Contents
pubmed.ncbi.nlm.nih.gov/37084148/
The Promise of Niacin in Neurology
要旨
ナイアシン (ビタミンB3)はペラグラを治療する必須栄養素であり、スタチンが登場する以前は、ナイアシンは脂質異常症によく使われていた。最近のエビデンスでは、ナイアシンはいくつかの神経疾患に対する有望な治療薬であると考えられている。
この総説では、NAD+の補充と代謝におけるナイアシンの恒常性維持の役割など、ナイアシンの生化学について述べる。 ナイアシンはまた、主にヒドロキシカルボン酸受容体2(Hcar2)に関与することにより、代謝以外の役割も担っている。
ナイアシンのこのような受容体を介した活性には、免疫応答の調節、 脱髄後のミエリン残屑やアルツハイマー病モデルにおけるアミロイドβの貪食、細胞からのコレステロール排出などがある。
ナイアシンが研究されてきた、あるいは候補薬として提案されてきた神経疾患について述べる。多発性硬化症、アルツハイマー病、 パーキンソン病、神経膠芽腫、筋萎縮性側索硬化症などである。最後に、ナイアシンが神経病理を改善する可能性のあるメカニズムについて述べる。いくつかの疑問は残るものの、神経障害を緩和する治療薬としてのナイアシンは有望である。
キーワード
ナイアシン治療 NAD+/NADP ヒドロキシカルボン酸受容体(Hcar)2神経疾患 貪食 免疫調節
はじめに
ビタミンB3としても知られるナイアシンは、食事からの摂取が不可欠な栄養素であり、肉、魚、穀物、野菜などが豊富に含まれている[1]。ナイアシンが欠乏すると、ペラグラという認知症、皮膚炎、下痢を特徴とする病気が発症し、最終的には死に至るため、ナイアシン濃度を適切に保つことの重要性は明らかである[2]。そのため、ナイアシンはペラグラの治療に使われる薬であり[3]、スタチンが登場する以前は脂質異常症[4]によく適応されていた。ヒトの場合、ナイアシンの推奨最低摂取量は15~20mg/日である一方[5]、脂質異常症に対しては3000mg/日までの薬理学的投与が行われており、幅広い用量にわたってその忍容性が実証されている[6]。
最近のエビデンスでは、ナイアシンは様々な神経疾患に対するエキサイティングな治療オプションであると考えられている。ナイアシンは、進行性多発性硬化症(MS) [7]の治療薬候補として3番目に有望視されており、MSの動物モデルにおいて、脱髄後の抑制性ミエリン残屑の貪食を促進し、再ミエリン化をもたらす。[8]。パーキンソン病[9]とアルツハイマー病[10]の動物モデルでは、ナイアシンは免疫調節やドーパミン補充などのメカニズムを通じて神経病理を改善する。さらに、ナイアシンは膠芽腫の動物モデル[11]において腫瘍の大きさと死亡率を減少させ、現在、膠芽腫患者を対象とした臨床試験[12]で研究されている。また、ナイアシンはパーキンソン病患者の運動症状を緩和し、[13]、ナイアシンの食事摂取はアルツハイマー病の発症率の低下と認知機能の低下に関連している[14]。
神経疾患におけるナイアシンの有用性が期待されているにもかかわらず、中枢神経系 (CNS)におけるナイアシンの作用に関しては、いくつかの未解決の疑問が残っている。 この総説では、ナイアシンの生化学と活性、受容体依存性および非依存性活性について述べる。そして、CNSにおけるナイアシンの作用機序を考察し、神経疾患の治療薬としての潜在的役割を探る。
ナイアシンの生物学と化学
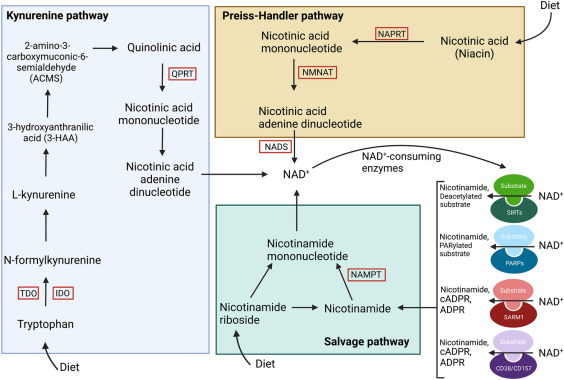
ナイアシンは、ビタミンのニコチン酸とニコチンアミドとして存在し、ナイアシン誘導体のニコチンアミドリボシドとニコチンアミドモノヌクレオチドを生成する。ナイアシンは、図1に要約した一連の代謝経路を経て、ニコチンアミドアデニンジヌクレオチド(NAD+)を生成する。Preiss-Handler経路では、ニコチン酸は、中間体であるニコチン酸モノヌクレオチドとニコチン酸アデニンジヌクレオチドを利用して、3段階でNAD+に変換される[15]。サルベージ経路は、NAD+の酵素活性の副産物であるニコチンアミドと食餌性ニコチンアミドリボシドを再利用してNAD+を生成する。最後に、NAD+のde novo生合成は、キヌレニン経路(図1 )を介して達成される。 キヌレニン経路では、食餌性トリプトファンが前駆体として機能し、一連の8つの酵素的段階を経てNAD+に変換される。デノボ経路は最も長く、最もエネルギーを消費する。その結果、トリプトファンは他の前駆体に比べてNAD+レベルを増加させる効率が低く、この経路はNAD+産生において控えめな役割を果たす[18,19]。対照的に、サルベージ経路は哺乳類細胞におけるNAD+の主要な産生経路であり、この代謝物の恒常性レベルの維持に大きく関わっている[18]。

図1 NAD+生合成経路。キヌレニン経路では、まず食餌性のトリプトファンがトリプトファン2,3-ジオキシゲナーゼ(TDO)またはインドールアミン2,3-ジオキシゲナーゼ(IDO)を介してN-ホルミルキヌレニンに変換される。 一連の4つの酵素的段階を経て、N-ホルミルキヌレニンはキノリン酸を生成し、キノリン酸ホスホリボシルトランスフェラーゼ(QPRT )が触媒する反応でニコチン酸モノヌクレオチドを生じる。最終段階で、ニコチン酸モノヌクレオチドはニコチン酸アデニンジヌクレオチドに変換され、NAD+が生成される。Preiss-Handler経路では、食餌性ニコチン酸(ナイアシン)がニコチン酸ホスホリボシルトランスフェラーゼ(NAPRT)を介してニコチン酸モノヌクレオチドに変換される。次にニコチン酸モノヌクレオチドは、ニコチンアミドモノヌクレオチドアデニリントランスフェラーゼ(NMNAT)を触媒とする反応でニコチン酸アデニンジヌクレオチドに変換され、NAD+合成酵素(NADS)を介してNAD+が生じる。サルベージ経路では、NAD+の酵素活性からリサイクルされたニコチンアミドが、ニコチンアミドホスホリボシルトランスフェラーゼ(NAMPT)を介してニコチンアミドモノヌクレオチドを生成するのに使われる。 食餌性ニコチンアミドリボシドは、ニコチンアミドモノヌクレオチド、またはニコチンアミドのいずれかを生成することができる。この経路の最終段階で、ニコチンアミドモノヌクレオチドはNAD +を生成する。いったん生成されたNAD+は、 サーチュイン(SIRTs)、ポリ(ADP-リボース)ポリメラーゼ(PARPs)、ステライルα・TIRモチーフ含有1(SARM1)、環状ADP-リボース(cADPR)合成酵素CD38とCD157など、いくつかの酵素によって消費される。 これらの酵素は副産物としてニコチンアミドを生成する。BioRenderで作成した図
一旦生成されたNAD+は、リン酸化体であるニコチンアミドアデニンジヌクレオチドリン酸(NADP)の前駆体として機能し、NAD+キナーゼを介して生成される[20,21]。NAD(P)のホメオスタシスは、生合成とNAD+を消費する酵素による利用のバランスを含んでいる。NAD +とNADPは酸化還元酵素にとって重要な補酵素であり、NAD +は細胞内の酸化還元に依存しない酵素プロセスの基質としても機能する。[22]。従って、NAD(P)の恒常性は細胞の適切な代謝機能にとって不可欠である。解糖や酸化的リン酸化などの酸化還元反応では、NAD+はプロトン受容体として機能し、還元型NADHを生成する。 NADHは次に電子伝達鎖で酸化され、ミトコンドリアのプロトン勾配に寄与し、 ATP合成酵素を介したATPの生成を可能にする[23]。
NAD+には酸化還元に依存しない機能もあり、NAD+を異化し、副産物としてニコチンアミドを生成するいくつかの重要な酵素の共基質として機能している(図1)[24]。これらの酵素には、サーチュイン(SIRTs)、ポリ(ADP-リボース)ポリメラーゼ(PARPs)、ステライルα、TIRモチーフ含有1(SARM1)、 CD38やCD157などの環状ADP-リボース(cADPR)合成酵素が含まれる [25]。サーチュインは、細胞代謝、炎症、酸化ストレスなどのプロセスに関与するタンパク質脱アセチル化酵素のファミリーである[26]。サーチュインが触媒する脱アセチル化の過程で、NAD+はニコチンアミドとADPリボース(ADPR)に切断される。ADPRはアシルアクセプターとして機能し、基質からアシル基を除去することができる[27]。PARPは、ポリADP-リボシル化と呼ばれるプロセスで、NAD+から標的高分子上への複数のADP-リボース基の転移を触媒する[28]。PARP活性はDNA損傷後にアップレギュレートされ、総NAD+レベルの大幅な低下とDNA修復シグナルの開始をもたらす[29]。神経細胞の損傷や傷害に応答して、SARM1が活性化され、Tol/インターロイキン-1受容体(TIR)モチーフを介して、NAD+をニコチンアミド、ADPR、cADPRに切断する。最後に、CD38とCD157はNAD+をニコチンアミド、ADPR、cADPRに加水分解し、後者はCa2+シグナルのセカンドメッセンジャーとして機能する[33,34]。これらの酵素の活性はNAD+レベルに依存している。したがって、NAD+の生合成が阻害されたり、ナイアシンのようなNAD+前駆体が不足したりすると、細胞活性が調節できなくなる可能性がある。
NADPは、ペントースリン酸経路において重要な役割を果たしている(図2)。ここで、NADPからNADPHへの還元は、DNAやRNAを含む多くの生体分子の前駆体として機能するリボース-5-リン酸の合成と結合する。NADPHはその後、脂肪酸、ステロール、ヌクレオチドなどの分子を生成する際の還元剤として使用される[35]。NADPHは酸化ストレスのバランスにも関与しており、 抗酸化物質であるグルタチオンの生成においてグルタチオン還元酵素の補酵素として機能し、[36]、活性酸素種の生成においてNADPHオキシダーゼの基質として電子を供与する [37,38]。このように、ナイアシンはNAD(P)を補充することで、適切な代謝機能を回復させる(図2)。

図2 NAD +の前駆体としてのナイアシンの恒常性維持役割。NAD+を消費する酵素の活性を通して、ナイアシンはDNA損傷応答やCa2+シグナル伝達などの細胞プロセスの維持に関与している。NAD+は還元されてNADHとなり、 電子伝達系でプロトン供与体として機能し、ミトコンドリアのプロトン勾配を生成してATPの産生につながる。NAD +はまたリン酸化されてNADPを生成する。NADPはリボース-5-リン酸の前駆体として機能し、DNAやRNAなどの核酸を生成する。最後に、NADPは還元されてNADPHを生成する。NADPHはその後、脂肪酸、ステロール、ヌクレオチドなどの生体分子を生成する際の還元剤として使用される。BioRenderで作成した図
内因性レベルでは、ナイアシンは受容体非依存的にNAD+とNADPの補充を通じて働き、適切な代謝機能を回復させる。腹部大動脈瘤のげっ歯類モデルにおいて、ニコチン酸とニコチンアミドは、ナイアシンの主要な受容体であるヒドロキシカルボン酸受容体(Hcar2)を活性化するのではなく、NAD+レベルとサーチュイン1活性を増加させることによって、動脈瘤の発生率と病理を減少させる。さらに、 ミトコンドリアミオパチーの患者は、筋力低下と疲労を経験し、ミトコンドリア活性の変化によって引き起こされると思われるNAD+レベルの減少を経験する。このような患者にナイアシンを補給すると、全身および筋肉のNAD+レベルが上昇し、筋力の増加とミトコンドリア生合成の改善につながる[40]。さらに、NAD+の網膜レベルの低下は加齢とともに観察され、緑内障などの網膜病態と関連している。緑内障のげっ歯類モデルにおいて、 ニコチンアミドの補給によるNAD+の減少防止は、 網膜神経節細胞の神経変性を防ぐと同時に [42]、緑内障に誘発される代謝の変化から保護し、ミトコンドリアの形態の完全性を維持する[43]。
なぜなら、ニコチン酸からNAD +を合成するのに必要な酵素であるニコチンアミドホスホリボシルトランスフェラーゼ(NAMPT)とニコチン酸ホスホリボシルトランスフェラーゼ(NAPRT)のレベルは、脳では他の組織に比べてはるかに低いからである[44,45]。その結果、中枢神経系におけるNAD+は、キヌレニン代謝経路など、他の供給源に由来すると考えられる[44]。したがって、NAD+の補給はナイアシンの正統的な役割かもしれないが、体内、特にCNSにおいてナイアシンの効果を媒介する重要な経路は他にもある。
ナイアシンのもう一つの注目すべき生化学的特徴は、体内からの排泄が速いことである。最大血漿中濃度は経口摂取後30~60分と報告されており、ヒトとマウスの両方で推定半減期は28~40分である[46,47,48]。
ナイアシン受容体
ヒドロキシカルボン酸受容体(Hcar)2
ナイアシンの主要な受容体は、GPR109Aとしても知られる抑制性Gi/oタンパク質共役型受容体(GPCR)Hcar2である[49,50]。GPCRとしてのHcar2は、リガンドと相互作用する細胞外N末端と、 ヘテロ三量体Gタンパク質との相互作用を介したシグナル伝達に関与する細胞内C末端を持つ7つの膜貫通ドメイン構造を特徴とする [51]。ナイアシンなどの細胞外リガンドに結合すると、Hcar2はコンフォメーション変化を起こし、Gi/o タンパク質αサブユニットに結合する。 脂肪細胞では、これが下流のシグナル伝達カスケードを刺激し、アデニルシクラーゼ活性を阻害するとともに、細胞内のサイクリックAMPレベルを低下させる[52,53]。免疫細胞では、Hcar2にリガンドが結合すると、細胞内Ca2+が一過性に上昇する(図3)[54]。

図3 脂肪細胞と免疫細胞におけるHcar2でのナイアシンの活性。脂肪細胞では、ナイアシンはヒドロキシカルボン酸受容体(Hcar2)を介してアデニルシクラーゼ活性を阻害する。 通常の状態では、アデニルシクラーゼはcAMPを生成し、プロテインキナーゼAを活性化する。プロテインキナーゼAはホルモン感受性リパーゼをリン酸化・活性化し、脂肪分解を促進する。アデニルシクラーゼを阻害することにより、ナイアシンはこの経路の活性を低下させ、脂肪分解の抑制につながる。免疫細胞では、ナイアシンがHcar2に結合すると細胞内Ca2+が増加する。 正確なメカニズムはまだ解明されていないが、一つのモデルとして、Hcar2アゴニズムがホスホリパーゼC(PLC)を活性化し、小胞体内の細胞内貯蔵物から Ca2+の放出を促進することが示唆されている。その後、Ca 2+はセカンドメッセンジャーとして作用し、炎症性NF-κB活性化の下流にあるp65のリン酸化を阻害する。逆に、Ca2+の一過性の増加は、細胞外ソースに由来すると考えられる。これは小胞体内にある細胞内Ca2+ 貯蔵量を安定化させ、細胞をストレスに対してより強くする。 細胞ストレスはNLRP3インフラマソームの活性化につながり、マクロファージ内のコレステロール蓄積を促進し、炎症性で有害な免疫細胞の表現型をもたらす。図では、ナイアシン/Hcar2相互作用に続く免疫細胞における炎症活性の抑制の段階が、赤いT記号で描かれている。図はBioRenderで作成
Hcar2の主な内因性リガンドは、短鎖脂肪酸の酪酸とケトン体のβ-ヒドロキシ酪酸である。[55,56,57]。薬理学的投与量では、ニコチンアミドではなくニコチン酸がHcar2のリガンドとして働く[58,59]。注目すべきは、MS治療薬であるフマル酸ジメチルの代謝産物であるフマル酸モノメチルもHcar2受容体を刺激することができ、これがMSのモデルにおける治療反応の少なくとも一部を媒介することである[60,61,62]。
Hcar2は全身に広く発現しており、ナイアシンのようなHcar2リガンドが幅広い生理学的・薬理学的効果を発揮するのはこのためであろう。白色および褐色脂肪組織の脂肪細胞上で、Hcar2は代謝物センサーとして働き、絶食時の脂肪分解を抑制し、ナイアシンの脂質修飾作用に寄与している[58,63]。Hcar2はマクロファージ、樹状細胞、好中球などの免疫細胞にも発現しており、ナイアシンの免疫調節作用の基礎となっている(図3)[49,64]。さらに、Hcar2は網膜、結腸、腸管の上皮細胞や皮膚のケラチノサイトにも発現している[55,65]。後者の細胞タイプでHcar2が活性化すると、 プロスタグランジンが放出され、ナイアシン補給に伴う顔面紅潮反応を部分的に媒介する。[66]。
ニコチン酸の内因性レベル(<0.3 nM)[67]は、Hcar2の活性に有意な影響を与えるには低すぎ、NAD+の補充を介して作用すると考えられるが、薬理学的用量は、Hcar2を活性化し、別の範囲の生物学的活性を媒介するには十分である[68]。実際、 脂質異常症のために1gのナイアシンを摂取しているヒトの平均血清中ニコチン酸濃度は、摂取後1~2時間で120~230μMである[69]。実験的研究によると、ヒトのHcar2に対するニコチン酸のEC50は0.13~1μMであり、最大応答は10~100μMである[50,70,71]。このことは、薬理学的なナイアシン補給によって得られるナイアシンの血漿中濃度が、Hcar2活性化のための最適なウィンドウ内にあることを示している。
Hcar2アゴニズム
前述のように、Hcar2は脂肪細胞に広く発現している。絶食時や運動時には、脂肪細胞上のHcar2は、その主要な内因性リガンドの一つであるβ-ヒドロキシ酪酸などのケトン体によって活性化される[72]。この活性化により、アデニルシクラーゼが阻害され、それに伴ってサイクリックAMPレベルとプロテインキナーゼA 活性が低下し、最終的に脂肪分解酵素であるホルモン感受性リパーゼの活性が低下する。[73,74]。野生型マウスから培養した脂肪細胞において、β-ヒドロキシ酪酸またはニコチン酸を投与すると、細胞培養液中に存在する遊離脂肪酸の量が減少し、脂肪分解が減少することが示される。注目すべきは、Hcar2の齧歯類オルソログであるPUMA-Gノックアウトマウスの脂肪細胞では、この結果が消失することである[75]。ヒトHcar2を過剰発現したトランスジェニックラットでは、血漿遊離脂肪酸レベルは野生型対照と比較して空腹時に減少する[76]。さらに、ニコチン酸を投与すると、野生型と比較して、これらのトランスジェニックマウスの遊離脂肪酸レベルがより強く低下することから、ナイアシンがHcar2を通じて脂肪分解を低下させるように働いていることが示される(図3)[76]。このように、Hcar2は絶食期間中の脂肪分解を抑制し、血液中に動員される遊離脂肪酸のレベルを低下させる。この活性は、ナイアシンが脂質異常症の治療に使用されるに至った脂質修飾的側面の根底にある。
マクロファージ、ミクログリア、樹状細胞、好中球を含む免疫細胞は、Hcar2を発現することが知られているもう一つの主要な細胞タイプである。正確なメカニズムはまだ不明であるが、免疫細胞におけるHcar2アゴニズムの作用に関しては、主に2つの仮説がある。アテローム性動脈硬化症のモデルにおいて、ニコチン酸によるマクロファージ上のHcar2の活性化は、 細胞外ソースからの細胞内Ca2+の増加につながり、小胞体からのCa2+の放出につながるホスホリパーゼC(PLC)/イノシトール三リン酸(IP 3)経路を阻害する [77]。特に、Hcar2によるPLCの阻害はCa2+依存性であり、細胞内Ca2+キレーターを作用させるとPLCの阻害は消失する [77]。したがって、細胞内のCa2+ 貯蔵量を安定化させることで、 細胞ストレスとインフラマソームの活性化を防いでいるという仮説が成り立つ (図3)。
上記の仮説とは対照的に、EAEモデルでの研究は、Hcar2の作動がPLC経路を活性化するGi/oシグナル伝達カスケードを引き起こすことを示唆している。この結果、細胞内ソースから細胞内Ca2+が上昇し、p65の過剰リン酸化と、多くの炎症性サイトカインの発現を制御する転写因子であるNF-κbの下流での活性化が阻止される(図3)[60,61]。このように、ナイアシンや他の化合物によるHcar2のアゴニズムが、細胞内Ca2+の増加と炎症の抑制をもたらすことは明らかであるが、その具体的な経路はまだ十分に解明されていない。
免疫細胞におけるHcar2の発現は、ナイアシンの抗動脈硬化作用と免疫調節作用に寄与している。実際、以前は、ナイアシンの抗動脈硬化作用は、遊離脂肪酸の血漿中濃度が低下するとVLDLやLDLの合成が減少し、HDLが増加するという抗脂肪分解作用に由来すると考えられていたが[78,79,80]、現在の研究では、ナイアシンの抗動脈硬化作用は、代わりに免疫細胞上のHcar2の活性化に関与していることが示されている。実際、アテローム性動脈硬化のアポリポ蛋白Eノックアウトモデルでは、マクロファージ上のHcar2の活性化がコレステロールの排出を促進し、アテローム性動脈硬化プラークの負担を軽減している[77]。
免疫細胞に発現するHcar2のアゴニズムは、炎症を調節する役割も果たしている。LPSで活性化されたヒト単球やマウスマクロファージにおいて、ナイアシン処理はTNF-αやIL-6などの炎症性サイトカインの発現を減少させるが、この効果はHcar2をノックダウンすると消失する[81,82]。リン酸化p-65やリン酸化NF-κBのようなNF-κB経路の中間体は、Hcar2アゴニズムによって減少することから、サイトカイン放出のこの減少は、おそらくNF-κBシグナル伝達の阻害によって起こると考えられる[81,82]。ナイアシンによるHcar2の活性化は、以前は主に抗炎症性であると説明されてきたが、最近の研究では、ナイアシンは単に免疫活性を抑制するのではなく、サイトカイン産生の変化や貪食能の亢進に代表される有益な表現型を促進するように作用することが示されている。この見解を支持するものとして、いくつかの神経疾患モデルにおける研究から、ナイアシンはミクログリア/マクロファージの活性を増強し、病理学的粒子の貪食作用の増強と病態の軽減を特徴とする有益な表現型を促進することが示されている[8,10,11]。さらに、脳卒中モデルにおいて、ナイアシン投与はHcar2を発現する神経保護マクロファージ集団を活性化し、虚血脳に浸潤して神経病理を改善する。[83]。
Hcar3
ナイアシンが介して作用することが示唆されているもう一つの受容体は、GPR109BまたはHM74としても知られるHcar3である。Hcar3もまた、主に脂肪細胞や免疫細胞に発現しており、主要な内因性リガンドである3-ヒドロキシオクタン酸やキヌレン酸を介して、脂肪酸代謝や炎症を制御している[84]。Hcar2と同様に、Hcar3はGi/o共役型GPCRであり、アデニリルシクラーゼ、サイクリックAMP、細胞内Ca2+レベルの調節を含む下流経路を通じて作用を発揮する[73,85]。以前の研究では、Hcar3が第二のナイアシン受容体であると報告されたが[86,87]、最近の研究では、Hcar2との相同性が高いにもかかわらず、Hcar3はナイアシンとの親和性が低いことが示唆されている[88]。実際、Hcar3はHcar2と比較して、ナイアシンとの親和性が約1000倍低い[50,71]。その結果、このレセプターで発揮されるナイアシンの作用は、Hcar2の作用と比較して最小である。
一過性受容体電位カチオンチャネルサブファミリーVメンバー1 (Trpv1)
ナイアシンもまた、非選択的カチオンチャネルであるカプサイシン受容体Trpv1に結合し、活性化することが示されている[89]。Trpv1は全身に広く発現しており、神経細胞、消化管の感覚細胞、免疫細胞、上皮細胞などに存在する[90]。熱感知受容体として、Trpv1の活性化は血管拡張を含む体温調節過程につながる。マウスにおいて、ニコチン酸を投与すると、ドップラー灌流イメージングによって検出されるように、皮膚血管拡張が起こる。この血管拡張は、Trpv1ノックアウトマウスでは改善され、[91]、Trpv1受容体の拮抗作用によっても改善された[92]。これらの結果から、ナイアシンによるTrpv1の活性化が、ナイアシンのサプリメントを摂取している人によく報告される顔面紅潮の副作用の一因であることが示唆される。
天然Trpv1受容体拮抗剤
- ルテオリン: フラボノイドの一種で、多くの植物に含まれています。ルテオリンはTRPV1活性を抑制することが示されており、抗炎症作用や鎮痛作用があると考えられています。
- レシチン: 大豆などに含まれる天然成分で、TRPV1受容体の活性を抑制する可能性が示唆されています。(by GPT-4)
神経疾患におけるナイアシンのメカニズムと応用
ナイアシンと血液脳関門
薬物が中枢神経系で直接的な薬理作用を発揮する前に、まず血液脳関門を通過しなければならないが、ナイアシンのこの性質は実証されている。実際、ニコチン酸とニコチンアミドの脳内取り込みは、健康なボランティアと神経変性疾患患者の両方において、静脈内投与後のポジトロン断層撮影によって検出されている。[93]。さらに、マウスでは経口投与により脳内ニコチン酸濃度の有意な増加が観察された[10]。
多発性硬化症
MSは、脱髄、炎症、進行性の神経変性を特徴とするCNSの慢性炎症性疾患である。MS患者では、再髄鞘化は臨床的障害のスコアの低下と関連しており、[94]、MSのモデルでは、再髄鞘化は機能回復を促進する[95]。MSの現在の治療法は、再発を予防するために炎症を調節することに重点を置いているが、承認されている治療薬の中で再髄鞘化を標的としたものはない。MSにおける脱髄は、ミエリン残屑の生成につながり、オリゴデンドロサイト前駆細胞の成熟を阻害することによって再ミエリン化を阻害する。[97]。その結果、ミクログリアと浸潤マクロファージの貪食能が向上し、抑制性のミエリン残屑の除去が促進されれば、MSにおける有望な治療戦略となるであろう。
コレステロールはミエリンの主要成分であり、ほとんどの哺乳類細胞では分解できない[98]。ミクログリアやマクロファージが脂質に富んだミエリンの残骸を貪食した後、コレステロールは逆コレステロール輸送を受けるか、エステル化され、脂質小滴に貯蔵されるか、リポタンパク質として放出される[99] ;これらのメカニズムは遊離コレステロールの毒性作用から細胞を保護する[100,101]。CNSでは、コレステロールのホメオスタシスは、 レチノイドX受容体と義務的ヘテロ二量体を形成する肝X受容体(LXR)によって制御されている。[102]。オキシステロールやデスモステロールなどのコレステロール誘導体は、LXRのリガンドとして機能し、細胞内コレステロールレベルの指標となる[103]。オキシステロールが結合すると、これらの受容体は転写因子として働き、アポリポタンパク質E(ApoE)やATP結合カセットサブファミリーメンバーであるABCA1およびABCG1の発現を促進する[103,104]。ABCA1、およびABCG1は、コレステロールの細胞外受容体ApoEへの輸送を仲介し、CNS全体に脂質を分散させるHDL様粒子の生成につながる[104,105]。マクロファージから排出されたコレステロールは、 オリゴデンドロサイトに取り込まれ、新しいミエリンの形成に再利用されるため、再ミエリン化が促進される(図4)[106]。

図4 CNSにおけるコレステロールのリサイクルとナイアシンの影響。 脱髄は、CNSの損傷や傷害の結果としてしばしば起こり、ミエリンの残骸を産生するが、この残骸はCNSのミクログリア/マクロファージによって貪食される。この残骸の取り込みは、ナイアシンによって促進される。[8]。 貪食後、ミエリン残骸はリソソームで部分的に分解される。分解できなかったコレステロールは、エステル化されて脂質滴に貯蔵されるか、細胞外に排出される。コレステロールプロセッシングの障害は、持続的なコレステロール蓄積とコレステロール結晶の形成を引き起こし、炎症性マクロファージの表現型を促進する。コレステロールの排出はABCA1とABCG1トランスポーターによって媒介され、遊離コレステロールを脂質に乏しいApoE粒子に移動させる。ナイアシンがABCA1とABCG1のmRNAレベル [107,108]とコレステロール排出[106]を促進するという証拠があるが、後者が受動的拡散によるものか、ABCA1依存的なメカニズムによるものかは未解決である。コレステロールとApoEは一緒になってHDL様粒子を生成し、CNS全体にコレステロールを分布させる。オリゴデンドロサイトは、この遊離コレステロールを受け取り、新しいミエリンの生成に利用する細胞タイプの一つである。核内では、コレステロール誘導体(オキシステロールなど )がLXRに結合する。LXRはRXRとヘテロ二量体を形成し、転写因子としてApoE、ABCA1、ABCG1の転写を促進する。BioRenderで作成した図
ナイアシンは、マクロファージからのコレステロールのリサイクルおよび排出を調節する。試験管内試験では、ナイアシンを投与した健康なヒトから採取したHDL粒子は、ABCA1およびABCG1依存的に泡沫状マクロファージからのコレステロール排出を促進する[107]。さらに、ApoEノックアウトマウスにナイアシンを投与すると、Abca1、Abcg1、LXR-αのmRNA発現が増加する[108]。ヒト単球をナイアシンとインキュベートすると、ABCA1のmRNA発現が増加し、脂質に乏しいHDL粒子へのコレステロール排出が促進される[109]。心血管疾患の既往歴のあるヒトにおいて、ナイアシンとアトルバスタチンの併用療法は、マクロファージのコレステロール排出能をスタチンのみの群で観察されたレベル以上に増加させるが、この研究ではABCA1特異的なコレステロール排出の増加は観察されなかった[110]。最後に、徐放性ナイアシンの効果を調べた試験管内試験の実験では、受動拡散を介するコレステロール流出能は増加するものの、ABCA1を介する流出には有意な効果は認められなかった。これらの研究を総合すると、ナイアシンはマクロファージからのコレステロール排出を促進するが、それがABCA1依存的な方法かABCA1非依存的な方法か、あるいはその両方かについては、まだ明らかにされていない。
コレステロールのリサイクルや排出の障害は、マクロファージの機能や表現型に悪影響を及ぼす可能性がある。例えば、コレステロールのリサイクル障害とそれに伴う細胞内コレステロール結晶の生成は、NLRP3インフラムソームの活性化につながる[112,113]。MSのリゾレシチンモデルでは、細胞内コレステロールのエステル化が障害されると、脂質滴の形成が低下し、再髄鞘形成に欠陥が生じる[114]。さらに、コレステロールの排出障害と貪食細胞内への持続的なミエリン断片の蓄積により、炎症性の泡沫状マクロファージの表現型が生じ[115]、破片を効果的に貪食する能力を失う可能性がある[116]。注目すべきは、脂質代謝異常がMSの病態に関与していることである。例えば、LXRノックアウトマウスでは、CNSの髄鞘形成が低下しており、[118]、低比重リポ蛋白コレステロールの血清レベルは、MRIで示されるように、MS患者の疾患活動性と正の相関がある[119]。
マクロファージにおけるコレステロールのホメオスタシスは、その貪食能に関係しており、脂質が蓄積したマクロファージは破片を効果的に貪食し続けることができず、最終的にはアポトーシスを起こす可能性がある[116,120]。したがって、ナイアシンは、おそらくコレステロールの排出を促進するナイアシンの能力と関連して、阻害性ミエリン残屑の貪食能の増加を通じて、MSの文脈で再髄鞘化を促進するように働くと考えられている[107]。ナイアシンは、コレステロールを豊富に含む膜を形成する再生オリゴデンドロサイトによって消費されるコレステロールの流出を促進することで、マクロファージ内での脂質の長期蓄積を防ぎ、そのファゴリソソームを解放して、飲み込まれた物質のさらなる消化を可能にすると同時に、有害な炎症性マクロファージの表現型を回避すると考えられる(図4)。
前臨床試験から有望なデータが得られており、ナイアシンがMSのモデルにおいて疾患を改善することが示されている[121]。リゾレシチン脱髄モデルでは、ナイアシン投与によりミクログリア/マクロファージによる抑制性ミエリンの残骸の貪食が促進され、病変部位のオリゴデンドロサイト前駆細胞の数が増加し、再髄化が改善した[8]。注目すべきは、このような貪食作用の亢進は、 ミクログリア/マクロファージ上のスカベンジャー受容体 CD36の発現が増加したことが一因であると考えられていることである [8]。ナイアシンを投与したEAEマウスでは、神経障害が改善し、炎症性浸潤の数が減少し、神経保護が強化された。[122]。さらに、ニコチン酸の豊富な餌を与えた若いラットは、対照と比較して、発生期の髄鞘形成が促進された[123]。このような有望なエビデンスの結果、進行性MS患者を対象とした臨床試験に再利用できる可能性のある認可薬を特定することを目的とした最近の研究では、ナイアシンが強く推奨される3つの候補薬の1つとして挙げられている[7]。今後の研究により、ナイアシンの再髄鞘形成作用や免疫調節作用の可能性を考慮し、ナイアシンがMS患者において治療効果を示すかどうかを明らかにすることができるであろう(図5)。

図5 神経疾患におけるナイアシンのメカニズム。 ナイアシンは、神経疾患や神経変性疾患の病態を緩和するために、さまざまなメカニズムで作用すると考えられる。前臨床試験に基づくこれらの推定される機序には、貪食能の増強と脂質の再利用、免疫調節、酸化ストレスの改善などが含まれる。BioRenderで作成した図
パーキンソン病
パーキンソン病(PD)は、主に黒質小体部におけるドーパミン作動性ニューロンの消失によって引き起こされる神経変性疾患である[124]。この神経病理は、大脳基底核におけるドパミン欠乏を引き起こし、患者に徐脈、振戦、および/または硬直、さらに認知機能の低下や抑うつなどの非運動症状として現れる[125]。パーキンソン病患者に対する治療の第一選択は、ドーパミンの前駆体であるレボドパである。レボドパは運動症状の治療に有効であるが、神経変性は防げず、基礎にある炎症にも影響を与えない。
ナイアシンがPDの病態を改善するメカニズムにはいくつかあるが、その第一は免疫調節である。異常な神経炎症は、PD発症の重要な促進因子として認識されつつある。パーキンソン病患者の死後脳、特に黒質などの患部では、活性化したミクログリアが濃縮されている[127,128]。さらに、健常対照者と比較して、パーキンソン病患者の髄液や末梢血ではIL-6、IFN-γ、IL-1βなどの炎症性サイトカインのシグナル伝達が増加している。[129]。そのため、ナイアシンが示すように、PDにおいて免疫活性を調節することは有益であると考えられる。ナイアシンを投与されたパーキンソン病患者から採取されたマクロファージは、有害な炎症表現型ではなく、むしろ有益な炎症表現型に極性を示す[130]。PDの1-メチル-4-フェニル-1,2,3,6-テトラヒドロピリジン(MPTP)モデルでは、ナイアシンの代謝産物であるNADPHを投与すると、 グリオーシスと炎症性NF-κBの活性化が抑制される。[131]。
ドーパミン作動性ニューロンの喪失により、PDはドーパミン受容体シグナルの減少を特徴とする。ナイアシンがPD病態を調節するもう一つのメカニズムは、ドーパミンレベルを補うことである。前述のように、ナイアシンはドーパミンの産生に関与するNAD +とNADHを産生する[132]。実際、げっ歯類の細胞をNADHでインキュベートすると、キノノイドのジヒドロビオプテリンからテトラヒドロビオプテリンへのリサイクリングが促進され、ドーパミン生合成が増加する。[133]。テトラヒドロビオプテリンは、ドーパミン産生の律速酵素であるチロシン水酸化酵素の合成に必要な補因子である[134,135]。PDのロテノンモデルにナイアシンを投与すると、ビヒクル対照と比較して脳内ドーパミン濃度が上昇することから、前臨床データはこの作用機序の支持を示している。[9]。線条体脳スライスでは、NADHの投与によりKCl誘発ドーパミン放出が増加する[136]。
酸化ストレスは、おそらく神経細胞喪失の根底にある、PDにおける神経変性の重要な推進因子として認識されつつある[137]。パーキンソン病患者の血液中には酸化ストレスのマーカーが観察され[138]、げっ歯類のドーパミン作動性ニューロンにおけるミトコンドリア複合体Iの遺伝子破壊は、ヒトに似た進行性のパーキンソニズムを引き起こす。[139]。PDモデルにおいて、ナイアシンはPDに関連した酸化ストレスを改善することが示されている。実際、ロテノンモデルにおけるナイアシン投与は、酸化ストレスの指標となるマロンジアルデヒドレベルを低下させ、抗酸化物質であるグルタチオンとスーパーオキシドジスムターゼのレベルを上昇させる[9]。MPTPモデルでは、NADPHの腹腔内投与によりグルタチオンレベルが回復し、活性酸素種の産生が減少する。[131]。
まとめると、ナイアシンは、炎症状態の変化、ドーパミンの補充、酸化ストレスの軽減など、PDの病態を修飾するいくつかの神経保護メカニズムを通して作用すると考えられる(図5)。予備的な臨床データは、ナイアシンがパーキンソン病患者において有望な治療法であることを示しており、いくつかの臨床試験が進行中である(表1)。ナイアシンを多く含む食品の摂取は、PDの発症リスクと負の相関があり[140,141]、パーキンソン病患者では、12カ月にわたる低用量ナイアシンの補充により、運動障害の指標であるUnifiedParkinson’s DiseaseRating Scaleが改善した。[13]。さらに、ある症例研究では、脂質異常症のためにナイアシンを投与したパーキンソン病患者において、硬直とブラジキネジアが軽減したことが示されている[142]。
表1 神経疾患におけるナイアシンの進行中および終了した試験
| 障害 | トライアルの詳細と状況 | ナイアシンの製剤と経口投与量 | 主要評価項目 | いくつかの副次的エンドポイント | データ |
|---|---|---|---|---|---|
| 膠芽腫 | 第I-II相試験(NCT04677049);募集中;試験終了予定日2026年1月 |
|
|
末梢単球数変化、QOL | 試験中であり、利用可能なデータはない |
| パーキンソン病 | 第II相試験(NCT03462680):2020年4月終了 | ニコチン酸;250mg/日 | MDS-UPDRS、レム睡眠パターン、睡眠パーセンテージ、MMSE、ストループテスト、疲労度 | CSFおよび血漿中サイトカインの変化、血漿中および尿中のナイアシン濃度、Hcar2発現、血漿中セロトニン濃度 | 運動機能に有意差なし[143] |
| 虚血性脳卒中 | 第II相試験(NCT00796887):2012年8月終了 | ニアスパン®;500または1000mg/日 | 有害事象 | 機能回復 | 機能回復に有意差なし。HDLコレステロールはナイアシン投与群で有意に増加した[144]。 |
| パーキンソン病 | 第II相試験 (NCT03808961)、実施中、募集中、試験終了予定日2024年4月 | ニコチン酸またはニコチンアミド;100mg錠、1日2回 | MDS-UPDRS、MMSE、Hcar2発現、血漿中サイトカイン濃度、血漿および尿中のナイアシン濃度 | VAFS、TMT、腕力、疲労度 | 試験中であり、利用可能なデータはない |
| アルツハイマー病または軽度認知障害 | 第II相試験(NCT03061474):実施中、募集中、試験終了予定日2022年8月 | ニコチンアミド、徐放錠;750mg錠、1日2回 | CSFのp-tau231の変化 | CSFのp-tau 181と総タウの変化 | 試験中であり、利用可能なデータはない |
| アルツハイマー病 | 第II相試験(NCT00580931);2014年7月終了 | Endur-amide®(ニコチンアミド);1500mg錠、1日2回 | ADAS-コグ | CIBIC-Plus、ADCS-ADL、CDR | 一次エンドポイント、二次エンドポイントともに有意差なし[145]。 |
| パーキンソン病 | 第I相試験(NCT05589766):未募集、試験終了予定日2024年12月 | Niagen™(ニコチンアミドリボシド);最大3000mg/日まで用量漸増 | 大脳および髄液のNADレベル、NRRP発現 | 有害事象、QOL、NAD代謝物レベル、MDS-NMS、MoCA、GIDS-PD、MDS-UPDRS | 試験中であり、利用可能なデータはない |
| アルツハイマー病 | 第I相試験(NCT05617508):未募集、試験終了予定日2024年12月 | Niagen™(ニコチンアミドリボシド);最大3000mg/日まで用量漸増 | 脳および髄液NADレベル、FDG-PET、31P-MRS | 有害事象、ADAS-Cog、CDR-SB、MoCA、TMT、IADL、PSMS、NPI-Q、MADRS | 試験中であり、利用可能なデータはない |
| パーキンソン病 | 第II相試験(NCT03568968);募集中;試験終了予定日2024年3月 | ナイアゲン™(ニコチンアミドリボシド);1000mg/日 | MDS-UPDRS | NAD代謝物レベル | 試験中であり、利用可能なデータはない |
| パーキンソン病 | 第I相試験(NCT05344404):2022年7月終了 | ナイアゲン™(ニコチンアミドリボシド);1500mg錠、1日2回 | 重篤な有害事象 | 軽度有害事象, NADメタボローム, MDS-UPDRS | 結果はまだ出ていない |
| アルツハイマー病または軽度認知障害 | 早期第Ⅰ相試験(NCT04430517);募集中;試験終了予定日2025年4月 | Niagen™(ニコチンアミドリボシド);250mg錠剤、1日4回 | 脳内NAD、脳内酸化還元状態 | ミトコンドリア機能、GSHレベル | 試験中であり、利用可能なデータはない |
| 軽度認知障害 | 第I相試験(NCT02942888):2021年8月終了 | Niagen™(ニコチンアミドリボシド);最大1000mg/日まで用量漸増 | モカ | 脳血流、血漿NAD値、SPPB、IADL、動脈圧、GDS、GAS、CLOX、EXIT、TAPS、握力 | 結果はまだ出ていない |
| 軽度認知障害 | 第II相試験(NCT04078178):試験終了予定日2022年9月 | ナイアゲン™(ニコチンアミドリボシド);1200mg/日 | RBANS | 該当なし | 試験中であり、利用可能なデータはない |
| パーキンソン病 | 第II相試験(NCT03816020):2020年2月終了 | ニコチンアミドリボシド;500mg錠、1日2回 | PDRPの変更 | MDS-UPDRS | 大脳NADレベルの有意な上昇と大脳代謝の変化[146] |
31P-MRS31Phosphorous-magneticresonance spectroscopy,ADAS-Cogアルツハイマー病評価尺度-認知サブスケール,ADCS-ADLAlzheimer’s Disease Cooperative Study-Activities of Daily Living Scale,CDRC臨床認知症評価尺度,CDR-SBC臨床認知症評価尺度Sum of Boxes、 CIBIC-PlusClinician’s Interview-Based Impression of Change Plus Caregiver Input、CLOXClock Drawing Task Protocol、CSFcerebrospinal fluid、EXITExecutive Interview、FDG-PETfluorodeoxyglucose-positron emission tomography、GASGeriatric Anxiety Scale、 GDS老年期抑うつ尺度、GSHグルタチオン、GIDS-PDパーキンソン病の胃腸機能障害尺度、 IADL日常生活動作評価尺度、MADRSモンゴメリー・アスバーグうつ病評価尺度、MDS-NMS運動障害学会非運動機能評価尺度、MDS-UPDRS運動障害学会統一パーキンソン病評価尺度、MMSEMini-精神状態検査、MoS-PDG 精神状態検査、MoCAMontreal Cognitive Assessment、NADNicotinamide adenine dinucleotide、NPI-QNeuropsychiatric Inventory brief questionnaire form(NPI-Q神経精神検査簡易質問票、NRRPニコチンアミドリボシド関連パターン、PDRPパーキンソン病関連パターン、PSMS身体的自己維持尺度、RBANS反復可能神経心理学的状態評価バッテリー、SPPB短時間身体能力バッテリー、TAPS聴覚処理能力テスト、TMTトレイルメーキングテスト、QOL生活の質、VAFS視覚的アナログ疲労尺度
アルツハイマー病
アルツハイマー病(AD)は最も一般的な認知症であり、その有病率と医療制度への負担は、人口の高齢化とともに増加する一方である。ADの病理学的特徴として、アミロイドβ斑および神経原線維変化[147]が挙げられ、AD患者は記憶、言語、実行機能を含む認知機能の低下を経験する[148]。AD患者に対する現在の治療法は限られており、主に神経病理学に影響を与えない症状管理に重点を置いている[149]。
ADにおけるナイアシンの作用機序は、MSにおける機序と類似しており、特に肝X受容体の発現と脂質のリサイクルを制御する(図5)。脂質動態の変化がADの病因に関与していることが示唆されている[150]。例えば、ApoE遺伝子はADの病態に強く関与している。ApoEε4対立遺伝子はAD発症の最も強い遺伝的危険因子であるが、ε2対立遺伝子を持つ個体はADから保護されている[151]。さらに、神経細胞のコレステロールレベルは、アミロイド前駆体タンパク質の切断を制御することにより、アミロイドβの産生を制御している[152,153]。さらに、ABCA1-およびABCG1を介したコレステロールの排出は、健常者および非AD認知症対照者と比較して、AD患者の髄液において障害されている[154]。したがって、ナイアシンがコレステロールの排出を促進し、中枢神経系のコレステロールと脂質のホメオスタシスを調節する(図4)。
ナイアシンは、その脂質修飾作用に加えて、若返ったミクログリア/マクロファージ表現型を促進することによってAD病態を変化させ、病的アミロイドBプラークの貪食を増強する。ADの5xFADトランスジェニックマウスモデルにおいて、徐放性ナイアシン(ニアスパンR)を投与すると、ミクログリアとプラークとの関わりが増加し、プラークの数と面積が減少し、貪食に関連するミクログリア遺伝子の発現が促進される[10]。このことは、 認知障害を減少させるという臨床的側面にも好影響を及ぼす。[10]。
ADにおけるナイアシンの新規作用機序を同定するために、遺伝子発現解析も行われた。ADのAPP/PS1トランスジェニックモデルにおいて、ナイアシンを補充されたマウスは認知機能が増強した。また、ナイアシンを補充したADマウスでは、Wntシグナル、翻訳後修飾、mTORシグナルの制御などのプロセスに関与するCtnnb1、Mdm2、Ptenなどの遺伝子の発現が上昇した[155]。
縦断的研究により、ナイアシンはアルツハイマー病患者において治療の可能性があることが示唆されている。例えば、これまでの研究で、食事からのナイアシン摂取量の増加が認知機能の改善[156] および後期ADリスクの低下[14]と関連することが立証されている。ナイアシンがアルツハイマー病患者において有効な治療法であるかどうかはまだ調査されていないが、今後の研究の有望な手段である。
膠芽腫
膠芽腫(GBM)は成人の中枢神経系で最も一般的な原発性腫瘍であり、10万人当たり約2.3人が罹患している[157]。現在の治療法は、外科的切除後に放射線療法とテモゾロミドによる化学療法を行うものであるが、GBMは最も死亡率の高い癌の一つであり、診断後の生存期間中央値は15カ月未満である。[158]。治療の進歩は、脳腫瘍発生細胞(BTIC)の自己複製能、免疫抑制性の腫瘍微小環境、BBBによる中枢神経系へのアクセス制限によって、一部止まっている[159,160]。実際、研究努力にもかかわらず 2005年にテモゾロミドが治療レジメンに導入されて以来、GBMの治療は改善されていない[161]。
BTICsは、自己複製能と増殖能により膠芽腫の増殖と発生を開始する癌細胞のサブクラスである[162] ;100個程度のヒトBTICsを移植するだけで、レシピエントマウスに同一の腫瘍を発生させることができる[163]。これらの細胞はDNA修復機構が非常に効率的であるため、二本鎖DNA切断を引き起こすことで細胞死を誘導する従来の放射線治療に対して抵抗性を示す[164]。BTICsは化学療法にも抵抗性を示すが、そのメカニズムはそれほど明らかではない。効果的なGBM治療は、免疫抑制性の腫瘍微小環境の存在によってさらに妨げられる。腫瘍関連マクロファージは、腫瘍微小環境において最も豊富な非形質転換細胞であり、抗炎症性サイトカインを放出し、T細胞応答を開始しないなど、明確な原腫瘍表現型を示す。[166]。さらに、腫瘍浸潤免疫細胞は、活性が低下して疲弊した表現型を示し、腫瘍細胞に対する適切な免疫応答を行うことができない[167]。したがって、GBMに対する潜在的な治療法は、BBBをうまく通過して免疫活性を刺激し、免疫抑制に対抗して免疫細胞によるBTICsの認識を促進することであろう。
齧歯類のGBMモデルにおいて、ナイアシン投与は有益な免疫調節を促進し、免疫抑制性の骨髄系細胞を若返らせ、腫瘍と闘う能力を高める(図5)。実際、ナイアシンは試験管内試験でGBM患者由来の骨髄系細胞を活性化し、TNF-αやIL-6などのサイトカインの放出を亢進させ、ナイアシン処理単球はGBM患者由来BTICsの増殖を抑制した[11]。さらに、BTICを移植したマウスにナイアシンを投与すると、脳腫瘍の増殖が抑制され、生存期間が延長する。このような有望な前臨床試験の結果を受け、標準治療に徐放性ナイアシン(niacinCRT)を追加することで、GBM患者を対象とした第I/IIa相臨床試験が開始された(clinicaltrials.gov NCT04677049)(表1)。現在進行中のこの臨床試験の結果によって、ナイアシンが臨床環境において抗腫瘍免疫機能を促進する可能性についての知見が深まるだろう。
筋萎縮性側索硬化症
筋萎縮性側索硬化症(ALS)は神経変性疾患であり、上下の運動ニューロンの喪失を特徴とする。現在の治療法は、筋痙縮、唾液漏出、疼痛などの症状を対象としているが、疾患修飾療法は存在しない。近年、腸内細菌叢がALSを含む多くの疾患の病因に関与していることが示唆されており[169]、特に微生物叢-腸-脳軸に関心が向けられている[170]。ALSの動物モデルでは、タイトジャンクションの完全性が損なわれ、腸の伝染性が亢進していることが観察され[171]、マイクロバイオームの異常は運動障害に先行する[172]。さらに、ALS患者は対照群と比べて腸内細菌叢の組成が有意に変化しており[173]、抗生物質の反復使用はALS発症リスクの増加と関連している[174]。特筆すべきは、以下に述べる最近の研究から、ナイアシンが腸内細菌叢の調節を通じて治療効果を発揮する可能性が示唆されていることである。
ALSのSod1トランスジェニックマウスモデルにおいて、Akkermansia muciniphilaの補充はニコチンアミドの血清レベルを上昇させ、運動機能と神経機能の改善につながることから、腸内細菌叢から放出されるニコチンアミドがALSの病態に有益な影響を及ぼすことが示唆される[175]。さらに、ALS患者ではニコチンアミドの合成に関与する分子の血清レベルが変化しており、血清ニコチンアミドの増加は機能状態の改善と相関している[175]。ALSに関する文献以外にも、肥満のヒトにおいて、食事からのナイアシン摂取量の低レベルは、α-ダイバーシティの低さとバクテロイデーテス(Bacteroidetes)の存在量の減少と相関している[176]。遅延放出型ニコチン酸を投与すると、バクテロイデーテス(Bacteroidetes)生息数が有意に増加することから、ナイアシン投与がマイクロバイオームの組成を調節できることが示された[176]。大腸炎を起こしやすくマイクロバイオームの生態系が変化するACE2ノックアウトマウスでは、ニコチンアミドを投与すると、マイクロバイオームの組成がコントロールレベルにまで回復し、大腸炎の胃腸症状が改善する[178]。このように、ナイアシンは腸内細菌叢を調節することで、ALSやその他の中枢神経系疾患に対する有望な治療選択肢となる可能性がある(図5)。
結論と残された疑問
結論として、ナイアシンは必須ビタミンであり、様々な疾患に対する忍容性の高い治療薬として長い間役立ってきた。ナイアシンは、NAD+/NADPの前駆体としての役割が一般的であるが、GPCR Hcar2における作動活性やマイクロバイオームの調節など、さらなる作用機序を有している。免疫細胞におけるHcar2の発現により、ナイアシンは免疫系の強力なモジュレーターとして浮上しており、有益な免疫細胞の表現型を促進し、有害な残骸の貪食を強化し、いくつかの神経疾患における神経病理学を軽減することが示されている。ナイアシンはまた、CNSマクロファージがミエリンなどの脂質を多く含む残骸を取り込む際に重要な役割を果たす、コレステロールのリサイクルを制御する役割も担っている。最近の研究により、ナイアシンはMS、アルツハイマー病、神経膠芽腫などの神経疾患における有望な治療選択肢として確立されつつある。
ナイアシンのメカニズムや有用性については、いくつかの疑問が残っている。ナイアシンの効果は、NAD+への変換のような代謝のみによるものなのか?Hcar2刺激が関与していると考えられる高用量の薬理学的投与が行われる場合、その恩恵はどの程度まで代謝メカニズムによってもたらされるのだろうか?ナイアシンの受容体はまだ特定されていないのか?ナイアシンは抗炎症性なのか、炎症促進性なのか、また、これらの潜在的に異なる活性を分離する濃度範囲はあるのか?MSのような慢性疾患での長期使用は、炎症性反応のリスクになるのか?ナイアシンをLXRの直接作動薬など他の治療薬と併用することで、より効果的な結果を得ることは可能か?ナイアシン治療が有効な神経疾患は他にもあるのだろうか?今後の研究により、神経保護剤としてのナイアシンの役割が解明され、臨床への普及が期待される。
謝辞
ナイアシンの前臨床および臨床研究に対するカナダ保健研究所の運営助成に感謝する。EWはMultiple Sclerosis CanadaよりPhD studentshipの支援を受けている。
必要な著者フォーム
著者から提供された情報開示書は、本論文のオンライン版で入手可能である。
