
Contents
Title: Ferrosenescence: the iron age of neurodegeneration?
受理した日 15-11-2017
著者。アドニス-スフェラ、ケルシー-ブロック、エイミー-プライス、ルズミン-インデリアス、カロリナ-オソリオ
ケルシー Bullock、MD -所属。ロマ リンダ大学、ロマ リンダ、カリフォルニア州
エイミー・プライス博士 -所属。オックスフォード大学、イギリス
Luzmin Inderias、MD -所属パットン州立病院、パットン、カリフォルニア州
ハイライト
- MicroRNA-29ファミリーは神経細胞の鉄保持を低下させる可能性がある。
- 鉄調節タンパク質-2を標的としたフェロトーシスの阻害
- BACE-1 セクレターゼを標的とした神経細胞の鉄の排出を促進する。
- MicroRNA-29ファミリーは、以下を介して強酸性化を防止する可能性がある。
- DNAメチル化保存によるゲノムの安定化
- p53の安定化によるDNA損傷修復システムの完全性の回復
要旨
老化は、神経細胞を含む多くの細胞タイプでの鉄の滞留と関連しており、フェロトーシスによる神経変性を促進している。過剰な細胞内鉄はDNAにダメージを与え、ゲノム修復システムをブロックすることで老化を加速させる。
新しい神経画像およびプロテオミクス技術は、鉄の保持と強老化の両方の指標をピンポイントで特定しており、それらの早期の修正を可能にし、潜在的に手の届く範囲で神経変性疾患の予防をもたらすことができる。
このレビューでは、栄養とマイクロバイオームの操作に基づく予防戦略に焦点を当てて、神経変性疾患における鉄の恒常性異常の初期マーカーを詳しく見ていく。
略語
トランスポッサブルエレメント(TE)鉄調節タンパク質-2(IRP-2)βセクレターゼ1(BACE-1)アミロイド前駆体タンパク質(APP)フェロポルチン(FPN)トランスフェリン(Tf)トランスフェリン受容体-1(TFR-1)鉄応答性エレメント(IRE)。ミトコンドリアDNA(mtDNA)DNAメチル化酵素3Aおよび3B(DNMT-3AおよびDNMT-3B)ヒストン-リジンN-メチル化酵素(SETDB1)定量的感受性マッピング(QSM)軽度認知障害(MCI)グルタチオンペルオキシダーゼ-4(GPX-4)。ホスファチジルエタノールアミン(PE)ミトコンドリア関連膜(MAM)内側ミトコンドリア膜(IMM)外側ミトコンドリア膜(OMM)TAR DNA結合タンパク質43(TDP-43)前頭側頭型認知症(FTD)ラパマイシンの哺乳類標的(mTOR)。核内因子赤血球2関連因子2(Nrf2)アニオンチャネル1および2(VDAC-1およびVDAC-2)アナンダミド(アラキドノイルエタノールアミド)(AEA)脂肪酸結合タンパク質3(FABP3)インスリン/インスリン様成長因子-1(IGF-1)。
はじめに
鉄は必須の微量栄養素であるが、潜在的に致命的であり、その欠乏と過剰の両方が病理学的にリンクされている。実際、貧血から毒性に至るまでの鉄の恒常性の異常は、人類の最も一般的な疾患の一つである(1)。しかし、これらの状態は「氷山の一角」に過ぎないのかもしれないが、新しい研究では、この生体金属が老化や神経変性疾患と関連していることがわかっている(2-3)。
老化は、神経細胞を含むほとんどの細胞型における鉄の滞留と関連しており、脂質の過酸化や小胞体(ER)内の不溶性タンパク質の凝集に寄与している。これらの変化は、細胞の抗酸化障害と相まって、フェロトーシスによる神経細胞の死につながり、やがて神経変性として現れる。
神経細胞内の鉄の滞留は、老年期や癌の特徴であるゲノムの不安定化を伴うDNA損傷を誘発し、早期老化を促進する(4)。さらに、過剰な細胞内鉄は、p53をダウンレギュレーションすることで、p53が介在するDNA修復機能を直接的に阻害する(5)。このような悪循環が生じ、加齢による細胞内鉄の滞留によりゲノムの崩壊が起こり、その結果、DNAの損傷や修復を阻害することで老化が促進され、これを強酸性化と定義している(図1)。ゲノムへの影響だけでなく、DNAのハイメチル化やトランスポーズ可能要素(TE)の動員を介してエピゲノムにもダメージを与える(6-7)。
MiR-29はゲノムおよびエピゲノムの損傷に応答してアップレギュレーションされ、老化組織や老化促進モデルで観察される現象である(8)。miR-29は最近、脳内鉄のマスターレギュレーターである鉄調節タンパク質-2(IRP-2)をコードする遺伝子IREB-2を標的とすることが報告された(9)。さらに、miR-29は、有害アミロイド種の生成に関与するβ-セクレターゼ1(BACE-1)を直接標的とする。βアミロイドは、鉄の輸出チャネルであるフェロポルチン(FPN)を不安定化させ、神経細胞内の鉄蓄積を促進することが実証された(10)(図2)。miR-29は、細胞内の鉄を低下させるだけでなく、加齢に伴うDNAメチル化の変化に対抗してエピゲノムを安定化させ、「エピゲノムの守護者」として機能していることが示唆されている(11)。さらに、miR-29は、p53タンパク質を間接的に活性化することで、DNA修復システムを修復する。このことは、抗神経変性および抗老化のp53/miR-29軸を生み出している可能性がある。この軸を強化することは、加齢に伴う疾患の新しい治療戦略になるかもしれない(12)。このことは、p53のネガティブレギュレーターであるヒストン-リジンN-メチルトランスフェラーゼ(SETDB1)を標的としたmir-29によってさらに強調されている(13-15)。
定量的感受性マッピング(QSM)を用いた新しい高分解能神経画像研究は、鉄マーカーの制御異常が神経変性疾患の臨床症状に先行している可能性を示唆しており、これらの疾患の予防を可能にしている(16 19)。
新しい実験室モデルや細胞培養研究では、脳の鉄沈着物が有毒なβアミロイド種と共局在化していることが観察されており、このバイオメタルを認知障害と結びつけている(20-21)。実際、過去20年間に得られた証拠は、鉄の欠乏と過剰の両方が軽度の認知障害(MCI)と関連しており、老化と認知における鉄の役割を文書化している(22-24)。
フェロトーシスは、脂質修復酵素グルタチオンペルオキシダーゼ-4(GPX-4)の非存在下で、リン脂質、特にホスファチジルエタノールアミン(PE)の過酸化によって引き起こされる制御された鉄依存性の細胞死である。このプロセスは小胞体(ER)のサブドメイン、おそらくミトコンドリア関連膜(MAM)やミトコンドリア内膜(IMM)で始まることが実証されている(25-27)。フェロトーシスはパーキンソン病(PD)、ハンチントン病(HD)、脳卒中、アルツハイマー病(AD)の神経変性と関連しており、病因的な役割を示唆している(28-30)。一方、フェロトーシスは、セレン供与体、リポキシゲナーゼ阻害剤、および化合物PBT434のような軽度の鉄キレート剤を介して防止することができ、神経変性疾患の早期介入のための機会の窓を開く(32-34)。
以上のことから、脳の鉄負荷と加齢と神経変性疾患のリスクとの関連性を示すエビデンスが増えてきており、鉄の不均衡を早期に是正することで、これらの晩期疾患を遅らせたり、予防したりする可能性があることが示唆されている。
図.1 細胞内鉄滞留の結果としての強酸性化。過剰な鉄分がDNA損傷の引き金となり、DNA修復が阻害され、さらに老化が進むため、ゲノムの崩壊につながるという悪循環が生じている。
原文参照
脳の鉄 クイックリマインダー
ほとんどの微量元素と同様に、過剰な遊離鉄は有毒であるため、トランスフェリン(Tf)その糖タンパク質トランスポーターによって保護されて循環しなければならない。脳が骨髄化とドーパミン合成(35)を含む鉄を必要とする生理機能を維持しているため、中枢神経系(中枢神経系)の鉄レベルはほとんどの臓器の鉄レベルを超えている。
それはチロシン水酸化酵素、ドーパミン生合成の速度制限酵素のために必要な補因子であるため、脳の鉄はドーパミン経路を影で示している。 nの結果として、PD、レストレスレッグス症候群、中毒、精神病や抗精神病薬の副作用を含むほとんどのドーパミン関連疾患は、鉄代謝異常を伴う(36-40)。
鉄は、脳微小血管の内皮細胞によって発現するトランスフェリン受容体-1(TFR-1)に結合することにより、Tfと一緒に中枢神経系にアクセスする。これらの細胞は、脂質ラフト依存性エンドサイトーシスによってTf-TFR-1複合体を取り込み、細胞質に放出する。細胞内では、代謝機能に利用された後、鉄はフェリチン(Ft)に貯蔵され、ミトコンドリアに運ばれたり、過剰になるとFPNチャネルを介して脳の間質液に輸出されたりする。
中枢神経系に移動した鉄は、セルロプラスミンによって酸化され、代謝のために神経細胞や非神経細胞に運ばれる。黒質体では、ドーパミン作動性ニューロンは、これらの細胞ではあまり発現していないFtの代わりに、ニューロメラニンに鉄を貯蔵することができる(41)。
ニューロンへの鉄の取り込みは二価金属トランスポーター-1 (DMT-1)とTFR-1受容体を介して行われるが、鉄はFPNチャネルを介してのみニューロンから出ることができる。したがって、これらのタンパク質は細胞内の鉄濃度を維持するために重要であり、FPNの制御異常は鉄の恒常性の障害や神経変性疾患と関連していた(42)。
生理学的には、FPN は肝ホルモンであるヘプシジンによってネガティブに制御されているが、ヘプシジンは血液脳関門 (BBB) を通過しない可能性があるため、脳は自らヘプシジンを合成することを余儀なくされる (43)。ヘプシジンのレベルが低いと神経細胞の鉄の排出が促進され、高いレベルでは鉄の滞留と関連している(44)。老化と炎症はヘプシジンをアップレギュレートし、老年期の人々の細胞内鉄濃度が高いことを説明しているのかもしれない(45)。興味深いことに、ヘプシジンのアップレギュレーションは最近、βアミロイド誘発性炎症を減少させることで神経保護効果があることが報告されている(46)。一方、いくつかの研究では、炎症を誘発することでヘプシジンの逆のアポトーシス促進効果が報告されている(45)。この問題を解明するためにはさらなる研究が必要であるが、アポトーシスよりもフェロトーシスの方が細胞の運命を決定している可能性があり、ヘプシジンの抗炎症作用は細胞の抗酸化システムの状態に依存している可能性がある。
細胞レベルでは、鉄の恒常性はmRNAの3′または5′の非翻訳領域に発現する鉄応答性エレメント(IRE)を介して制御されており、それゆえに鉄タンパク質の転写を許可するか否かを決定している。IRP-2は、IREB-2遺伝子によってコードされる脳内鉄レベルのマスターレギュレーターであり、それぞれのIREを介してTFR-1,FPN、Ftを調節することが示された(図2)。IRP-2の制御異常は、AD、PD、HD、脳卒中と関連しており、これらの疾患の病態における鉄の保持の役割を強調している(47-50)。
図2
原文参照
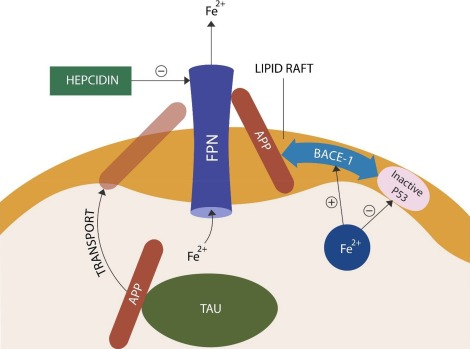
機能不全IRP-2が加齢に伴う神経細胞の鉄保持を説明する可能性がある。適切に処理されたアミロイド前駆体タンパク質(APP)はFPNチャネルを安定化し、鉄の流出を促進する。BACE-1が誤処理されたAPPはFPNの安定化に失敗し、鉄の滞留を引き起こす。IRP-2の機能不全は、TFR-1やFtからの鉄解離を介して神経細胞の鉄蓄積を促進する可能性がある。 MiR-29は、BACE-1とIRP-2の両方を直接標的とすることで、細胞の鉄の恒常性を回復させる可能性がある。
鉄と生体膜
FPN タンパク質は,脂質ラフトやカベオラ (42)(56) と呼ばれる特殊なサブドメインを介して生体膜に固定されている。コレステロールとスフィンゴ脂質を豊富に含むこれらの構造は、ほとんどの既知の受容体や神経変性に関連したタンパク質を収容しているため、神経伝達に重要な役割を果たしている(51-55)。高い脂質含量、ラフトおよび特殊なERラフトのために、ミトコンドリア関連膜(MAM)は過酸化、神経伝達障害および神経変性に対して脆弱である。実際、ラフト/MAM鉄関連脂質過酸化は、いくつかの神経変性疾患の共通分母である可能性がある(図3)。
ヘプシジンに反応するためには、FPN MANUSCRIPT分子が細胞膜に強固に固定されていなければならない(56)。FPNの安定化と固定化には、細胞質から細胞表面へのタウ関連APPの輸送が必要である(57)(図3)。ラフト過酸化やタウの機能不全はこの過程を阻害し、FPN を不安定化させ、その結果として鉄の滞留を引き起こす可能性がある(58-61)(Fig.3)。
APPとタウに加えて、MAMはPD、筋萎縮性側索硬化症(ALS)前頭側頭型認知症(FTD)の分子アクターであるαシヌクレインとTAR DNA結合タンパク質43(DP-43)を有しており、脂質過酸化は神経変性の震源地に位置している(62)。
ラフト/MAMsの過酸化とともに、生体膜は歪みと損傷を受け、神経伝達に障害を与え、マルチレセプターの障害を引き起こす可能性がある(63-65)。これらの障害は、いくつかの神経精神疾患にまたがる臨床症状の重複を説明するかもしれない。実際、脂質過酸化とラフト病理は、正常な老化からAD、PD、脳卒中、統合失調症、不安、うつ病、自閉症、てんかん、プリオン病に至るまでの幅広い病態と関連している(66-67)。
図3
原文参照
アミロイドパチーとタウオパチーの鉄のつながり ヘプシジンと相互作用するためには、FPNはAPPによって細胞表面に固定されていなければならない。APPは、細胞表面に到達するためには、タウによるトラフィッキングを必要とする。BACE-1で切断されたAPPはFPNを認識しない可能性が高く、FPNを不安定化させ、神経細胞の鉄の脱出を妨げる。また、鉄はBACE-1をアップレギュレートし、ゲノム損傷を促進するp53を不活性化する。また、BACE-1はPDに関連してタウの過リン酸化(図示せず)を誘発する (68)。
鉄性骨粗鬆症の神経画像および細胞膜マーカー
加齢は神経変性疾患の重要な危険因子であり、神経細胞の鉄滞留は正常な加齢と晩期の神経変性の両方で実証されている。QSMを含む新しい高分解能MRI技術は、脳の様々な核における鉄の非侵襲的な可視化と定量化を可能にし、臨床症状との相関を示し、潜在的なバイオマーカーを構成している(69~70)。
神経変性における最近の研究では、脳内鉄沈着と、他の研究者は、晩年の認知能力は幼少期の脳鉄レベルによって予測できることを観察することによって、鉄の蓄積と異なる年齢における認知状態との間の相関関係を確立した(71-74)。
いくつかのAD神経画像化研究は、海馬の鉄鉱床を測定し、ミニ精神状態試験(MMSE)のスコアに関連付けることに焦点を当て、進行性ADにおけるこれらの方法の予測値を示唆している(75)。他の研究者や臨床家は、診断可能な認知障害に必要な組織損失の量を推定することを目的として、海馬の体積と鉄沈着物を測定した(20)(76-77)。最近のQSM研究では、血管疾患における皮質下鉄沈着物と認知障害との間の相関関係を確立し、血管性認知症の早期診断における鉄マーカーの役割を示唆した(17)。
実験室モデルや細胞培養物を用いた非神経画像研究では、鉄に関連した有毒なアミロイド種の蓄積が観察され、このバイオメタルとプロテオスタシス不全との関連が示されている(20-21)(24)(78)。別の証拠は、MCIを鉄欠乏と鉄過剰の両方と関連させ、認知と鉄の恒常性障害との密接な関連を指摘している(79)(48)。この関連性は、認知障害のないAPOE-e4キャリアで脳脊髄液のFt値が高く、7年以内に記憶障害を発症した者を対象とした最近の研究によってさらに強調され、このマーカーが症状前のADに対する予測値を示すことが示された(80)。
新しいPD神経イメージング研究では、この疾患の前運動期における信頼性の高い鉄のバイオマーカーが同定され、予防の機会が開かれた(16)(81-82)。同様に、最近のPD研究では、症状と若年期の鉄機能障害が関連している(83)。さらに、PBT434という化合物を用いた鉄キレートはαシヌクレインの毒性を逆転させることが示されており、早期PDの可能性を示唆している(31)。他のグループは、変異したαシヌクレインを、依存症、精神病、およびその他の非PD性ドーパミン障害における鉄-ドーパミン輸送の障害と関連づけた(39)(41)(84)。
これらを総合すると、新しいプロテオミクスマーカーと高解像度の神経イメージング技術により、研究者や臨床医は、脳の鉄イメージングと鉄タンパク質の定量化により、早期の神経変性の信頼性の高い予測因子を導き出すことができるようになった。
フェロセネセンスとは何か?
鉄老化は、細胞内鉄の増加によって促進される早期老化の表現型である。DNA損傷、ゲノム修復不全、メチル化欠損がこの表現型の特徴である(4)(85-87)。
DNA損傷
培養内皮細胞を対象とした最近の研究では、鉄は1錠の鉄剤を摂取した後、数分で開始する直接的なDNA損傷を誘発する可能性があることが報告されている(4)。p53-DNA修復系の活性化を測定したこの研究では、循環鉄の一過性の上昇がゲノム修復系を活性化するのに十分であることが示された。このデータは、ゲノムの完全性を維持する上でp53が重要な役割を果たしていることを示唆しており、このタンパク質に与えられた「ゲノムの守護者」という称号を正当化するものである。
新しい研究では、p53の鉄結合部位が占有されるとこのタンパク質が不活性化されることが報告されている(5)。同様のメカニズムがミトコンドリアでも報告されており、鉄-p53の不活性化は鉄硫黄クラスター組立酵素(ISCU)を刺激して鉄の蓄積を促進し、最終的にはミトコンドリアDNA(mtDNA)の損傷を引き起こすことになる(88)。p53は「mtDNAの保護者」でもあるので、p53の不活性化は、いくつかの神経変性疾患で報告されているこれらの小器官の障害の原因となるかもしれない(89)。
ゲノム修復不全
多くの癌は、p53遺伝子の変異によって増殖し、その抗腫瘍特性の喪失だけでなく、このタンパク質を癌遺伝子または感染性プリオンへと変化させる(90)。実際、鉄はp53の構造変化を促進し、発がんにつながることが報告されている(91-92)。
他の研究では、癌に関連したp53アミロイド生成を実証し、このタンパク質をADの病理学と結びつけ、おそらく老年人口における癌の発生率の増加を説明している(93-95)。別の最近の研究では、認知障害のある高齢者の小脳に不溶性のp53アミロイド凝集体が存在することを示すことで、p53アミロイド新生と神経変性とを関連づけている(97)。他にも、AD初期のp53タンパク質の変異が報告されており、この生体分子のマーカーとしての価値を示唆している(96-98)。実際、p53を無力化することに加えて、鉄はBACE-1をアップレギュレートすることが実証されており、おそらくこの酵素とp53アミロイドとの関連性が示唆されている(99)。
MicroRNA-29はBACE-1を直接標的としており、おそらく神経変性といくつかの癌の両方の保護機構を構成していると思われる(100-101)。実際、組換えプレmicroR-29は、ADにおいて有益な効果を有することが実証された(100)。さらに、miR-29は、その負の調節因子のうち2つをサイレンシングすることで、間接的にp53をアップレギュレーションすることが示されており、このmiRの抗アミロイド的な役割を実証している(102)。
DNAのハイポメチル化
ゲノムの変化とは別に、フェロセネセンスはDNAメチル化とクロマチン密度を変化させることでエピゲノムの損傷を発生させる(86-87)(4)。グローバルなハイメチル化は、癌、老化、脂質病理と関連していた(87)(103)。実際、脂質過酸化は、DNAメチルトランスフェラーゼ3Aおよび3B(DNMT-3AおよびDNMT-3B)をアップレギュレーションすることにより、グローバルおよびプロモーター特異的なメチル化変化を誘導し、細胞の老化を誘発することが報告された(104-105)。
MiR-29はDNMT-3Aと3Bを直接標的とすることが示され、したがって、老化や癌に関連するDNAメチル化変化に対抗することが示された(106)。実際、通常の状況下では、miR-29はDNAメチル化酵素、メチラーゼ、ヒストンメチル化酵素SETDB1の発現をサイレンシングすることで、ゲノムメチル化の現状を維持していることが報告されている(11)(13)。これらの一見矛盾した作用は、ゲノムを安定化させ、メチル化の景観を微調整すると考えられている(11)。MicroRNA-29はメチル化に対しても反対にも機能せず、むしろ「抗de novoメチル化」剤として機能している。実際、DNMT-3Aと3Bの両方が、老化や癌に有利なエピゲノムの再配線を行うde novo methyltransferaseであることが示されている(107-109)。同様のことが、miR-29のもう一つの直接の標的であるSETDB1ヒストンメチルトランスフェラーゼについても報告されており、DNMT-3Aとde novo methylationの調整を行っている(110)。SETDB1は、そのエピゲノム的役割とは別に、p53遺伝子を直接標的とし、DNA修復を不能にすることが実証された(111)。
トランスポーザブルエレメント
ゲノムの低メチル化は、老化や癌の特徴として確立されているトランスポゾンやトランスポゾン可能要素(TE)の動員と関連していた(112-113)。TEは、二重らせんから抜け出し、その後、異なるゲノムアドレスに自己再挿入する能力を持つDNAのセグメントである(114)。加齢に伴うTEの動員は、最近、DNAのハイメチル化とヒストンの脱縮合を介したTEの活性化に基づく「老化のトランスポゾン理論」を提唱した。逆に、TEの動員はヘテロクロマチンの安定化によって止めることができ、これは加齢に伴う疾患への応用が期待されている(115)。
TE動員は、ALS、FTD、PD、海綿状脳症および統合失調症を含む神経変性病理と関連している(116-120)。
microRNA-29は、ゲノムの安定性を促進し、DNAメチル化を維持し、p53-DNA修復の完全性を回復させることで、TE動員を打ち消す可能性がある(122-123)(図4)。この点から、miR-29/p53は抗老化/抗神経変性の軸を構成している可能性があり、これは治療のモダリティとして利用される可能性がある(8)(121)。実際、両親媒性ペプチドであるLK-L1C/K6W/L8Cはすでに合成されており、miR29bの発現を増強し、p53活性を促進することを目的としている(124)。さらに、miR-29/p53の抗老化作用は、最近の前臨床研究で、p53-FOXO-4分離によりいくつかの老化現象が逆転したことからも強調されており、癌と老化の境界線を維持する上でのmiRの役割が示唆されている(125)。
図4 鉄と対照的なmiR-29のゲノムおよびエピゲノム機能の模式図
原文参照
p53の安定性を守り、DNAメチル化を維持することで、TEを不活性化する可能性がある。一方、鉄はゲノムやエピゲノムを不安定化させ、p53-DNA修復を無効化し、ハイメチル化を促進することで、TEの動員に有利な環境を作り出している。また、miR-29はDNMTやSETB1の発現を抑制していることも注目に値する。
以上のことから、miR-29/p53は、p53の安定性を守り、DNAメチル化を維持し、神経細胞内の鉄を低下させ、鉄による脂質過酸化を減少させることで、ゲノムおよびエピゲノムの安定化を達成する可能性があると考えられる。
神経発生のMAM
フェロセネセンスに伴うゲノムの崩壊は、最終的にはフェロトーシスまたはアポトーシスによって神経細胞を死滅させることになる。フェロトーシスとミトコンドリア関連膜(MAM)との関連はまだ強調されていないが、最近の重要な研究では、このプロセスの開始はERサブドメイン、おそらくMAMにあると考えられている(25) (126-127)。実際、この領域は最近、いくつかの加齢性疾患と関連した老化と神経変性のハブであることが確認された(128)(62)(図5,6)。
前述したように、MAMは特殊な脂質ラフトであり、細胞の「脂質工場」であるERに関連しており、脂質過酸化やタンパク質のミスフォールディングに対して脆弱である(62)。神経変性関連タンパク質および神経伝達関連受容体が共通の位置にあることは、いくつかの神経変性疾患における臨床症状およびタンパク質マーカーのミスフォールディングの重複を説明するのに役立つかもしれない。例えば、誤って折り畳まれたαシヌクレインは、PD、AD、ALS、F Dおよびレビー小体型認知症を含むいくつかの神経変性疾患で遭遇する(129-132)。さらに、うつ病、不安、妄想または幻覚を含む症状が、同じ条件で遭遇することがある。
フェロトーシスは、抗酸化物質であるセレノプロテインGPX-4の非存在下で、細胞膜PEの鉄過酸化によって引き起こされる制御された細胞死である(25)。最近、フェロトーシスは、15-リポキシゲナーゼの酵素作用に由来するアラキドン酸(AA)脂肪酸アシルアラキドノイルAAとアドレノイル(AdA)を介して実行されることが実証された(25)(図5)。他の研究では、フェロトーシスとミトコンドリア内膜(IMM)における12/15リポキシゲナーゼとカルジオリピン(CL)の酸化を関連づけている(133-135)(図5)。
フェロトーシスの分子アクターがアポトーシスを誘発し、多くの神経変性疾患における制御された細胞死とMAMを結びつけていることは注目に値する(127)(136-137)。フェロトーシスはPEの酸化のみが引き金となるが、アポトーシス経路は他の複数の基質によって活性化される可能性がある(25)。また、前臨床研究では、GPX-4欠損や12/15リポキシゲナーゼの過剰発現とアポトーシスやADとの関連性が示されており、フェロトーシスとアポトーシスの密接な関連性が示唆されている(138-139)(27)(30)(図5)。
細胞死や神経変性とは別に、MAMは長寿にも関連していた。例えば、MAMの住人であるPEと哺乳類のラパマイシン標的(mTOR)経路の構成要素であるmTORC2は寿命の増加と関連していた(140)(図6)。最近では、TORブロッカーであるラパマイシンが長寿、癌発生率の低下、認知機能の低下を逆転させることが前臨床研究で明らかになり、TOR経路への関心が高まっている(141-142)。興味深いことに、最近の研究では、miR-29が間接的にmTOR経路をダウンレギュレートし、げっ歯類の寿命を延ばすことが示されており、このmiRが長寿の役割を果たしていることが示唆されている(143-144)。さらに、別の研究では、MAMテザリングタンパク質であるアニオンチャネル1および2(VDAC-1およびVDAC-2)を直接標的とすることで、miR-29のアンチエイジング的役割が強調されているが、VDAC-3は標的としていない(145)。前臨床研究では、VDAC-1とVDAC-3のスワップが寿命の増加と関連していることが示された(146)。さらに、VDAC-1およびVDAC-2の機能不全は、AD、PD、ダウン症候群およびてんかんにおける神経変性と関連していた(147-150)。
興味深いことに、VDACは神経細胞のフェロトーシスと関連しており、がん細胞ではフェロトーシスはシスチン/グルタミン酸アンチポーターと関連していた(151-153)。このシステムは成熟ニューロンでは発現が乏しいため、VDACがニューロン細胞でフェロトーシスを実行している可能性が高いと考えられる(154)。
MAMと鉄の恒常性との直接的な関係は、MAMとミトコンドリアのテザーである小胞体-ミトコンドリア遭遇構造(ERMES)を介して下等生物で確立された(155)。ERMESは哺乳類では発現していないので、VDACは高等生物ではそれに相当するものであり、外側ミトコンドリア膜(OMM)を横切る鉄輸送に関与している可能性がある。
エンドカンナビノイド系とMAMsとの関連については、これまで検討されていなかったが、カンナビノイド受容体-1(CB-1)の過剰発現がERストレスと関連していることから、神経変性に関与していることが示唆された(156)。また、エンドカンナビノイドであるアナンドアミド(アラキドノイルエタノールアミド)(AEA)は、15/リポキシゲナーゼによって代謝されることが報告されており、おそらくフェロトーシスと関連していると考えられている(157)。さらに、生化学的研究では、AEAがAAリポキシゲナーゼ経路を活性化し、アナンドアミドをフェロトーシスやアポトーシスによる細胞死に結びつけることが報告されている(158)。実際、AEAの認知への悪影響や、AD、PD、統合失調症への関与は、多くの研究によって立証されている(159-163)。さらに、FAAH阻害薬(シナプスのAEAを増加させる)の臨床試験は、一連の副作用のために最近中止され、おそらくフェロトーシスに関連していると思われる(164)。
以上のことから、MAMが神経変性疾患と関連していることは多くの研究で明らかにされているが、その理由は、MAMが脂質過酸化と神経変性疾患で見られるタンパク質のミスフォールディングに関連しているからである。
図5
原文参照
強誘電体化とアポトーシスは同一の分子アクターによって引き起こされるが、強誘電体化はPE過酸化のみが実行する。AAがFtに結合すると、マスター抗酸化調節因子Nrf2が活性化されることは注目に値する。FAB3はAAをFt結合部位まで輸送すると考えられる。その結果、Nrf2はFtとGPX-4をアップレギュレーションする。Nrf2はまた、DNAの脱メチル化によって活性化され、GSKβによって不活性化される。GSKβ阻害剤であるリチウムは、間接的にNrf2をアップレギュレートする。
細胞の酸化剤と抗酸化剤
強精症は、失敗した細胞性抗酸化物質、特にGPX-4の結果であり、核内因子エリスロイド2関連因子2(Nrf2)による活性化を必要とする。Nrf2は細胞性抗酸化物質のマスターレギュレーターであり、抗酸化応答エレメント(ARES)を介して複数の抗酸化遺伝子の転写を制御している(165)。Nrf2のダウンレギュレーションをADおよびPDと関連させた研究の新たなボディは、細胞性抗酸化物質がこれらの障害において重要な役割を果たしている可能性を示唆している(168-170)。さらに、加齢に伴うDNAのハイポメチル化はDNMTを介してNrf2を活性化することが示され、加齢や加齢に伴う疾患とリンクしている(171)(図5)。実際、Nrf2の障害はGPX-4および/またはFtの不活性化を介してフェロトーシスを引き起こす可能性がある(166-168)。
MiR-29は、Nrf2遺伝子の負の調節因子であるDNMTとグリコーゲン合成酵素キナーゼ-3β(GSK-3β)を標的としているため、間接的なNrf-2調節因子である可能性がある(172-175)(図5)。さらに、最近の研究では、Nrf2-miR-29との直接的な関連が報告されている(176)。
複数の研究でGSK-3βとDやPD、さらには双極性障害、うつ病、不安、統合失調症、薬物依存症などの精神疾患との関連が報告されている(177-179)。GSK-3遮断薬であるリチウムは、おそらくNrf2のアップレギュレーションによって、ADモデルにおいてACCEPTEDdemonstratedtohavebebeicialeffectsを示した(180-181)。実際、リチウムの予防的使用は最近健康な人に推奨されているが、新しい集団ベースの研究では、飲料水に含まれるリチウムの天然レベルが高くなるとADの発生率が低下すると関連している(182-183)。興味深いことに、以前の研究では、自殺の発生率の低下と飲料水のリチウムとの関連があった(184)。さらに、動物モデルでは、リチウムはBACE-1とαシヌクレインの発現を減少させることが実証され、神経変性における有益な効果を強調している(185-186)。
主要な細胞抗酸化物質であるFtは、鉄貯蔵機能のためにNrf2-AREの活性化を必要とするため、フェロトーシスや神経変性のマーカーとなっている(187)。実際、脳脊髄液のFtレベルはすでに初期のADマーカーとして報告されており、我々の例では4年後の血漿Ftレベルと認知機能障害との密接な関連を示唆している(80)。FtはAAと結合することで抗酸化作用を発揮し、過酸化基質としての12/15リポキシゲナーゼを拒否し、フェロトーシスを防止している可能性がある(188)(図5)。一方、Ft分解(フェリチノファジー)は、Ft-鉄の解離を増加させることでフェロトーシスを促進し、鉄過剰と抗酸化不全を結びつけることが示された(189)。脂肪酸結合タンパク質3(FABP3)は、AD、レビー小体型認知症、PDに関連する新しい神経変性マーカーとして浮上している(190-191)。この細胞内脂質トランスポーターは、おそらくAAをFt結合部位まで運び、抗酸化機能の低下を防ぎ、フェロトーシスを回避していると考えられる(図5)。
図6 ミトコンドリア関連膜(MAM)とその外側ミトコンドリア膜(OMM)への結合を模式的に示したもの
原文参照
このレベルでのmiR-29の直接の標的はBACE-1,DNMT3A、VDACsである。ホスファチジルエタノールアミン(PE)とカルジオリピン(CL)は、鉄過酸化、フェロトーシスの開始、神経変性病理と関連していた。
フェロセネセンス介入
MicroRNA-29は、AD、脳卒中、およびいくつかの精神疾患において有益であることが報告されている。
疾患では、おそらくMANUSCRIPTBACE-1と鉄保持を低下させる(100)(192-193)。さらに、このmiRは疾患の進行を反映していることから、最近ではPDのバイオマーカーとして示唆されている(194-195)。さらに、げっ歯類では、miR-29は寿命の延長や生殖率の低下と関連していた(196)(9)。しかし、その神経保護的な役割はさておき、新しい研究では、腸管上皮の完全性を維持し、循環系への細菌の移動を防ぎ、miR-29の腸管保護機能があることが明らかにされている(197)。最近の研究では、NOD2遺伝子は敗血症性腸内フローラを調節し、病原性微生物の出現を抑制することで、腸管保護の役割を果たしていると考えられている(198)。興味深いことに、NOD2はmiR-29をアップレギュレートすることが明らかになり、このmiRの腸管保護的役割が強調された(199)。
鉄キレート剤
最近の前臨床研究では、デスフェリオキサミン(DFO)のような鉄キレート剤はタウリン酸化を低下させることでADの認知機能低下の進行を遅らせることが報告されている(200)。これに関連して、DFOの経鼻投与は、ADの動物モデルで記憶喪失を低下させることも実証された(201)。
低親和性の鉄キレート剤は、このバイオメタルの有益な効果を阻害しない可能性があるため、高齢者の鉄滞留の治療に適していると考えられている。より穏やかで植物由来の化合物は、そのレベルを劇的に変化させることなく、効果的に過剰な鉄を除去することができるかもしれない。PBT 434は、新規のキナゾリノン化合物であり、最近、PDの潜在的な適応症として記載された(31)。PBT 434は、げっ歯類において細胞内の鉄を選択的に低下させ、FPNレベルを上昇させることが示された(31)。
表1 神経変性疾患における潜在的な利益を有するハーブの鉄キレート剤
植物ベースの鉄キレート剤
参考文献
- クルクミン
- コラビロン
- フローラノール
- プロシアニジン
- バイカライン
- テトラメチルピラジンとフェルラ酸
- ポリフェノール
- カテキン
- エピガロカテキン-3-ガレート。
- フィテート
- エピメジウム
- アストラガルス
- 螺鈿女
- アザディラクタ・インディカ
セレンと鉄のリンク
ヒトおよび動物実験では、miR-29のようなセレンは、ゲノム修復にこのマイクロエレメントをリンクして、グローバルなDNAハイポメチル化とDNMT活性に逆に関連付けられていることが示されている(214)。さらに、がん患者では、セレンは、DNAに損傷を与える化学療法(215)から保護することが実証された。例えば、レジメンにセレンを追加すると、より高い化学療法用量の患者の忍容性を増強することが報告された(215)。さらに、セレン代謝物であるセレノジグルタチオンは、抗がん治療の有効性を高め、p53合成を促進することが示された(216)。
いくつかの研究では、おそらく鉄誘導脂質過酸化とGPX-4セレンタンパク質(217)の損失の両方を占めて、鉄レベルの上昇とセレン欠乏をリンクしている。ADでは、これらの要素のフェーズ特異的なマーカーの役割を示唆し、後期段階では低セレンによってマークされたが、より高い鉄レベルでは、初期段階で報告された(218)。さらに、他の人は、さらに老化にこの微量栄養素をリンクして、低セレンとセレンタンパク質のレベルと細胞の老化を関連付けている(219)。
新しい研究では、敗血症微生物によるセレンの隔離は、ホストのセレンレベル(220-221)を低下させる可能性があることを示し、セレンと腸内微生物の間のリンクを探索した。興味深いことに、重要な研究では、血漿中のセレン濃度は必ずしも神経変性疾患における脳の抗酸化状態を反映していないことが報告され、新規の中枢神経系抗酸化マーカーの必要性を示している(222-223)。
セレン化合物、ebselenは、最近、神経変性疾患のための潜在的な投与範囲を示し、1日800 mgまでの用量で難聴のために有益であることが判明した(224)。別の有望なセレンドナーは、セレノネイン、また、そのヘムキレート特性(32)(225)を介して鉄を低下させるクロマグロに由来する化合物である。興味深いことに、エルゴチオネインは、セレノネイン姉妹分子は、最近、神経変性疾患におけるこれらの化合物の役割を示す、ADマウスモデルで有益であることが示された(226)。
12/15 リポキシゲナーゼ阻害剤
12/15 膜リン脂質の酸化に直接関与する酵素であるリポキシゲナーゼは、脳卒中や神経変性の新たなターゲットである(227)。新しく記載された化合物99089は、この酵素を標的とした薬剤開発の出発点となる可能性がある(228)。興味深いことに、12/15リポキシゲナーゼとGPX-4の両方が腸管膜に存在し、細菌の血流への移動に影響を与えることが報告されている(229)。
12/15リポキシゲナーゼの基質であるオメガ3とオメガ6ポリ不飽和脂肪酸(PUFA)は、その比率(オメガ6/オメガ3)がAD、過敏性腸症候群(IBD)関節リウマチだけでなく、うつ病や統合失調症(230-232)を含むいくつかの慢性疾患のマーカーとして浮上しているので、最近精査されている。オメガ6(AAを含む)は、腸内細菌やクロストリジウムなどの炎症を起こす腸内微生物を促進することが示され、オメガ3(エイコサペンタエン酸を含む)は、乳酸菌やビフィズス菌などの有益な微生物を促進することで炎症を逆転させることが示されている(233)。
インスリン/IGF-1軸
インスリン/インスリン様成長因子-1(IGF-1)シグナル伝達経路は、線虫やメラノガスター(D. melanogaster)などの単純な生物において長寿と関連していた(234)。
動物モデルを用いた別の研究では、インスリン/IGF-1操作により寿命が2倍に増加したことが報告されている(235-236)。ヒトでは、食物繊維の発酵により腸内で生成される短鎖脂肪酸(SCFA)がIGF-1シグナル伝達を促進し、消化管と寿命を結びつけることが報告されている(237)。興味深いことに、インスリン/IGF-1軸がTFR-1を介して鉄のホメオスタシスを制御していることが新しい研究で明らかになり、老化における鉄の役割が改めて証明された(238)。また、鉄はtristetraprolinMANUSCRIPTexpression.ristetraprolinisadirect target of miR-29を介してmTOR経路を調節している。
大規模なマイクロバイオーム研究では、腸内細菌株の加齢に関連した変化が実証されている。例えば、老齢と疾患は、ルミノコッカス科、ラクノスピラセ科、バクテロイデス科の菌株の優位性の低下と関連していた(243)。一方、健康な老年期には、Akkermansia、Bifidobacterium、Christensenellaceae (243)の存在と関連していた。興味深いことに、腸内微生物は、AD、PD、および不安などの一部の精神疾患を含む神経変性疾患の発症を促進することが実証された(244-246)。さらに、腸内微生物の分類および真菌株は、中枢神経系のアミロイド生成に関連していることが示されている(247-248)。他の鎖はALS関連の神経毒を分泌することが示されており、この神経変性疾患と腸との関連を示唆している(249)。さらに、過敏性腸症候群に関連するPDは、腸から発生し、二次的に脳に広がる可能性があるという仮説が立てられた(250-251)。実際、PrevotellaおよびEnterobacteriaを含む特定の腸内微生物がPDと関連していた(252)。他の研究では、細菌性サイトカインがERタンパク質のミスフォールディングと酸化ストレスを誘発し、結果として神経変性を引き起こす可能性が示唆されている(253)。同様に、腸内細菌群集は、SCFAのような化合物を介して宿主のDNAメチル化と遺伝子発現を変化させ、宿主の寿命と健康寿命に影響を与えることが実証されている(254-255)。例えば、線虫の長寿の増加は、最近、変異大腸菌株によって産生される細菌性多糖体コラン酸と関連しており、マイクロバイオームの操作が寿命に影響を与える可能性があることを示唆している(256-257)。
一方、大腸に到達した未吸収鉄は、血流移動で知られる病原性微生物種の活性化と関連していた(258)。一方、宿主鉄欠乏は、鉄の隔離が可能なシデロフォア含有微生物株によって引き起こされ得る(259)。例えば、鉄結合性のビフィズス菌やストレプトコッカス・サーモフィルスは、病原菌にこのバイオメタルを拒否し、それらを制御下に維持することができる(258)(260)。
最近の研究をまとめると、マイクロバイオームの操作が長寿や病気の進行に影響を与え、加齢に関連する病気を遅らせたり予防したりする可能性があることが示唆されている。
結論
検出可能な鉄および鉄関連タンパク質は、早期老化および神経変性のマーカーとして浮上している。しかし、将来の神経変性に関する事前知識は、これらの疾患の進行や転帰に影響を与えることができるのだろうか?言い換えれば、ライフスタイルの変化は、ADやPDのような状態の予後に影響を与えることができるのであろうか?
我々は、食事介入やライフスタイルの変化が、有益なエピジェネティックな変化を促進することで、老化や神経変性疾患を遅らせる可能性があることを主張している。例えば、高齢者のアルコール消費は、老化の2つの可逆的なマーカー、鉄の保持とDNAのハイメチル化と関連している(261-262)。逆に、適切なオメガ6/オメガ3比の食事は、高齢者のDNAメチル化と無傷の認知と関連していた(263-264)。