行し、そこで神経細胞およびグリア細胞に分化し、軸索伸長抑制分子の発現を減少させ、神経炎症を抑制し、成長因子の放出を促進し、それに伴って神経学的機能の実質的な改善をもたらすことが明らかにされた(Das et al 2019)。興味深いことに、蓄積された証拠は、MSCの保護効果が、切断された神経細胞の分化および置換に完全に起因するのではなく、生理活性分子のパラクライン放出または直接的な細胞間相互作用を介した常駐細胞の生存および増殖の促進を介してもよいことを示唆している(Chen et al 2002;Mahmood et al 2004a)。この点で、MSCから放出されるエクソソームは、これらの有益な効果を媒介する有望な候補として浮上している。MSCsから放出された無細胞エクソソームの全身投与は、ラットTBIモデルにおいて、神経血管リモデリング、歯状回における神経新生、および神経炎症の減少と相まって、認知機能および感覚運動機能の回復を促進することが見出された(Zhangh et al 2015)。また、MSCから単離されたエクソソームの静脈内注入は、TBI後のマウスにおいて、神経炎症を抑制し、認知機能や空間学習機能を改善することができる(Kim et al 2016)。エクソソソームは、直径50~200nmの小さな膜小胞である(Trams et al 1981;Schneider and Simons、2013)。それらは、タンパク質、RNA、microRNA、脂質を運び、リガンド-レセプター結合および内部化を介してこれらのカーゴを他の細胞に転送することにより、細胞間シグナル伝達機能を発揮する(TaylorおよびGercel-Taylor、2014)。例えば、損傷を受けた感覚ニューロンから放出されるエキソソームは、マクロファージによって貪食されると炎症反応を促進するノンコーディングマイクロRNAであるmiR-21に富む。miR-21に対するアンタゴミルの投与は、神経障害性過敏症および傷害部位への炎症性マクロファージのリクルートを減少させる(Simeoli et al 2017)。対照的に、損傷マウス脳抽出物によって以前にプライミングされたニューロンから放出されたエキソソーム中のmiR-21は、最近、ニューロンのオートファジーの活性を阻害することが示されている(Li et al 2019)。さらに、miR-17-92クラスターに富むエキソソームは、脳卒中による切断された中枢神経系において、神経新生、オリゴデンドロゲネシス、および軸索伸長を促進することが示されている(Xin et al 2017)。 エキソソームが運ぶmiR-132は、脳血管の完全性を調節するための細胞間シグナルとして作用する(Xu et al 2017)。つまり、神経細胞およびグリア細胞由来のエクソソームは、遺伝子発現およびmiRNA活性を自己分泌的に調節することができ、一般的には、神経新生を促進し、炎症を抑制し、血管新生を増加させ、組織リモデリングを促進することにより、神経保護および神経回復効果を媒介しているのである。
治療薬の脳への送達
生理的障壁の克服
中枢神経系と末梢循環を隔てる内皮細胞によって維持されているBBBや血液-脳脊髄液バリアのような生理的バリアは、脳を保護する上で非常に重要である。これらの界面は低分子の中枢神経系への移行を強固に制御しているため、TBI治療における薬物送達に課題を投げかけている。しかし、TBIの直接的な結果として、しばしばBBBの無傷性が損なわれることに留意すべきである。BBB機能障害は、TBI後の二次的損傷の長期化に大きく寄与しているが、BBB機能障害は、経鼻送達のような他の侵入経路を介して投与された治療用タンパク質またはペプチドが、緊密な内皮接合を越えて損傷部位に到達することを可能にしている(Habgood et al 2007; Lotocki et al 2009; Ligade et al 2010)。実験的TBIでは、治療薬の脳室内投与は、脳脊髄液への直接送達によってこれらの障壁を克服する一般的で実行可能な方法である(Temsamani et al 2000)。TBIの臨床管理においては、頭蓋内圧および浮腫を緩和するために外科的介入が必要とされることが多く、これもまた直接薬物送達の機会を提供する。
浸透圧ポンプを介した持続的かつ制御された薬物送達
上述した治療薬は、試験管内試験および生体内試験でのTBI研究において様々な神経保護効果を示しているが、二次的な脳損傷に関連した副作用が長期にわたって持続することから、TBIからの回復を促進するためにこれらの候補治療薬の潜在能力を最大限に発揮させるために、一定の持続的かつ制御された放出を可能にするデリバリーシステムの開発が必要とされている。ラットにおけるTBIの実験モデルにおいて、浸透圧ミニポンプを用いてNGFおよびS100B神経栄養タンパク質を一定の速度で脳の側脳室に送達することに成功しており、その結果、認知機能が促進されている(Dixon et al 1997; Kleindienst et al 2004)。これらのミニポンプは移植可能であり、ポンプ内の浸透液と体内の間質液との間の浸透圧差から開発された圧力によって駆動されるため、外部電力を必要としない。浸透圧ミニポンプは、タンパク質やペプチドのように半減期の短い薬剤の生体内試験での有効性や毒性を評価するための強力なツールであるが、感染を最小限に抑え、被験者の自由な動きを可能にするためには、ポンプの皮下埋め込みが必要である。
ナノキャリア
浸透圧ポンプに加えて、マイクロまたはナノ粒子への薬物のカプセル化は、TBI研究において治療薬の持続的かつ制御された送達を可能にする有望な方法として浮上してきている。天然ポリマーと合成ポリマーの両方が、生体適合性、生分解性、一般的に不活性であり、低分子やタンパク質に付着またはカプセル化することが可能であるという共通の特徴を持つ、薬物デポとしての使用に成功している(Orive et al 2009)。バイオポリマーベースの薬物送達システムは、多くの組織および器官に適用されているが、TBI治療におけるそれらの使用の報告は限られている(Heile and Brinker, 2011; Guan et al 2013; Khalin et al 2016)。Turkogluら(2010)は、ラットのTBI誘導後にシクロスポリンAを負荷した天然キトサン微小球を脳室に投与した。それはミトコンドリア損傷を減少させ、脂質過酸化を低下させることに成功したが、有益な効果は、実際には、シクロスポリンA単独で腹腔内注射した対照群と同等であった(Turkoglu et al 2010)。これは、キトサン微小球の亜最適な配合、薬剤の投与量、および投与経路に起因している可能性がある。薬物カプセル化に一般的に使用される他の天然バイオポリマーには、アルギン酸およびゼラチンが含まれる(Orive et al 2009)。薬物送達目的でナノキャリアとして使用される最も一般的な合成バイオポリマーの一つは、ポリ(D,L-ラクチド-コ-グリコリド;PLGAポリ乳酸(PLA)およびポリグリコール酸(PGA)のファミリーである。特筆すべきは、これらのポリマーは、米国の食品医薬品局によって承認されており、神経系との適合性が確認されていることである。用途に応じて、PLGAポリマーは、特定の技術を用いて、異なる剤形で調製することができる(Anderson and Shive, 1997; Soppimath et al 2001)。乳化溶媒蒸発法は、例えば、広くPLGAマイクロスフィアの製造に使用されている(Jain, 2000)。最近では、エレクトロスピニング技術がナノファイバーを製造するために開発されている(Li et al 2002)。これらの方法論の両方とも、製造プロセス中に薬物の取り込みの高効率を可能にする。Tanら(2007)は、水-油-水-中-乳化法を用いて、細胞内に自由に入り込む強力なRhoA阻害剤である組換えタンパク質Tat-C3トランスフェラーゼをPLGAマイクロスフィアにカプセル化することで、80%以上のローディング効率を実証している(Tan et al 2007)。あるいは、薬物は、予め作製したポリマー粒子に吸着させることもできる。
PLGAベースのデポからの薬物放出は、水素および共有結合が水によって加水分解されて乳酸およびグリコール酸を形成し、体内のクレブスサイクルによって代謝され得るポリマーの段階的な分解を伴う(Park,1995)。ポリマー中のラクチドモノマーとグリコリドモノマーの比率を操作することで、分解プロファイルを調節することができ、その結果、薬物放出率を調整することができる。例えば、グリコリドの含有量が高いと、より速い加水分解および薬物放出と相関する。他の要因としては、溶解度、多孔性、分子量などのポリマーの物理化学的特性が挙げられる(Anderson and Shive, 1997)。さらに、エステルでエンドキャップされたポリマーは、遊離カルボン酸でエンドキャップされたポリマーよりも加水分解に対してより耐性がある。Tanら(2007)による試験管内試験研究では、カプセル化された組換えタンパク質Tat-C3の最適な放出プロファイルを決定するために、キャップされていない(遊離カルボキシル)末端基とキャップされた(ラウリルエステル)末端基を有するPLGAポリマーを様々な比率でブレンドした。放出速度解析の結果、30%キャップされた70%キャップされていないPLGAの配合では、28日間にわたって一定のタンパク質放出速度を維持しながら、穏やかな初期バーストが可能であることが明らかになった。タンパク質放出特性は、キャップされたPLGAとキャップされていないPLGAのバランスのとれた分解速度の結果であり、また、走査型電子顕微鏡(Tan et al 2007)によって明らかにされたように、既存の気孔内の新しい内部気孔の形成に起因する微小球体の気孔率のそれに伴う漸進的な増加であった(Tan et al 2007)。
バイオポリマーベースの薬物送達システムの生体内試験応用は、組織との直接かつ長時間の接触を伴うので、主要な懸念事項の一つは、それらの生体適合性であり、これは、線条体、側脳室、前頭葉および黒質などの脳の様々な部位への移植後に誘導される炎症反応に応じて決定され得る(Fournier et al 2003;Lampe et al 2011)。PLGAポリマーは一般に生体適合性があることが知られているが、いくつかの研究では、機械的外傷の非特異的な結果である可能性はあるが、アストロサイトの免疫組織化学的染色によって検出されるように、急性炎症反応を誘発することが報告されている(Emerich et al 1999; Lampe et al 2011)。PLGAポリマーの既知の問題は、カプセル化されたタンパク質またはペプチドの安定性に対する悪影響である。制御放出プロセス中のタンパク質活性または完全性の損失は、タンパク質のポリマーへの吸着、またはPLGAポリマーが乳酸およびグリコール酸に分解する際の酸性化によるタンパク質の変性の程度が大きいことに起因し得る。カプセル化された生理活性剤の安定性は、カプセル化プロセス中に炭酸カルシウムまたは水酸化マグネシウムなどのpH調整剤を組み込むことによって改善され得る(Houchin and Topp, 2008)。同様に、1-8-ビス-(ジメチルアミノ)ナフタレンのような塩基性アミンであるプロトンスカベンジャー/スポンジを賦形剤として添加することもできる(Houchin er al)。 さらに、最近の研究では、その反応性アミンとPLGAのエステル結合とのアシル化反応によるカプセル化ペプチドの不活性化が報告されている(Domb et al 1994)。カプセル化に先立ってペプチドをPEG化することは、PLGAとのこれらの望ましくない共有結合相互作用を防止することができる(NaおよびDelluca、2005)。結果として得られるPEG化ペプチドはまた、減少した免疫原性を示す。
薬物送達のための細胞外ベシクル
エクソソソームは、ほぼすべての細胞タイプで放出される脂質二重層膜小胞である。エクソソソームが運ぶ貨物は、主にエンドソーム由来の分子であり、mRNA、microRNA、タンパク質から脂質に至るまで、細胞由来によって様々である(Chopp and Zhang, 2015)。最近では、MSC由来のエクソソームがTBIsの動物モデルにおける機能回復を促進する効果があることから注目されている(Zhang et al 2015)。その根底にあるメカニズムは完全には解明されていないが、エクソソームからトランスフェクトされたmiRNAは、有益な効果を媒介する上で極めて重要な役割を果たしているように思われる(Taylor and Gercel-Taylor, 2013)。重要なことに、高い生体適合性、低い免疫原性、BBBおよび細胞膜を横切る効率的な透過性を示す天然の脂質ベースのナノベシクルとしてのエクソソソームのユニークな特性は、エクソソームを中枢神経系のための新規薬物送達システムとして開発される有望な候補としている(Xiong et al 2017)。蓄積された証拠は、エクソソームがリガンド-受容体結合および内部化を介して膜を横切ることを示唆している。マクロファージエクソソームは、例えば、表面にインテグリンリンパ球機能関連抗原1(LFA-1)を発現し、これは、炎症を起こした脳のBBBの内皮細胞上の高度にアップレギュレーションされた細胞内接着分子1(ICAM-1)と相互作用する。BDNFを予め装填したマクロファージエクソソームの静脈内投与は、このタンパク質を脳に送達することに成功していることが示されている(Yuan et al 2017)。脈絡叢上皮細胞由来のエクソソームは、葉酸受容体α(FRα)を発現し、これは上衣細胞と相互作用し、脳内のアストロサイトおよびニューロンによって取り込まれる前に、脳脊髄液-脳関門を横切って媒介する(Grapp et al 2013)。これらの観察は、受容体を介したエクソソームのトランスサイトーシスが、中枢神経系への薬物送達のための有望な方法であり得ることを示唆している。意図された受信者細胞に結合するタンパク質または脂質リガンドを本質的に発現する天然のエクソソソームを使用することとは別に、エクソソームは、特定のリガンドまたはホーミングペプチドの異所性発現によって、特定の細胞タイプまたは組織を標的とするように設計することもできる。Alvarez-Ervitiら(2011)は、樹状細胞から単離したエクソソソームにおいて、エクソソーム膜タンパク質Lamp2bとニューロン特異的RVGペプチドとの融合タンパク質を強制的に発現させた。精製されたエクソソソームにGAパーキンソン病Hに対するsiRNAを負荷し、静脈注射でマウスに導入した。驚くべきことに、エクソソソームを介したこれらの siRNA の送達は、脳内のニューロン、ミクログリア、およびオリゴデンドロサイトにおいて、標的 mRNA のダウンレギュレーションに成功したことが判明した (Alvarez-Erviti et al 2011)。エクソソソームは安定であり、タンパク質や核酸のコンフォメーションと生理活性を維持することができるため、中枢神経系への標的薬物送達のための理想的な天然の乗り物として機能する。
薬物の細胞侵入を促進するための細胞貫通ペプチド
薬物の持続的かつ制御された送達の問題は、上述の様々なアプローチによって解決され得るが、細胞内標的に向けられたペプチドやタンパク質のような治療薬は、しばしば、それらの低い膜透過性のために、細胞内へのアクセスを得ることが困難に遭遇することが多い。細胞侵入の効率を改善するために、これらのタンパク質は、生物学的膜を横断することができ、細胞送達ビヒクルとして機能する、細胞貫通タンパク質(CPPs)として知られる特異なクラスのタンパク質に融合させることができる(KorrenおよびTorchirin,2012;Guidottiら,2017)。タンパク質伝達ドメインとしても一般的に知られているCPPは、アルギニンおよびリジンのような主に正に荷電したアミノ酸を含む小さな両親媒性ペプチドである。これらのCPPの異なるユニークな特性および性質は、共役ペプチドまたは低分子の、形質膜を介した非侵襲的な内部化を可能にする(Gupta et al 2005;FogedおよびNirelsen、2008)。これらのCPPの広範な特徴付けにもかかわらず、これらのCPPが細胞膜を透過する正確なメカニズムについてはまだ議論の余地があり、決定されていない。細胞内への内在化の複数のメカニズムがCPPにおいて提案されており、トランスロケーションの効率は個々のCPPの性質に依存しているようである(Koren and Torchilin, 2012)。例えば、標的ペプチドと共役したCPPは、膜に孔を形成することにより、脂質二重層を横切って直接トランスロケーションすることができる。あるいは、CPPが介在する内在化は、エネルギー依存性エンドサイトーシスを介して行うことができる。最後に、CPPP-カーゴ融合タンパク質は、細胞膜を不安定化させることが可能なベシクルおよび倒立ミセルを形成することができ、それによって共役タンパク質を細胞内に放出することができる。特定のカチオン性CPPは、カーゴの内部化のために細胞表面のプロテオグリカン(ヘパリン硫酸塩)に結合することができる(Foged and Nielsen, 2008; Sebbage, 2009)。中枢神経系損傷の試験管内試験および生体内試験の両方の研究は、トランス活性化転写(Tat)因子、ペネトレータチン、膜トランスロケーション配列、トランスポータンおよびPep-1を含む様々なCPPPにコンジュゲートすることにより、異なるタンパク質の細胞トランスロケーションが成功したことを実証している(Lindggren et al 2000)。それにもかかわらず、これらのCPPの細胞毒性および特異性に関する懸念は依然として論争の的となっている。大半の研究では、低濃度での CPP の毒性は低いレベルであることが示されているが、ラットの神経細胞培養では高い細胞毒性が報告されている(Antoniou and Borsello, 2010)。したがって、CPPの生体適合性のさらなる検証が必要とされている。
考察
中枢神経系における外傷の研究は、その基礎となる病態生理と分子機構の理解を大きく広げてきた。TBIにおける一次損傷は大部分が不可逆的であるが、数ヶ月から数年をかけて進行する二次損傷は治療的介入の対象となる。この損傷の遅延期には、興奮障害、アポトーシス細胞死、軸索再生の阻害、神経炎症、酸化ストレスなど、多くの事象が関与しているため、効果的な治療戦略を考案するためには、長期間にわたって複数のメカニズムを標的とした治療戦略が必要となる。細胞膜を透過して細胞内に入ることが可能な治療薬の調節された持続的な送達のためのデポシステムの利用可能性は、既存の薬剤のバイオアベイラビリティーをさらに向上させることを可能にすると思われる。さらに重要なことは、薬物を投与可能な標的に対する新規薬剤の治療の可能性を探る機会を提供することである。実際、この治療アプローチは、アルツハイマー病、ハンチントン病、パーキンソン病などの多くの神経変性疾患の治療に適用されている(Popovic and Brundin, 2006; Saraiva er al)。 TBIの管理におけるこの戦略の実現可能性はまだ確立されていないが、TBIにおける二次的損傷の間のイベントの進行が遅いことから、有望と思われる。TBIは世界中で大きな健康問題、社会経済問題となっており、より効果的な治療法が求められている現代社会に大きな医療負担を強いている。また、現代の技術によって致死からよりよく保護されるようになると、軍人の間で微妙な中枢神経系の損傷が劇的に増加するため、防衛科学における有効な問題を表している。