pubmed.ncbi.nlm.nih.gov/36139084

Iron in Alzheimer’s Disease: From Physiology to Disease Disabilities
オンライン公開 2022年9月6 日
PMCID: PMC9496246
PMID:36139084
要旨
活性酸素種(ROS)は神経変性の過程で重要な役割を果たしている。酸化ストレスの増加は、脳組織の脂質、タンパク質、核酸に損傷を与え、生体恒常性の喪失に結びつく。このため、活性酸素を生成するいくつかの生化学反応に関与する遷移金属に注目が集まっている。
アルツハイマー病(AD)の根底にある毒性経路の生成における金属の役割が、多くの証拠によって明らかにされてきたにもかかわらず、アミロイド・カスケード仮説の出現によって、この問題は脇に追いやられてきた。しかし、金属とADとの関連は、ADに重要であることが知られている脳領域での局所的蓄積に焦点を当て、過去20年間に研究されてきた。最近の証拠により、鉄とADの関係、特にフェロプトーシスを通じてADのリスクを増大させる鉄の能力が明らかになった。
本総説では、我々の体内における鉄の機能を特徴づける主要なポイントを簡潔にまとめ、鉄が我々の生活に不可欠であるにもかかわらず、特にADのリスクをコントロールしたいのであれば、その機能障害を監視しなければならない理由を強調する。
キーワード アルツハイマー病、フェロプターシス、鉄、セルロプラスミン、活性酸素種
1.はじめに
アルツハイマー病(AD)は最も一般的な認知症であり、一連の症状として定義される[1] 。主要な神経変性認知症は、満たされていない重大な医療負担であり、世界的な健康上の課題となっている。認知症は世界中で約5,500万人を苦しめており、毎年約1,000万人が新たに発症している(WHO 2021)。ADは思考、記憶、自立に影響を及ぼす。認知症の全タイプの60~80%を占め(アルツハイマー病協会、2021 Alzheimer’s Disease Facts and Figures)、アミロイド斑を形成する細胞外アミロイドβ(Aβ)ペプチド形成と、神経原線維のもつれを作るリン酸化タウタンパク質(P-tau)の蓄積によって典型化される[2,3]。2019年には、AD患者数は5,000万人に上ると推定されている[4]。臨床症状の発現に基づき、ADは早期発症または後期発症(それぞれ65歳未満または65歳以上)と呼ばれ、また、多因子複合疾患の病因を持つ家族性(5%未満)と散発性に区別される。早期発症の家族性ADは、アミロイド前駆蛋白(APP)、プレセニリン1(PSEN1)、またはプレセニリン2(PSEN2)遺伝子に変異がある場合に同定される[5] 。
高齢、親族にAD患者がいること、遺伝(アポリポ蛋白E4をコードするAPOE4遺伝子)が晩発性ADの最も重要な危険因子である[1] 。認知機能の緩やかな低下が病気の経過を特徴付け、最終的には認知症と死に至る。2018 National Institute on Aging-Alzheimer’s Association (NIA-AA) Research Framework[6]によると、認知機能未障害(正常)、軽度認知障害(MCI)、認知症を含む症候学的認知病期分類が区別される。認知症の最終段階は、さらに軽度、中等度、重度に分けられる。国際ワーキンググループ(IWG)と米国老化研究所-アルツハイマー病協会は2014年、神経画像とバイオマーカーの最近の進歩に基づいて、AD診断のための新しい基準を定義した[7] 。ADの特徴は、脳内のアミロイド斑と神経原線維のもつれの沈着である。神経原線維性もつれとは、細胞内の修飾蛋白質タウが高リン酸化(P-tau)を受けて凝集したもので、蛋白質が凝集して神経細胞の構造を破壊する。
アミロイド斑は、神経細胞やグリア細胞の周囲の細胞外腔で不溶性の凝集体を形成するタンパク質線維の凝集体である。その核は主に、大脳皮質のシナプスで高発現する膜貫通タンパク質であるAPPに由来する、39から43アミノ酸からなる4kDaのペプチドであるAβでできている。APPはαセクレターゼとγセクレターゼという酵素によって細胞外で切断されるが、βセクレターゼによっても切断され、Aβペプチドが細胞外に放出され、老人斑に凝集する[8]。
この20年間で、酸化ストレスが脳の機能性に影響を及ぼすことがよく知られるようになった。この一連の研究は、フリーラジカルを生成するいくつかの生化学反応に関与する遷移金属の重要な役割を明らかにしてきた。鉄と銅はフェントン型反応に関与することでよく知られており、この反応は活性酸素種(ROS)を発生させ、細胞区画を損傷し破壊する[9]。ADの根底にある毒性経路の生成における金属の役割は、多くの証拠によって明らかにされている。しかしながら、金属はADの潜在的な促進因子として、1990年代から横並びになってきた。アミロイド・カスケード仮説[8,10] の出現により、AD患者の少なくともサブセットを特徴づける最も重要な欠損や異常のいくつかを説明できる可能性があるにもかかわらず、他のすべての研究前線が見えなくなってしまったのである[11] 。ここでは、ADの病態において鉄が果たす基本的な役割、特に銅との相互作用について簡潔に説明し、ADのリスクとの関連において鉄の恒常性をモニターすることを研究者に促している理由を論理的に要約することを目的として、ADにおける鉄の関与について議論する。
2.鉄の本質
鉄は、Fe2+(2価の第一鉄)とFe3+(3価の第二鉄)の酸化状態に代表されるd-ブロック遷移金属である。体内に最も多く存在する遷移金属であり、私たちの生命維持に不可欠ないくつかの機能に関与している。鉄はヘモグロビン(細胞に酸素を供給するのに重要)、ミオグロビン、シトクロムの構成成分である。ミオグロビンは筋肉組織に酸素を供給し、そこで酸素を貯蔵する。さらに鉄は、酸素代謝を包括するカタラーゼやペルオキシダーゼの働きや、ミトコンドリアの呼吸鎖や電子輸送におけるチトクロムの働きに関与している。鉄はミエリンと神経伝達物質の生合成に関与し、免疫系とDNA合成と遺伝子制御に関与している。この金属は、生物学的利用能が低いにもかかわらず必須である。さらに、毒性を持つ可能性もある。その結果、細胞は鉄の吸収を処理する複雑なメカニズムを進化させてきた。ヘプシジンはペプチドホルモンであり、細胞表面上のフェロポルティンのレベルを調節することによって、哺乳類における食餌性鉄吸収と全身性鉄輸送を制御し、細胞からの鉄排出を促進する。細胞レベルでは、鉄の取り込み、貯蔵、利用は、鉄応答性エレメント/鉄制御タンパク質(IRE/IRP)によって制御されている。ここでは、全身レベルおよび細胞レベルで作用する、より関連性の高い調節経路について簡潔に報告する(専門的な総説は[12]を参照)。
ADは、鉄タンパク質、ヘプシジン、フェリチン、およびインターロイキン-1β(IL-1β)、IL-6、ApoE 4対立遺伝子などの炎症性バイオマーカーと関連している。ヘプシジン、フェリチン、IL-6は、ADにおける神経炎症に関連する宿主防御機構に大きく関与している。ヘプシジンはAD患者の血清で上昇し、疾患との相関を示している[13] 。
2.1.鉄の吸収と分布
ヒトの生物には鉄を排出する生理的メカニズムは存在せず、腸細胞の死滅、皮膚の剥離、女性の月経時のような少量の出血によって、最低限排出される。
一般的な欧米の食事では、1日あたり約15mgの鉄が摂取できるが、このうち私たちの体が吸収するのは1~2mg/日である。これが必要量である[14]。人体には常に3~5gの鉄が含まれており、そのうち2.5gがヘモグロビンに、約600mgが細網内皮マクロファージに、さらに300mgが特定の細胞プロセスを実行するためにタンパク質に利用され、3~4mgがトランスフェリンに結合し、残りはフェリチンとして貯蔵されている[15]。
血清フェリチンは鉄貯蔵タンパク質であり、個人差が大きい。生理機能に影響を及ぼすのは、主要な欠乏症のみである。フェリチンはヘモクロマトーシスやその他の過剰鉄貯蔵障害のある人では高値になる。フェリチンは急性期反応物質であり、炎症状態で増加することがある:フェリチンは鉄を隔離し、微生物による鉄の消去を阻害する。フェリチンの合成はサイトカインに反応し、主に肝細胞とマクロファージにおいて転写と翻訳の両レベルで制御されている。TNF-αやIL-1α、IRP1やIRP2によって制御されている[16]。
一般に、循環鉄はトランスフェリンと結合している。そのレベルはヘプシジンによって制御されている。ヘプシジンは肝臓で生合成されるホルモンで、フェロポルチンの分解速度を制御する:(1)腸細胞膜、ひいては腸細胞から血液中への鉄輸出速度、(2)肝細胞、貯蔵鉄輸送の制御、(3)マクロファージ、リサイクル鉄輸送の制御、そして全体として、全身の鉄ホメオスタシスを制御する。
身体は巧みな還元と酸化のプロセスで鉄の吸収を調節している。食事から摂取される鉄は、事実上すべて第二鉄の酸化状態(Fe3+)にあり、十二指腸の腸管細胞の先端表面を通って輸送されるためには、まずFe2+に還元されなければならない。実際、二価の鉄のみを結合する二価金属トランスポーター1(DMT-1)は、Fe2+の輸送を促進する[17]。
腸管細胞の先端表面には、様々な第二鉄還元酵素が発現しており、例えば、Cytochrome b558第二鉄/銅還元酵素[18] やSteap2[19] がこの還元を行っている。さらに、プリオンタンパク質(PrPC)がフェリレダクターゼとして機能することが報告されている。全身の鉄ホメオスタシスが異常であるPrPKOマウスで示されているように、PrPCは十二指腸腸細胞を介した鉄輸送を促進しているようである。特に、鉄代謝におけるPrPの機能的役割が報告されており、プリオン病における鉄ホメオスタシスの不均衡を、PrPの機能喪失の結果として説明できるかもしれない[20]。還元後、DMT-1は、H+の電気化学的勾配(細胞の外側から内側へ)のおかげで、Fe2+を腸管にチャネルする[17,21](表1、図1)。

図1 腸細胞からの鉄吸収
還元酵素は、腸管に入る前にFe3+を還元する。DMT1(二価鉄トランスポーター)は、電気化学的勾配を利用するFe2+トランスポーターである(図示せず)。二価の鉄は、腸管細胞の先端表面にも存在するヘムキャリアタンパク質1(HCP1)を介して、ヘムの一部として吸収されることもある。ヘムオキシゲナーゼがヘムを切断して鉄2+を放出し、フェリチンに貯蔵される。フェロポルティンは、基底側腸細胞表面の門脈血漿への鉄の移行を促進する。その後、フェロキシダーゼであるヘファエスチン(HP)がFe2+をFe3+に再酸化し、アポトランスフェリン(アポTf)と結合してホロトランスフェリン(Tf)を形成し、血液中の鉄を輸送する。過剰な鉄は、ヘプシジンによってフェロポルティンが内在化・分解され、腸から排出される。
表1 鉄の吸収と分布
| エンテロシテ | 鉄 | |
|---|---|---|
| 酵素/タンパク質 | タンパク質の機能 | |
| 細胞頂上皮 | 非ヘム CYBRD1 シトクロムb558鉄/銅還元酵素 ステープ2 |
Fe3+のFe2+への酸化還元 |
| 細胞膜 | 非ヘム DMT-1 |
電気化学的勾配を介したFe2+のチャネリング |
| ヘム HCP-1 |
ヘム鉄の輸入 | |
| サイトソール | 非ヘム フェリチン |
ストレージ |
| ヘム ヘムオキシゲナーゼ |
ヘムの切断部分 | |
| 細胞基底膜 | フェロポルチン | Fe2+は細胞質から血液循環に輸送される。 |
| 細胞基底面外側上皮 | ヘファスチン | の酸化還元 Fe2+からFe3+へ |
| コントロール | ペプチドホルモン ヘプシジン | 鉄過剰:ヘプシジンはフェロポルティンの分解と鉄貯蔵を制御する。 |
| プラズマ | トランスフェリン(ホロ/アポTf) | Tfは2原子のFe3+と結合し、輸送する。 |
| ヘモグロビン | 赤血球生成 | |
また、完全には解明されていないが、主に食肉由来の鉄が、腸管細胞の先端表面にも存在するヘムキャリアタンパク質1(HCP-1)を介して、ヘムの一部として、つまり二価の鉄として腸管細胞に入る二次的なメカニズムもある。この時点で、鉄はヘムオキシゲナーゼの作用によって放出され、DMT1によって供給されるFe2+プールに加わる[22](表1、図1)。
腸管細胞内に入ると、鉄はさらに吸収制御過程を経る。細胞内で「過剰」になった鉄は、450 kDaの巨大なタンパク質であるフェリチンに貯蔵され、最大4500原子の鉄を貯蔵することができ、微絨毛を通って再利用されるか、部分的に分泌される腸細胞が死滅するまでそこに留まる[23]。その代わりに、制御された量の鉄が腸細胞の基底側面に移動し、フェロポルチンによって門脈血漿に移行する[24,25,26]。吸収される鉄はこの部分だけである。基底側外表面では、フェロキシダーゼであるヘファエスチンがFe2+をFe3+に再酸化し、これがトランスフェリンと結合できる唯一の鉄形態であり、最終的に循環に取り込まれる[27](表1、図1)。
重要な調節の役割を果たすのは、ヘプシジンというペプチドである(詳細な総説は[28]を参照)。肝臓は、トランスフェリンに結合した鉄の過剰を検出すると、ヘプシジンの合成を増加させることで反応する。ヘプシジンは腸細胞の基底側面にあるフェロポルティンに結合し、フェロポルティンを細胞内に取り込ませ、そこでフェロポルティンが急速に分解される[29] 。これにより、より多くの鉄が腸管細胞内に留まり、フェリチンに貯蔵されるため、トランスフェリンによる輸送に利用できる鉄の量が減少し、一般循環に放出される。フェリチンの減少(低フェリチン血症、鉄レベルの低下)は、炎症が引き起こすヘプシジン濃度の上昇に関連して、炎症状態でも起こりうる[30] 。ヘプシジンは、フェリチンなどの鉄結合タンパク質の急性期増加にもかかわらず、鉄の吸収と利用可能性を低下させ、機能的鉄欠乏をもたらす。鉄欠乏の状態は、鉄濃度が低いこと、トランスフェリン/鉄結合能(TIBC)が高いこと、トランスフェリン飽和率が低いことでも特徴づけられる。実際、トランスフェリンは炎症時に減少する負の急性期タンパク質である。
ヒト恒常性鉄制御タンパク質(HFE)は、腸細胞を含む様々な細胞種の膜内に存在するタンパク質で、細胞膜上のTfRとの親和性を制御することにより鉄の吸収を制御している。HFE遺伝子の変異は、食事からの鉄の吸収を促進し、その結果、肝臓、脳、その他の臓器に過剰に沈着することにより、HFE遺伝性ヘモクロマトーシスを引き起こす。ヘモクロマトーシスはメンデル型遺伝をする。ヘモクロマトーシスは常染色体劣性遺伝し、6番染色体にある主要組織適合複合体と関連している[31] 。
腸管細胞の外で鉄が再酸化されてFe3+になると、鉄は肝臓で合成された80kDaの糖タンパク質であるトランスフェリンと結合し、循環輸送される。トランスフェリンはさらなる調節ステップを提供する。各トランスフェリンタンパク質は、2つのFe3+イオン(ホロトランスフェリン)と非常に高い親和性で結合するが、pH依存性もある[32]。肝臓のトランスフェリン産生は、吸収された鉄のうち身体が必要とする割合だけを結合し輸送するように調整される。特殊な状況が生じた場合、肝臓はトランスフェリン合成を調節する。例えば、鉄欠乏状態では、肝臓はより多くのトランスフェリンを産生し、血液中の総TIBCを増加させる。逆に、ヘモクロマトーシスのように鉄が多い場合、肝臓はトランスフェリンの産生を減らして血液中のTIBCを減らす。
結合鉄の割合を評価する効果的な方法は、いわゆるトランスフェリン飽和度(TSAT)を測定することで、血清中に存在する鉄の量とTIBCの比(100倍)を表す。TIBCの測定は、トランスフェリンを直接測定するよりも簡単で、後者の正確な測定値を提供する。正常なトランスフェリン飽和度の範囲は20~45%である。トランスフェリンはヘプシジンの上流制御因子でもある[33]。鉄代謝におけるトランスフェリンの役割については、[34]を参照。
吸収された鉄の大部分は、トランスフェリンによって直ちに骨髄に運ばれ、酸素運搬タンパク質であり赤血球の構成成分であるヘモグロビンを作り、赤血球造血に関与する。
2.2.鉄代謝制御ユニット:肝細胞
赤血球造血に関与しない鉄は肝臓に到達し、肝細胞と網状赤血球内皮細胞に加わり、古い赤血球を摂取してヘモグロビンを分解し、再利用される鉄分を放出することで鉄のリサイクルを可能にする[35]。
肝細胞による鉄の取り込みは、受容体を介したエンドサイトーシスによって起こる。この機構は、細胞表面に存在するトランスフェリン1受容体(TfR1)[36,37]が、高い親和性を持つホロトランスフェリン[38]と結合することで起こる(表2、図2)。

図2 肝細胞における鉄代謝鉄は、トランスフェリン受容体1(TfR1)を介したエンドサイトーシスによって肝細胞に取り込まれる:
TfR1は細胞表面にあり、ホロトランスフェリン(Fe-TfA)プロトンATPaseポンプと結合し、エンドサイトーシス後にエンドソームを酸性化し、ホロTfからのFe3+の放出を可能にする。その後、Fe3+は還元酵素によってFe2+に還元され、Fe2+はDMT-1を通過することができる。鉄はエンドソーム内に入ると、クエン酸アデノシン三リン酸(ATP)、アデノシン一リン酸(AMP)、または他のペプチド(不安定なプール)と不安定な結合を形成し、ミトコンドリアに到達すると考えられている。ミトコンドリアでは、鉄はヘムや鉄硫黄クラスターの生合成に使われる。また、フェリチン(予備鉄の主な供給源)に貯蔵されることもある。フェロポルティンは、過剰な鉄をFe2+として血液中に運び、そこでセルロプラスミン(ホロCp)が鉄をFe3+に酸化し、トランスフェリン(Tf)への負荷を促進する。
表2 肝臓における鉄代謝
| 肝細胞 | 鉄 | |
|---|---|---|
| 酵素/タンパク質 | タンパク質の機能 | |
| 頂部表面 | TfR1 ホロTf |
鉄はTfR1/Holo-Tfと複合体を形成して内在化する: 酸性化エンドソームの生合成 |
| サイトソール | 還元酵素 DMT1 |
電気化学的勾配を通してFe3+はFe2+に還元される。 エンドソーム外へのFe2+の移動 |
| フェリチン クエン酸、AMP、ATP |
貯蔵 不安定プール |
|
| 胆管との界面 | ||
| 基底側外膜 | フェロポルチン | Fe2+は細胞質から血液循環に輸送される。 |
| セルロプラスミン | Fe2+はFe3+に酸化される。 | |
| コントロール | IRE/IRP | 鉄は利用、貯蔵、輸送のために細胞内に取り込まれる。 |
| サーキュレーション | トランスフェリン(ホロ/アポTf) | 2原子のFe3+の結合と輸送 |
| フェリチン | 鉄貯蔵タンパク質 | |
複合体エンドサイトーシスの後、アデノシン三リン酸(ATP)プロトンポンプがエンドソームを酸性化し、ホロトランスフェリンからのFe3+の脱離を可能にする。その後、Fe3+は還元酵素によってFe2+に還元され、DMT1を通過できるようになる[16,39]。いったん内部に入ると、鉄はクエン酸、ATP、アデノシン一リン酸(AMP)、あるいはその他の化合物とゆるやかに結合すると考えられている[40,41]。これらの複合体において、鉄2+はミトコンドリアに到達し、そこでヘムと鉄硫黄のクラスターが生合成されるか、フェリチンに貯蔵される[23,42,43,44](表2、図2)。
すべての細胞タイプにフェリチンの貯蔵があるが、骨髄、肝臓、脾臓に最も多い。特に肝臓の貯蔵は、最も豊富な鉄の貯蔵庫である。女性は月経による鉄の喪失を統合する必要があるため、貯蔵量をより多く使う。過剰なFe2+はトランスフェリンによって血液に移行しやすくなる。その後、Fe2+はセルロプラスミン(腸管細胞でヘファエスチンと同じ働きをする)によってFe3+に酸化され、トランスフェリンに取り込まれる。鉄代謝は複雑で、IRE/IRPシステムを含む転写後プロセスによって厳密に制御されている。IRE/IRP系は、細胞内への鉄の取り込みだけでなく、利用、貯蔵、輸送も制御するため、極めて重要である[45](表2)。IREは進化的に保存されたmRNA配列で、25-30ヌクレオチドのステムループ構造を持つ。鉄欠乏状態では、IRPは標的IRE領域に結合し、TfR1のmRNAを安定化し、フェリチンmRNAの翻訳を立体的に阻害する。TfR1の合成は、循環トランスフェリンからの細胞内鉄取り込みを促進する。
逆に、フェリチンの生合成が阻害されると、鉄の貯蔵が妨げられ、金属が動員されるようになる。鉄が過剰になると、IRPが不活性化され、TfR1 mRNAの分解とフェリチンmRNAの翻訳が起こる。この制御により、TfR1を介した鉄の追加的な内在化が減少し、細胞質フェリチンへの鉄の蓄積が促進される[46]。
2.3.全身循環から脳への鉄の取り込み
生命維持と脳機能にとって極めて重要であるにもかかわらず、ヒトの中枢神経系(CNS)における金属獲得のメカニズムは、いまだにほとんど解明されていない。トランスフェリンは、循環中、そして脳の毛細血管を通してFe3+を輸送する。毛細血管では、脳毛細血管内皮細胞(BCEC)が緊密に結合し、血液脳関門(BBB)を構成している。BCECの内腔表面では、トランスフェリンと鉄の複合体がレセプターTfR1によって取り込まれ(図3A,B)、トランスフェリン/TfR1はエンドソームに内包される。エンドソーム内で鉄は複合体から切り離され、TfR1が細胞質内を移動する間、そこに留まる。その後、エンドソームはBCECの内腔側に到達し、細胞膜と融合する。これによりFe3+が露出し、細胞外の間質空間に放出される。アポトランスフェリンはTfR1に付着したまま、再びエンドサイトーシスを受け、BCECの内腔側に戻る。ここでアポトランスフェリンは毛細血管に放出され、Fe3+イオン結合の次のサイクルを開始する準備が整う[47,48,49,50,51]。また、マクロファージが脳実質と末梢循環の間の鉄タンパク質の通過に関与し、鉄のホメオスタシスに重要な役割を果たしていることも報告されている[13]。


図3 中枢神経系における鉄の代謝
(A):鉄3+はトランスフェリンによって脳毛細血管に輸送される。脳毛細血管内皮細胞(BCEC)は血液脳関門(BBB)を形成している。BCECの内腔表面では、トランスフェリンR1受容体(TfR1)が鉄を負荷したトランスフェリンを捕捉し、トランスフェリン/TfR1はエンドソームに内在化される。エンドソームは細胞内空間を移動し、BCECの内腔側に到達する。その後、エンドソームは外膜と融合する。これによりFe3+が露出し、間質空間に放出される。アポ-トランスフェリンはまだTfR1と結合しており、再びエンドサイトーシスを受け、内腔側のBCECに戻る。この時点でアポTfは放出される。(B):Fe3+トランスフェリンの複合体は細胞外空間を移動し、神経細胞に到達する。ニューロン表面のTfR1がエンドサイトーシスを開始する。神経細胞膜はBCECとの差でDMT1を示す。これがエンドソームに巻き付き、一緒に侵入する。シナプス前小胞にはフェロポルティンが存在する(図示していない)ことから、Fe2+はシナプス小胞を経由してシナプス間腔に移動し、小胞融合後にFe2+が放出されることが示唆される。このように、シナプス前後の間隙には遊離鉄が存在する。アストロサイト先端の外側では、グリコシルホスファチジルイノシトール(GPI)アンカー型セルロプラスミン(Cp-GPI)がFe2+をFe3+に酸化するのを促進し、アポトランスフェリンへの鉄のアップロードを可能にしている。細胞内と細胞外のメカニズムが相乗的に働いて、神経細胞への鉄の移動が可能になる。
ほとんどの場合、鉄はこのようにして脳に入るようである。しかし、低トランスフェリン血症マウスを使った実験では、鉄はトランスフェリンに依存しないメカニズムでも脳に移動することが証明されている[52]。この機序の詳細については、Millsらの研究[53]を参照。
BCECの内腔側はアストロサイトと接触しており、ニューロンとは直接つながっていないようである。アストロサイトとBCECの間の空間では、Fe3+はアポトランスフェリンやATP、クエン酸塩、その他の低重量元素と結合することができる。これらの複合体は脳の細胞外腔のどこにでも存在するが、主にアストロサイトとBCEC側の間の腔に集中しているように見える。
トランスフェリン鉄は、細胞外液を介して、TfRが膜上に存在するニューロンの領域に容易に到達することができる。さらに、ニューロン表面を取り囲むアストロサイトの足突起は、フェロポルティン[54] とCp-GPI-[55] の両方を発現しており、別の経路があることを示唆している。ATPまたはクエン酸に結合した鉄3+は、アストロサイトに内在化される可能性がある。この時点で、鉄はアストロサイト内を輸送され、まだ同定されていないフェリレダクターゼによって還元され、フェロポルティンを介してアストロサイトの末端から外部に輸送される可能性がある。ここで、セルロプラスミンがFe2+のFe3+への酸化を触媒し(下記参照)、アポトランスフェリンへの鉄のアップロードを促進するかもしれない。説明したメカニズムは相互に排他的ではないが、神経細胞に鉄を運ぶために、間違いなく一緒に働くことができる(図3B)。
BCECと同様に、ニューロンの膜にもTfRがあり、再びエンドサイトーシスを開始することができる。BCECと神経細胞表面の決定的な違いは、後者にはDMT-1があり、エンドソームに飲み込まれて一緒に沈むことである[51](図3B)。
さらに、フェロポルティンはシナプス前小胞にも発現している[54]。これは、Fe2+がシナプス前小胞を介してシナプス間腔に放出されるのではないかという仮説を支持するものである。シナプス間腔の遊離Fe2+は、シナプス前膜とシナプス後膜の界面で酸化ストレスを引き起こす可能性がある。
2.4.銅と鉄のクロストーク・タンパク質としての重要なセルロプラスミン機能
すでに述べたように、セルロプラスミンの生合成は主に肝細胞で起こる。銅ポンプATP7Bタンパク質は、6原子の銅をアポ-セルロプラスミンに転移する。これにより、セルロプラスミンは活性のあるホロセルロプラスミンとして折り畳まれ、一般循環に送り出される。セルロプラスミンはまた、CNSの細胞によって内因性に合成され、ニューロン近傍のアストロサイトによってCp-GPI形態で強く発現される[55] 。セルロプラスミンは、以下のような生理学的に重要な反応を介して、Fe2+をFe3+に酸化する触媒となる:
4Fe2++ 4 H++O2→ 4Fe3++ 2H2O (1)
この反応は、Fe2+の生物学的利用能を低下させる効果がある。この金属は、Fe3+のみを運搬するトランスフェリンによって、様々な組織や器官に運搬される[56,57]。式1の副産物は単なる水であるため、ほとんどの場合、セルロプラスミンは潜在的に有毒な鉄を無毒化する。
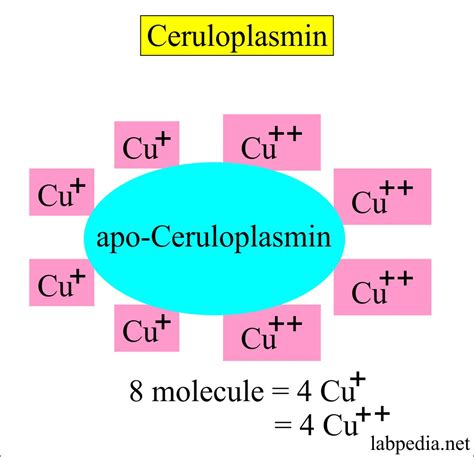
セルロプラスミンにはもう一つ重要な役割があり、それはH2O2[58]を消去し、酸素ラジカルの発生を防ぐことである。これは、生理的pHで起こりうるFe2+の非酵素的酸化が、危険なフェントン反応を引き起こす可能性があるためである。セルロプラスミンは、脂質の過酸化も防ぐことができる。
肝臓で利用可能な銅の量は、ホロセルロプラスミンの合成を調節する。銅が少ないほど、セルロプラスミンは少なくなる。[59].さらに、銅のバイオアベイラビリティが低下すると、フェロキシダーゼ活性を持たず、血漿中ですぐに分解されるアポ-セルロプラスミンが一定量生成される。ATP7B遺伝子の突然変異は、非セルロプラスミン銅中毒の典型的な疾患であるウィルソン病や、 肝臓と脳の両方に銅が蓄積するウィルソン病に見られるように、ホロセルロプラスミン濃度が低くなる[60,61]。加えて、ATP7B遺伝子の機能的な一塩基多型は、セルロプラスミン活性の変化や、主にADに関連する特異的な活性低下をもたらす可能性がある。代わりに、セルロプラスミンをコードするCP遺伝子に変異があると、アセロプラスミン血症になる([62]に総説あり)。
セルロプラスミンの減少により、フェロキシダーゼ活性が低下し、フェントン反応を触媒する遊離鉄が増加する。このため、アセロプラスミン血症は重度の鉄過剰症を引き起こす。アポセルロプラスミンは鉄ホメオスタシスの障害と考えられている。症状はヘモクロマトーシスと共通する。アセロプラスミン血症ではトランスフェリン飽和度が低く、ヘモクロマトーシスではトランスフェリン飽和度が高いので、誤診を避けるために、トランスフェリン飽和度の測定が有用であろう。アセロプラスミン血症の合併症として、脂質過酸化の亢進と脂肪酸の酸化障害がある。
セルロプラスミンは、血清の鉄酸化率を決定する因子である。酸化は、鉄を組織貯蔵から全身循環に運ぶために必要である[63,64,65] 。従って、セルロプラスミンは鉄代謝と銅代謝の間のクロストーク蛋白である。60年代に行われた灌流肝標品の実験では、セルロプラスミンを添加すると血液循環中の鉄の放出が著しく促進されることが示された。このことは、セルロプラスミンが鉄の動員にとって重要であることを示唆している[65]。このように、セルロプラスミンは抗酸化防御に重要な役割を果たす複合タンパク質であり(図4)、その結果、肝疾患や神経変性疾患の分子メカニズムに関与している[66,67]。

図4 セルロプラスミンは鉄と銅の輸送をつなぐクロストークタンパク質である。ホロセルロプラスミン(ホロCp)は、鉄と銅の代謝をつなぐクロストーク・タンパク質として黄色で図示されている。肝細胞のATP7B銅ポンプは、6個の銅原子をアポ・セルロプラスミン(アポCp)にアップロードする。これにより、フェロキシダーゼ活性に必要な正しいホロCpのフォールディングが可能になる。
2.5.鉄はいつ、どのようにして有毒になるのか?
これまで見てきたように、身体は鉄の吸収を厳密に調節するために、高度で多様な一連のプロセスを経ている。生理的バランスが少しでも崩れると、フリーラジカルがすぐに生成され、その結果、必須タンパク質プロセスの破壊、組織損傷、異常蓄積を引き起こし、全体的な酸化ストレスが生じるからである[68,69]。フリーラジカルは不対になった価電子を持つ分子で、反応性が高い[70] 。酸素は最も活性の高いラジカル、すなわち活性酸素を生成する。酸化ストレスとは、細胞環境における活性酸素と抗酸化物質のバランスが失われた結果、細胞の機能や生存能力に有害な影響を及ぼすことと定義される。過剰な活性酸素は、タンパク質、DNA、ヌクレオチドプールなどの生体分子に損傷を引き起こす。8-オキソ-7,8-ジヒドロ-2′-デオキシグアノシン(oxo8dG)は、DNAに組み込まれた2′-デオキシグアノシン(dG)またはヌクレオチドプールに組み込まれたグアニンのC8位のヒドロキシルラジカルとの反応によって生成される塩基修飾の一般的な酸化型であり、三リン酸型(dGTPまたはGTP)である。最も重要な活性酸素は、代謝過程によって自然に、あるいは放射線照射によって人工的に生成されるスーパーオキシドアニオンO2-である。O2-は、酵素や金属触媒を介したプロセスで他の分子と相互作用し、さらに活性酸素を発生させるので重要である。例えば、私たちの免疫システムは、炎症プロセスに反応してスーパーオキシドアニオンと一酸化窒素を生成する。この2つは互いに反応しやすく、脂質の酸化やDNAの断片化を引き起こすことで知られるペルオキシナイトライトアニオンを生成する。
no-+o2-→onoo- (2)
スーパーオキシドアニオンもまた、反応によってFe3+をFe2+に還元する:
Fe3++O2-→Fe2++O2 (3)
ここで、Fe2+はフェントン反応に速やかに移行し、活性酸素の中でも最も反応性の高いもののひとつであるヒドロキシルラジカル-OHを生成するため、非常に危険である。
Fe2++H2O2→Fe3++-OH+OH- (4)
さらに、上記の反応は次のように容易に進行する:
Fe3++H2O2→Fe2++-OOH+ H+ (5)
これは再び式(3)のように進行する。言い換えれば、Fe2+と過酸化物との出会いは、悪質な活性酸素種の周期的産生を引き起こす可能性がある。これが、前段落で述べた吸収と分布のメカニズムから読み取れるように、私たちの身体が効果的にFe2+の形の鉄の循環を最小限に抑えようとする理由である。
注目すべきは、銅も同じようにフェントン反応に関与していることである:
Cu++H2O2→Cu2++-OH-+OH- (6)
OHのようなラジカルが悪質である理由の一つは、多価不飽和脂肪酸の過酸化を促進することである:開始段階は、不飽和脂質から水素を除去し、脂質ラジカルR(-)を生成する。
r(h) +-oh→ r(-)+h2o (7)
として伝播する。
r(-)+o2→ r(oo-) (8)
これは別の不飽和脂質と反応し、脂肪酸の過酸化を終了させ、さらに脂質ラジカルを生成する:
r(oo-)+ r(h) → r(ooh) + r(-) (9)
しかし、脂質ラジカルR(-)は、この時点で再び反応に参加することで、鎖の過酸化に関与することができる(8)。
2.6.神経疾患における金属
過去20年間で、金属に関する神経学的研究の数は非常に増加した。遷移金属は、細胞環境の酸化還元状態を変化させ、生物学的に酸化還元反応[71] を触媒し、神経細胞の構造を破壊する[69] ため、神経生物学において不可欠な機能を示している。特に、鉄、銅、亜鉛は、エネルギー代謝(鉄、銅)、抗酸化防御(銅、亜鉛)、髄鞘形成(鉄)、DNA合成(鉄、亜鉛)、神経細胞骨格の完全性(亜鉛)、神経伝達物質合成(鉄、銅、亜鉛)など、多様な神経学的プロセスに寄与している[72]。遺伝性金属代謝異常の遺伝学的研究により、脳機能障害をもたらすいくつかの金属関連経路の変化に関する洞察が得られており、脳機能における金属ホメオスタシスの役割の理解に役立っている[64,73] 。
鉄代謝に関連する遺伝性疾患は、銅に関連する疾患よりも一般的で数も多く、その中には神経学的表現型を示すものもある。アセロプラスミン血症では、進行性の認知症、構音障害、ジストニアがみられるが、これは大脳基底核の鉄蓄積による二次的なものである[74] 。HFE遺伝性ヘモクロマトーシス患者に関する研究では、大脳皮質、視床下部、レンズ状核、歯状核、基底核、黒質、赤核における鉄蓄積の存在が強調され、HFE変異が神経変性疾患の発症リスクを変化させることが示唆された[75] 。神経フェリチノパチー患者では、空洞変性を伴う基底核、神経細胞の喪失、鉄とフェリチンを含むミクログリアが認められる。 HFE遺伝子の遺伝子変異、特にH63D変異は、AD患者の潜在的な危険因子として作用することが報告されている[76,77] 。Lehmannらは、HFE282YとTF C2、そしておそらくHFE63HHとTF-2AAの組み合わせが、ADの前臨床段階において鉄過剰、ひいては酸化ストレスの発生につながる可能性があることを明らかにした[78]。APOE4と年齢が、それぞれ前者と後者の組み合わせに影響を及ぼすことが提唱されている。注目すべきことに、細胞レベルでは、HFEH63D遺伝子変異体から形成されるHFE変異タンパク質が、鉄のホメオスタシス異常、酸化ストレスの亢進、グルタミン酸放出、タウのリン酸化、炎症反応の変動と関連していることが判明しており、これらは神経変性疾患発症の一因と考えられている。
フリードライヒ失調症は、神経細胞の生存に対するミトコンドリア鉄ホメオスタシスの障害の影響により、感覚神経細胞や小脳の変性と関連している[79] 。他の制御タンパク質(すなわち、PANK2、PLA2G6、FA2H、ATP13A2、DCAF17)をコードする遺伝子の変異は、異なる神経学的症状(例えば、ジストニア、構音障害、色素性網膜症)をもたらす基底核の鉄蓄積と相関している[64] 。
神経変性疾患、例えばAD、パーキンソン病(PD)、多発性硬化症(MS)をこれらの遺伝性金属代謝異常症との関連で、特にその主な神経学的特徴との関連で研究すると、それらの典型的な徴候のいくつかは遺伝性精神疾患の徴候と類似しているように思われる。言い換えれば、神経変性疾患は複雑な病因を持ち、遺伝性金属代謝異常症と遺伝的類似性を想定するのに十分な特徴を共有している(表3)。
表3 遺伝性金属代謝疾患と神経変性疾患の共有形質
| メタル | ジーン | 鉄代謝に関連する単発性疾患 | 鉄代謝に関連した複雑な病因を持つ神経変性疾患 |
|---|---|---|---|
| CP | アセロプラズマ血症 | パーキンソン病 | |
| 鉄 | エッチエフイー | 遺伝性ヘモクロマトーシス | アルツハイマー病 パーキンソン病 アミロイド側索硬化症 |
| フェリチン | H-フェリチン関連鉄過剰症 | – | |
| TF | アトランスフェリン血症 | アルツハイマー病 | |
| DMT-1 | DMT-1欠損症 | アルツハイマー病 アミロイド側索硬化症 |
|
| IREB2 | – | アルツハイマー病 パーキンソン病 |
|
| TFR | 非HFE遺伝性ヘモクロマトーシス | – |
金属生物学と鉄蓄積の役割もPDに関与していると主張されている[80] 。CP遺伝子の変異はPDと関連している[81] 。具体的には、経頭蓋超音波検査によって、脳黒質に鉄沈着物の増加(高エコー源性)が認められることを示した著者もいる。これらの沈着はCP遺伝子の変異と関連していた。しかしながら、ヒトを対象とした研究では、PD患者と健常対照者との間で、金属における全身的な変動に関する結果の間に高い不均一性があることが示された。
神経変性疾患における金属再配列の関与は、早期診断[75,82,83]、環境暴露[84,85,86]、予防戦略、治療介入[87]に関する研究の新たなターゲットとなっている。
2.7.ADと鉄の役割
ヘプシジン、フェリチン、IL-6は、ADにおける神経炎症に関連する宿主防御機構に大きく関与している。ヘプシジンはAD患者の血清で上昇し、疾患との相関を示している[13] 。いくつかの研究で、AD患者の脳、血液、髄液における金属ホメオスタシスの異常が報告されている。参加者94人のコホートにおいて、血漿中と髄液中のフェリチンは、認知機能障害の前の前臨床段階で、新皮質のAβ負荷が低いグループと高いグループに分類されたADを識別する可能性が示された[88] 。フェリチンレベルの上昇は、CSFと脳における鉄レベルの上昇を示唆しており、フェロプトーシス(過酸化脂質の蓄積に起因し、グルタチオンペルオキシダーゼ4によって制御される鉄依存性の細胞死)に関連する可能性がある(総説は[89])。さらに、トランスフェリンレベルはADのリスクを増加させることが明らかにされており、ADの12%のリスクと関連するタンパク質が減少している[90]。
さらに、セルロプラスミンは、CSFで評価した場合、あるいは血清中のセルロプラスミン特異的活性を測定した場合 [91] 、Aβ病態を有する人の認知機能低下と脳萎縮を予測することが示されている[90] 。これらの知見と一致するように、われわれの以前の結果では、内側側頭萎縮の重篤なAD患者では、前頭側頭葉変性症患者とは異なり、鉄の全身濃度が上昇し、TSATの割合が増加していた[92]。さらに、AD患者はセルロプラスミン-トランスフェリン(Cp-Tf)系の活性化を示した[93] 。1980年代初頭の研究では、Cp-Tf系が血漿中で作用する主要な抗酸化系であると主張された[94]。Cp/Tf比は、酸化状態の銅(Cu2+)を結合するセルロプラスミン[95] とアポトランスフェリン[96] の複合抗酸化活性を反映することが、電子常磁性共鳴(EPR)分析によって明らかにされた。濃度単位の測定から得られるCp/Tf比は、EPRスペクトロメトリーでモニターするには高価なCp-Tfシステムの機能性を合理的に表すことができる[94]。実験的な高コレステロール血症におけるいくつかの研究では、Cp-Tf系の活性化が脂質過酸化を減少させることが示された[94]。Cp-Tf系の活性化は、全身状態として患者を苦しめているAD患者における酸化ストレスの増加を示唆している。これらの結果と一致して、セルロプラスミン、過酸化物、Cp/TfはAD患者で上昇し、Mini-Mental State Examination(MMSE)スコアと逆相関していた。また、AD患者は健常対照者よりもアルブミン値が低く、プロトロンビン時間が長く、トランスアミナーゼ比(アスパラギン酸/アラニントランスアミナーゼ、AST/ALT)が高いことが報告されている。これは、トランスフェリンの減少と血清フェリチン値の上昇とともに、肝機能低下を示している[82] 。HFEの H63D変異のAD保因者は、鉄濃度の上昇とトランスフェリンおよびセルロプラスミンの低下を示し、ヘモクロマトーシスに似ているが、これはH 63D非保因者のAD患者では認められなかった。むしろ、肝機能障害の状態、H63D遺伝子変異、鉄の増加という、より多くの要因が相乗的に作用して、ADのリスクを高めている可能性がある[82] 。これらの知見は、セクション2.9で述べるように、鉄のホメオスタシス異常がADリスクの中心的なドライバーであるとする[98] 、ADにおける鉄の関与に関する最近の仮説の定式化と一致している。
2.8.ADにおける鉄の神経画像研究
詳述したように、脳内のフェリチンやヘモシデリンに貯蔵された鉄は、重要な生理的プロセスに非常に不可欠であるが、鉄過剰はフリーラジカルの発生や酸化的損傷を促進する。一貫して、加齢に伴う貯蔵鉄の増加は、脳細胞内の酸化還元活性鉄プールを拡大することによって、AD発症の可能性を高める糖尿病や血管疾患などの一般的な病状を促進することが実証されている。AD脳における鉄の取り込みの増加は、TfR1 mRNAの安定性を高めると予想される、より安定なIRE/IRP1複合体の生成によるのかもしれない[99]。さらに、AD脳ではフェリチン中の細胞内鉄貯蔵量が変化している可能性がある[80,99,100,101] 。
高鉄負荷による貯蔵鉄の増加と鉄の貯蔵/解毒の障害は、ADの初期変化の重要な徴候である、傷つきやすい神経細胞におけるフェントン反応を介した酸化ストレス/フリーラジカル損傷を悪化させる可能性がある。さらに、鉄による酸化ストレスと細胞シグナル伝達の障害は、微小管関連タンパク質タウのリン酸化を促進する可能性がある。神経斑と神経原線維のもつれは、ADの主な組織学的特徴と考えられている。同時に、鉄レベルの上昇は、神経斑の基本成分であるAβペプチドの形成増加と関連している。このように、脳鉄レベルの上昇はADリスクの上昇と相関しているが、脳内鉄沈着がADの主要な原因であるかどうかは明らかではない。この事実に一致して、ADの病態を理解するために、鉄レベルの空間的分布や変化をモニターしようとする研究がいくつかなされている。これまでの研究では、脳切片の磁気共鳴画像法(MRI)による横緩和[102] や組織化学[103] を用いた死後研究において、鉄とプラークとの関連が観察されている。
同様の結果が、蛍光X線を用いたAPP/PS1マウスの脳でも得られている[104] 。Aytonらは、MRIとPETを用いた混合モダリティ研究により、高濃度の脳鉄が、生きたAD患者における認知機能の経時的低下やAβ負荷と関連していることを示した[105]。同じ研究グループが最近、バイオマーカーでAD病態が決定された被験者において、CSFフェリチン値が認知機能の低下と縦断的に関連し、Aβ沈着を促進する可能性があることを示した[106]。フェリチン値はアポリポ蛋白E4[107] の影響を受けている可能性があり、これはAβと鉄[108] の相互作用の増加を示唆しており、酸化ストレスとAβ凝集の原因となっている[109,110] と提唱されている。MoonらとDuらも、MRIと定量的帯磁率マッピング(QSM)を用いて、AD患者の被殻と尾状核における脳内鉄沈着の増加を報告しており、ADとの関連に特異的な関心を示している[102,111]。
その理由は、神経原線維のもつれやAβ線維と密接に関連する脳内の鉄蓄積に関係している可能性がある。加えて、内因性の鉄と亜鉛がAβ線維の凝集を促進したり、APPをコードするmRNAに鉄応答性エレメントが存在したりするのかもしれない。in vivoの研究では、異なる形態と磁気/酸化状態を持つ鉄沈着がプラーク様領域に不可欠であるようであることが示された。高度電子分析顕微鏡の統合的セットを用いて、AD死後症例のアミロイドプラークコア(APC)内に鉱化した鉄が酸化鉄(Fe3O4)マグネタイトナノ粒子として存在し、ADにおける鉄蓄積とAβ凝集の関連を示す証拠となった[112]。
これと同様に、Tellingたちは、高度なX線顕微鏡技術を応用して、ADのAPP/PS1モデルマウスの皮質において、アミロイド構造と共局在する大量の還元鉄(Fe2+)とマグネタイト沈着物の存在を明らかにした[113]。注目すべきは、野生型マウスの皮質切片には鉄沈着物はほとんど見られなかったことである。鉄2+の濃縮は、フェントン反応による反応性の高いヒドロキシルラジカルを含むフリーラジカル生成の潜在的な源として働く可能性がある。プラーク中のAβは、Fe3+を純粋なFe2+に酸化還元サイクリングさせ、それによってミネラルの沈着にダイナミックに関与すると考えられている[114]。さらに、鉄の沈着はAβの凝集を促進する可能性があり、これはすでにin vitro[114]やin vivoの研究[115,116]で示されている。Sternbergらによって行われたパイロット研究では、臨床的認知症に基づいて患者を層別化した後、鉄および鉄関連タンパク質の濃度が対照群と比較して上位50%で高いことが明らかにされ、鉄の恒常性異常が認知障害を悪化させる可能性が示された。さらに、純粋なAD患者は対照群と比較して血清ヘプシジン濃度が3倍高い。したがって、ヘプシジンと鉄関連タンパク質の両方が、ADの診断と進行に関係する血清バイオマーカー群と考えられている[117]。最近の研究では、超伝導量子干渉素子(SQUID)磁力計を用いて、イングランド北部で採取されたADと認知機能が正常な脳で、不均一な磁鉄鉱/マグヘマイトの分布パターンが観察された[118] 。
2.9.AD発症における鉄の遺伝学
鉄、銅、亜鉛などの生体金属は、脳内で細かく調節されている。それらの神経毒性は、単に曝露量の増加によるものではなく、むしろそれらのホメオスタシスの乱れや、酸化ストレスや興奮毒性の過程における関連するコンパートメント化によるものである。脳内金属代謝の調節不全は、遺伝的要因だけでなく、非遺伝的要因によっても引き起こされると考えられている。また、異なるレベル(すなわち、取り込みと放出、貯蔵、細胞内代謝、調節)で生じることもある[119] 。
ヒトの遺伝的変異に関する知識が深まったことで、金属代謝の調節タンパク質をコードする遺伝子にいくつかのLoF(Loss-of-Function)変異体を発見することができ、生化学的基準によって最初に説明された神経変性疾患の金属生物学への関心を検証することができた[120] 。実際、複雑な病因を持つ神経疾患の中には、先に述べたように、遺伝性金属障害を典型的に区別する特徴を共有しているものがあるようである。この段落では、この問題について、特に金属遺伝子、その遺伝性疾患、ADの話題を取り上げて議論する。鉄代謝に関しては、過剰症も欠乏症も神経細胞の機能障害を引き起こすと主張されている。脳において、鉄のホメオスタシスに主に関与するタンパク質は以下の通りである:HFE、フェリチン、トランスフェリン、TfR、IRP、DMT1、セルロプラスミンである。遺伝学の力を利用して、多くの研究が神経変性疾患、特にADにおけるこれらの鉄関連LoF変異体の役割を探求してきた[75,121] 。最も研究されている仮説の一つは、ADで報告されている鉄増加沈着の原因であるHFE遺伝子の変異に関するものである。HFEは鉄の吸収を調節するタンパク質である。HFE遺伝子に変異があると、食事からの鉄の吸収が亢進し、脳を含む組織や臓器に鉄が蓄積するヘモクロマトーシスを引き起こす[31] 。HFEタンパク質は脳のさまざまな部位に発現し、脳の鉄の取り込みに影響を及ぼしているが、HFE突然変異と神経変性疾患との関係は2000年まで注目されなかった。この仮説に従って、HFE変異とADリスクに関する最初の報告が発表された[122]。著者らは、家族性ADに罹患した男性やAPOE4対立遺伝子の非保有者において、健康な高齢男性に比べてHFE変異の頻度が増加していることを示した。その後、ADリスクにおけるHFE変異の役割を探求する多くの研究が続き、ADの特徴、例えば発症[123,124]、認知症状[125]、臨床的欠損の重症度[126]、髄液中のマーカー[76]、軽度認知障害(MCI)からADへの転換[125]などとの有意な関連が報告された。しかし、多様な研究結果は、HFE対立遺伝子頻度の民族間差や、遺伝的関連を混乱させる可能性のある遺伝的、環境的、人口統計学的因子との相互作用に関連すると思われる相反する像を描いており、一義的な結果には至っていない[75]。
他の鉄関連遺伝子(TFR、DMT1、IREB2)を調査した追加研究では、そのうちのいくつかとAD表現型との正の関連が認められた[78,127] 。さらに、HFEの一般的なタンパク質変異であるH63D HFEは、複数の神経疾患の修飾因子として認識されている[128]。とはいえ、HFE遺伝子に関しても、これらの制御遺伝子に関しても、関連性を確認し、解析された遺伝子変異が鉄の貯蔵と沈着に機能的な影響を及ぼすかどうかを決定するためには、さらなる研究が必要である。最後に、マウスモデルを用いた研究では、FTH1(フェリチンをコードする)、CP、TFR遺伝子の欠損が、神経障害の典型的な徴候の表現型と関連していることが示された[129]。これらの結果は、鉄関連遺伝子のLoFがADの病因に影響を及ぼす可能性を強調しているが、その役割を確認するためには、ヒトを対象としたさらなる遺伝子関連研究が必要である。
神経変性疾患や特にADに関しては、血清、血漿、髄液、脳、肝臓など、様々なヒトのマトリックス中の銅濃度を分析した研究がいくつかあるにもかかわらず[130]、ADにおける銅関連遺伝子の直接的な役割に関する情報はまだ乏しく、ATP7B遺伝子に限られている[131]。
現在までのところ、ADに関するゲノムワイド関連研究(GWAS)において、金属調節[132] や金属依存性経路[133] に関連する遺伝子に有意な変異が見つかった例はない(www.alzgene.org、2022年8月29日閲覧)。このような状況は、遺伝的要素と環境的要素が互いに強く影響し合う金属ホメオスタシス制御の複雑さに起因している可能性がある。したがって、生化学的および環境的調査と組み合わせた候補遺伝子アプローチは、AD発症における金属調節遺伝子の役割を理解するのに役立つ可能性がある。これを裏付けるように、鉄のホメオスタシスとAPPプロセシングの制御との間に直接的な関連があることが明らかになった。APP転写産物の5′-非翻訳領域には、鉄に反応してAPPの翻訳を促進するIREがある[134] 。
2.10.ADに関与する新たな神経変性過程としてのフェロプターシス
ADにおける鉄の関与に深く焦点を当てた最近の縦断的研究[98] では、ADの脳における鉄を評価した結果、バルク組織における鉄負荷とAβ斑との関連は示されなかったものの、少なくとも下側頭皮質において、組織の鉄負荷がNFT形成に影響を及ぼす可能性があることが確認された。ADでは主に前帯状皮質、中前頭皮質、下側頭皮質の3つの脳領域が影響を受け、小脳皮質も比較的影響を受けなかった。合計645の死後AD脳を調査し、脳鉄とADの病理学的変化および死前臨床変化との関連を検証した[98] 。この研究により、脳内鉄負荷とADの臨床診断および神経病理学的変化との関連は緩やかであったが、患者の死亡前10年間の認知機能低下率との関連は厳密であった。この結果は、タウやアミロイドの病態とは別に、神経変性のエフェクターとしての鉄の下流での役割を論証するものである。
これらのデータは、鉄がAPPとタウの産生を促進し、AβとP-タウの凝集を促進することによって、プラークともつれの形成を促進するという、ADの金属仮説の新たな定式化に影響を与えた。新しい仮説では、鉄はフェロプトーシスによる神経細胞の死滅しやすさに影響を与えることで、プロテインパチーの下流でさらなる役割を果たすと仮定している。フェロプターシスは、グルタチオンの枯渇と脂質の過酸化を特徴とする、アポトーシスを起こさないユニークな細胞死である。フェロプターシスの発生は、鉄が毒性レベルまで増加することとは関係なく、鉄がフェロプターシス中に脂質過酸化によって活性化され、毒性を誘発する。したがって、鉄は感受性の調節因子として働き、フェロプターシスが活性化すると細胞死が起こる。最近の研究では、フェロプトーシスがAD、PD、筋萎縮性側索硬化症(ALS)など多くの疾患の病態生理学的過程と密接に関連していることが示されている[136,137] 。
これまで、ADにおける鉄に関する研究では、特徴的な病態を促進する鉄の役割について研究されてきた。フェロプトーシスの発見は、タンパク症が出現した後に起こるADの神経変性に新たな説明を与えた。
現在、ADにおけるフェロプトーシスの役割に関する報告が出始めている[138] 。さらなる臨床的証拠によって確認されれば、この仮説は、ADにおける鉄の病態生理の理解だけでなく、何よりも鉄経路を標的とする新たな治療機会を提供する可能性という点で、重要な意味を持つことになるであろう[139,140]。
哺乳類で唯一同定された非ヘム鉄排出因子であるフェロポルチン1(Fpn)は、アルツハイマー病およびAD患者のモデルマウスであるAPPswe/PS1dE9マウスの脳で制御が低下している。Fpn(fl/fl)マウスとNEX-Creマウスを交配させ、大脳新皮質と海馬の神経細胞でFpnを遺伝的に欠失させると、海馬の萎縮と記憶障害というADの表現型が出現した。フェロプトーシスはFpn(fl/fl/NEXcre)マウスとADマウスの両方で観察された。フェロプトーシス関連RNA-seqデータの遺伝子セット濃縮解析(GSEA)により、フェロプトーシスで差次的に発現する遺伝子は、AD関連遺伝子セットに高度に濃縮されていることが示された。濃縮遺伝子の主な機能は、活性酸素代謝過程とアミロイドβ形成としての機能に関連して、いくつかの重要な経路に分布しており、AD発症におけるフェロプトーシスの重要な役割を裏付けていた[138] 。鉄キレート剤は、グルタチオンとグルタチオンペルオキシダーゼ4が関与する脂質修復システムを誘導することによって、フェロプターシスを抑制したり[140] 、脂質ヒドロペルオキシドを脂質アルコールに変換することによって、フェロプターシスを予防したりすることができるという新しい証拠が示唆されている[141] 。
2.11.鉄とAPP/Aβ系の代謝
AD脳の特徴として、神経細胞の外側と内側にそれぞれAβ斑と神経原線維変化が存在することはよく知られている。冒頭で述べたように、アミロイド凝集塊の主成分はAβであり、APPが切断されてできる39-43アミノ酸のペプチドである。より具体的には、APPは非アミロイド原性経路とアミロイド原性経路の2つに分けて処理されることが報告されている。非アミロイド化経路では、APPは細胞外ドメインと膜貫通ドメインの2つの部位で切断され、それぞれαセクレターゼとγセクレターゼによって切断され、切断されたP3断片が放出される。アミロイド形成経路では、APPはまず細胞表面外の部位でβセクレターゼによって切断され、C末端断片が生成され、続いて膜貫通ドメイン内の部位でγセクレターゼによって切断される。この切断の産物がAβである[総説8](図5)。APPは事実上どこにでも存在し、細胞膜上のフェロポルティンと結合することによって鉄の輸出に関与していることが報告されている[142]。

図5 Aβ産生の経路:
金属との関連。アミロイド凝集体の主成分はAβであり、膜貫通タンパク質であるAPPが切断されて生じる39-43アミノ酸のペプチドである。より具体的には、APPは2つの経路に分かれて処理されることが報告されている。アミロイド生成経路では、まずAPPが細胞表面外の部位でβセクレターゼによって切断される。これによってC末端断片が生成され、続いて膜貫通ドメイン内の部位でγセクレターゼによって切断される。この切断の産物がAβである。非アミロイド生成経路では、APPは最初にαセクレターゼによって切断され、次にγセクレターゼによって切断され、切断されたP3断片が放出される。APPには酸化還元活性を触媒する特異的な銅と亜鉛結合ドメインがあり、最小濃度でもプラーク中のAβ沈殿を決定することが判明した。Aβペプチド中の銅と亜鉛のドメインは、等モル量の2つの金属(Me2+)と結合する。しかし、アシドーシス条件下では、Aβ亜鉛は銅によって完全に置換される。
さらに、APPのmRNA転写産物は5′-UTRにIRE配列を持っている。細胞の鉄の状態が低いと、IRP1はAPP mRNAのIREに結合する。その結果、APP mRNAはAPPタンパク質に翻訳されず、APPを介したフェロポルティンの安定化が抑制され、鉄の排出がさらに阻害される[143]。この点に関する追加的な証拠は、APPノックアウトマウスが脳鉄の蓄積を示すというADの前臨床モデルから得られており[144]、健康な脳では、APPが鉄のホメオスタシスに関与している可能性を示している。
現在文献上では、鉄はAPPとタウの産生を促進し、Aβとリン酸化タウの凝集を促進することによって、プラークともつれの形成を促進するという仮説が立てられている。したがって、645例の死後脳から得られた新たな証拠は、少なくとも下側頭皮質では、組織の鉄濃度が神経原線維のもつれ形成に影響を及ぼす可能性があることを確認した。組織全体の鉄濃度に関するこの調査では、組織全体の鉄濃度とアミロイド形成の関係は支持されなかった。さらに、タウの機能喪失が神経細胞の鉄蓄積を引き起こす可能性も示唆されている[145] 。
病的なアミロイド沈着に大きく寄与しているのが酸化ストレスであり、過剰な活性酸素活性[146] や間接的に脂質過酸化産物[147] によって生じることが明らかになると、AD研究者の関心は遷移金属に向けられるようになった。遷移金属は制御不能な活性酸素を発生させる様々な化学反応に関与しており、分子や細胞のコンパートメントを損傷したり破壊したりすることができる[9] 。現在では、APPは銅と亜鉛の結合ドメインを持つ銅タンパク質であり、酸化還元活性を媒介し、Aβ凝集体の形成にも関与していることが立証されている[148,149]。Aβにも選択的な銅と亜鉛のドメインがあり、通常の状態では等モル量の二つの金属が結合する。しかしながら、Aβの亜鉛はアシドーシス時には完全に銅に置換される[150]。
Aβ斑の存在に関連して鉄の濃度が上昇していることを示したいくつかの研究によると、(1)Aβ斑や神経原線維変化内では、鉄と銅の濃度がともに上昇していることが報告されている:(1)Aβ斑および神経原線維のもつれ内では、鉄と銅の濃度が上昇していることが報告されている[151,152]; (2)基底核[153,154]、髄液[155]、およびADで影響を受ける他の脳領域[98]で、鉄の沈着または濃度の上昇が認められている; (3)主に海馬、二次的に大脳皮質がAβ-金属毒性の影響を受ける。
現在では、この見解は、Aβが獲得した毒性特性の可能な説明として受け入れられており、ADと、AD発症の可能な経路として遷移金属によって引き起こされる酸化ストレス現象との間に関連性が存在するという一般的な合意を支持している(ADの金属仮説[136])。
3.ADにおける鉄キレート化を標的とした治療選択肢の現在の展望
その証拠に、全タンパク質の約半分が、効果的な機能を発揮するために、金属と結合して金属タンパク質を形成する必要があることが報告されている。先に述べたように、脳内の金属イオンは、酵素の適切な機能、神経伝達、老化に必要である[72]。一方、生体金属(鉄、銅、亜鉛)のホメオスタシス異常や金属-アミロイド相互作用を、ADを含む様々な神経変性疾患に関連付ける研究も蓄積されている[112] 。したがって、ADはタンパク質の凝集による疾患であると示唆されている。しかし、ADは金属恒常性維持異常の疾患であるとも考えられている。銅、鉄、亜鉛のAPPへの結合には共通の結果がある。銅(および鉄)はアミロイドタンパク質に結合し、神経細胞内の抗酸化防御システムを直接打ち負かす活性酸素を発生させることによって、酸化的損傷のリスクを高める可能性がある[156]。さらに、亜鉛はAPPとAβの両方に結合することが知られている。亜鉛のAPPへの結合はLys16で起こり、αセクレターゼの切断活性に影響を与え、可溶性APPαの集合を減少させ、Aβの産生を促進する可能性がある[157]。しかし、脳の亜鉛レベルは非常に厳密にコントロールされており、亜鉛療法の過剰投与(亜鉛中毒)や過剰な亜鉛の長期摂取の主な結果は銅欠乏である。一方、AD脳の神経細胞における鉄レベルの上昇は、活性酸素の発生を介して病態と強く関連している[156]。
しかし、ADの発症と進行における生体金属の役割は非常に複雑であるため、ADが鉄の過剰と関連するか欠乏と関連するかはまだ議論の余地がある。金属量の減少と過剰の両方が、ADを含む神経疾患と関連している[158,159] 。AD患者のアミロイド斑では、銅、鉄、亜鉛などの生体金属が、正常脳と比較してそれぞれ最大5.7倍、2.9倍、2.8倍過剰に蓄積している[160] 。逆に、Brewerらは、AD患者では対照群と比べて血清中の亜鉛濃度が低いことを観察している[158]。また、鉄、フェリチン、トランスフェリンの変化を含む、ADにおける鉄の恒常性異常の主張を示す証拠もある[89,90] 。脳の鉄レベルの低下は、運動ニューロン障害、ドーパミン活性の変化、ミエリン形成の異常と関連している[161] 。文献から明らかなように、鉄はリン酸化が進んだタウと結合して凝集を促進し、NFTの形成につながる[145] 。対照的に、いくつかのin vitro研究では、鉄がAβの凝集と神経斑の形成を減少させることが明らかにされている[162]。しかしながら、鉄の過剰レベルは、特にその酸化還元活性によって脳に脅威をもたらす。すでに述べたように、鉄はフェントン反応に関与し、タンパク質、脂質、DNAと相互作用するヒドロキシルラジカルやスーパーオキシドラジカルの生成を促進する[68,69] 。したがって、脳の鉄ホメオスタシスを維持するためには、極めて進化的に保存されたメカニズムが必要である。
生体金属イオンの役割とアミロイドとの相互作用は、ADの病態形成に極めて重要であるため、高親和性金属-神経タンパク質相互作用を評価することは、ADや他の神経変性疾患の治療のための新規治療アプローチの同定に役立つ可能性がある。近年、キレート剤が金属イオンとタンパク質間の相互作用を解除し、酸化ストレスを軽減し、認知機能を改善する可能性が報告されている[163]。最近、銅、鉄、亜鉛のキレート剤がAβ線維の凝集を効果的に抑制することが明らかにされた[163,164,165,166]。確かに、金属キレート治療は、急性だけでなく時には慢性の酸化的損傷に対する防御に対処し、タンパク質の凝集を阻害する。Fasae博士らは、ADにおける金属(銅、鉄、亜鉛)キレート剤の臨床使用について論じている[166]。デフェロキサミン(DFO)はトリヒドロキサム酸の一種で、他の金属と比較して鉄(III)に対して高い親和性を示すキレート剤であり、サラセミアなどの鉄過剰症のための正確なキレート剤と考えられている。デフェリプロン(L1)(1,2-ジメチル-3-ヒドロキシピリド-4-オン)およびデフェラシロクス(4-[3,5-ビス(2-ヒドロキシフェニル)-1,2,4-トリアゾール-1-イル]-安息香酸(Exjade、ICL-670)は、DFOと比較して、輸血鉄過剰症を効果的に治療する高い血漿寿命、より多くの腸耐性、および患者のコンプライアンス[167]を有するDFOの代替品を提供している[168]。以前の研究によると、DFO(125mgを1日2回/週5回、2年間)を投与した後、プラセボ群と比較してAD患者の顕著な改善が観察された[169] 。しかし、ADにおける有能な鉄キレート剤に関する最近のデータはない。様々な研究により、経口鉄キレート剤(デフェロキサミン、デフェリプロン、デフェラシロクス)の薬理作用と治療可能性が、ヒトとラットの両モデルで明らかにされている[170,171]。多座キレート剤であるEDTA(エチレンジアミン四酢酸)は、カルシウム、マグネシウム、鉄、亜鉛、マンガンなどの2価および3価の金属イオンに高い親和性を持つ。Klangたちは最近、線虫にEDTA二ナトリウムカルシウム(CaNa2EDTA)を投与すると、鉄、銅、亜鉛によって引き起こされるタンパク質の凝集と神経毒性が逆転し、線虫の寿命も改善することを観察した[172]。さらに、DFO、EDTA、DPAの組み合わせは、鉄、銅、マンガンに暴露されたハエの生存率と運動活性の改善を示した[173]。多官能性化合物(MC)は、そのキレート基部位に基づ いて、二座キレーターまたは三座キレーターに分類される[174]。例えば、トリス(DOP)誘導体L1h6(ベンゼン-1,3,5-トリカルボン酸トリス-2-(3, 4-ジヒドロキシフェニル)-エチル]-アミド)は、Fe3+イオンへの配位において有望な幾何学的配置を有している[175]。トリス(DOP)誘導体化合物は抗酸化特性を示し、酸化ストレスが介在する毒性を抑制する[176]。化学的に修飾された親油性フェリクロムやトリスヒドロキサメ ートキレーターは、鉄と結合し、鉄過剰症をDFOの25倍減少させる ことが報告されている[177,178]。フラボノイドのような天然化合物とは別に、天然の抗酸化物質も鉄や銅をキレートする能力を持つことが知られている。ケルセチン[179]やクルクミン[180]は、動物モデルやAD患者の治療だけでなく、キレート化プロセスにも構造的に有利であることが観察されている[181]。神経保護ペプチド(NAP)のようなネイティブの神経細胞ペプチドなどの他の化合物は、ヒドロキサメートまたは8-ヒドロキシキノリン部分に結合し、水中の生理的pHでFe2+、Cu2+およびZn2+と複合体を形成する[182]。さらに、ナノ粒子は、受容体を介したシステムによってBBBを通過する能力も発見され、薬物送達における実績もある[183]。キレーター・ナノ粒子は、キレーター・鉄複合体を脳から運び出し、鉄イオンの毒性を軽減することも確認されている。ナノ粒子-キレーター複合体が、ADの脳組織から鉄またはその貯蔵タンパク質であるフェリチンを隔離することが示されたという証拠もある[185]。
ほとんどのキレート剤は、機能的必須金属イオンを保持/置換し、金属酵素の機能を変化させ、その結果、臨床的回復が乏しくなり、また酸化促進活性もあるため、いくつかの軽度または重篤な副作用と限界を示す。しかし、もう一つの金属である銅はADとも強く関連しており、亜鉛療法は有益な効果をもたらすかもしれない。186]で広範に議論されているように、亜鉛療法は銅の競合物質として作用し、メタロチオネインの発現を刺激することで体内の銅濃度を低下させ、金属の直接的なキレートを防ぐ。ウィルソン病に関しては、亜鉛療法は、キレート療法による重篤な有害事象を起こすことなく、認知機能低下を抑制する効果が期待できる。軽度認知障害を対象とした臨床試験(ZINCAID、EudraCT 2019-000604-15)は現在も進行中である。
4.結論
以上の研究から、ADでは鉄の乱れが起こると結論づけられる。鉄の調節は、ADのアミロイド形成とタウ蛋白質に関連している。この金属は、フェントン反応を介してフェロプトーシスや酸化ストレスを促進することによって、また、生理的な鉄のホメオスタシスに関与しているタウ、APP、APOE代謝と関連することによって、疾患発症の原因因子となりうる。正常範囲内の鉄濃度がADのリスク上昇と関連し、すでにタンパク症が存在する場合には疾患の進行と関連するという知見は、この金属が神経変性の下流過程にも関与している可能性を示唆している。ADの発症と進行を抑制する新たな治療的介入を確立するために、これらの問題を探求する追加研究が必要である。
略語
| アルツハイマー病 | (AD) |
| アポリポ蛋白質E | (APOE4) |
| アミロイド・ベータ | (Aβ) |
| アミロイド前駆体タンパク質 | (APP) |
| 脳アミロイド血管症 | (CAA) |
| 磁気共鳴画像法 | (MRI) |
| 活性酸素種 | (ROS) |
| 二価金属トランスポーター1 | (DMT-1) |
| シトクロムC酸化酵素アセンブリーホモログ | (COX17) |
| 抗酸化タンパク質1ホモログ | (ATOX1) |
| SODの銅シャペロン | (CCS) |
| スーパーオキシドジスムターゼ1 | (SOD1) |
| 鉄応答性エレメント/鉄調節タンパク質 | (IRE/IRP) |
| トランスフェリン受容体 | (TfR) |
| 遺伝性血色素症 | (HFE) |
| トランスフェリン飽和度 | (TSAT) |
| メタロチオネイン | (MT) |
| Znトランスポーター | (ZnT) |
| 亜鉛制御および鉄制御トランスポータータンパク質 | (ZIPファミリー) |
| パントテン酸キナーゼ2 | (PANK2) |
| ホスホリパーゼA2グループVI | (PLA2G6) |
| 脂肪酸2-水酸化酵素 | (FA2H) |
| 13A2型ATPアーゼ | (ATP13A2) |
| DDB1およびCUL4関連因子17 | (DCAF17) |
| 内皮細胞 | (BCEC) |
| 血液脳関門 | (BBB) |
| 血液脳脊髄液関門 | (BCB) |
| 脳脊髄液 | (CSF) |
| 中枢神経系 | (CNS) |
| 多発性硬化症 | (MS) |
| パーキンソン病 | (PD) |
| ウィルソン病 | (WD) |
| 機能喪失 | (LoF) |
| ミニ精神状態検査 | (MMSE) |
| 軽度認知障害 | (MCI) |
| セミカルバジド感受性アミン酸化酵素 | (SSAO) |
| 血管性認知症 | (VaD) |
| 抗酸化能力 | (TAS) |
| 長期増強 | (LTP) |
| ゲノムワイド関連研究 | (GWAS)。 |
| 一塩基多型 | (SNPs) |
| メチレンテトラヒドロ葉酸還元酵素 | (MTHFR) |
| 脳波検査 | (脳波) |
ファンディング・ステートメント
本研究は、イタリア保健省(Ricerca Corrente; RS)の支援と資金提供を受けた。また、本研究はアルツハイマー病協会パート・ザ・クラウドの助成を受けている:Translational Research Funding for Alzheimer’s disease (PTC) PTC-19-602325, EudraCT 2019-000604-15(RS)。
利益相反
著者らは利益相反がないことを表明している。
