
コンテンツ
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8357117/
2021年8月11日オンライン公開
キーワード:クエイン、クイイン、クエウイン、ケウイン、キューイーン、ココナッツウォーター
概要
マイクロバイオームを構成するヒト腸内細菌が、いくつかの神経変性疾患と関連していることを示唆する証拠が増えつつある。パーキンソン病(PD)およびアルツハイマー病(AD)患者の腸内細菌集団に不均衡があることが、いくつかの研究で検出されている。このような腸内細菌の異常は、人体を保護する微生物由来分子や有害な微生物由来分子をそれぞれ減少または増加させ、その変化はいわゆる「腸脳軸」を通じて脳に伝達される可能性が非常に高い。微生物由来の分子であるQueuineは、脳に濃縮された超修飾核酸塩基であり、バクテリアによってのみ生産され、ヒトは腸管上皮を通じてサルベージされる。Queuineは、tRNAのウォブル位置(34位)のグアニンをGUNアンチコドンと置換し、細胞質およびミトコンドリアのmRNAの効率的な翻訳を促進する。Queuineの枯渇は、マウスやヒトの細胞において、タンパク質のミスフォールディング、小胞体ストレスやアンフォールドドタンパク質応答経路の活性化をもたらす。タンパク質の凝集とミトコンドリアの障害は、しばしば神経機能障害や神経変性と関連している。Queuineがタンパク質のフォールディングを促進し、タンパク質の凝集やミトコンドリア障害を防ぐことができるかどうかを解明するために、化学的に合成されたQueuine、STL-101の効果をいくつかのin vitro神経変性モデルで試験した。STL-101で神経細胞を前処理したところ、PDのシヌクレイン障害モデルではαシヌクレイン凝集のマーカーである過リン酸化αシヌクレインの著しい減少が、ADの急性および慢性モデルではタウの過リン酸化が減少することが観察された。さらに、STL-101で前処理した細胞では、両ADモデルおよび神経毒性PDモデルにおいて、神経細胞の生存率が増加することが確認された。神経学的に健康な180人の血漿中のQueuineを測定したところ、健康なヒトは保護レベルのQueuineを維持していることが示唆された。我々の研究は、神経保護におけるQueuineの新しい役割を明らかにし、神経疾患におけるSTL-101の治療の可能性を明らかにするものである。
はじめに
パーキンソン病およびアルツハイマー病(PDおよびAD)は、最も一般的な神経変性疾患であり、世界でそれぞれ700万人および4400万人が罹患している。PDは、古典的な運動障害と、診断の何年も前に発症する非運動症状によって特徴付けられる。便秘などの消化器症状は、PDの主な非運動症状のひとつであり、発症の10年以上前に発症することが多い [1] 。最近、レビー小体などの神経変性の特徴が腸管神経系(ENS)に見られること [2] や、パーキンソン病患者が腸内環境の異常に耐えていることがいくつかの研究で示された [3, 4]。アルツハイマー病は、認知症の主要な原因であり、記憶、思考、言語および学習能力の進行性の低下を含むいくつかの臨床症状によって特徴付けられる。また、アルツハイマー病患者は、健常者と比較して微生物叢に変化が見られる[5-7]。PDおよびADのマウスモデルにおける腸内細菌叢の移植は、細菌組成と神経変性の間の機能的な関連性を強調した[8, 9]。これらのデータを総合すると、腸と脳の間のコミュニケーション、すなわち腸脳軸[10]が疾患に関連しているという理解が広まっている。
真核生物と共進化した細菌は、腸と脳を含むいくつかの臓器をつなぐ多くのユニークな分子の供給源であり、人間の健康や脳機能に影響を与えている。Queuineは、ピロロピリミジンを含むグアニンのアナログであり(図1)、バクテリアによってのみ合成され、ヒトを含むほとんどの真核生物に存在する。ヒトは、自身の腸内細菌叢とこのバクテリア由来の分子を含む食事からQueuineを獲得する [11].腸内細菌叢によって産生されたQueuineは、ヒトによって腸管上皮を通して救い出され、脳を含む幅広い組織に分布し、そこで濃縮されている[12]。Queuineは、G34U35N36(N = 任意のヌクレオチド)のアンチコドン配列を含む細胞質およびミトコンドリアの転移RNA(tRNA)のウォブル位置(ヌクレオシド34)に組み込まれる [13, 14]。一方、その対応するヌクレオシドであるqueuosine は、アミノ酸であるアスパラギン、アスパラギン酸、ヒスチジン、チロシンの4つの特異的なtRNAのアクセプターに存在する(図1) [11, 15, 16].tRNA(Q-tRNA)のQueuine修飾は、触媒的なQTRT1サブユニットと付属のQTRT2サブユニットからなる真核生物のヘテロ二量体tRNAグアニン転移酵素(TGT)により、塩基対塩基の交換反応(Queuineによるグアニン置換)を通じて起こる不可逆的事象である(図1) [18]( ※1). tRNAのアンチコドンループのほとんどの転写後修飾と同様に、特に34位のqueuosine は翻訳プロセスにとって重要である[19]。tRNAへのqueuosineの組み込みは、翻訳速度を調節し、精度を維持する[14, 20-22]。ヒトおよびマウスの細胞において、queuosine枯渇に伴う翻訳の制御不能は、小胞体(ER)ストレスおよびUnfolded Protein Response(UPR)の活性化を引き起こす、未変化タンパク質の形成につながる[14, 21]。QTRT2 KO細胞におけるミトリボソームプロファイリングにより、34位のQueuine(Q34)がミトコンドリアにおける伸長速度を細胞質におけるものと同様に制御していることが最近明らかになった[14]。ミトコンドリアで翻訳される13の同定されたタンパク質はすべて酸化的リン酸化装置の構成要素であり[23]、ミトコンドリアタンパク質の恒常性に欠陥があると、細胞の代謝が劇的に損なわれると考えられる。ミトコンドリアの機能不全は、PDやADを含むいくつかの神経疾患と実際に関連している[24]。
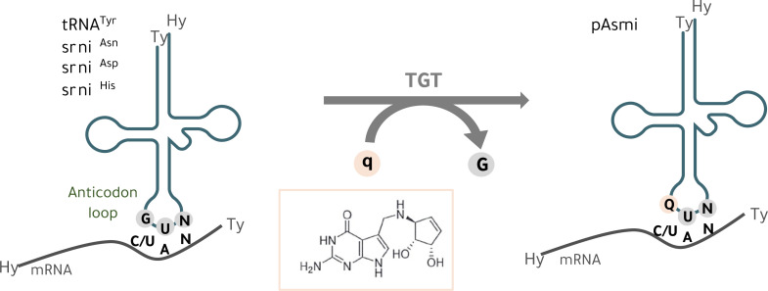
図1 tRNAへのQueuineの組み込み
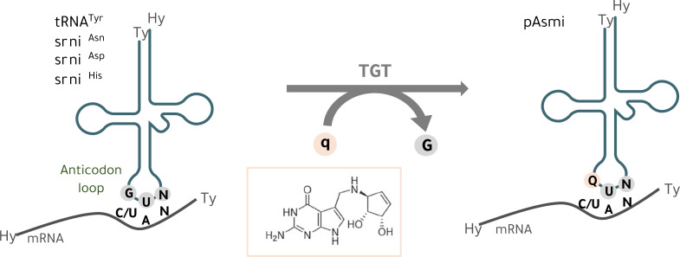
真核細胞では、tRNAのグアニントランスグリコシラーゼ(TGT)酵素が、グアニン(G)を核酸塩基のQueuine(q)とウォブル位置(34.1位)で交換する
GUNアンチコドン配列(N=任意の塩基)を持ち、チロシン(tRNATyr)、アスパラギン(tRNAAsn)、アスパラギン酸(tRNAAsp)、ヒスチジン(tRNAHis)に対するtRNAアイソアクセプターに特異的なtRNAの、アンチコドンアンシートの1塩基目のウォブルポジション(34位)で、グアニン(G)を核酸塩基であるQueuine(Q)と交換する。Qはqに対応するヌクレオシドであり、Q-tRNAとNAYコドン(Y = CまたはU)との塩基対形成は、mRNAの翻訳速度および忠実性に影響を与える[14, 20-22]
もう一つ興味深いのは、フェニルアラニン水酸化酵素(PAH)やチロシン水酸化酵素(TH)といった芳香族アミノ酸水酸化酵素の必須補因子であるテトラヒドロビオプテリン(BH4)を正常に保つために、QueuineとPDが必要であるという点である。PAHはフェニルアラニンからチロシンを合成し、それがTHによって水酸化され、ドーパミンの前駆体であるレボドパ(L-DOPA)の生成を触媒する[25]。PDは、黒質(SN)のドーパミン作動性ニューロンの死によって特徴付けられ、その結果、運動障害の一因となるドーパミン欠乏を引き起こす。Queuineを欠くマウスは無症状に見えるが、チロシンとQueuineの両方を欠く飼料を与えた無菌マウスは神経異常を起こし、18日前に死亡する[26]。さらに、TGT欠損マウスでは、BH4が減少し、その酸化体であるBH2(ジヒドロビオプテリン)のレベルが上昇し、チロシン産生が損なわれることから、チロシン代謝におけるQueuineの重要な役割が示唆された [27]。BH4はまた、PD患者でも欠損している神経伝達物質であるセロトニンの産生につながるトリプトファン水酸化酵素(TPH)の中心的な補因子である [28].queuosineはBH4の酸化をブロックするようであるが、queuosineはPDにおける神経細胞変性のもう一つの既知の要因である酸化ストレスからHeLa細胞を保護することも示されている [29, 30]。これらの知見は、QueuineがBH4代謝および酸化ストレスの防止を通じてドーパミン合成に重要な役割を果たすことを示唆している。
Queuineは脳内に豊富に存在し、いくつかの神経疾患の特徴であるタンパク質凝集を防ぐのに必要な最適な細胞質およびミトコンドリアmRNA翻訳を促進することから、いくつかの神経変性モデルにおいて合成されたQueuineの保護効果の可能性について検討した。
結果
合成されたQueuineは、神経毒を用いたPDモデルにおいてαシヌクレインの過リン酸化を低下させ、神経保護作用を示す。
Queuineは最適な翻訳を促進し、腸内細菌の変化はいくつかの研究でPDと関連していることから [4]、まずシヌクレイン障害のin vitroモデルで合成Queuine(以下、STL-101)[31]の効果を検討した。初代神経細胞を組換えヒトプレフォームドα-シヌクレイン(α-syn)フィブリル(huPFFs)に暴露すると、内因性α-synがPDの特徴であるレビー小体に凝集する[32]。我々は、野生型(WT)マウス初代皮質神経細胞をSTL-101添加または無添加で培養した後、huPFFsに3週間暴露し、デノボα-syn凝集の代替マーカーであるセリン129でリン酸化したα-synの免疫蛍光染色(IF)によりα-syn凝集をモニターした[33](図2)。Syn-211 抗体はヒトのα-syn を特異的に検出し、EP1536Y 抗体はヒトとネズミのα-syn のセリン 129 リン酸化型(α-syn pSer129)を特異的に検出する。huPFFsを3週間暴露した後、EP1536Y染色は、huPFFsに暴露したWTニューロンの神経細胞体にレビー神経突起とα-syn凝集体を検出したが、コントロール条件ではバックグラウンド信号しか観察されなかった(図2A)。STL-101の投与により、α-syn pSer129は有意に減少し、α-synの凝集が強く減少した(図2B)。EP1536Y抗体のシグナルを定量すると、STL-101を100nMで前処理した細胞では、STL-101で処理しなかった対照細胞と比較して、α-syn pSer129が約40%減少し、STL-101を10μMで処理すると最大で約50%減少した(Fig 2C)。
図2
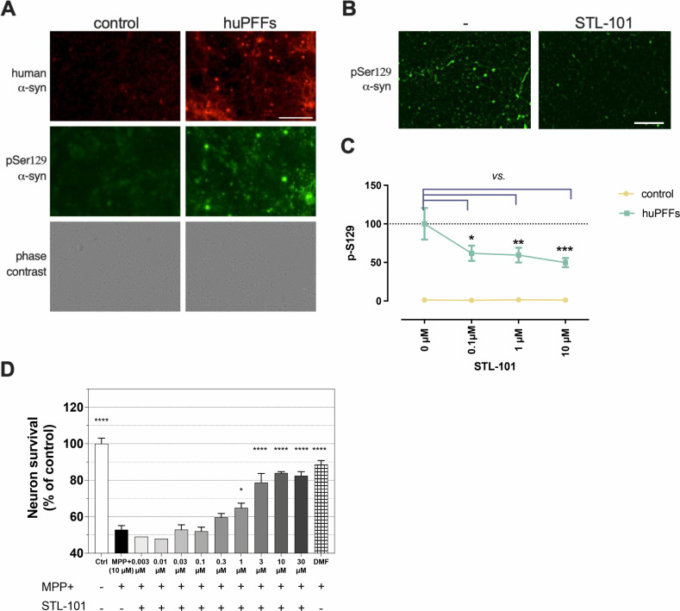
STL-101は、シヌクレイン障害のinvitroモデルにおいてα-syn pSer129を減少させ、ドーパミン作動性(DA)ニューロンをMPP+損傷から保護する
(A)コントロールおよびhuPFFs処理したマウス皮質ニューロン上のヒトα-syn(syn211抗体で赤色表示)およびα-synのリン酸化/凝集型(α-syn pSer129、EP1536Y抗体で緑色表示)のIF。スケールバー=200μm。(B)未処理(-)およびSTL-101で0.1μMで処理したマウス皮質ニューロンにおけるhuPFFs曝露の3週間後のα-syn pSer129のIF(EP1536Y抗体)。スケールバー=200μm。(C)0、0.1、1および10μMでPBSで希釈したSTL-101で処理したコントロール(huPFFsなし)およびhuPFFs曝露マウス皮質ニューロンにおけるBで示したようなα-syn pSer129の定量化。データは平均値+/- SEMで表し、二元配置分散分析に続いてダネットの多重比較を用いて分析した;***p<0.001 cf. huPFFs + [STL-101] = 0μM.。(D)培養液で希釈したSTL-101を、10μMのMPP+中毒の1日前に、ラットDAニューロン培養液に表示濃度で添加した。MPP+傷害の48時間後に、DAニューロンの生存をTH+ニューロンカウントにより評価した。統計的有意性は、一元配置分散分析、ダネットの多重比較検定を用いて算出した(*p<0.05、***p<0.0001はMPP+処理のみとの比較)。n = 3生物学的複製。陽性対照としてフマル酸ジメチル(DMF)を10μMで使用した。
STL-101の効果の頑健性を検証するために、ドーパミン作動性(DA)ニューロンの変性を誘発する神経毒MPP+(1-メチル-4-フェニルピリジニウム)を用いて、異なるPDのモデルでテストを行った。MPP+ はミトコンドリア複合体 I を阻害し、ミトコンドリアによる活性酸素の産生とそれに続く DA ニューロンの死を引き起こす[34]。ラット DA ニューロンを 6 日間体外培養し(DIV)、最終濃度 10μM の MPP+ を添加する 1 日前に、いくつかの濃度(0.003μM から 30μM)の STL-101 で処理した。2日後、DAニューロンの生存をTH+ニューロンを数えることによって評価した(図2D)。MPP+処理では50%のDAニューロンが死滅したが、STL-101を1μM以上処理するとDAニューロン死が有意に回復し、TH+ニューロンが65-85%生存することが分かった。この結果を確認し、同じモデルでα-syn凝集をテストするために[35]、我々はラットDAニューロンをMPP+(4μM)とSTL-101をいくつかの濃度(0.01μMから10μMまで)で共処理して同様の実験を行った(S1図)。前処理なしで、STL-101はMPP+中毒後の3μMおよび10μMで依然として神経細胞生存率の有意な増加を示した(S1A Fig)。MPP+傷害がDAニューロンにおけるα-synの増加を誘発することは、このモデルにおいて以前に観察されており、それは主に凝集したα-synであることが確認された[35]。重要なことに、細胞を1μM以上のSTL-101で処理したときに、IFによって検出されるα-synシグナルの有意な減少を測定し[未処理細胞(MPP+単独)と比較して〜30%減少]、STL-101がin vitro MPP+モデルにおいてα-syn凝集に影響を及ぼすことを示す(S1B図)。
これらの結果は、STL-101がPDの研究に広く用いられている2つの異なるシステムにおいて神経保護作用とα-syn凝集を減少させることを示し、プロテインパシーと神経変性に苦しむヒトにおける治療の可能性を明らかにするものである。
STL-101は、ADのin vitroモデルにおいて、タウの過リン酸化と細胞死を抑制する
アルツハイマー病は、アミロイドβ(アミロイドβ前駆体タンパク質(APP)から切断された約40アミノ酸長のペプチド)の細胞外への蓄積によるアミロイド斑、細胞内の神経原線維変化(NFT)を形成するタウの高リン酸化および凝集、神経炎症が特徴である。Aβ(Aβ1-40およびAβ1-42が最も多い)の増加は、AD患者および動物モデルの脳で認められ、AD発症における原因または少なくとも毒性に寄与すると考えられているのは、Aβオリゴマーである[36]。ADはまた、疾患進行の初期に観察されるミクログリアと呼ばれるミクログリアの活性化とも関連している。まず、STL-101 の Aβ 毒性低減能力を評価するために、AD の本質的な神経病理学的特徴と神経炎症反応を再現する急性 Aβ1-42 オリゴマー損傷モデルを用いた [37]。Aβペプチドは可溶性のモノマーとして生成され、核形成依存性の重合プロセスを経てアミロイド線維の形成とともにオリゴマー化を受ける[38]。STL-101 をラット皮質ニューロンの初代培養に播種後 10 日目、Aβ1-42 傷害の 1 日前に 20 μM で添加した。Aβ1-42傷害の24時間後に、MAP-2(微小管関連タンパク質2)および過リン酸化タウ(p-tau)のIF染色を行い、皮質ニューロンの生存、神経突起ネットワークおよびp-tauを評価した(Fig 3A)。Aβ1-42損傷はコントロールと比較してp-tauの有意な増加をもたらしたが、STL-101の投与は用量依存的にp-tauを強く減少させた(Fig 3B)。STL-101はまた、Aβ1-42損傷によって通常変化する神経突起ネットワークも保護した(図3C)。重要なことは、30nM以上の濃度のSTL-101の前処理が神経保護を示し、0.3μMと1μMの濃度で細胞生存率を約70%から80-85%に増加させたことであった。
図3 STL-101は、ラット皮質ニューロンにおけるAβ1-42による急性傷害から保護する
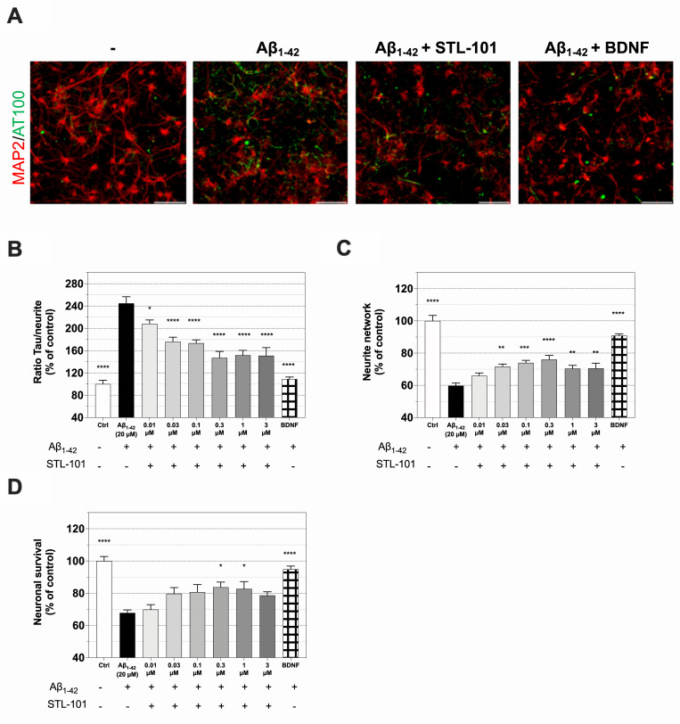
(A)コントロール(-)、Aβ1-42傷害神経細胞、STL-101を0.3μMで24時間前処理したAβ1-42傷害神経細胞、BDNFを50ng/mLで処理したAβ1-42傷害神経細胞のMAP2(赤)および p-tau(AT100, 緑)についてのIFの代表図である。Aβ1-42は20μMで使用した。スケールバー=100μm。(B)A.に示したように、および示した濃度のSTL-101前処理を行ったMAP2/AT100 IFにおけるp-tauの定量化。(C)Aに示したような神経突起ネットワークの測定(D)Aβ1-42損傷後の神経細胞生存率定量化。STL-101前処理を指示濃度で行い、神経栄養因子BDNFを50ng/mLで陽性対照として使用した。コントロール(Ctrl)=0.1%DMSOを含む培養液。結果は対照条件に対するパーセンテージで表し、平均 +/- SEM (n = 4-6 wells/condition) を示した。統計解析は、一元配置分散分析後にDunnettの多重比較検定を行った(Aβ1-42との比較では、*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 )
さらに、ラット初代神経細胞培養液とAβ1-42を5μMで72時間処理したミクログリアの両方でSTL-101の効果を検証した(S2 Fig)。急性損傷後の効果と同様に、慢性損傷では、STL-101の濃度が1μM以上で神経突起ネットワークが有意に増加し(S1A Fig)、STL-101の濃度が0.3μM以上でp-tauが減少した(S1B Fig)。STL-101の関連する神経保護効果を観察したが、統計的に有意ではなかった(S1C図)。興味深いことに、Aβ1-42傷害によって誘導された培養液中のTNF-α放出は、コントロールと比較して、STL-101で前処理した細胞(0.3μM以上)で有意に低かった(S1D Fig)。これらのデータは、STL-101が神経保護特性を示すだけでなく、ADの慢性モデルにおいて抗炎症効果を示すことを示す。
血漿中Queuine濃度は年齢依存性はないが、女性で高い
神経変性疾患は、加齢と関連することが非常に多い。クィーネレベルの年齢差や性差を明らかにするために、50歳から90歳までの神経学的に健康な男性80名と神経学的に健康な女性80名の血漿中のクィーネを測定した。LC-MS/MSによるクィーンレベルの測定では、血漿中のクィーンの年齢上昇に伴う有意な減少は認められなかった(Fig 4A)。このデータは、健常者が保護的なクワイヌレベルを維持している一方で、クワイヌの利用可能性の低下が、ディスバイオシスを経験している患者で起こっていると考えられることと完全に一致する。予想外なことに、女性の血漿中では、男性に比べて有意に高いレベルのQueuineが検出された(図4B)。興味深いことに、45週齢(〜1歳)のマウスの血漿中のQueuineレベルを解析したところ、雌は雄よりも高いレベル(3倍以上)であった(S3 Fig)。
図4 血漿中のQueuineレベルは年齢による変化はないが、男性よりも女性で高い
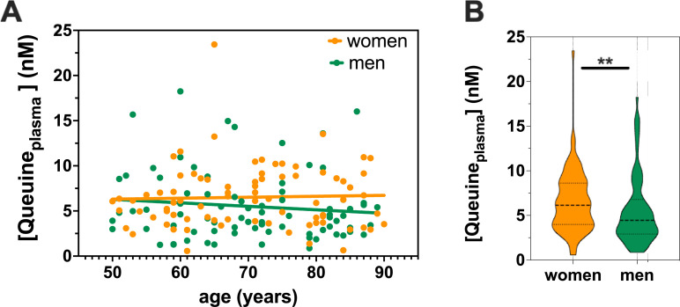
(A) 50歳から90歳までの神経学的に健康な女性80名と神経学的に健康な男性80名の血漿試料について、LC-MS/MSによりクワイヌレベルを測定した。(B)Aで示したQueuineレベルの男女比較 **p<0.01 (Mann Whitney test)
考察
我々は、天然にバクテリアによってのみ生産される分子である化学合成されたQueuineの保護特性を、いくつかのタンパク質異常症と神経変性症のin vitroモデルにおいて示した。まず、化学合成されたQueuine(本研究ではSTL-101と命名)でhuPFFs曝露神経細胞を前処理すると、α-syn pSer129が約50%減少することが示された。これは、外因性huPFFによって通常引き起こされるα-synの凝集を強力に抑制・防止していることを示している。我々はまた、PDのMPP+神経毒モデル、ADの急性および慢性モデルの両方において、STL-101の神経保護的役割を示した。STL-101の前処理を行うか行わないかで、MPP+に曝露されたDAニューロンの約15-35%を死から救い、α-syn凝集を30%防止/減少させた。急性期ADモデルで確認された神経保護効果は、リン酸化タウの減少に関連しており、おそらくADの特徴であるNFTの形成に関連していると思われる。重要なことは、慢性Aβ1-42傷害モデルでは、STL-101処理した細胞で炎症性サイトカインの産生が減少することが明らかになったことだ。我々は、有意な保護に必要なSTL-101の濃度は、他のモデルよりもMPP+モデルで高いことに注目している(それぞれ1μM vs. 0.1-0.3μM)。STL-101は、huPFFsの曝露の4日前に添加されたが、これは、損傷前の前処理なしまたは1日前処理(MPP+またはAβ1-42)より優れた保護に寄与する可能性がある。また、MPP+によって引き起こされたミトコンドリアストレスがSTL-101の機能を変化させ(例えば、分子の酸化、細胞摂取量の減少)、保護に達するにはより多くの分子を必要とする可能性がある。興味深いことに、1μM以上のSTL-101を前処理なしで添加した場合、MPP+モデルにおけるα-syn凝集の有意な減少が見られ、3μMのSTL-101では有意な神経細胞の生存が見られる。これは、あるMPP+誘発の表現型は生理的濃度付近のSTL-101によって救済され(α-syn凝集)、他の表現型は薬理学的濃度のSTL-101を必要とする(細胞生存)ことを示す。これはまた、STL-101処置が細胞生存に影響を与える前にα-syn凝集を減少/防止することを示す可能性がある。mRNAの翻訳を最適化し、新しく合成されたタンパク質のミスフォールディングを防ぐことによって、STL-101は、(huPFFsのような播種モデルや、ミトコンドリア機能障害(例えば、MPP+中毒)によって凝集が引き起こされるかもしれないモデルで)神経細胞の損失を防ぐ前に一定のレベルに達する必要のあるタンパク質凝集を遅らせたり、防いでいるという可能性がある。これらのデータを総合すると、STL-101による治療は、タンパク質の凝集が共通の特徴である様々な神経変性モデルにおいて、いくつかの有益な効果をもたらすことが示される。我々は、STL-101を予防および併用治療としてテストしたが、次に重要なのは、STL-101を損傷後に投与した場合に同様の効果があるかどうかをテストすることだ。
興味深いことに、健常者の血漿中のQueuineの濃度は年齢によって変化せず、男性よりも女性で高いことが示された。神経変性疾患はしばしば性差を示すことが知られており[39]、例えばPDや筋萎縮性側索硬化症(ALS)は女性よりも男性に多く、ADや多発性硬化症ではその逆である。これらの性差が、Queuineの分布とレベルの違いによって説明できるかどうかを調べるのは興味深いことだ。また、STL-101治療のような特定の治療から最も恩恵を受ける可能性の高い部分集団を選択するために、患者を層別化することの重要性が浮き彫りになっている。神経学的に健康な人の生体液や組織と、ADやPDなどの神経変性疾患に罹患した患者の生体液や組織におけるQueuineレベルを比較することは、神経変性がQueuine不足に関連しており、おそらくディスバイオシスによって引き起こされるという我々の仮説を立証するために特に重要であろう。重要なことは、マウスとヒトの血漿中のQueuine濃度に同様の性差が見られたことから、マウスモデルが薬力学的および薬物動態学的研究をさらに行うのに適していることが示されたことだ。
tRNA修飾の欠陥に関連する多くの疾患が報告されており、代謝性疾患、呼吸器系障害、筋疾患、およびいくつかのミトコンドリア障害が含まれる[40]。平均して、tRNA には 13 の転写後修飾があり、合計で約 100 の修飾が同定されており、その多くは mRNA の解読に重要なウォブル位置またはその近傍に存在している[41]。ヒトの脳はtRNA修飾の欠陥に非常に敏感であり、いくつかの神経障害はtRNAの転写後修飾を担う遺伝子の変異に関連している[42]。グアノシンに代わるqueuosine のような保存状態の良い複雑な修飾が重要な役割を果たすことは予想できたが、神経細胞におけるその有利な利点と保護作用は予想外であった。
Q-tRNAがmRNAの翻訳を制御するもう一つの方法は、翻訳を妨害するtRNA断片への切断から保護することだ。実際、Queuine修飾を持たないtRNAは、リボヌクレアーゼであるアンジオジェニン(ANG)によるストレス誘発性の切断を、アンチコドンループで特に受けやすい[43, 44]。実際、tRNAはいくつかのエンドヌクレアーゼによって切断され、様々な配列とサイズの安定したtRNA断片(tRF)が多数生成される[45]。これらの tRF のうち、5′ tRNA の半分と 3′ tRNA の半分(tRNA-derived stress-induced RNA または tiRNA とも呼ばれる)は、ANG 切断の生成物として、ストレス環境下で非常に多く存在し [43, 46]、タンパク質合成を阻害することが分かっている [47, 48]。興味深いことに、tRNAハーフはPD患者の血清やCSF(脳脊髄液)中にも検出され、バイオマーカーとしての利用が提案されている[49]。このように、tRNAの切断阻害や翻訳制御におけるQueuineの役割は、S. pombeで示されたtRNAAspのC38のメチル化など、他のtRNA修飾を刺激する役割にも拡大することができる[50]。Q34と同様に、C38はRNAエンドヌクレアーゼによるストレス誘発性tRNA切断を阻害することが示されている[44, 51]。Queuineは、特定のtRNAのプールの安定性を調節するだけでなく、ストレス条件下でtRFsの合成を通じてタンパク質合成の抑制に貢献することができる[47, 48]。しかし、tRNAの切断に対するQueuineの保護的役割は、ANGの神経保護的役割を示す研究[52-54]や、ANGの変異がPDやALSに関連しているという事実[55]とは対照的である。しかしながら、PDのモデルマウスの黒質におけるANGの過剰発現は、対照と比較してドーパミン神経細胞死をわずかに悪化させる[56]。
Q-tRNAは細胞質及びミトコンドリアのmRNAの翻訳速度及び忠実性に機能しているが、神経細胞に対する保護効果につながるメカニズムは不明である。TGT KOマウスは生存可能でほぼ無症状であるが、フェニルケトン尿症(PKU)と同様の症状を示し、フェニルアラニンからのチロシン生成が欠損し、血漿中のBH4レベルが著しく低下し、BH4酸化産物のジヒドロバイオプテリン(BH2)は上昇する[27]。PKUはフェニルアラニン水酸化酵素の変異に起因し、フェニルアラニンの蓄積とそれに伴う神経学的な問題を引き起こす。BH4レベルの低下はPDに関連しているが [25, 57]、Queuine/queuosine がBH4に影響を与える正確なメカニズムはまだ解明されていない。
ミトコンドリア機能の異常は、PD [58] とAD [59] の両方の分子病態に寄与する可能性がある。ミトコンドリアは、酸化的リン酸化に不可欠な電子輸送チェーンの13のサブユニットの翻訳 [60] と、広範な細胞機能に不可欠な鉄硫黄クラスター(ISC)の生合成 [61] を制御している。AD患者の大脳皮質ではミトコンドリアのUnfolded protein response(UPRmt)が活性化されており、ミトコンドリア機能とプロテオスタシスの強化はAβの凝集と毒性の減少を示すことが分かっている[62]。α-syn,タウ,あるいはAPPの適切な翻訳を潜在的に保証するために,queuosine がミトコンドリアの翻訳と恒常性の品質管理を保証している可能性が非常に高い。最近の研究では、queuosine 欠乏がミトコンドリア機能障害を促進し、HeLa細胞におけるプロトン漏出率の増加およびATP産生の減少をもたらすことが見出された[63]。
また、Queuineの欠乏は、いくつかのヒトの癌の進行の増加と関連している[64]。興味深いことに、Queuineの類似体は、自己免疫疾患である多発性硬化症のマウスモデル(EAE)において、疾患の特徴を逆転させることにより、すでに顕著な効果を示している[65]。これらの報告および今回紹介する我々の研究は、化学的に合成されたQueuine(STL-101)が、一般的および特異的なmRNAの翻訳品質管理のディレクターとして、おそらく治療上の可能性を示し、最適な脳機能および特にタンパク質異常症からの保護に重要な役割を果たす可能性があることを示している。この仮説を確認し、このような機能の背後にある分子メカニズムをさらに理解するために、我々はin vivo神経変性モデルマウスを含むSTL-101の実験を継続する予定である。
材料と方法
省略