
Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two Potential Targets for COVID-19 Treatment
www.hindawi.com/journals/mi/2020/7527953/
要旨
COVID-19は、主に呼吸器系に影響を及ぼす新型コロナウイルスSARS-CoV-2によって引き起こされるパンデミック疾患である。結果として生じる炎症は、ウイルスをクリアすることができない。持続的な過剰な炎症反応は、管理が非常に困難で、潜在的に致命的な臨床像を構築することができる。
免疫反応を調整することは、病気と闘う上で重要な役割を果たす。主な防御システムの一つは、オートファジーの刺激の下で好中球の細胞外トラップ(NET)を放出する好中球の活性化である。
様々な分子がネット症およびオートファジーを誘導することができる;いくつかの強力な活性化剤は、ダメージ関連分子パターン(DAMP)および、特に、高可動性グループボックス1(HMGB1)である。この分子は、損傷を受けた肺細胞によって放出され、頑健な自然免疫応答を誘導することができる。
HMGB1およびNETosisの増加は、SARS-CoV-2感染による持続的な炎症につながる可能性がある。したがって、これらの分子をブロックすることは、COVID-19治療に有用である可能性があり、標的治療の文脈でさらに研究されるべきである。
1. はじめに
COVID-19は、現在、世界中の多くの医療システムに大きなストレスを与えている新型コロナウイルスSARS-CoV-2[1]によるパンデミック課題である。コロナウイルスは、重症急性呼吸器症候群コロナウイルス1型(SARS-CoV)および中東呼吸器症候群(MERS-CoV)ウイルスを含むコロナウイルスファミリーに属している。コロナウイルスは肺細胞に優先的にトロピズムを持つ[2]。SARS-CoV-2は、SARS-CoVと同じ受容体、すなわちアンジオテンシン変換酵素II(ACE2)を用いて宿主細胞に侵入することが知られている[2]。急性SARS-CoV-2患者は、無症状または軽度の症状(感冒)から重症化し、しばしば致命的な疾患に至るまで、幅広い臨床症状を呈する。後者は通常、両側性間質性肺炎、中等度から重度の酸素脱飽和症および低酸素症を呈する。多くの患者が呼吸不全(RF)および急性呼吸窮迫症候群(ARDS)[3]を発症し、集中治療室(ICU)への迅速な入院を必要とする。通常の ARDS とは異なり、これらの患者は正常またはわずかに肺のコンプライアンスが向上しており、ほとんどの場合、高流量酸素または持続的陽圧換気(CPAP)が必要である [4]。SARS-CoV-2肺炎の換気成績は、間質性肺疾患の呼吸不全で報告されているものと類似している [5]。
SARS-CoV-2は多くの免疫学的問題を提起している。報告 [6] や中国のガイドライン [7] では、肺胞障害が確認されている。同じファミリーのウイルスに基づく以前の報告では、サイトカインストームを示している。中国での最初の報告は、これらの患者におけるIL-6の増加を同定している[8]が、重症例でピークを迎える。主な治療戦略は、炎症を調節するためのサイトカイン遮断に基づいている。適応外の適応でSARS-CoV-2の治療に最も一般的に使用されている薬剤には、クロロキン(クロロキン)および/またはヒドロキシクロロキン( ヒドロキシクロロキン)がある。これらの薬剤は、複数の抗炎症作用を発揮し、ループスおよび関節リウマチなどの慢性炎症性疾患の治療に有効であることがよく知られている。抗炎症作用のメカニズムは完全には解明されていない。しかし、液胞pHを上昇させることでオートファジーを阻害し、DNA修復やリソソーム形成を阻害することが確立されている[9、10]。クロロキン/ ヒドロキシクロロキンはまた、好中球の細胞外トラップ(NET)やダメージ関連分子パターン(DAMP)の分泌を減少させる[11]。COVID-19の剖検から得られた最近のデータでは、肺の空域に好中球が浸潤していることが記載されている[12]と血管にも記載されている[13]。さらに、健康なボランティアの血液サンプルと比較して、COVID-19の血液サンプルでは、Zuoらは、無細胞DNA、ミエロペルオキシダーゼ(MPO-)DNA、およびシトルリン化ヒストンH3(Cit-H3)[14]として定量化されたNETの増加を発見し、臨床バイオマーカーと相関していた。臨床症状は、免疫系に対するDAMPの作用の結果であると思われる。COVID-19の臨床データとクロロキン/ ヒドロキシクロロキンの使用に関する経験的データから、これら2つのメカニズムが免疫調節とSARS-CoV-2感染症の宿主障害のキープレイヤーである可能性が推測される。本レビューでは、SARS-CoV-2感染による肺障害におけるNETとDAMPの役割の可能性に焦点を当て、疾患治療のターゲットとなりうる免疫学的な示唆を与えている。
2. 好中球細胞外トラップ(NETs)と呼吸器ウイルス感染症
NETsは、細胞外空間の好中球から放出された大規模な細胞外、ウェブのような構造である。彼らは病原体と戦うために採用された好中球の武器の一つである。これらの構造は、分解されたクロマチンと細胞質および顆粒タンパク質[15]で構成されている。NETのDNAは、核とミトコンドリア材料に由来する。NETの2つの形態が知られている。1つは自殺性NET症である。これは、好中球が核内のクロマチンと細胞質内のDNAを分解する数時間の時間枠のプロセスである。次に、クロマチンとDNAは、顆粒由来の抗菌ペプチドと混合する。最後に、この混合物は活性酸素種(ROS)の拡散とともに細胞外空間に放出される [16]。第二の形式は、NETsが細胞死なしでリリースされているバイタルNETosisであり、したがって、細胞が生き残ることができるし、貪食を含む正常な機能がまだ可能である。自殺NETosisとは異なり、バイタルNETosisは、活性酸素の生成もRaf/MERK/ERK経路の活性化を必要とせず、細胞が刺激された後、通常5〜60分以内に、迅速に発生する[17、18]。好中球刺激は、トール様受容体(TLR)またはC3タンパク質リガンド結合のための補体受容体を介して起こる。これらの経路の活性化は、核膜の形態の変化を誘導する。ベシクルの出芽が始まる。したがって、核内DNAを含む小胞は細胞質を移動し、形質膜と合体し、その負荷を細胞外に放出する [17-19]。
NETは、その中和と殺傷機能のおかげで、病原体の拡散を防ぐために有用である[20]。NET症は細胞外真菌ハイパーによって直接誘導されるが、大規模な細菌とその集合体である細胞内細菌は、NETs [20]を形成することができないことに注意してほしい。
ウイルスのような小さな病原体の場合は、NETは両刃の役割を果たしている。1つのメカニズムは、合胞性呼吸器ウイルス(RSV)感染[21]やインフルエンザ[22]で観察されるように、NETによるウイルスの巻き込みであるが、生体内での抗ウイルス的役割は発見されていない[23]。これらのウイルスはいずれも、SARS-CoV-2と同様にRNAウイルスであり、年間300万~500万人の重症患者と25万~50万人の死亡者を世界中で引き起こしている[24]。これらのウイルスは呼吸器上皮細胞で複製し、壊死性の組織障害を引き起こす。インフルエンザウイルス感染では、NET遺伝子の活性化と発現の大幅な増加が認められているが、サイトカイン産生の増加は見られない[25]。これは、疾患の重症度に応じたウイルスによる好中球活性化のモードに関係している[25]。実際、軽度のインフルエンザではNETは形成されないが[26]、重症化したインフルエンザ[27]や致死性疾患[28]ではNETが形成されることが多い。さらに、ウイルス感染では、ウイルスを感染部位でブロックし、DNAの網に巻き込む効果がある。他の免疫細胞へのウイルス曝露は、マクロファージによる切断を達成する可能性がある[29]。
同時に、インフルエンザ肺炎で死亡した人の血中好中球の増加が報告されている[27]。NETに関連した抗菌因子は宿主にとって有害である可能性がある。NETとともに放出された抗菌性タンパク質は、組織に直接毒性を持つ可能性があり、その大量生産は組織障害を引き起こす可能性がある[30]。実際、NETの形成はエラスターゼとミエロペルオキシダーゼを放出し、これらは感染部位で宿主タンパク質を切断するだけでなく、組織傷害を引き起こすのに有用である[31, 32]。
NET形成はTLR-4の活性化に依存しており、NETosisはウイルスタンパク質がTLR-4に結合することによって引き起こされる[21]。実際、RSV融合タンパク質は、TLR-4シグナル伝達活性化により、細胞外空間で好中球DNA放出を誘導することができる。これは、NADPHオキシダーゼ複合体のアセンブリと活性酸素の形成を活性化するシグナル伝達カスケードを介してNETosisを媒介する[33]。TLR-4に対する中和抗体を用いることで、細胞外のDNA産生を大幅に抑制することができる。コロナウイルス融合タンパク質も同様のメカニズムを誘導する可能性がある[34, 35]。
インフルエンザウイルス感染後の急性肺炎では、好中球の過剰な浸潤が肺に起こる[22]。しかし、好中球は肺で保護および有害な役割の両方を果たす[22、36]。これらの細胞は、インフルエンザA H1N1ウイルスに反応して過剰なNETを生成する。NETの形成は、プロテインアルギニンデアミナーゼ4(PAD4)によるヒストン脱アミノ化に依存している[23]。NETには、感染細胞のプロテインキナーゼC(PKC)を阻害することでH1N1ウイルスの複製を直接阻害することができる抗菌タンパク質であるα-デフェンシン-1も存在している[37]。α-デフェンシンはH1N1ウイルス感染時に増加し[38]、NET線維に隔離されたウイルスを不活性化し、肺の標的細胞への侵入を防ぐ。しかし、α-デフェンシンは細胞毒性を持つため、宿主細胞や組織にダメージを与えることも可能である[36]。
また、肺ではNETタンパク質が毛細血管の破壊や漏出を引き起こし、肺上皮の破壊や死と同様に内皮細胞死や血栓症を誘発する[39-41]。一方、NET症は、インフルエンザウイルス感染後、ダメージ関連分子パターン(DAMP)を介して肺上皮から直接誘導される[27]。これらの現象は最終的に感染の進化につながるので、好中球の活性化とNETの形成は呼吸不全(RF)と急性呼吸窮迫症候群(ARDS)の予測因子である[25, 27, 40]。NETの有害な役割は、ウイルスへの長期暴露に関連している可能性がある。それは、NETの形成がウイルスに対する防御の最初のラインであると仮定することができる。同じことがインフルエンザやシンチウイルスで起こるように、小さな病原体のサイズは、過剰なNETosisにつながる。正味の結果は、宿主組織のために有毒である分子を放出する活性化好中球の蓄積である。
また、NETosisが異なる設定で増加することも考慮しなければならない。実際には、これらの違いは、NETの形成に影響を与えることができる。実際には、NETの増加は、脂肪の多い食事[42]、肥満[43、44]、糖尿病[45、46]、年齢[47]、および性差[48、49]に関連している。注目すべきは、女性ではプロゲステロンの放出によりNETosisの活性化が低下することである[50]。
SARS-CoV-2感染における病理学的所見[6]、生化学的所見[8、51]、および臨床的所見[1、3、51]は、肺上皮の損傷および疾患の重症度によって誘発されるNET反応に関連しているように思われる。他のウイルス肺障害におけるこれまでの所見や、COVID-19の血清NET増加、サイトカインプロファイル、臨床的進展に関するデータは、NETがSAR-CoV-2感染に関連する重症肺炎の主役であるという考えを支持するものである。患者は、最初の無症状または軽度の症状期から重症肺炎に至るまで、COVID-19の臨床的進化を経験する。COVID-19では健康な対照群と比較してNETが増加していたが、換気を行った患者では換気を行わなかった患者と比較してNETが増加していた[14]。これらのデータは、ネットがCOVID-19での形成に継続的に強い刺激の結果として、臨床的な進化のキープレーヤーである可能性を強調している。このメカニズムは、異なる要因に関連している可能性がある。第一に、ウイルスの急速な拡散は、肺組織におけるウイルス負荷の急速な増加を誘発する。上で議論したように、ウイルスタンパク質はNETosisの強力な活性化剤と考えられるかもしれない。さらに、損傷を受けた組織および好中球自体は、両方とも好中球活性を高めるサイトカインを産生する。実際、サイトカインの結果は、SARS-CoV-2刺激後に強く産生される。その作用は、好中球の化学戦術的リクルートで役割を果たし、活性酸素およびNET形成を増強する。また、NET症の誘導は、ダメージ関連分子パターン(DAMP)のような損傷組織から分泌される他の分子によるものである。
3. ダメージ関連分子パターン(DAMPs)とウイルス感染
細胞は、微生物の侵入やストレス因子に続く予定外の細胞死を自然免疫系に警告する内因性の危険信号としてDAMPを放出する [52]。これらの内因性自己抗原、例えば高可動性グループボックス1(HMGB1)やヒートショックプロテイン(HSP)は、マイトジェン活性化プロテインキナーゼ(MAPK)やB細胞における核因子κ光ポリペプチド遺伝子エンハンサー(NF-κB)のシグナル伝達を活性化し、炎症反応を誘発する [52, 53]。HMGB1 は、9 アミノ酸ループと高度に乱れた負に帯電した C 末端尾で連結された 2 つのドメインで形成されたクロマチン関連タンパク質である [54, 55]。HMGB1は、細胞内での局在に応じて異なる機能を果たす。核内タンパク質として、DNA修復、転写、およびゲノムの安定性に関与している[55-57]一方、細胞死や炎症の際には細胞外に放出され、アラーミンとして分類される[58, 59]。DAMPは、腫瘍壊死因子(TNF)、インターロイキン(IL-)1β、IL-12などの炎症性サイトカインの放出を誘導し、急性組織障害を悪化させる。
DAMPsはNETosisとオートファジーの両方の経路に関与している [53]。核内および細胞質環境は、完全に還元された形態(fr-HMGB1)でHMGB1を維持する負の酸化還元電位によって特徴づけられる。この形態は、肺胞上皮細胞上で構成的に高度に発現しているが、他の組織では基本的な条件の間にほとんどまたは全く発現していない受容体である高度な糖化最終生成物(RAGE)のための受容体に結合する[60]。
HMGB1-RAGE相互作用は、壊死後の好中球介在性傷害増幅を誘発する [61]。最近では、RAGEがHMGB1のエンドサイトーシスをエンドリゾソームコンパートメントに仲介することが実証されている[62, 63]。
この経路は、危険な細胞外環境を細胞に警告する重要な経路である。HMGB1は、リソソーム系内部の酸性条件のために、リソソーム膜内で洗浄剤として作用する[63、64]。HMGB1が輸送されたパートナー分子は、予想されたリソソームの分解を回避し、細胞質受容体に到達するために細胞質に漏れ出し、炎症反応を刺激する。HMGB1-RAGEもまた、オートファジーによって制御される重要な経路である。実際、atg7オートファジー遺伝子欠損マウスでは、HMGB1-RAGE軸はパラクリンループでマクロファージ活性化を促進する[65]。
このメカニズムの生物学的含意は、高い構成性細胞表面RAGE発現に起因する重症肺炎の病態形成において基礎的なものである可能性がある。HMGB1放出の増加は、原炎症性刺激下での急性肺損傷(ALI)中に観察されている[66]が、同じモデルではオートファジー刺激がHMGB1放出とALIの減少に有効であった。実際、前臨床研究および臨床研究では、RSVが肺の炎症時に細胞外HMGB1放出を発生させ、HMGB1特異的アンタゴニストがこれらの状態を改善することが実証されている[67、68]。
炎症の間、活性酸素を豊富に含む細胞外空間は、TLR-2およびTLR-4を活性化するジスルフィド結合(ds-)HMGB1の形成につながり、自然免疫および適応免疫の両方を活性化する前炎症性ケモカインおよびサイトカインの放出を誘導する [69, 70]。CXCリガンド12(CXCL12)は、恒常性と炎症性の両方の条件下で多くの組織で発現しており、CXCケモカイン受容体タイプ4(CXCR4)を活性化することで細胞のリクルートを刺激することができる[71]。CXCL12/HMGB1によって形成されるヘテロコンプレックスは、主に単球の遊走を促進し、関節リウマチ[72]などの多くの炎症性プロセスの病態形成において極めて重要な役割を果たしている[73]。CXCL12はHMGB1をfr-HMGB1としてのみ結合する [74]。ヘテロコンプレックスの形成は細胞外空間で起こり、そこでは活性酸素がfr-HMGB1ではなくds-HMGB1につながる。この事象を回避するために、細胞は活性酸素に対してグルタチオン還元酵素およびチオレドキシン系の酵素を放出し、したがって、fr-HMGB1の形態を維持する [72, 74, 75] と、炎症および敗血症におけるその機能を維持する。急性肺疾患では、HMGB1は自然免疫の活性化因子として作用し、IL-1β、IL-6、およびTNF-αの産生につながる。敗血症またはARDSはHMGB1を誘発するため、自然免疫を強力に活性化する。しかし、HMGB1はNET形成も誘発する[76]。前節で説明したように、HMGB1誘導NET症はまた、組織障害を引き起こす可能性がある。最近の研究では、LPS誘発性急性肺損傷におけるHMGB1は、プロテインキナーゼR(PKR)を刺激するTLR-2およびTLR-4の結合を介してマクロファージの炎症性分極を引き起こした[77]。男性では、テストステロン産生の結果、大量のTLR-4が発見されている[78]。テストステロンはDAMP放出を誘導し、その結果、男性では女性に比べてTLR4シグナル伝達が増加する。最終的な結果は、女性よりも男性の方が炎症性サイトカイン(IL-1βやIL-18など)の産生が高くなることである。COVID-19患者では、特に重症例では、IL-6およびIL-8を含む多くのサイトカインの増加が認められている[8]。これらのサイトカインは肺損傷でも増加する。実験的な生体内試験(in vivo)モデルでは、HMGB1の増加がオートファジーによって媒介される可能性があることが実証されている[79]。HMGB1はまた、PI3K/Akt/mTOR経路の活性化において化学吸引剤として重要な役割を果たすサイトカインであるIL8 [80]の産生を刺激する [81]。実際、ラパマイシンを投与したマウスでは、ビヒクルを投与したマウスと比較して肺損傷が減少した。しかし、ラパマイシンはIL-6産生に影響を与えなかったため、この効果は免疫抑制とは関係がないようであった[79]。IL-6はまた、インフルエンザA肺炎に感染した女性と比較して男性でも増加している[82]。
これは、女性よりも男性の方がSARS-CoV-2病の重症度が高いという疫学的データと一致している[3、83]。以前の文献と一致して、COVID-19重症患者におけるIL-6の増加[8]はHMGB1マクロファージ放出に関連しているはずである[80]が、さらなる調査が必要である。HMGB1とTLR-4との関連もまた、この受容体の血小板刺激を介して凝固因子として作用する[84]。SARS-CoV-2病のいくつかの側面はインフルエンザのそれと重複しており、特に自然免疫の過剰な活性化である。野坂らは、抗HMGB1 mAbを投与したH1N1病マウスはウイルスから保護され、対照群よりも長い全生存期間を示した(抗HMGB1群では15匹中1匹が死亡したのに対し、対照群では15匹中8匹が死亡した)[85]。
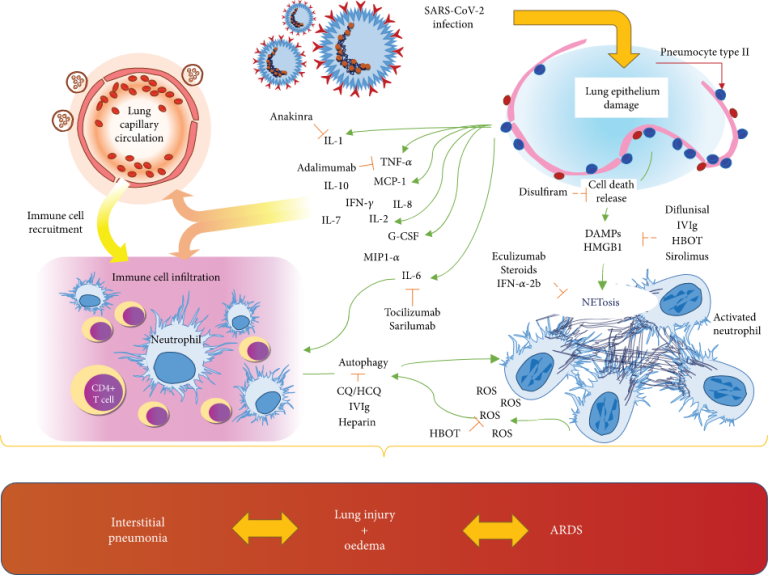
最近、抗HMGB1 mAbとノイラミニダーゼ阻害剤ペラミビル®を併用することで、H1N1病のマウスにおいて優れた治療活性が示された[86]。これは、ウイルスクリアランスの増加によっても説明できる[87]。Entezariら[88]は、嚢胞性線維症のマウスに抗HMGB1 mAbを使用し、肺水腫および肺損傷の減少、および緑膿菌に対するマクロファージの貪食能の向上を示した。インフルエンザウイルスと細菌の組み合わせによる肺感染症に罹患した患者のデータでは、HMGB1およびIL-6の血清濃度の上昇が示されている[89]。これらのデータは、SARS-CoV-2患者における細菌による重感染の間であっても、抗HMGB1治療の重要性を示唆している(図1)。実際、HMGB1は肺損傷の促進および持続に重要な役割を果たしている可能性がある。しかし、SARS-CoV-2の場合、他の呼吸器ウイルスとの違いには注意しなければならない。実際、最近のデータでは、インフルエンザウイルスと比較して、このウイルスが肺血管障害を引き起こす上でより大きな役割を果たしていることが示されている[90]。血管損傷は、組織の低酸素化を促進する重要な因子の一つである。COVID-19肺標本に見られる肺毛細血管血栓症は、デッドスペースを増加させ、ROS産生に伴う虚血性傷害を誘発する。活性酸素はHMGB1活性化においても重要な分子である。実際には、低酸素[53]は、オートファジーとNET活性化を介して肺の損傷につながるHMGB1を活性化する。HMGB1が以前に特定の抗体[91]によってブロックされている場合と同様に、高酸素肺損傷は、生体内試験(in vivo)のマウスモデルで減衰される。換気重度のCOVID-19患者[14]で増加したネットの所見は、結果と危険な自己永続ループでHMGB1リリースのトリガーの両方である可能性がある。低酸素は、細胞外HMBG1の増加の結果として細胞死を誘発する可能性がある。同時に、活性酸素は、ネットとオートファジーを誘導する。DAMP放出と組み合わせた後者のメカニズムは、白血球の誘致と組織浸潤を強化し、堅牢な炎症反応を維持することができる。活性化された免疫細胞は、炎症や感染症の間にユビキタスに放出されるIL-6をはじめとする複数のサイトカインのパラクリン放出を介して、このプロセスに参加している。これらの経路はすべて、宿主組織の損傷をもたらし、最終的には、SARS-CoV-2感染の臨床的重症度が高くなり、肺不全と死を誘発する可能性がある。
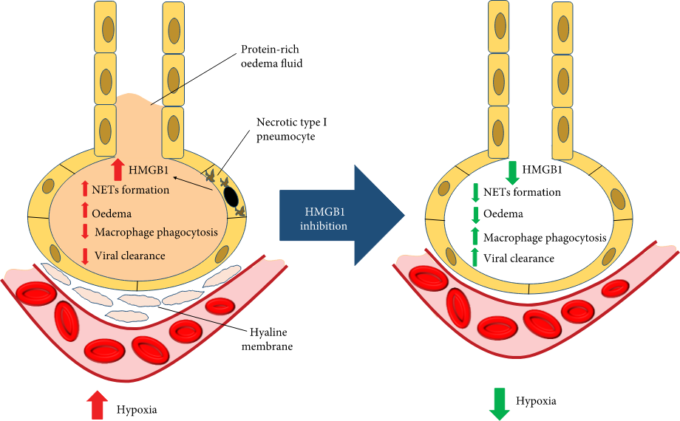
図1
HMGB1の効果。壊死性肺上皮はHMGB1を放出し、その増加は肺水腫、マクロファージ貪食、NETosisを誘導し、ウイルスクリアランスを低下させ、低酸素症を引き起こす。HMGB1遮断薬は、これらの作用を逆転させ、低酸素症の軽減に影響を及ぼす可能性がある。HMGB1:高可動性グループボックス1;NETosis:好中球細胞外トラップの放出。
4. インターロイキン-6、NETs、およびウイルス
IL-6ファミリーは、共通のシグナル伝達受容体サブユニット糖タンパク質130 kDa(gp130)を使用するサイトカインのグループである[92]。IL-6ファミリーは、B細胞刺激や急性期タンパク質の誘導、代謝および神経栄養機能など、多数の細胞機能を担う多数の分子で構成されている [92]。ウイルスは、それ自体は IL-6 の強力な誘導因子ではないようである [93]。実際、IL-6は大型病原体の感染によって大部分が誘導される[94]が、その際には急性期タンパク質産生を強く刺激する。IL-6のアップレギュレーションは、Tヘルパーリンパ球(Th)17分化のためのナイーブT細胞のプライミング、調節性T細胞(Tregs)の免疫抑制機能の阻害、およびTh17細胞のTregsへの変換の防止において重要な因子である[93, 95]。Th17応答は、抗菌ペプチドの発現および好中球のリクルートを介して粘膜バリア機能を高めるIL-17およびIL-22の産生および放出によって特徴づけられる[95、96]。Th17応答は、抗アポトーシス分子のウイルスアップレギュレーション、細胞傷害性Tリンパ球(CTL)による標的細胞の破壊をブロックし、感染細胞の生存率を高めることにより、ウイルスの持続性に寄与する可能性がある[97]。このように、インターフェロン(IFN-)γの放出を介した強力なTh1応答の設定では、Th17応答は、細胞内細菌やウイルスなどの細胞内病原体の殺傷をブロックするのに非常に有効である可能性がある[98]。Th17応答を得るために、TLR刺激下で樹状細胞(DC)によりIL-1β、IL-12、TNF-α、IL-6が産生され[99-101]、Tリンパ球のTh1分極を誘導する[102]。サイトカインDC産生もHMGB1によって調節される。好中球性喘息モデルでは、HMGB1はTh17の分化を調節し、DCによって放出される炎症性サイトカイン産生を刺激する [103]。著者らはまた、特定のHMGB1阻害剤を用いた活性化DCの遮断を介したTh17活性化の低下も実証した。また、インフルエンザ感染においても、HMGB1はIL-6およびIL-8産生を誘導し、TLR-4経路を介してDCを活性化する可能性がある[104]。このインフルエンザ感染の生体内試験(in vivo)モデルでは、ヒトDCのTLR-4刺激に対する特異的な阻害剤の使用は、インフルエンザウイルス関連の致死率、肺における炎症性サイトカイン遺伝子発現、および急性肺損傷(ALI)を減少させる結果となった。この効果は、HMGB1によって媒介されるTLR-4依存性サイトカインストームを減少させることによって誘導される[104]。
文献データは、老化の間にTh1がTh2サイトカイン応答にシフトすることを示している[105]。Th2リンパ球は炎症反応を調節するために必要であり、喘息や自己免疫疾患では増加する。高齢者では、TLR誘導性Th1細胞分化がワクチン接種後の抗原特異的CTL活性化を刺激し、その結果、ワクチンに対する応答が増加し、抗体が殺菌免疫を提供し、感染を防ぐことができない場合の重症疾患に対する臨床的保護の改善に寄与している[98、105、106]。特に、TLR-4が高齢者におけるIL-6産生の重要な経路であることが実証されている。実際、高齢者では、特定のアゴニストを介してTLR-4を刺激した後、DCはIL-6の重要な供給源であり、若年者よりも高いレベルを示している[107]。したがって、高齢者では、抗原刺激の結果として、若年者と比較してIL-6産生が有意に増加している[107]。高齢者におけるCOVID-19の死亡率および重症度の増加は、Th1からTh2への免疫学的シフトの結果であると考えられる。この反応はウイルス感染に対して効果がないことが示されていたため、DCサイトカイン産生はTh17応答を介してそれを維持した。
生体内試験(in vivo)モデルでは、低酸素は呼吸器系および血液中のHMGB1、TLR-2およびTLR-4、ならびにIL-6、IL-8およびIL-10の増加を誘導する[108]。同時に、肺組織および肺胞マクロファージの両方で低酸素によるHMGB1刺激の結果として、IL-6および他の炎症性サイトカインの産生も増加する[53]。これらの刺激は、C反応性タンパク質(CRP)やプロカルシトニン(PCT)などの急性期タンパク質の強い増加には効果的ではない。実際、細菌感染とは異なり、ウイルス感染では、CRPおよびPCTの増加はほとんど見られないが、これはおそらく、IL-6およびTNF-αがTh1分化を刺激した結果、IFN-γ産生が増加したためであろう[94]。SARS-CoV-2患者において低酸素状態の間に見られる有意なIL-6の増加は、[109]これらのデータと一致している。IL-6機能における有害な役割は、ほとんどが誘導された宿主の肺損傷によるものである。免疫浸潤は、ウイルスの切断を効果的に得るのではなく、ウイルスをNETに巻き込むことによってウイルスをブロックしようとする。その結果、感染細胞の浸潤が増加する。SARS-CoV-2誘導性低酸素および組織浸潤は、細胞壊死およびHMGB1放出を誘導し、免疫応答の指数関数的な増強においてDC IL-6産生を促進する。
IL-6は、肥満者、高齢者、および男性において増加することが明らかになっている。肥満患者は基底性炎症状態を呈している[110]。肥満におけるIL-6、IL-8、およびTNF-αのレベルの上昇は、脂肪組織の放出によるものであることが実証されている[111]。炎症性サイトカインもまた、肥満におけるインスリン抵抗性に関連している[111、112]。IL-6は、高齢者では主要な慢性変性疾患の発症に関連した低悪性度の炎症状態を呈するため、若年者に比べて高齢者では増加する[113, 114]。性もまた、サイトカインプロファイルに影響を与える。実際、男性では基底状態ではIL-6の産生が増加し、女性では感染症時にサイトカイン産生が強く増加する[113]。逆に、男性は出血性ショックの低酸素状態の間、持続的に増加したIL-6を示す[115]。ヒトの体の老化は、IL-6の増加の原因と結果である可能性がある。Aniszewskaら[116]は、IL-6欠損マウスが野生型マウスよりも活性が高いことを実証した。別の研究では、非虚弱患者と比較して、IL-6は虚弱患者において増加し、ヘモグロビンとは逆に関連していることが示された[116]。
宿主の特性に加えて、IL-6および他のサイトカイン産生の増加は、ほとんどの場合、ウイルスの病原力に関連している。実際、高病原性インフルエンザウイルスは低病原性ウイルスに比べてIL-6およびTNF-αの産生を増強することが実証されている[22]。実際、ヒトマクロファージおよびDCでのウイルスの活発な複製が記述されており[22]、このプロセスの間、NETおよびHMGB1の両方が役割を果たしている可能性がある。実際、Sabbioneら[117]は、肺細胞のNET刺激の結果としてIL-6およびIL-8の発現が増加していることを発見した。両方のサイトカインの発現は、敗血症抗原およびHMGB1の存在下で増加した。著者らはまた、NETが肺細胞/NET相互作用で喫煙者に何が起こるかを模倣し、煙の抽出物によって刺激された場合、サイトカインの発現が増加したことを発見した[117]。これは、積極的な喫煙がCOVID-19の重症度の増加と相関しているという知見と一致している[118]。この結果は、喫煙者の患者で経験した活性酸素の増加と低酸素の結果であると推測される。
コロナウイルスはCOVID-19におけるこれらの効果のすべてを説明しているかもしれない。実際、IL-6およびTNF-αの発現結果は、生体内試験(in vivo)の糖尿病モデルにおいて、特にMERS-CoV感染に対する初期反応として改変されている[119]。これらの証拠はすべて、IL-6の増加に依存してSARS-CoV-2における重症化のリスクを説明することができ、一方、糖尿病におけるTh2への分極は、サイトカイン産生の障害の結果であるはずである。さらに、プレプリントは、SARS-CoV-2感染の結果におけるIL-6レベルがT細胞の減少と逆に関連していることを示している[120]。COVID-19は、最終的にサイトカインストームを引き起こす可能性がある[121]。ウイルスは、ウイルス切断を得ようとして、HMGB1放出およびNETosisを誘導する。効果のない結果は、DCによる強力なサイトカイン放出に関連した過剰なTh17-Th2応答に起因する免疫系の機能不全を引き起こし、これは重度のCOVID-19病を誘発し得る。IL-6は、このプロセスにおける主要なサイトカインの一つである。男性性、肥満、虚弱性、および高齢のような宿主の特徴は、嵐を増強する補因子である。COVID-19の管理では、サイトカインのストームの予防が必須であるかもしれないが、投薬や換気もサイトカインの放出に影響を与える。このシナリオでは、免疫応答を調節するためにオートファジーの調節が考慮されるかもしれない。
5. オートファジー、免疫、およびウイルス感染
オートファジーは、エネルギー供給が不足しているときに細胞が使用する生存メカニズムであり、自身の代謝性廃棄物を消化することで栄養素を回収する [122]。代謝性廃棄物をカプセル化した後、二重層膜は徐々に崩壊して閉じたオートファゴソームを形成し、リソソソームと融合してオートリソソームを得る [123]。消化された内容物は細胞質に放出され、生合成に利用される。このメカニズムはAtg遺伝子[123]によって誘導され、オートファジーの負のレギュレーターである哺乳類のラパマイシン標的(mTOR)で交差するAMPK、PI3K/Akt、およびMAPK/ERK経路を含む多数の細胞内シグナル伝達経路によって制御される[122, 124]。オートファジーは、免疫応答および細胞死において重要な役割を果たしている。細胞はこのメカニズムを利用して病原体の侵入に抵抗する。オートファジーは、自然免疫応答と適応免疫応答の両方に参加している[125, 126]。しかし、過剰な免疫応答とオートファジーによって媒介される過剰な抗体の産生は、組織の損傷を引き起こし、自己免疫疾患を誘発する可能性もある[127]。
NET形成によって特徴づけられる細胞死は、オートファジーと活性酸素の発生の両方を必要とする[123, 128]。チェンら[129]は、オートファジーと活性酸素がミトゲンミリスチン酸エステル(PMA-)誘導NETosisのために重要であることを証明したが、活性酸素は独立してオートファジーとは無関係にNETosisを誘導することができる。オートファジー駆動NETosisはまた、mTORによって制御される。mTOR経路の薬理学的阻害剤であるラパマイシンを用いて、オートファジーを増加させ、低酸素誘導性因子1α(HIF-1α)活性によるNETosisを促進させることが実証された[130]。このように、HIF-1α活性による低酸素はNETosisの重要な誘導因子である。
また、サイトカインもオートファジーやNETosisを誘導する。Phamら[131]は、IL-8が好中球のオートファジーとNETを強く誘導することを実証し、それらが相互に相関していることを示した。オートファジーはNET形成に積極的に参加するだけでなく、NETの過剰放出を抑制する。緑膿菌に感染して肺炎に罹患したマウスを対象とした研究では、自然免疫の活性化により、オートファジーを亢進させてNETを迅速に排除する抗菌剤を介して病状が改善され、重度の組織損傷を防ぐことが示されている[132]。オートファジーを必要とするNETの生成と放出を促進することは、抗菌薬治療の重要なプロセスであり、NET症におけるオートファジーの二重の役割を証明するものである。
研究は、バフィロマイシンA1(Baf-A1)およびクロロキン(クロロキン)などの最終期のオートファジー阻害剤の使用が、敗血症の設定でオートファジーの減少に続いてNET産生を減少させることを示した[133, 134]。
上記のように、NETは感染に応答してホストに有益であるが、オートファジーがない場合には、NETの過剰生産は、重度の組織損傷を引き起こす可能性がある[27、36、39]。これは、オートファジーによって媒介される損傷において特に当てはまる[123]。
SARS-CoV-2は血栓形成を誘導し、特に肺検体で実証されている [6, 13, 90]。この病理学的所見は、オートファジーの活性化とNET形成によって説明される可能性がある。Manfrediら[135]は、炎症性疾患ではオートファジーが常にNETの生成と放出を伴うことを証明した。彼らは、オートファジーフラックスの阻害剤の使用は、NETの生成を防ぐことを観察した。同様に、彼らは、試験管内試験(in vitro)で低分子ヘパリンで前処理された好中球は、細胞がIL-8またはHMGB1によって刺激された場合でも、有意に減少したオートファジー活性とNETosisの過剰を示すことを発見した。後者の分子は、これらのメカニズムのいずれもHMGB1遺伝子欠損血小板[84]によって誘導されなかったが、血小板を活性化することによってオートファジーおよびNET形成を刺激することが示されている。最後に、健康なボランティアにパルナパリンを予防的に単回投与した場合にも、同様の有意な抗炎症作用が認められた。別の研究では、抗ウイルス性サイトカインIFN-λ1/IL-29は、オートファジーの減少を介して、mTORに対するポリリン酸塩の抑制効果を打ち消すために、無機ポリリン酸塩(ポリリン酸塩)によって誘導されたネットの形成を逆にすることができることを示した[136]。
自己炎症性疾患や自己免疫疾患に関する研究では、オートファジー/NET/IL-1βシグナル伝達軸内のオートファジー媒介NET形成において、ストレスの重要なメディエーターであるDNA損傷応答1(REDD1)タンパク質の発生とDNA損傷応答1(REDD1)の調節役割が示唆されている。これらの研究は、オートファジーによって媒介されたNETの過剰な放出はまた、その後の線維化[137、138]を刺激する可能性があり、プロ炎症性の効果を持っていたことを示している。したがって、オートファジーとNETは、ヒトの線維化性疾患の潜在的な治療標的である可能性があるが、それらは、免疫寛容性を破壊し、同様に自己免疫疾患を誘導する上で重要な役割を果たしている[139]。このことが、 ヒドロキシクロロキンなどの薬物の抗炎症作用を説明する可能性がある。しかし、 ヒドロキシクロロキンの副作用はオートファジー阻害の結果である可能性もある。オートファジーは細胞の恒常性維持に必要であるため、心臓QT伸長[140、141]および貧血[142]は、オートファジー阻害に関連する一般的なクロロキン/ ヒドロキシクロロキンの副作用であり、注意して使用する必要がある。さらに、クロロキンはゴルジ体の無秩序化[143]や肺血管拡張[144]のようなオートファジーに依存しない作用を誘導し、その臨床活性には賛否両論がある。これらの作用を回避し、サイトカイン活性化を減少させるためにオートファジーの制限を得るために、IL-6R mAbは様々な設定で有効であることが判明している[145, 146]。
オートファジーにおける機能障害および調節障害は、多くの疾患において、肥満[147-149]、年齢および男性性[150]と関連している。特に、肥満の人は、炎症の増加とオートファジーの最終段階の阻害との組み合わせにより、オートファゴソームの数が増加していることを示す[147、148]。高齢者では、オートファジーの欠乏は、酸化ストレスがこのメカニズムの最も強力な活性化因子の1つであるにもかかわらず、活性酸素で損傷したタンパク質の分解の減少をもたらす[151、152]。
しかしながら、オートファジーはまた、ARDSにおける過剰なサイトカインの放出を減少させる保護的役割を有しており、重度の炎症反応の調節における最終段階を表している。実際、重度の肺損傷の生体内試験(in vivo)モデルでは、オートファジー誘導剤であるラパマイシンは、クロロキンのような阻害剤ではなく、肺水腫の減少と酸素化の増加に効果的であることが実証されている[153]。また、クロロキン/ ヒドロキシクロロキンとは逆の作用を持つmTOR阻害剤もCOVID-19の抗ウイルス薬として提案されているが[154]、治療した患者のデータは限られており、臨床試験の結果は得られていない。
SAR-CoV-2感染におけるオートファジーの役割については、実験的に確立する必要があり、データはまだ得られていない。SARS-CoV-2感染における仮説として、これまでのコロナウイルスのデータに基づくオートファジー調節障害が提案されている[155]が、SARS-CoV-2感染におけるクロロキン/ ヒドロキシクロロキンの使用に関する決定的なデータも、他のオートファジー調節因子に関するデータもまだ得られていない。
6. SARS-CoV-2:炎症および肺疾患
炎症性活性化は SARS-CoV-2 感染の自然経過において極めて重要な役割を果たしている.免疫系の調節障害はSARS-CoV-2感染の重症度と関連しており、重症症例では多くのサイトカインの増加を示す。IL-1、IL-2、IL-6、IL-7、IL-8、IL-10、顆粒球コロニー刺激因子(GSF)、IFN-γ、誘導性タンパク質10、単球化学吸引性タンパク質1、マクロファージ炎症性タンパク質1-α、およびTNF-αである[8、51]。中国のデータは、高レベルのフェリチンおよび低リンパ球数が致死の予測因子であることを示している [3, 8]。
肺生検では、免疫細胞の浸潤(主にマクロファージとCD4陽性Tリンパ球)によって引き起こされる肺胞損傷が明らかになり、ヒアリン膜とII型肺胞上皮の増殖が並行して見られる。局所肺線維症は、肺上皮およびマクロファージにおけるSARS-CoV-2抗原と関連している[6]。血管のうっ血とともに、微小血管ではヒアライン血栓がしばしば観察される [6]。SARS-CoV-2は、コロナウイルスによる肺の致死的感染による最初のパンデミックである。以前のコロナウイルス(SARS-CoVおよびMERS-CoV)は同様に致死的であったが、SARS-CoV-2のように世界的にパンデミックすることはなかった。以前のSARS患者では、上皮成長因子受容体(EGFR)シグナル伝達の過剰活性化が急性肺損傷に応答して示されている[156]。SARS-CoVに感染したマウスでは、シグナル伝達物質および転写活性化因子1(STAT1)が不十分なためにIFN産生が不足した結果として、Th2が調節不能になる。これらのマウスは、EGFRの増加に続いてIL-8の増加および好中球のリクルートが起こるために線維化しやすい[156]。別の試験管内試験(in vitro)研究[157]では、SARS-CoV感染はTLR9を介して肺交通のケモカインであるIL8およびIL17を活性化し、これが症状の原因となり、単球-マクロファージの活性化および凝固アップレギュレーションを引き起こすことが確認されている。MERS-CoVに関する研究では、Aboagyeら[158]は、ウイルス核カプシドが抗ウイルス遺伝子、すなわちTNF、IL-6、IL8、CXCL10の過剰発現を引き起こし、その発現は経時的に安定していることを示した。SARS-CoV-2損傷の発症機序はまだ不明であるが、他のSARSとは異なり、SARS-CoV-2は炎症を抑制し、Th1過剰反応のバランスをとるTh2サイトカインIL-4およびIL-10の初期増加を示す[51]。
SARS-CoV-2の発症と肺障害では、免疫応答が主役のようである。しかし、免疫反応は両刃の作用をもたらす:免疫反応は最初にウイルス感染を制限しようとするが、継続的なSARS-CoV-2の刺激はそれをサイトカインストームに変えてしまう[159]。このコーナーポイントは、症状の少ない疾患をびまん性の重症間質性肺炎に変える[8, 51](図2)。サイトカインは免疫細胞の化学吸引に役割を果たす。IL-8はこのメカニズムにおいて中心的な役割を果たしており、免疫応答を調節することで、DCsの遊走における方向転換を促進し、NETosisを誘発する[160, 161]。ウイルス性呼吸器感染症の間、気道分泌物中にIL-8の増加レベルが存在する。これは好中球数、好中球エラスターゼレベル、および臨床重症度スコアと正の相関がある [162, 163]。IL-8の増加は、呼吸器上皮および内皮のHMGB1刺激に由来する効果の1つである[164、165]。オートファジーはNF-κB活性化を介してIL-8放出を増強する[166]。同時に、IL8は、自己調節機構において、PI3K/Akt/mTOR経路[81]の活性化において重要な役割を果たす。
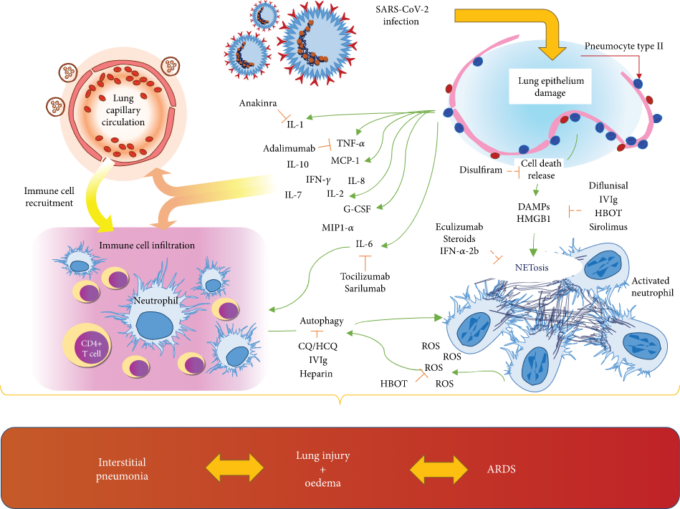
図2
SARS-CoV-2によって媒介される肺上皮の感染は、複数のサイトカインおよびDAMP(HMGB1)の細胞放出をもたらす。これらの分子は、常駐細胞への直接的な作用として、また、血液循環におけるサイトカイン放出の結果として、免疫細胞のリクルートを誘導する。活性化された好中球は、活性酸素の放出を伴うオートファジーおよびNETosisの増加を示す。その結果、間質性肺炎から急性呼吸窮迫症候群(ARDS)までの間質性肺炎を引き起こす肺の損傷や水腫につながるメカニズムである。DAMPs:damage-associated molecular patterns、G-脳脊髄液:granulocyte colony stimulating factor、HMGB1:high-mobility group box 1、IL:interleukin、IFN-γ:interferon-γ、MCP1:monocyte chemoattractant protein 1、MIP1-α:macrophage inflammatory protein 1-α、NETosis:release of neutrophil extracellular traps、ROS:好中球細胞外トラップの放出。活性酸素種;TNF-α:腫瘍壊死因子-α。
免疫宿主の損傷は様々なパターンで現れる。肺および毛細血管損傷は、COVID-19の主な焦点である[6, 12, 13, 90]。肺塞栓症はしばしばSARS-CoV-2を合併することがある[167]。小肺毛細血管内の微小トロンビは、非閉塞性肺セグメントの過灌流を介して小血管血管障害につながる。最初の報告では、最初の併存疾患の一つとして心血管疾患が示されている[83]。また、最近のデータでは、急性心障害を呈する患者に急性凝固障害とリウマチ因子が認められ、死亡リスクが上昇することも示されている[168]。
内皮炎は、SARS-CoV-2感染の重要な因子であるように思われる。免疫細胞の浸潤および毛細血管壁の結果としての損傷は、毛細血管漏出、肺水腫の増加、凝固カスケードの活性化、および微小血栓形成につながる。Vargaら[13]は、内皮細胞へのウイルスの直接感染およびびまん性内皮炎症の証拠を発見した。ウイルスは、宿主に感染するために肺胞上皮細胞で発現するACE2受容体を用いるが、ACE2受容体は内皮細胞にも広く発現している。内皮への直接のウイルス感染または免疫媒介による免疫細胞応答は、アポトーシスを伴う広範な内皮機能障害を引き起こし、多数のサイトカインおよびDAMPを放出する。凝固カスケードの結果としての活性化は、微小血栓形成につながると考えられる。この現象は、密接にNETに関連している可能性がある。ネットと血小板の相互作用は、HMGB1-TLR4軸を介した血小板の活性化と同様に、凝固カスケードの活性化に影響を与え、びまん性小血栓形成につながる可能性がある。
COVID-19で回復した患者からの最近のデータは、体液性免疫に関連した細胞性免疫の存在を示した。実際、Niら[169]は、回復した患者においてSタンパク質結合ドメインに特異的な抗体の産生を発見し、また、これらの回復した患者におけるウイルス無力化活性も記載している。この観察は、急性治療だけでなくワクチンの観点からも、これらの患者の将来の治療の礎となるであろう。SARS-CoV-2感染中に発生する宿主損傷に対する免疫の影響は、COVID-19の病態および進化のマイルストーンである。免疫応答の調節は、COVID-19患者を治療するための基本的な戦略の一つであるように思われる。
7. SARS-CoV-2感染における炎症制御の治療的課題
議論されたすべてのデータは、SARS-CoV-2におけるサイトカインストーム症候群の上昇の可能性と一致している。実際、これらの患者で記載されたIL-6の増加[8]は、重症患者の有望な治療のための新たな展望を開くかもしれない。しかし、多くの免疫調節治療法がすでに臨床試験で提案され、評価されている(表1および図2)。
薬物またはカクテル 状況とメカニズム NETs DAMPs オートファジー 臨床試験(試験掲載日
Adalimumab 抗TNF-αヒト化mAb 自己免疫疾患や免疫介在性疾患における炎症反応の一部であるTNF-αに対する生理的反応を抑制することが可能 データなし データなし 自己免疫疾患において、オートファジーを阻害し、調節機能を強化する。
アナキンラ IL-1受容体拮抗薬はサイトカインのストームを鎮めると仮説されている SARS-CoV-2の併用療法としてのデータはない NET形成を阻害する役割が考えられる 抗IL1RはDAMP放出を阻害する役割が考えられる 抗IL1Rは調節機能を回復する役割が考えられる 抗IL1Rは単剤または併用で12の臨床試験が欧州で実施されている
ベバシズマブ 血管内皮増殖因子(VEGF)と内皮受容体Flt-1およびKDRとの結合を阻害する組換えヒト化mAb 米国で複数の癌の承認を取得 データなし 活性なし オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を阻害 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を促進 オートファジーがベバシズマブに対する抵抗性を阻害
クロロキンまたはヒドロキシクロロキン エンドソーム酸性化融合阻害剤 オートファジー遮断の結果としてのNETosisの阻害 敗血症の生体内試験(in vivo)モデルにおけるHMGB1活性の阻害 オートファゴソームの融合と分解を阻害する
SARS-CoV-2感染から回復した回復期患者の血漿は、SARS-CoV-1、MERS、エボラ、H1N1インフルエンザと同様に使用され、成功を収めている。
サリチル酸塩に含まれる非ステロイド性抗炎症薬(NSAID)であり、関節疾患における逍遥症の予防及び疼痛抑制を 特有の適応症としている。 データなし 免疫細胞のリクルートを阻害し、HMGB1/CXCL12の活性化を阻害する。
ジスルフィラム チウラム誘導体 アルコールの酸化を阻害するチウラム誘導体であり、SARSのパパイン様プロテアーゼを競合的に阻害する能力を示すが、臨床データはない。データなし アポトーシス細胞におけるDAMPsの放出を減少させる ジスルフィラムの二重の役割 癌におけるジスルフィラムのメカニズムが研究されている 該当なし
Eculizumab 補体タンパク質C5に結合し、膜攻撃複合体(MAC)の形成を阻害するヒト化IgG mAb 補体活性化を介したNET形成を阻害 データなし データなし SARS-CoV-2を対象とした臨床試験(NCT04288713)において、免疫反応を抑制することが評価されている。単剤で新規臨床試験(NCT04346797)を募集中
ヘパリン/低分子ヘパリン ヘパリンは酵素阻害剤アンチトロンビンIII (AT)に結合し、構造変化を引き起こし、トロンビン、第Xa因子、および他のプロテアーゼの活性化とその結果としての不活性化をもたらす。免疫調節における役割 オートファジーの減少によるNETosisの減少 免疫細胞表面へのHMGB1結合の遮断による活性阻害 好中球活性化オートファジーの減少 SARS-CoV-2の合併症予防における有効性を評価するために、少なくとも40の臨床試験が計画されている。
高気圧酸素 高気圧酸素療法とは、大気圧よりも高い周囲圧力で酸素を使用する医療用の治療法である。炎症を抑制し、サイトカインの放出を調節し、活性酸素の産生を増加させ、アポトーシスを減少させ、白血球の活性化と接着を調節する。 活性酸素依存性ネット放出の減少 HMGB1経路の活性化の減少 オートファジー活性化の抑制 活性酸素の減少 ARDSや肺炎に対する有効性を評価するための臨床試験が進行中。
インターフェロンα-2b単独または他の薬剤との併用 インターフェロンα-2bは抗ウイルス作用を有する組換えサイトカインである;リバビリンはグアニン誘導体である;上記のようにタイプI IFNはNETosisを強化する HMGB1は炎症を維持する上でIFNαと関連している 抗ウイルスオートファジーを強化する役割を果たす可能性がある SARS-CoV-2の単剤または併用として登録された60の臨床試験
静脈内免疫グロブリン(IVIg) IVIgは、米国では深刻な国家的不足のままである。SARS-CoV-2 肺炎患者における有効性は明らかではない IVIg はネット形成を抑制する HMGB1 誘発細胞死からの保護 TLR および RAGE 発現を調節する IVIg は調節機能を調節する役割を持つ可能性がある SARS-CoV-2 肺炎における有効性を評価するために、世界中で 250 以上の臨床試験が計画されている
メチルプレドニゾロン 核内受容体に結合して炎症性サイトカインを抑制する 合成コルチコステロイド 試験管内試験(in vitro)および生体内試験(in vivo)モデルでのNET形成の抑制 HMGB1の放出およびTLR4との相互作用の抑制 オートファジー制御のモジュレーション COVID-19病で25件の臨床試験が米国および中国で単剤または標準治療薬またはmAbとの併用で実施されている
レムデシビル アデノシン類似体 ウイルスRNA鎖の早期または遅延終結をもたらす データなし データなし データなし COVID-19の治療薬として初めて承認された
サリルマブ IL-6 受容体拮抗剤 関節リウマチを適応症としてFDAに承認され、NET形成を阻害する役割が期待される データなし オートファジーの調節機能を阻害する役割 SARS-CoV-2病を対象とした15の臨床試験が単剤または併用で実施中
シロリムス ラパマイシンの市販薬形態で、mTORとそれに続く経路を遮断し、サイトカイン(IL-6、TNF-αなど)の発現を低下させる 外部刺激を受けずにNETosisを増強 HMGB1の発現を低下させる mTOR遮断の直接的な効果としてオートファジーを刺激 SARS-CoV-2病の患者を対象に、単剤で4つの臨床試験が実施された
Tocilizumab IL-6を標的としたヒト化mAbで、NET形成を阻害する役割を期待 データなし 低酸素誘導オートファジーを阻害する役割 各国で最大60件の臨床試験が実施されている。
最終検索日は 6 月 06 日で、https://clinicaltrials.gov と www.chictr.org.cn を使用している。この表には、SARS-CoV-2感染者の管理のために研究および/または理論的に検討されている薬剤が記載されている。現時点では、これらの薬剤を推奨することはできない。一般的に、これらの薬剤は、追加のエビデンスがない限り、使用を避けるべきである。
表1
SARS-CoV-2の治療のために臨床開発中の免疫調節薬。この表には、患者管理のために研究中および/または理論的に検討中の薬剤を列挙している。これらの薬剤の中には適応外のものもあるが、現時点では推奨することはできない。
現在のところ、SARS-CoV-2感染による肺不全では、酸素と人工呼吸が必要な治療法である。十分な組織酸素化を確保するためのこれらの必須治療にもかかわらず、文献データは、増加するNETosisの役割の可能性を示している[36, 170]。
実際、過去のコロナウイルス感染症や試験管内試験(in vitro)のデータを考慮すると、アダリムマブ、アナキンラ、ディフルニサル、ジスルフィラム、バリシチニブ、シロリムス、ヘパリン、静脈内免疫グロブリン(IVIg)(表1)、ステロイド、INFなどの免疫調節療法が提案されているが、これらは臨床試験で有効性が評価されている[171-174]。
7.1. レムデシビル
レムデシビル(RDV)は、SARS-CoV-2感染症に使用される抗ウイルス薬の一つである。RDVは、ウイルスRNA鎖の早期または遅延終結を誘導するアデノシン類似体である。エボラ感染症を治療するために最初に開発された[175]が、コロナウイルスを含む広範なRNAウイルスに対する有望な抗ウイルス薬と考えられていた。この薬剤は、ウイルスが標的細胞に侵入した後、重要な酵素であるRNA依存性RNAポリメラーゼ(RdRP)を阻害することで作用する[176]。RNA鎖の特定の位置に取り込まれると、RDVは、薬剤が取り込まれた部位までの5ヌクレオチド下でRNA合成の阻害を引き起こし、鎖の終結を遅らせる[176]。これまでのコロナウイルス感染データに基づき、RDVは、臨界炎症反応を有するMERS-CoV感染マウスにおいて、疾患転帰を改善し、ウイルス負荷を減衰させる。RDVはまた、げっ歯類動物において、好中球浸潤を減少させることにより、急性肺損傷(ALI)に対する保護効果を示し、これはIFNsの媒介と関連していた[177]。非ヒト霊長類を対象とした研究では、MERS-CoVのような他のコロナウイルス感染症における本剤の予防的および治療的価値が示されている[178]。別の最近の試験管内試験(in vitro)研究では、SARS-CoV-2感染におけるRDVの役割が報告されている。研究者らは、試験したいくつかの薬剤の中で、クロロキンに関連するRDVのみが、SARS-CoV-2に感受性のあるヒト細胞におけるウイルス感染を抑制できると結論づけた[179]。最近、米国食品医薬品局は、2020年5月1日にレムデシビルをCOVID-19患者の治療薬として承認した。これは現在、SARS-CoV-2感染症の治療薬として承認されている唯一の薬剤である。RDVはまだ調査中であるが、予備的なデータでは、本剤で治療されたCOVID-19患者の生存率が上昇していることが示されている。主なS3およびS4の副作用は貧血または急性腎障害であった。COVID-19患者におけるSARS-CoV-2の拡散を抑制するレムデシビルの有効性を証明するデータがあるにもかかわらず、免疫反応をコントロールできるという証拠は見つかっていない。SARS-CoV-2負荷の低下がNETやDAMPの形成に影響を与えているのではないかと推測されるが、まだデータはない。実際、本レビューで指摘しているように、NET症とDAMPsは2つの有望な治療ターゲットであるように思われる。このように考えると、COVID-19患者における炎症のより良いコントロールを得るためには、免疫系に作用する薬剤を追加する必要があるはずである。
7.2. クロロキン/ヒドロキシクロロキン
SARS-CoV-2で最も広く使用されている治療法の一つは、抗マラリア薬であるクロロキン(クロロキン)/ヒドロキシクロロキン( ヒドロキシクロロキン)である。この薬剤には複数の機能がある。最もよく知られているのは、ファゴリゾソームをアルカリ化する能力であり、これはその抗ウイルス機序の根底にあるメカニズムの一つである[180]。敗血症では、細胞死の減少におけるクロロキンの役割が実証されており、HMGB1の放出を防止している。特に、SARS感染では免疫調節作用が提案されている[180, 181]。IL-2はSARS-CoV-2で増加し、T細胞のTh2分化のための「プライミング」に重要な役割を果たしている[8, 182]。クロロキンは、IL-2の産生および応答性を低下させることにより、T細胞の増殖を阻害する[183]。Th2応答は、SARS-CoV-2感染における炎症の抑制に役割を果たしている可能性があり、クロロキン/ ヒドロキシクロロキンはウイルスに対する免疫応答に影響を与えている可能性がある。それにもかかわらず、その抗炎症特性を考慮すると、クロロキン/ ヒドロキシクロロキンはSARSに対して何らかの効果を有する可能性がある[184]が、特にプロ炎症性サイトカイン(TNF-α、IL-6)の産生を阻害することにより、それゆえARDSにつながる経路のカスケードをブロックする[184]。初期の報告では、クロロキン/ ヒドロキシクロロキン使用後の臨床的有用性が示されている。しかし、クロロキン/ ヒドロキシクロロキンは敗血症において二重の役割を持つことがわかっている。COVID-19に対するこれらの薬剤の明確な有用性を示す質の高い臨床データや臨床試験の結果が報告されていないため、注意が示唆されている[185]。COVID-19は、生体内試験(in vivo)の敗血症モデルではオートファジーを介して致死的な症例を予防するが[187]、重症症例では同様の効果が疾患の悪化をもたらす[153]。この二重の役割は、長時間の低酸素に関係しているはずである。実際、文献データによると、低酸素の初期段階では、クロロキンはオートファジー、HMGB1、IL-6、臓器損傷の減少を誘導し、一方、低酸素状態が長期化すると、さらなる損傷を促進し、アポトーシスとオートファジーを介した細胞損傷を増強することが示されている[188]。しかしながら、SARS-CoV-2感染に対する体液性反応におけるT細胞免疫の役割を考慮すると[169]、クロロキン/ ヒドロキシクロロキンは保護免疫グロブリンの形成に有害な役割を果たす可能性がある。
このレビューの時点では、標準化された用量レジメンはクロロキン 500 mg b.i.d.または ヒドロキシクロロキン 200 mg b.i.d.である[189]が、SARS-CoV-2に対するクロロキンおよび ヒドロキシクロロキンベースのレジメンに関するいくつかの臨床試験がまだ進行中であり(表1)[173]、そのような使用はまだ適応外である。
Yaoら[190]は、SARS-CoV-2の阻害における ヒドロキシクロロキンの試験管内試験(in vitro)活性を報告している。オープンラベルデータは、クロロキン/ ヒドロキシクロロキンで治療された患者における臨床的有益性を示している [191]。上記の証拠、無視できるほどのコスト、および世界中での広範な使用により、クロロキン/ ヒドロキシクロロキンは、SARS-CoV-2の影響を受ける非重症患者において有用な薬剤となる可能性があると考えられている[190-193]。クロロキンは、亜鉛イオノフォアとして作用し、濃度依存的に亜鉛細胞の取り込みを促進することが実証されている[194]。亜鉛はRNA依存性RNAポリメラーゼを阻害し、以前のSARS-CoVに対して試験管内試験(in vitro)でこれを行うことが示されている[195]。Carlucciら[196]は最近のプレプリントで、標準的な ヒドロキシクロロキン投与に加えて亜鉛治療を受けた場合、患者の死亡率、疾患重症度、ICUまたは人工呼吸の必要性が減少したことを実証した。軽度のCOVID-19を対象とした二重盲検ランダム化臨床試験の予備データでは、 ヒドロキシクロロキン投与は軽度から重度への移行を予防するのに有効であるようである [197]。
しかしながら、対照的なデータが発表されている。最近、Rosenbergらは、 ヒドロキシクロロキン投与を受けた入院COVID-19患者の死亡率がアジスロマイシン投与と比較して増加したことを報告していた[198]。さらに、 ヒドロキシクロロキンおよびアジスロマイシンを服用した患者では、他の治療と比較して死亡率が高かった[198]。これらの患者の主な死因は心停止であった[198]が、 ヒドロキシクロロキンの一般的な副作用である心電図で示されるQT間隔[195]の延長によるものであろう。最近のデータは、国際的な文献[199]で議論されたり、撤回されたりしており、エビデンスに基づいたランダム化データが必要であることを示唆している。臨床試験の結果から、クロロキン/ ヒドロキシクロロキン使用の決定的な適応が明らかになるはずである。
7.3. トシリズマブおよび他のIL-6R阻害剤
トシリズマブと呼ばれる最初のIL-6受容体(IL-6R-)中和mAbは、自己免疫疾患の治療のために100カ国以上で承認されている[200]。IL-6カスケードを遮断することは、炎症反応を調節する根拠となりうる。トシリズマブは、SARS-CoV-2の治療において実験的に静脈内投与され、心強い結果が得られた[201]。
予備データはトシリズマブの有効性を示している。重症患者に使用された最初の中国での報告では、体温とCRPの低下とそれに伴うSaO2%とリンパ球率の上昇が報告されており、臨床転帰が改善されたとされている[202]。また、イタリアの入院患者を対象とした適応外使用例では、トシリズマブが発熱や炎症マーカーに対して迅速に有益な効果を発揮し、体温やCRP値の劇的な低下、リンパ球数の有意な増加、臨床的に重篤な疾患での有益性が示されている[203, 204]。
しかし、高血糖はIL-6R阻害剤に影響を及ぼす可能性がある。実際、高血糖血症患者と正常血糖血症患者の最近のデータでは、高血糖血症時にIL-6レベルが5倍高いことが明らかになっている[205]。さらに、高血糖症患者では、より高いIL-6血漿レベルがTCZの効果を減少させた[205]ことから、糖尿病患者または非糖尿病患者のいずれにおいても、高血糖症時にはTCZによる最適なCOVID-19感染管理が達成されないことが示唆された。
中国人及びイタリア人集団におけるこれらの最近の適応外データに基づき、他のIL-6R阻害剤と同様にトシリズマブの世界的な多くの試験が開始されている(表1)。
7.4. シロリムス
シロリムスはラパマイシンの市販薬である。mTORとそれに続く経路をブロックし、サイトカイン(IL-6やTNF-αなど)の発現を低下させる。外部刺激がなくてもNETosisを増強し、mTOR遮断の直接的な効果としてHMGB1の発現を低下させ、オートファジーを刺激する。mTOR経路は、細胞の代謝、増殖、増殖、生存の中心的な調節因子として機能している。mTOR はタンパク質合成、脂質代謝、オートファジー、転写を制御するために細胞内および細胞外のシグナルを感知している[206]。mTORC1は主に栄養/エネルギー/酸化還元センサーとして機能し、タンパク質合成、脂質代謝、オルガネラの生合成を制御しており、ラパマイシン感受性である[206]。一方、ラパマイシン感受性の mTORC2 はアクチン細胞骨格、代謝、細胞生存の調節因子として機能している[206]。さらに、mTOR経路は生殖中枢におけるB細胞の発生にも重要な役割を果たしている[208]。これらの経路のために、病原体はDCおよびマクロファージ内でこの経路を標的とする戦略を進化させ、免疫逃避を促進してきた[40]。mTORC1の阻害は樹状細胞(DC)のT細胞刺激活性を高め、マクロファージのオートファジーを促進する。このように、SARS-CoV-2感染の初期段階にあるハイリスク患者では、免疫増強過程を回避し、重篤な症状を緩和することができると推測される。mTOR阻害剤ラパマイシンはまた、マカクにおいてワクチン接種に対するウイルス特異的CD8+ T細胞応答の大きさと質を高める[209]。これらの研究により、mTOR阻害剤の新たな機構的特徴が解明され、免疫抑制剤としての役割を超えた免疫学的応用が示唆されている。一方、試験管内試験(in vitro)での研究では、mTOR阻害剤ラパマイシンがMERS-CoVの複製を阻害することが示されている[210]。mTOR阻害薬とコルチコステロイドによる補助療法は、H1N1インフルエンザウイルス感染のICU患者の転帰を有意に改善する可能性がある[210]。現在、COVID-19患者を治療するためにmTOR阻害剤シロリムスを使用した2つの臨床試験が進行中である。
7.5. ステロイド
コルチコステロイドは炎症や自己免疫疾患の治療薬としてよく知られている。ステロイドは核内受容体に結合し、炎症性サイトカインの放出を減少させる。ステロイドは試験管内試験(in vitro)および生体内試験(in vivo)モデルでNET形成の減少を誘導する[211]。ステロイドはまた、HMGB1の放出とTLR4との相互作用を減少させ[212]、オートファジーを調節し、アポトーシスを抑制し、調節性オートファゴソームを増強する[212, 213]。
しかし、SARS-CoV-2感染症では、ステロイド治療は文献で明確な結果を示しておらず、適応症の記載もない。
異なる病因のARDSを呈する患者におけるコルチコステロイドの使用については、まだ議論の余地がある。動物実験では、炎症を減少させ、ALIを減少させ、生存率を改善するために、重症疾患の急性期にグルココルチコイドを使用することの証拠が示されている[214]。文献上の結果は、主に観察研究から得られたものであるが、結論が出ず、時には矛盾することもあった。世界的には、高用量のグルココルチコイドはARDSで最も頻繁に使用される薬剤の一つである[215]。全身性コルチコステロイドは、炎症性メディエーターの循環レベルを低下させる役割があることから、ARDSを呈する重篤な患者に長い間使用されてきた[216, 217]。さらに、グルココルチコイドの適切で長期的な補充は、内因性のコルチコステロイド不足を緩和することが証明されており、その結果、肺および全身の炎症の解決を促進することが証明されている[218]。ある系統的レビューでは、プラセボと比較して、グルココルチコイドの長期投与が臨床転帰を改善したことが明らかになった[214]。最近のメタアナリシスでは、ARDSに対するメチルプレドニゾロンの長期投与を評価した4つのRCTを組み合わせて、無呼吸日数の増加(13日対7日)を伴う死亡率の有意な減少が報告されている[219]。しかし、他の研究では、ARDSの死亡率を低下させるためのコルチコステロイドの有効性についての説得力のある証拠は得られておらず、グルココルチコイド療法はこの疾患では必要なく、疾患の臨床経過を悪化させる可能性さえあることが示唆されている。
以前のコロナウイルス感染症であるSARSやMERSについても、ステロイドの使用はまだ議論されている。
コルチコステロイド療法は重症SARSの治療に使用されており、初期の逸話的経験に裏付けられている[220, 221]。2003年3月、中国は、SARSに感染した患者が3日以上発熱が持続している場合、または放射線学的所見が肺病変の持続または進行性の悪化を示唆する場合には、高用量のグルココルチコイドを使用すべきであると提案した[222]。
SARS-CoV感染に関するある系統的レビューでは、標準治療に加えてグルココルチコイドを使用することの役割について、25の研究で結論が出ておらず、4つの研究ではSARS患者における全身性グルココルチコイドの使用が害をもたらす可能性があることが示されている[223]。
重症のMERS患者にもグルココルチコイド療法が用いられた。MERS-CoV肺炎の低酸素血症患者にステロイドが投与されたが、改善の兆候が見られなかった[224]。この研究では、90日死亡率に差はなく、これらの患者ではMERS-CoV RNAクリアランスの遅延が関連していたと報告されている。
最近のエビデンスは、重度のCOVID-19患者のサブセットが、肺病変(ARDSを含む)[121]および多臓器不全に頻繁に関連する状態であるサイトカインストーム症候群[51]を有している可能性を示唆している。免疫抑制を誘導してウイルス主導の高炎症に拮抗させるために、高サイトカイン血症の臨床検査パターンが確認された患者では、トシリズマブによる治療が進行中である。これらの患者では、コルチコステロイドの治療的役割も仮説として立てられる [225]。
COVID-19におけるコルチコステロイドの使用に関するデータはまだ決定的なものではない。中国人患者542人を含むシステマティックレビューでは、著者らはSARS-CoV-2感染症における非ステロイドの使用について決定的な証拠を見いだせなかった[226]。特に、2つの研究では、これらの薬剤に関して否定的な知見が報告されている[227、228]。1つの研究では、コルチコステロイドと臨床転帰との間に有意な関連はないと報告されているが[229]、もう1つの研究では、ARDSを発症したCOVID-19肺炎患者の死亡率を減少させると結論づけている[230]。また、最近のデータでは、早期の短期コースのメチルプレドニゾロンは、死亡率と呼吸不全、ARDS、ICU入院への進行を減少させるのに有効であることが示されている[231]。武漢で行われた SARS-CoV-2 肺炎を対象とした別の研究では、メチルプレドニゾロンの投与により末梢酸素が早期に回復し、ICU 入院期間が短縮されたことが示されている[232]。これらのデータから、メチルプレドニゾロンの早期短期投与は重症COVID-19肺炎患者のより良い臨床転帰と関連しており、ARDSの発生前に検討すべきであると主張することができる。
7.6. ヘパリン
ヘパリンは分岐グリコサミノグリカンの不均一な混合物で、未分画ヘパリン(UFH)として定義されている。酵素阻害剤アンチトロンビンIII(AT)と結合すると、ヘパリンは構造変化、活性化、その結果としてトロンビン、第Xa因子、および凝固カスケードに関与する他のプロテアーゼの不活性化を引き起こする[233]。同じ効果は低分子量分子ヘパリン(LMWHs)によって媒介される。これらは多硫化グリコサミノグリカンであり、UFHの約3分の1の分子量である[233]。両方とも免疫調節に役割を果たす可能性がある。LMWHは、好中球活性化オートファジーの減少に起因するNETosisを減少させることがわかっている[135]。好中球活性化の調節はまた、免疫細胞表面へのHMGB1結合の阻害の結果であり、好中球をDAMP刺激に対して難治性にする[84、135]。
ヘパリンは、SARS-CoV-2感染症において最も有用な治療法の一つであるように思われる。以前のSARS-CoVのデータを分析すると、凝固因子Xaが宿主細胞に侵入するために使用されるコロナウイルスプロテアーゼの一つであることが判明している[234]。さらに、凝固カスケードの活性化はCOVID-19のメインフレームの一つであり[90]、おそらくNETosisとDAMPの放出をもたらす。このような観点から、ヘパリンはSARS-CoV-2感染者において有用であると考えられる。凝固活性化を確認するために、マーカーとしてDダイマーが提案された。COVID-19患者では、抗凝固薬が禁忌とされている患者を除き、高Dダイマーが検出された場合[235]や固定入院患者[236]には抗凝固療法が推奨されている。しかし、D-ダイマーは特異的なマーカーであり、その増加は炎症そのものにも関連している可能性があることを考慮しなければならない。
Tangら[237]は、ヘパリンで治療されたCOVID-19患者において、血栓活性化マーカーの大幅な改善と28日死亡率の低下を報告している。LMWHの推奨治療用量は、少なくとも5日間、皮下注射により12時間あたり体重kgあたり100Uを皮下投与することであるが、複数の危険因子が検出された場合には予防的なLMWHを考慮しなければならない[236]。
7.7. 回復期血漿と静脈内免疫グロブリン
SARS-CoV、MERS-CoV、エボラ、H5N1鳥インフルエンザ、およびH1N1インフルエンザのような他のウイルス感染症に関連する以前の経験に基づいて、PCR陰性で回復した患者から得られたIgG抗SARS-CoV-2(高免疫IgG含有血漿(HIgCP))を含む血漿血清の輸液は、新たに感染した対象者における治療的アプローチである[238-241]。
同様に、静脈内免疫グロブリン(IVIg)は、免疫応答の調節に役割を持っていた。IVIgの抗炎症/免疫調節的役割はまた、対応するFcγ受容体(FcγR)とのそれらのFc領域の相互作用に依存している。FcγRは、自然免疫(食細胞)および適応免疫(T細胞、B細胞)に関与する細胞や抗原提示細胞に発現しており、自然免疫と適応免疫の橋渡しに必要である。この相互作用は、FcγRを介したシグナル伝達を調節し、最終的に強力な抗炎症効果を誘導する可能性がある[242, 243]。IVIgはまた、炎症を制御し、T細胞の活性化[244]、TNF-α産生、IL-6およびマトリックスメタロプロテアーゼ9活性を抑制するのに役立つTregの数と機能に影響を与える可能性がある。また、IVIgはNET形成を抑制し[245]、制御的オートファジーの調節にも役割を果たしていると考えられる。さらに、FcフラグメントおよびIVIgは、サイトカインおよびDAMP産生を減少させ[246、247]、TLRおよびRAGE発現を調節してHMGB1誘発細胞死から保護する可能性がある[248]。
これらの特性は、ARDSの原因となるサイトカインが介在する間質性および肺胞壁水腫を予防し、打ち消すために、SARS-CoV-2感染におけるHIgCPおよびIVIgの使用を示唆する根拠となっている。
HIgCPの投与は、SARS-CoV-2誘発性ARDSの治療または予防に有用であると考えられる。Igの投与はウイルスクリアランスを促進する可能性があり、SARS-CoV-2回復患者で中和性Igが認められていることを考慮すると、[169, 249]。しかし、ドナーとレシピエントの間で血液型が一致する必要があり、また他のウイルス感染のリスクもあるため、HIgCPは大規模な投与にはIVIgよりも不向きである。予備的な臨床経験は有望であるが [250] 、決定的なものには程遠い。現在進行中の臨床試験では、COVID-19患者におけるIVIgの真の有効性が評価されている。
7.8. インターフェロン
インターフェロンは、抗ウイルス特性を有する組換えサイトカインである[251]。それは、単剤として、またはRNAウイルスのRNA依存性複製における致死的変異を誘導するグアニン誘導体であるリバビリンとの組み合わせで投与される。
I型IFNは、NETossisを増強することが実証されている。この経路は双方向のメカニズムを示す。試験管内試験(in vitro)では、NETはDC上のTLRを誘発し、インターフェロンα(IFNα)の産生をもたらす [252, 253]。ターンでは、IFNαは好中球がNETを放出するためのプライミングを行い[252、253]、その結果、NET放出およびIFNα産生の永続的な病原性サイクルが生じる。HMGB1は炎症を持続させる上でIFNαと関連している。特に、IFNは、グラム陰性感染時に血流中のHMGB1放出を促進するのに有効である[254]。HMGB1とIFNの両方とも、特に低酸素時の炎症性サイトカインの放出に関連しており[255]、TLR4/MyD88/NF-κB炎症性シグナル経路を介した炎症の持続に関与している[256, 257]。IFNは、ウイルスの切断に必要なオートファジーの調節にも役割を持っている[258]。これは、IFNの抗ウイルス特性の一つである。実際、ウイルスの複製は、呼吸器ウイルスで実証されたオートファジー依存性のメカニズムである[259]。オートファジーの調節は複製を妨害し、ウイルスの切断を促進する[258、259]。
以前のコロナウイルス疾患におけるインターフェロンの役割については、相反するデータが報告されている。インターフェロンはSARS-CoVに有効であることが判明し、コルチコステロイドとの併用試験も行われた[260]。インターフェロンは、インターフェロン受容体1型に結合することで抗ウイルス活性を示す。さらに、二量体化を促進し、ヤヌスキナーゼ1(Jak1)およびチロシンキナーゼ2(Tyk2)のリン酸化を活性化する。STAT1およびSTAT2は、リン酸化されたIFN受容体に結合し、プロテインキナーゼR(PKR)を含む免疫調節剤および抗ウイルスタンパク質の発現を誘発する[261]。
32人のMERS患者を対象としたレトロスペクティブな観察研究では、インターフェロンα2aによる死亡率は85%であったのに対し、インターフェロンβ1aによる死亡率は64%であった[262]。349人の重症MERS患者を対象とした多施設観察研究では、インターフェロンとリバビリンの併用は、死亡率やウイルスクリアランスに何らの利益をもたらさなかった[263]。コロナウイルスに対するIFNの抗ウイルス活性については複数の報告があり、これらの薬剤はSARS-CoV-2に対しても有効である可能性がある。現在のところ、COVID-19に対するインターフェロンの有効性を支持する証拠はない。中国での使用を示す多くの報告があるが[264-267]、データは決定的なものには程遠く、臨床試験の結果が必要である。SARS-CoV-2治療薬として承認するためには、追加の臨床試験が必要である。
7.9. その他の治療法
アナキンラは、RA患者への使用が承認されている改良型ヒトIL-1受容体拮抗薬(IL-1RA)である。受容体のIL-1ファミリーは自然免疫応答を誘発し、有害な炎症と関連していた[268]。NETosisは、IL-1放出の刺激に関与する免疫経路の一つである[269]。IL-1の増加は、結果的にIL-8の増加を誘導し、自己永続的なループでNET形成を促進する[269]。このため、IL-1RAはNET形成を阻害する役割を持つ [269]。抗IL1Rは、DAMP放出をブロックする役割を持つ可能性がある。実際、IL-1αは呼吸器炎症モデルにおけるHMGB1の増加に関連しており、IL-1RAは気管支肺胞液中のHMGB1を減少させるのに有効である[270]。さらに、抗IL1Rは調節性オートファジーを回復させる可能性がある[271、272]。その作用機序により、サイトカインストームの緩和が仮説される可能性がある。アナキンラ治療を含む臨床試験が進行中であり、そのうちのいくつかは比較のためにトシリズマブを使用している。逸話的な症例 [273, 274] を除き、SARS-CoV-2 の臨床使用例および予備データはない。
Eculizumabは、補体タンパク質C5に結合し、膜攻撃複合体(MAC)の形成を阻害するヒト化IgG mAbである。補体活性化によるNET形成を阻害することができる[275]。COVID-19では、いくつかの無作為化臨床試験で検討中である。予備的な報告では、重度の肺炎またはARDSで集中治療室に入院したCOVID-19患者のサルベージに有効であったことが示されている[276]。
高気圧酸素療法(HBOT)は、大気圧よりも高い周囲圧力での酸素の医療利用である。炎症を軽減し、サイトカイン放出を調節し、活性酸素産生を増加させ、アポトーシスを減少させ、白血球の活性化と接着を調節する[277]。この治療は、ROS依存性NETs放出[278]、オートファジー[279]およびHMGB1経路活性化[280]を減少させるのに有効であるように見える。COVID-19患者の小規模な症例シリーズでは、HBOTは、治療後24時間後に呼吸数を減少させ、血中酸素濃度を改善するのに有効であるように見える[281]。しかし、思いやりのために与えられたものであり、より大規模なプロスペクティブ臨床試験が必要である。
8. 結論
SARS-CoV-2感染は重症化をもたらし、最良の治療法の選択を支持するエビデンスに基づくデータはまだない。免疫系は、病気と宿主の損傷を悪化させる主なカウンターパートである。NETは、肺障害の進行を防ぐための有望な標的となり得る。同時に、HMGB1 は、進行予防と肺障害治療の両方のターゲットとなる重要なポイントであるべきである。さらなるエビデンスを得るためには、大規模な多中心臨床試験が必要である。
略語
ARDS:急性呼吸窮迫症候群
Baf-A1:バフィロマイシンA1
クロロキン:クロロキン
COVID-19。 コロナウイルス病19
CPAPのこと。 連続的な正気道圧
CRP:C反応性タンパク質
CXCL12:CXCケモカインリガンド12
CXCR4:CXCケモカイン受容体4型
DAMPs. ダメージ関連分子パターン
DCである。 樹状細胞
ds-HMGB1. ジスルフィド結合HMGB1
EGFR:上皮成長因子受容体
fr-HMGB1。 還元型HMGB1
G-脳脊髄液:顆粒球-コロニー刺激因子
ヒドロキシクロロキン:ヒドロキシクロロキン
HIF-1α。 低酸素誘導因子1α
HMGB1:高機動グループボックス1
IL:インターロイキン
HSP:ヒートショックプロテイン
IFN:インターフェロン
IVIg. 静脈内免疫グロブリン
mAb. モノクローナル抗体
MAPKs. ミトゲン活性化プロテインキナーゼ
MCP:単球化学吸引性タンパク質
MERS-CoV-2:中東呼吸器症候群コロナウイルス2型
MIP:マクロファージ炎症性タンパク質
mTOR:哺乳類のラパマイシン標的
NETs. 好中球の細胞外トラップ
NF-κB:B細胞におけるκ光ポリペプチド遺伝子エンハンサーの核因子
PAD4:プロテインアルギニンデアミナーゼ4
PCT:プロカルシトニン
PKC:プロテインキナーゼC
PKR:プロテインキナーゼR
PMA:ミトゲンミリスチン酸エステル酢酸エステル
polyP:ポリリン酸塩
RAGE. 高度な糖化最終生成物の受容体
RDV:レムデシビル
REDD1. 発生とDNA損傷応答で制御される1
活性酸素種(ROS)。 活性酸素種
RSV:Syncytial respiratory virus
SARSである。 重症急性呼吸器症候群
SARS-CoV:重症急性呼吸器症候群コロナウイルス1型
SARS-CoV-2:重症急性呼吸器症候群コロナウイルス2型
STAT1: Signal transducer and activator of transcription 1
Th. Tヘルパーリンパ球
TLR:Toll様受容体
TNF:腫瘍壊死因子
トレッグス 調節性T細胞。