
Suppressed anti-inflammatory heat shock response in high-risk COVID-19 patients: lessons from basic research (inclusive bats), light on conceivable therapies
要旨
COVID-19患者における致死的転帰の主な危険因子は、高齢であること、および既存の代謝性および心血管疾患(心血管疾患)であることであり、欠陥ヒートショック応答(HSR)に関連した炎症性の慢性変性疾患であるという特徴を共通して有している。炎症の生理的解決につながる主要な代謝経路であるHSRの分子成分は、恒常性を脅かすストレス状況(例えば、熱ストレス、酸化ストレス、代謝ストレス)の間、ヒートショックプロテイン(HSP)ファミリーの分子シャペロンが関与する抗炎症性生化学経路である。
一方、標的細胞へのSARSコロナウイルスの侵入は、この特定のグループの患者のHSRを既に悪化させる。さらに、ウイルスに対する細胞の反撃は、インターフェロン(IFN)介在性の炎症反応を伴う。したがって、HSRが障害された個体は、致命的な「サイトカインストーム」につながる炎症の悪化した形態の影響を受けやすく、ウイルスによって誘発された炎症のバーストを生理学的に解決することができない。
興味深いことに、SARS-CoV-2を含む人獣共通感染症ウイルスの自然な保存場所であるコウモリのいくつかの種は、永続的に活性化されたIFNベースの抗ウイルス性炎症反応を持っているが、病気やサイトカインストームの兆候を示していない。これは、コウモリが人間のそれよりもはるかに(何百倍も)強くて速い構成的なHSRを提示し、高いコア温度に関連付けられているために可能である。
同様に、ヒトでは、解熱剤は炎症の初期段階をブロックするが、解熱剤はHSRを介して炎症の解決段階を損なうのに対し、発熱はHSRの生理的誘導剤である。これらの知見は、COVID-19を含むSARSにおける患者ケアと発熱抑制の再評価の根拠となるものである。
キーワード
COVID-19、重症患者、発熱、ヒートショック反応、炎症、SARS-CoV-2
担当科目:循環器・血管生物学、糖尿病・代謝疾患、免疫・炎症、健康・疾患の分子基盤、トラン
COVID-19によるパンデミックと致死的転帰のリスク
2019年末、武漢市衛生委員会は、中国湖北省武漢市の水産物卸売市場と関連した「¨原因不明の肺炎」の患者群に注目した[1]。2019年12月30日に武漢市金仁潭病院で採取した気管支肺炎サンプルを分析したところ、患者で観察された重症急性呼吸器症候群(SARS)の原因となった病因は、Coronaviridae科のRNAウイルスと一致した[2]。興味深いことに、これまで知られていなかったβ-CoV(ベータコロナウイルス)のゲノム配列を解析したところ、コウモリSARS様コロナウイルスbat-SL-CoVZC45(MG772933.1)との同一性は85%であり、コウモリCoVのRATG13との全ゲノム配列同一性は96.2%であった[3]。この新規CoVはSARS-CoV-2ウイルスと命名され、その後WHOは新規感染性肺炎¨コロナウイルス病2019 「」またはCOVID-19と命名した[4]。
COVID-19は高い感染力を持つことが証明されている。2020年1月21日までに、中国のみに限定して278例が発生しており[5]、4週間という短い期間で、中国国内で1114人が死亡、中国国外で441例が発生する44,730例の大パンデミックとなった[6]。2020年3月11日までにWHOは世界的に11万8,319人の確定症例と4,292人の死亡を集計し、COVID-2019をパンデミックと宣言した[7]が、瞬く間に世界中に広がり、2020年3月31日までに世界的に750,890人(米国では140,640人)の症例と3万6,405人の死亡を記録し、前回の24時間の間に世界的に57,610人の新たな症例と3,301人の新たな死亡を記録した[8]。最近、WHOは、2020年6月30日までに世界で過去24時間の間に39,46人が死亡した503,862人の累積データを通知した[9]。2020年7月31日現在、世界では約1,730万人の症例が確認され、67万4,000人以上の死亡が確認されている:米国では15万2,000人以上、ブラジルでは約9万2,000人の死亡が確認されており、後者は現在のCOVID-19の震源地です[10]。
驚くべきことに、COVID-19の発生が全体として中国に限定されていた場合でも、小児での発生が報告された症例はわずかであり、その半数近くが60歳以上の成人であった[11,12]。全体として、COVID-19に関連した症例の31%、入院の45%、集中治療室(ICU)入院の53%、および死亡の80%は65歳以上の成人であり、重篤な転帰の割合は85歳以上の人が最も高くなっている [13]。80歳以上の症例死亡率は、イタリアや中国のような特徴の異なる集団を比較しても、非常によく似ている(約20%)。すべての年齢の子供はCOVID-19に感受性があることが明らかにされているが[15]、COVID-19による重篤な転帰や致死率は成人よりも子供の方が低い傾向にある[15,16]。全体として、年齢はCOVID-19による死亡リスクを示す変数であり、60歳以上では調整オッズ比(OR)が少なくとも18.82(CI95 %: 7.20-41.55)と最も高い関連性を示しているようである[17]。これは、米国(および世界の他の地域でも)のCOVID-19入院患者の治療に強く求められている経験豊富な看護師や医師が45歳以上であることが、当惑と懸念の理由となっている[18]。
興味深いことに、ヨーロッパにおけるSARS-CoV-2パンデミックの震源地であるイタリアとスペインの7つの病院でCOVID-19と重篤な疾患(呼吸不全と定義)を有する患者1,980人を対象としたゲノムワイド関連研究(GWAS)に基づく最近の年齢と性別を補正したメタアナリシス[19]では、血液型Aの患者では、他の血液型の患者よりも重篤な疾患のリスクが高いことが示された(オッズ比、1.45;95%CI、1.45、2.5%CI、1.5%CI、1.5%CI、1.5%CI、1.5%CI、1.5%CI)。 45; 95 % CI, 1.20-1.75; P=1.48×10-4)、血液型O群は他の血液群と比較して保護効果があることが示された(オッズ比、0.65; 95 % CI, 0.53-0.79; P=1.06×10-5)。このような所見の説明はまだ不明であるが、ABO血液群は以前にも他のSARS(SARS-CoV-1感染症)への感受性に関与していることが示唆されている[20]。
また、中国で最初の症例が報告されて以来 [12,21,22]、入院患者の約半数では、高齢のほかに既往症が顕著に見られ、高血圧が最も多く(30%)、次いで糖尿病(19%)、冠動脈性心疾患(8%)、慢性閉塞性肺疾患などの肺疾患の既往がある患者の3%であることが明らかになっている[22]。同様の有病率は、ニューヨーク市の大規模コホート研究でも報告されており、糖尿病患者におけるCOVID-19の重症度は、糖尿病患者ではない場合と比較して明らかになっている[23]。COVID-19入院患者では、以前に合併症を持っていた患者も死亡率が最も高く、高血圧患者では7.42(95%CI:6.33-8.79)、糖尿病患者では9.03(95%CI:7.39-11.35)、冠動脈性心疾患では12.83(95%CI:10.27-15.86)、慢性閉塞性肺疾患では7.79(95%CI:5.54-10.43)の調整後ORであった[17]。
最後に、高齢と併存疾患の有病率が非常に印象的な観察は注目すべきである。非生存者では、この差はさらに大きく、約70%が男性である[22]。イタリアのロンバルディア州では、集中治療室(ICU)に入院した患者の82%が高齢の男性であった[24]。中国では、このような差は喫煙に関連しているとの仮説が立てられているが(女性に比べて男性の方が高い)、COVID-19患者に見られる性差と男性の喫煙率との具体的な関連はまだない[25,26]。また、2014年4月から5月にかけてサウジアラビアで発生したMERS-CoVによる中東呼吸器症候群(MERS)のアウトブレイクのピーク時には、男女比は似通ってた:症例の62%が男性で、死亡率は男性の方が高く(52%対女性の23%)、年齢に関連したハザード、すなわち60歳以上の人が最も感染して死亡する可能性が高いことがわかった[27]。2003年3月から9月にかけて中国でSARS-CoV-1によるSARSがパンデミックした際にも、ほぼ同様の特徴が観察されたが、その際の死亡率は女性よりも男性の方が高かった [28]。
このような男女間、また若年者と高齢者の間の格差の原因は、マウスの研究から得られている。C57BL/6およびBALB/cマウスに致死量のマウス適応SARS-CoV(MA15)株を感染させたところ、23歳のヒトに相当する生後6週齢の若いマウスはCoV感染に対して完全に抵抗性を示した[29]が、マウスが高齢になるにつれて病気や死亡に対する感受性が増加することが示された[30]。驚くべきことに、ヒトのSARS様疾患(例えば、COVID-19)と同様に、メスのマウスは、81歳の閉経後の女性に相当する高度な年齢(約20ヶ月)であっても、ほとんどの場合、MA15 CoVから保護されている。この性差はエストロゲン受容体に依存しており、Tリンパ球やBリンパ球の反応とは無関係であることが明らかになった[30]。卵巣摘出術とエストロゲン受容体拮抗薬は、非常に老年期の女性でもこの保護を無効にするのに対し、卵巣摘出術や抗アンドロゲン治療は男性マウスではこのシナリオを全く変更しないというのは興味深いことである。さらに、雄マウスで観察された疾患の重症化には、より高いウイルス力価、肺の炎症性単球/マクロファージおよび好中球浸潤の増加が寄与していることが明らかになった[30]。
上記の証拠は、他のコロナウイルス疾患と同様にCOVID-19患者における好ましくない転帰の有病率につながる年齢、性別、および併存疾患との共通の関連性について疑問を投げかけている。これらの関連する問題は次のトピックで議論され、ウイルス誘発性炎症との関係、および高リスクのCOVID-19患者が炎症を生理的に解決することができないために炎症亢進を回避することができないことを強調している。コウモリにおけるウイルス感染の非常に珍しい病態生理についても、最後のセクションで議論した。最後に、これらのポイントを明らかにし、COVID-19患者に対する代替治療法を検討するための新しい臨床試験を提案した。
心血管疾患患者におけるACE2受容体の役割
SARS-CoV-2は、他のコロナウイルスと同様に、通常、スパイク(S)タンパク質に位置する受容体結合ドメイン(RBD)を用いて、宿主受容体に結合した際に細胞感染を開始し[31]、宿主細胞プロテアーゼによるSタンパク質のプライミングを行い、侵入を完了させる[32]。SARS-CoV-2 Sタンパク質の構造をクローニングしてアミノ酸配列を解析した後、分子ドッキング研究により、SARS-CoV-2 RBD Sタンパク質がアンジオテンシン変換酵素2(ACE2)と緊密に複合化していることが明らかになった[33,34]。この知見は、ウイルス粒子が侵入できないACE2の発現を欠いたヒト細胞でさらに確認された[3]。また、SARS-CoV-2の侵入因子は、嗅覚障害や中枢神経系(中枢神経系)感染に関与していると考えられる鼻上皮細胞[36]や角膜・腸上皮細胞[37]でも高発現している[36]。さらに、ACE2 mRNAは、事実上すべての臓器や動脈・静脈内皮細胞に存在しており、例えば、肺肺胞上皮細胞、口腔粘膜、鼻咽頭、胃、結腸、皮膚、リンパ節、胸腺、骨髄、脾臓、肝臓、腎臓、脳などにも存在している[38]。
一方、ACE2は、アンジオテンシンII(Ang II)からアンジオテンシン1-7(Ang 1-7)を生成する役割を担っており、したがって、血漿中で慢性的に上昇した場合に心血管系に損傷を与える可能性があるAng IIの血管収縮、内皮および血管壁リモデリング効果を打ち消す[39]。COVID-19患者のかなりの割合が高血圧性であるか、または冠動脈性心疾患の既往があるため(上記)、ACE阻害薬(ACEI)またはAng II受容体拮抗薬(ARB)などの降圧薬の使用が、そのような患者におけるCOVID-19の進化に影響を与えうるという懸念があった[33,34]。SARS-CoV-1感染から回復した患者の12年間の追跡調査では、68%が高脂血症、44%が心血管系異常、60%がブドウ糖代謝障害を有していたが、SARS-MERS-CoV感染から回復した患者でも同様のパターンが観察された[34]。
700の肺トランスクリプトームを対象とした最近のメタアナリシスでは、COVID-19に関連する合併症を持つ患者ではACE2が高レベルで発現しており、これはエピゲノムの改変に関連している可能性があることが明らかになった[40]。さらに、驚くべきことに、Zieglerと共同研究者らは、ヒト(マウスではない)肺Ⅱ型肺細胞、回腸吸収性腸球、鼻杯分泌細胞においてもACE2がヒトインターフェロン(IFN)刺激遺伝子であることを発見した[41]。IFN経路はウイルス感染に応答して強く活性化されるので(以下に詳述)、COVID-19患者における抗ウイルスIFNに基づく応答が実際にSARS-CoV-2感染を増強する可能性を排除することはできない。しかしながら、SARS-CoV-2感染またはその重症度のためにACEIまたはARBによる治療を中止すべきであることを示唆する臨床的または基礎科学的証拠はまだ存在しない[33,34]。さらに、少なくとも中国では、COVID-19患者のうち、レニン-アンジオテンシン-アルドステロン系(RAAS)阻害薬で以前に治療を受けたことがある患者はごく一部にすぎないと推定されている[42]。
RAAS阻害薬がACE2受容体の「可能性がある」発現亢進によりSARS-CoV-2が細胞内に侵入する可能性を高めるとすれば、ACE2の発現亢進は肺や心血管系に全く影響を及ぼさないことが知られている[33,43]。実際、ACE2/Ang1-7系のカウンターレギュレーション作用は、COVID-19患者で観察される一般的な合併症である心不全を妨げる駆出率を維持するために重要である[39]。このため、現在の理解では、RAAS阻害剤治療は、リスクの高いCOVID-19患者の治療薬に追加したり、削除したりすべきではないということになっている[44]。したがって、既存の心血管疾患(心血管疾患)を有する患者が、COVID-19の致死的な形態を発症しやすい理由は、依然としてつかみどころのないものである。ヒト膵β細胞および肝臓オルガノイドを用いた研究では、SARS-CoV-2感染に対する高い寛容性と関連するACE2の発現が特に高いことが示されており[45]、SARS-CoV-2感染が新規発症糖尿病を誘発する可能性があることが示されている[46]が、糖尿病に関しても同様である。
ヒートショック応答(HSR)の欠損によるヒートショックプロテイン(HSP)を介した炎症の生理的解決の慢性的な抑制
COVID-19における年齢、男性性、および特定の既往症との関連を説明する1つの可能性は、炎症を生理学的に解決するHSRの欠乏である。堅牢なHSRの欠乏は、心血管疾患や肥満関連疾患など、多くの(すべてではないにせよ)慢性炎症性疾患の予後不良の根本的な理由である可能性が高い[47]。COVID-19の高リスク群においても同様のことが疑われる。
例えば、男性と女性の間の致死的転帰の不一致は、エストロゲンがHSRの強力な誘導因子であり[48]、エストロゲン受容体シグナル伝達の遮断は女性のSARS-CoV-1に対する保護を廃止するため、男性のHSRの低下と関連している可能性がある[30]。さらに、男性の心筋細胞はエストロゲンとプロゲステロンに反応して熱ショック因子1(HSF1)の活性化と70 kDa熱ショック蛋白質(HSP70)の発現を増加させるが、同様の保護効果を促進しうるアンドロゲン受容体を持っていない[49]。
つまり、男性はエストロゲン-HSR軸を中心とした心保護作用を持っていない(図1の抗炎症性HSRの例示的な要約を参照してほしい)。したがって、炎症性の合併症を合併している男性は、炎症関連の合併症のリスクが高い可能性が高いと考えられる。一方、女性は炎症性の老化関連サイトカイン[50]から構成的に保護されているようであり、これは次に述べるように、女性がCOVID-19のほとんどの重篤な形態を発症しにくくなる可能性がある。
興味深いことに、ウイルス誘発性の直接的な細胞病理学的効果はSARSの劇症的な臨床症状に役割を果たすことができるが、ウイルスに対する自然免疫反応の亢進が、炎症反応の悪化を伴う死亡率の高さの原因であると考えられている[30]。この点では、肺浸潤性炎症性単球/マクロファージを減少させることで、致死的なSARSからマウスを部分的に保護することができた[30]。
2019年のアウトブレイク中の武漢の患者から得られたデータはまた、COVID-19の死亡率が、劇症型心筋炎と同様にウイルス活性化された「サイトカインストーム症候群(別名:サイトカイン放出症候群)」に起因する可能性があることを示唆している[21]。
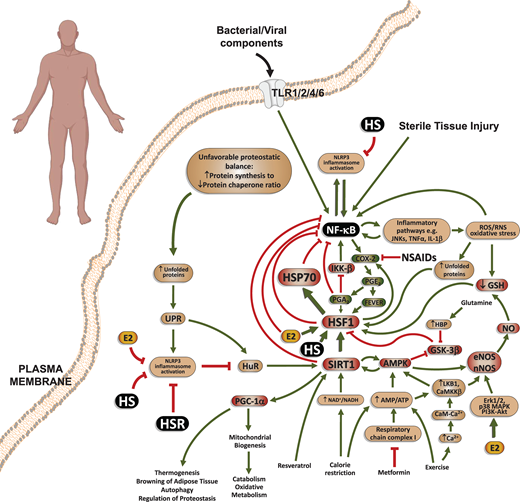
図1
ヒートショック応答(HSR)生化学経路の抗炎症枝
HSRは、熱ショック転写因子-1(HSF1)を中心とした生化学的経路であり、70 kDaの熱ショック蛋白質ファミリー(HSP70)の大量産生をもたらす。HSF1は、PGE2による体温上昇(発熱)、ヒートショック(HS)、エストロゲン(E2)、および抗ウイルス性シクロペンテノンプロスタグランジン(cyPG)、例えばPGE2由来のPGA2などによって直接活性化され、その生理的産生は炎症の末期に増強される。炎症反応中のこの時期(刺激後約48時間)に、第二波シクロオキシゲナーゼ-2(COX-2)の発現が起こり、プロ炎症性PGの産生がcyPGの産生に置き換わる。
HSP70やHSF1を含むHSRの様々な成分が、直接的にも間接的にも、マスター炎症核因子であるNF-κBの活性化と転写活性を阻害するため、HSRはそれ自体が抗炎症性である。一方、解熱剤・非ステロイド系抗炎症薬(NSAIDs)の使用は、炎症に伴う不快感を和らげる一方で、HSRを介した炎症の解決段階を著しく阻害する。
注目すべきことに、HSF1は、遺伝子制御レベルでプロ炎症性サイトカイン(例えば、IL-6、IL-1β、TNF-α)の産生を抑制する(本明細書には示されていない)。アンフォールドされていないタンパク質の構造変化は、直接的に(例えば、酸化ストレスおよび活性酸素および窒素種(ROS/RNS)の形成後)、グルタチオン(GSH)/タンパク質スルフィドリルの酸化還元状態の変化を介して、または最終的に、サーチュイン-1(SIRT1)経路を介してHSF1を活性化するアンフォールドされていないタンパク質応答(UPR)プロトコルの活性化後のいずれかで、HSF1に頼ることができる。
HSRのこの分岐は、SIRT1 mRNAおよびその発現を安定化するRNA結合タンパク質であるHuR(ELAV1としても知られている)のUPRを介した活性化を介して行われる。SIRT1の下流シグナルはまた、代謝変化(例えば、↑NAD+/NADHまたは↑AMP/ATP比)およびその結果としての5′-AMPキナーゼ(AMPK)の活性化を介してHSR経路に伝達され得る。
グリコーゲン合成酵素キナーゼ-3β(GSK-3β)は、HSF1およびHSRを構成的に阻害するが、AMPKおよびグルタミン(ヘキソサミン生化学的経路、HBPを介して)によって阻害される可能性があり、したがって、エネルギー制限状況下、運動下、または抗糖尿病薬メトホルミンを介して薬理学的に抗炎症性HSRを増加させることができる。
HSP70はタンパク質の凝集体の形成を阻害するタンパク質シャペロンであるため、HLP3の慢性的な活性化を回避し、結果として低悪性度の慢性炎症性疾患を予防するためには、HSRが重要であると考えられる。
その他の略語。NF-κB、κB(κB)ファミリーのκ軽鎖エンハンサーの核内因子転写因子;NLRP3インフラマソーム、NLR[ヌクレオチド結合オリゴマー化ドメイン(NOD)-ロイシンリッチリピートおよびピリンドメイン(LRP)含有タンパク質]-3インフラマソーム;NOS、一酸化窒素合成酵素;PGC-1α、ペルオキシソーム増殖因子活性化受容体-1αサブタイプ;TLR、Toll様受容体。緑の矢印は直接的な刺激経路を示し、赤線は阻害経路を示す。線の太さは、他の経路と比較した場合の刺激/阻害の相対的な程度を示す。
「サイトカインストーム 」は、汚染された病原体、エンドトキシン、または任意の基礎疾患が存在しない場合には、若い被験者であっても起こり得る生命に関わる状況である[51]。重要なことに、サイトカインストームは、2002年から 2003年にかけてのSARS-CoV-1のパンデミックにおいて、死亡率の主要な誘因であることが明らかになった[52]。サイトカインストームはCOVID-19患者で再び一般的であり、インターロイキン6(IL-6)およびIL-6誘発C反応性蛋白(CRP)の血清レベルの上昇は、呼吸不全、急性呼吸窮迫症候群(ARDS)および不良な臨床転帰と相関している[53]。急速で爆発的な炎症性症候群に関連して、COVID-19非生存者の100%が敗血症を経験しているという事実がある[22]。
サイトカインストームに関連した炎症は、特にウイルス誘発性のものやIFN誘発性のものであれば、通常、特定の部位から始まり、全身循環を介して全身に広がる[52]。サイトカインストームは、局所の炎症が全身循環に波及し、持続的な低血圧、高熱または低体温、白血球症または白血球減少症、およびしばしば血小板減少症によって定義される全身性敗血症を引き起こす重度の肺感染症に最もよく例示される [52]。
さらに、サイトカインストームの結果としてSARSを発症して死亡したCOVID-19患者の剖検サンプルから得られた最近の所見では、好中球細胞外トラップ(NET)の関与が示されている[54]。すなわち、肺では、NETが嚢胞性線維症患者の気道の粘液の蓄積を駆動するのに対し、血管系のNETは、臓器機能に壊滅的な効果を持つアテローム性動脈硬化症や大動脈瘤だけでなく、血栓症(特に微小血栓症)を駆動する[54]。このことから、炎症を解決するための合併症が既往している人は、プロ炎症性サイトカインストームの影響を受けやすいと予想される。
ヒートショック応答(HSR)、発熱、炎症の生理的解決
炎症は、動物の進化の間に、病原体から生物全体を保護し、無菌状態または病原体によって誘発された侮辱の後であるかどうかにかかわらず、損傷を受けた組織の修復を刺激する迅速で自己解決的な反応であるように選択された。従って、そのような傷害が検出されると、自然免疫系の細胞は、数分から数時間以内に(好中球)から数時間以内に(単球/マクロファージ)傷害/侵入部位にリクルートされる。このような刺激に応答して、活性化 B 細胞 (κB) ファミリーのκ軽鎖エンハンサー (NF-κB) の核内転写因子を中心とした誘導性タンパク質のきめ細やかな発現が初期段階で炎症を引き起こす [55] と同時に、約 48 時間後には炎症が収束するようになる [47,56,57]。これらの誘導性タンパク質の一つがシクロオキシゲナーゼ-2 (COX-2) であり、炎症性アラキドン酸由来のプロスタグランジン (PG) やその他の脂質メディエーター、血管透過性を高め、炎症性細胞の到着と活性化、組織修復を可能にする血管活性化合物の産生に関与している [58]。
損傷刺激の性質およびサイトカインの量に応じて(例えば インターロイキン1β、IL-1βなど)の量に応じて、COX-2由来のプロスタグランジンE2(PGE2)は、視床下部前部の視床前野での熱感知情報の処理をブロックすることで発熱を誘導する(図1)が、これは、体温を上昇させるための協調交感神経/副交感神経の熱温存メカニズムの活性化につながる[48]。約2~3℃のコア温度の上昇はHSRを開始する[59]。これは主にヒートショック因子-1(HSF1)依存性のHSPおよび他の抗凝集性タンパク質シャペロンの発現を中心とした抗炎症プログラムである[60,61]。これは、そうでなければ「超生理学的」温度のために生体に課せられるであろうタンパク質の変性を回避する。発熱の確立中の形質膜の構造変化もまた、HSF1活性化に関与している[62]。
HSF1活性化は、HSPの転写を駆動するだけでなく、重要なサイトカインおよび早期応答遺伝子の発現を調節する[63]が、HSF1による炎症性サイトカイン発現の相互調節およびサイトカイン転写因子による抗炎症性HSPの相互調節を提供する方法である[47]。さらに、熱で活性化されたHSF1はCOX-2の転写を直接制御するため、炎症のマウンティング中に高スループットのPGE2産生を可能にする(図1)[64]。しかし、後の段階では、HSR は核内因子κB (NF-κB) や他の下流のプロ炎症性シグナルを遮断することで急性炎症を強力に解決する [47,56,57]。
炎症反応の過程で、同じ発熱誘発性PGE2が脱水を経て、α,β-不飽和シクロペンテノンPG(cyPG)であるPGA2へと変化することは注目に値する。さらに、炎症を誘発する刺激の48時間後には、COX-2発現の第二のピークがあり、これは現在、最小のPGE2産生と関連しているものの、2時間後のピークよりも350%大きい。後期になると、PGE2 の産生は、NF-κB 活性化を直接阻害することによってそれ自体が抗炎症性である cyPG の産生に置き換わる [66] したがって、炎症の生理学的な解決を助けます (図 1)。また、HSF1 の活性化に応答して産生される HSP70 は、NF-κB とその阻害剤(IκB)が形成する複合体と結合し、NF-κB の核内への移動を阻害する [67]。したがって、HSP70ファミリータンパク質(すなわち、HSP72、HSP73、HSP78)は抗炎症性シャペロンであり、HSP70誘導性抗炎症性cyPGの薬理学的利用は、一連の炎症状態を改善することが証明されている[68,69]。その結果、炎症反応の初期段階では、選択的COX-2阻害剤(COXIB)および従来のデュアルCOX-1/COX-2非ステロイド性抗炎症薬(NSAID)の両方が、この初期段階の進行を抑制することができる(図1)。しかし、このような阻害剤は、炎症の解離期を阻害することで、後期の炎症を強く悪化させる[57]。このシナリオは、炎症状態を永続させ、生理的に解決できない重度の急性炎症反応を悪化させる傾向がある。補論として、HSRの主要な生理的誘導因子である発熱は、その性質上抗炎症性であり、NSAIDsによる薬理学的に発熱を抑制することは、一部の急性炎症においては非常に不適切であるかもしれない。
発熱(その「広義の」意味での)は、約6億年前に現代の動物で進化した可能性があり[59]、また、下等脊椎動物、節足動物、および無脊椎動物を含む多くの吸熱性動物もまた、感染または傷害に反応して体温を上昇させるので、恒温性の哺乳類および鳥類の特権ではない[63]。それにもかかわらず、発熱の発生は複雑な反応であり、代謝的には非常にコストがかかる。したがって、ヒトでは、発熱には代謝率の6倍の上昇が必要であるが、発熱レベルで体温を維持するためには、1℃上昇につき約12%の代謝率上昇が必要であると推定されている[59]。しかし、この単純化されたアプローチでは、発熱がもたらす可能性のある利益を考慮に入れていない。もし発熱(または高熱の維持)が(低HSR能 力に関連した)有害な状態や心不全に至らないように注意深く監視できるならば、発熱は重症敗血症患者の増悪したサイトカイン産生を阻止することができる最後の資源であり、自然な治療手段であるかもしれない。
ICUでの解熱剤の使用は議論の余地があるが、重症患者では解熱剤で対処するのが一般的である [70-72]。実際、NSAIDsまたはアセトアミノフェンによる治療は、敗血症患者の28日死亡率を独立して増加させる(調整オッズ比:NSAIDs:2.61、P=0.028;アセトアミノフェン:2.05、P=0.01)が、非敗血症患者では増加しない [71]。実際、頑健な発熱反応がないことは、菌血症患者における死亡リスクの増大と関連している可能性がある[72]。ラットの敗血症誘発性急性肺損傷において、熱誘発性HSRが死亡率および臓器損傷を顕著に減少させることは以前から認識されていた [73]。システマティックレビューのメタ解析では、少なくとも実験動物(ヒトの研究は包括基準に達していない)において、アスピリン、パラセタモール、ジクロフェナクによる治療がインフルエンザ感染症における死亡リスクを増加させることが示されている[74]。重要なことは、Oxford COVID-19 Evidence Service [75]が主張するように、現在のエビデンスは、急性呼吸器感染症およびCOVID-19の発熱を治療するためのルーチンの解熱剤投与を支持していないことである。
武漢のCOVID-19患者において、発熱は致死的転帰とは相関しておらず(P=0.94)、入院患者の94%(191人中180人)が発熱(37.3℃以上)していたのに対し、死亡に至った患者の74%(69人中51人)、生存者の94%(137人中129人)も発熱していた[22]。しかし、2020年7月現在、COVID-19患者を対象とした最近の研究では、病院に到着した大多数の入院患者に使用されていると思われる解熱剤の使用についての詳細な情報を提供しているものはなかった。我々の知る限りでは、2020年5月25日にオンラインで発表された論文 [76] のうち、COVID-19の解熱剤としてイブプロフェンを市販のものを使用すると、発熱が抗ウイルス性であるために病勢が悪化する可能性があるとの見解を示したものは1件のみであった。もしそうだとしたら、COVID-19の患者が適切なケアを受けていても解熱剤治療を受けていない場合の転帰はどちらになるのだろうか。これは特に重要な点であり、高熱が抗ウイルス性であるために有益であるだけでなく、重症COVID-19患者の大多数は炎症性の慢性疾患を呈しているためである。言い換えれば、入院患者が背景にある慢性炎症を生理的に解決することが困難に直面している場合、抗炎症薬は状況を悪化させるであろう。ヒトにおける解熱薬の使用を決定的に保証する対照臨床試験はないが、「発熱は有害である」[77]という考えが主流であり続けている。
上述したように、SARS-CoV感染における致死的転帰の主な原因の1つは、ウイルス誘発性サイトカインストームである[52,78]。対照的に、HSP70はHSRの基本的な抗炎症性シャペロンであり、完全な炎症反応および最終的にはサイトカインストームに必要とされるNF-κBのp65サブユニットとの結合/分解を介してSARSウイルスによって誘導されるサイトカインストームを阻害する可能性がある[79]。一方、Chionhら[80]は、コウモリにおけるHSPの巨大な基底発現(以下で詳細に論じる)が、長時間の熱処理に対する細胞の生存および酸化ストレスに対する防御に関連していることを示している。他の観点から見ると、コウモリはハイパーインフレ ーション誘発性サイトカインストームに悩まされることなく、恒常的に活性化された IFN 応答 [81] を実装することができる。ウイルス感染における高炎症に対するHSRの保護的役割の支持としては、IFN-γがHSP70の産生および細胞からの輸出を増強することができるという事実がある[82]。さらに、2003年に発生したSARS患者のデータセットを解析した最近のプレプリント[83]は、薬剤の再配置を行うためのプラットフォームで、90 kDa熱ショックタンパク質(HSP90)阻害剤、主にゲルダナマイシンおよびその誘導体がCOVID-19患者の治療に有用である可能性を示唆している。この観察は、HSP合成が増強された条件下でのウイルス複製の阻害を記述した膨大な文献と対照的に見えるかもしれない。しかしながら、この矛盾は、ゲルダナマイシンおよび他のHSP90阻害剤がHSF1依存性HSRを活性化し、それらの投与はHSP90自体と同様にHSP70のレベルの上昇をもたらすので、明らかなだけである[84]。したがって、HSRの誘導剤は、生理学的(例えば、発熱、ホットタブ)であれ、薬理学的(例えば、cyPG)であれ、以前に他のウイルスについて報告されたように、炎症を生理学的に解決することによってCOVID-19サイトカインストームを打ち消すことが想定されている[84]。残念ながら、MERS-CoV、SARS-CoV-1またはSARS-CoV-2に感染した人のHSRを対象とした対照無作為化臨床試験(RCT)はまだ存在しない。
特に、重度の肺生まれの全身性炎症を発症しやすいCOVID-19患者の場合には、SARS-CoV-2陽性患者が発熱を含むCOVID-19の最初のシグナルを示した直後に解熱療法の有効性を確認するために、プラセボ対照臨床試験(少なくともデータ収集者、アウトカム判定者および/またはデータ解析者を盲検化)を実施することが重要であると考えられる。COVID-19の解熱療法を受けていない患者では、解熱療法を受けていない患者よりもHSRが温存されているために、より良い反応が得られる可能性がある。実際、一般集団に影響を及ぼすこの疾患の重症度の低い症例についても同様のことが予測される。
低悪性度炎症性慢性退行性疾患における細胞老化とHSRの抑制
COVID-19の重篤な症状を呈する前の健康な患者もいると報告されているが、中国で報告された最初のCOVID-19症例[21,22]では、病院に入院した患者に見られる合併症の発生率の高さ(約50%)と、そのような合併症を持つ患者の死亡率の高さ(約70%)が報告されている。既往の心血管疾患と糖尿病が最も多くみられた。中国とイタリアの死亡率の差は肥満関連疾患が原因であると考えられるが、これは後者の方が高齢者や肥満の成人の有病率が高いためである [85]。重度の肥満と糖尿病は、コロナウイルスによるすべてのタイプのSARSにおける死亡率の増加に関連していることが再発して発見されている[86]。これらの併存疾患には共通して、その病態の根底にある慢性炎症がある[47]。この点は、高リスクCOVID-19患者における病変の発生とHSRの潜在的な役割を理解するために、さらに詳細な検討が必要である。
この点で、低悪性度炎症性の慢性変性疾患はすべてHSRが顕著に抑制されていることが特徴である。実際、抗炎症性HSRの確立に重要なHSF1-HSPs軸は、代謝組織におけるHSP70とHSF1の両方の発現が徐々に抑制されることにより、徐々に鈍化していく(図2)。糖尿病および肥満では、HSF1およびHSP70発現の低下は、患者の脂肪組織[87]、肝臓[87]、骨格筋[88,89]および血管床[90,91]で常に観察される。この欠点は、神経変性疾患を呈する高齢者[92]および更年期に関連した代謝機能障害[48,93]においても見られる。同様のことが、肥満およびインスリン抵抗性のげっ歯類モデルで遭遇することができる[94-96]。これらの場合、HSP70の生理的、薬理学的またはトランスジェニックな誘導は、インスリン抵抗性を回復させる[47,94]。
図2
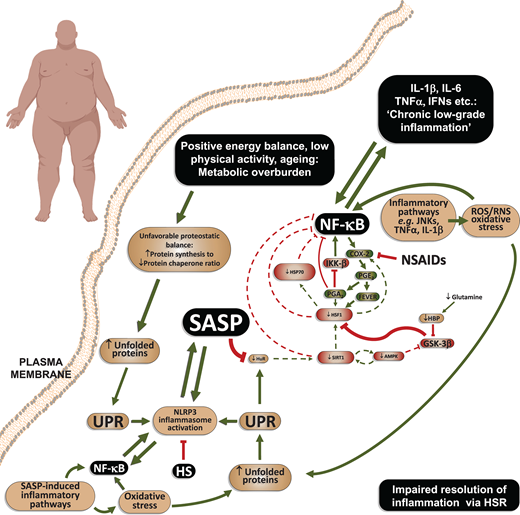
慢性炎症性疾患におけるHSRの抑制
炎症性のすべての慢性変性疾患(例えば、肥満、2型糖尿病、低エストロゲン状態、神経変性疾患および心血管疾患)に共通の特徴があり、それは、低いタンパク質シャペロンの利用可能性に直面してタンパク質合成のための過剰な需要によって引き起こされる好ましくないタンパク質の安定性のバランスである。この不均衡は、低身体活動、正のエネルギーバランス、脂質の高血漿レベル、動脈性高血圧、老化などのさまざまな要因の組み合わせによって引き起こされる。その結果、標的組織では、HSP70などのタンパク質シャペロンの発現を増加させながらタンパク質合成を停止させる細胞戦略であるアンフォールドタンパク質応答(UPR)につながるアンフォールドタンパク質の蓄積が見られる。しかし、UPRは炎症性分岐を持っており、損傷刺激(高脂血症、高血圧など)を除去しないと、NLRP3の炎症性サイトカインの大量産生で炎症が永久化し、SASP(senescence-associated secretory phenotype)として知られている状態につながる炎症性サイトカインの大量生産を伴うような方法でNF-κBとNLRP3の炎症ソームの活性化につながる。一方、SASPはHuR-SIRT1-HSF1軸の障害につながるため、HSRと炎症の生理的解決を阻害し、慢性炎症状態に陥る。逆にヒートショック(HS)はNLRP3インフラマソームを分解することができる。略語や表記は図1の凡例の通りである。
HSR経路の抑制は、脂肪組織におけるc-Jun N末端キナーゼ(JNK1)およびJNK2発現の亢進の程度と強く相関している[47,87]。これに続いて、JNK(p-JNK1およびp-JNK2)の活性化型およびプロ炎症型であるThr183/Tyr185二リン酸化の量も同様に上昇する。ストレスによって誘発されるHSP70はJNK依存性のプロ炎症性シグナル伝達を阻害する[97]ので、HSRが弱くなると慢性炎症の解消が困難になる。このような場合、これらの慢性疾患におけるHSRの抑制はどのように説明できるのだろうか。細胞の老化と老化に伴う分泌表現型(SASP)がこの問題の答えかもしれない。
生物が正のエネルギー不均衡(例えば、肥満および2型糖尿病)および/または継続的な脂質入力と乱流せん断ストレス(例えば、動脈硬化性心血管疾患および高血圧症)に直面すると、細胞小胞体(ER)の利用の過負荷とERストレスが存在する[98-105]。ERストレスの継続は、インスリン抵抗性、非アルコール性脂肪性肝疾患(NAFLD)、膵β細胞機能障害の発症に因果関係があることが明らかになった[102]。不適切な食事、低運動、制御されていない高血圧[48]のように、有害な刺激が消えないためにERストレスが解消されないため(図2)、ERストレスはアンフォールドタンパク応答(UPR)[106]へと進化する。これは、シャペロン合成を増加させながら細胞機能を停止させ、その結果として生じるアポトーシス細胞死および組織傷害を伴うタンパク質凝集体の蓄積を回避するための細胞戦略である[106]。しかしながら、UPRは様々な潜在的にプロ炎症性の経路を活性化するので、ERストレス誘発因子が除去されなければ、UPRは無期限に炎症性になる[48]。無限に続くUPRの下では、細胞は、不活性なプロカスパーゼ-1およびプロインターロイキンの活性型への切断を媒介するヌクレオチド結合オリゴマー化ドメインロイシン-リッチリピートおよびピリンドメイン含有プロテイン-3(NLRP3)インフラマソームの活性化に絶えずシグナルを送る [107]。この活性化は、酸化ストレスとその結果として生じるDNA損傷と並んで、SASPと細胞の老化を引き起こす [47,48]。SASPは、インターロイキン(例:IL1β、IL-6、IL8、IL18)を含む一連の炎症性因子で構成されており、組織機能を局所的に阻害し、炎症性シグナルを全身に拡散させる[47,108]。
持続的なNLRP3炎症アソーム活性化の結果の一つは、NLRP3依存性のカスパーゼ-1媒介によるRNA結合タンパク質(HuR/ELAVL1)の切断であり、これはサーチュイン-1(SIRT1)経路を介してHSF1の細胞内発現および活性を高く維持する役割を果たしている[47,48]。SIRT1はHSF1の発現と転写活性の両方を増強するため、堅牢なHSRをサポートする[47,109,110]。すなわち、NLRP3とSASPの長期的な増加はHSRを徐々に抑制し、細胞の老化と低悪性度の炎症を全身に拡散させる(図2)。この状態は、一部のコロナウイルスに見られるように、NLRP3のインフラマソーム活性化を誘発するウイルスの存在によって劇的に悪化する[111]。さらに、がん治療に用いられるプロセンスDNA損傷誘発剤であるドキソルビシンは、脂肪組織におけるケモカインの発現を増加させ、炎症性マクロファージや好中球の浸潤を促進し、脂肪細胞のインスリン抵抗性を誘導する[112]。これは、癌を有するCOVID-19患者と、HSRが低下した状態でのサイトカインストームによる死亡との間の別の関連を提供する可能性がある。
老人性細胞の蓄積は、加齢に伴う慢性疾患、酸化ストレス、ホルモン環境や発生因子の変化、慢性ウイルス感染、一部のがん化学療法薬、HIVプロテアーゼ阻害薬、電離放射線への曝露など、様々な機能不全状態で見られる[113]。効果は二重である。細胞の老化は、アポトーシスを回避するUPRに対する代替反応である[47]。一方で、それは不可逆的な成長停止をもたらし、細胞ストレスに応答してアポトーシスによってそのような細胞を排除することへの抵抗性をもたらす[114-116]。
老人性細胞は、アップレギュレートされたプロ生存経路を有しており、それは彼ら自身のプロアポトーシスSASPから彼らを保護する[116,117]。アポトーシスに対する抵抗性があるため、老化細胞は炎症に関連した細胞機能不全とSASPを永続させる[47]。さらに、放射線誘発性の老化前腺細胞で観察されるSASPは、老化後の内因性の老化細胞や特発性肺線維症で観察されるSASPと類似している[118]。
肥満では、SASPは脂肪組織から始まり、肝臓、膵臓のランゲルハンス島、骨格筋、血管などの全組織に広がり、他の代謝組織、そして最後に脳に到達する [92]。脂肪組織の老化は、インスリン抵抗性および耐糖能不耐症の前にも起こりうる[112]。げっ歯類では、比較的少数の老化細胞であっても、長期的な細胞機能障害を引き起こし、他の組織へと広がる可能性がある[118]。ヒトでは、肥満は脂肪組織由来の間葉系間葉系間葉系間質/幹細胞における早期老化プログラムを誘発する[119]。さらに、老化と糖尿病の両方は、動脈硬化、冠動脈疾患、インスリン抵抗性および関連する代謝異常、NAFLD、神経変性疾患、骨粗鬆症、慢性腎臓病、末梢血管疾患、歯周炎、免疫応答の低下、虚弱体質およびサルコペニアなどのERストレス依存性の老化関連機能不全を共有している[87,113,120,121]。
重要なことに、高齢者の組織と慢性炎症性疾患に罹患している(若年者または高齢者)ヒトの組織を比較すると、老化細胞の蓄積は区別がつかない。自然免疫応答と適応免疫応答の両方に関連した免疫老化もまた、免疫の不均衡が観察される老化や加齢に関連した疾患において役割を果たしている可能性がある [122-124]。実際、細胞の老化は、免疫細胞の適切な機能に必要なHSRを減衰させる[125-128]。
SARS-CoV-2感染者の場合、細胞老化とSASPは、IL-6の循環レベルが上昇し、慢性炎症性疾患の既往がある高リスクのCOVID-19患者で確認された [22]。COVID-19患者におけるERストレス、UPRおよび老年期関連慢性炎症性合併症との関連に関連して、高齢男性の肺のACE2発現肺胞上皮細胞ではTRIB3(別名SKIP3)の発現が低下しているが、女性では低下していない [129]。さらに、TRIB3 は UPR の間、HuR 媒介による NF-κB への負のフィードバックを提供している[130]。高齢男性におけるTRIB3発現の低下は、これらの患者が重度のCOVID-19に罹患しやすい理由を説明するのに役立つかもしれない。これらの所見は、慢性炎症性合併症とCOVID-19患者の未病的な免疫炎症状態との間に強い関連性があることを少なくとも部分的に説明することになるだろう(図3)。
図3
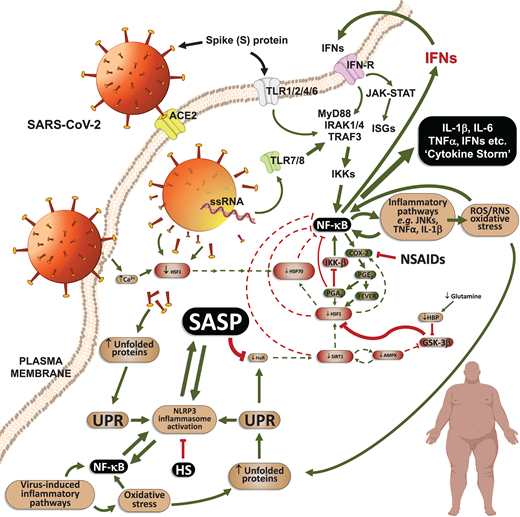
SASP関連低悪性度炎症を呈する高リスクCOVID-19人におけるサイトカインストームの起源
SARS-CoV-2 粒子とその成分の細胞処理は、インターフェロン(IFN)および他の NF-κB 依存性サイトカインの産生を悪化させる正のフィードバックルートのいくつかのループを持つ高揚した炎症反応を生成する。SASP媒介性慢性炎症性疾患(図2)を呈する人は、HSRを介して生理的に炎症を解消することができないので、生命を脅かすようなサイトカインストームが起こり、この高リスクのCOVID-19患者にとっては致命的であるかもしれない。この意味で、解熱剤やNSAIDs全般は、HSRを介した炎症の解消を阻害することで、シナリオを悪化させる可能性がある。また、ICU患者では非常に一般的なグルタミンの体内レベルが低いことも、グルタミン関連のヘキソサミン生化学経路(HBP)がHSRを構成的に阻害するGSK-3β活性を低下させるため、この状況を悪化させる可能性がある。略語および表記は図1の凡例の通りである。
ウイルス誘発性小胞体ストレスとHSRの抑制
ウイルスは宿主細胞の代謝を変化させるだけでなく、細胞シグナル伝達経路と転写機械をハイジャックしてウイルスに有利に制御する [131]。例えば、単純ヘルペスウイルスは、宿主の NF-κB 下流経路を破壊し、ウイルス遺伝子の NF-κB 依存性の転写を指示する [132]。宿主タンパク質シャペローニング、UPR、および翻訳機械の制御は、侵入ウイルスが宿主細胞の運命を操作するための強力なツールである[84]。ポジティブセンス一本鎖RNA SARS-CoV-2のようなRNAウイルスは、宿主細胞の翻訳装置を制御するためにいくつかの戦略を進化させ、通常、宿主細胞のタンパク質合成の劇的なシャットオフを誘発する[84]。ERストレスの誘導もまた、異なるコロナウイルスに感染した細胞の共通の特徴であるが[133]、ERストレスがウイルスの複製や宿主の保護に有利なUPRにどのように影響を与えるかは、コロナウイルスに依存する。
SARS-CoV-2がどのようにして宿主のタンパク質機械を引き継ぐかについての完全なパノラマをまだ持っていない。一般に、コロナウイルス(例えば、SARS-CoV-1、MERS)によるウイルスタンパク質の産生は、UPRが細胞内でのタンパク質合成を停止するように進化するため、ウイルスを脅かす傾向のあるER応答を誘発する。しかしながら、コロナウイルスは、適切なウイルスタンパク質合成を確保するためのトランスレーショナル・シャットダウンを克服するために、UPRを迂回させることができる[134]。例えば、SARS-CoV-1のSタンパク質は、目立つERストレスを誘導し、その後PERK経路を介してUPRを誘導してウイルスの複製を促進する[135]一方、感染性気管支炎コロナウイルスエンベロープ(E)タンパク質は、プロ炎症性サイトカインの誘導に関連するERストレスを誘導し、ウイルスの病原性を高めた[136]。Murine hepatitis coronavirusはERストレスを誘発し、UPRを改変して宿主タンパク質合成のシャットダウンを持続させると同時に、ウイルスタンパク質の翻訳を促進する[137]。一方、感染性胃腸炎ウイルス(α-コロナウイルス)は、試験管内試験(in vitro)および生体内試験(in vivo)の両方で、感染細胞においてERストレスおよび強力なUPRを誘導するが、UPRのPERKアームはNF-κB活性化を誘発してタイプIインターフェロン(IFN-αおよびIFN-β)の産生を可能にする[133]が、これらは発火性であり、強力な自己分泌系およびパラクリン系抗ウイルス応答を誘発する[138]。
IFNに基づくものを含むヒト細胞の異なる抗ウイルス戦略は、強力な炎症反応を誘発することができるので、以前の慢性低悪性度炎症性疾患の存在は、炎症を解決するために必要な欠陥HSRのためにウイルス誘発性炎症を悪化させる可能性がある。このことは、ウイルス誘発性炎症が既存のSASP介在性慢性炎症に加わることで、COVID-19の老年期合併症を呈する患者の臨床状態を悪化させることに寄与するであろう。このように増悪した状態は、避けられないサイトカインストームを引き起こすことになる(図3)。
女性の生理的抗ストレスプログラム
女性の生理は、男性とは異なるホルモン介在性の抗産生プログラムを発達させた。例えば、炎症様に駆動される分娩反応を含む妊娠の段階では、SASP成分が産生される[50,139]。それにもかかわらず、これは胎児の健康や陣痛に影響を与えず、また、分娩後の雌では細胞老化やSASP関連の慢性炎症性疾患の徴候は見られない[50]。逆に、細胞老化は、妊娠中の適切な恒常性を確保し、有益なプロセスとして胎盤および胎児の発達に重要な役割を果たしている[139]。生殖年齢に沿って、女性はエストロゲン抗老化作用によって慢性炎症から保護されている[48]。エストロゲンによる保護作用は、細胞の老化によってHSF1の利用可能性が低下する悪循環を遮断することによって、強力なHSR(図1)を維持する能力に依存している[48]。しかしながら、SARS-CoV感染マウス[30]およびコロナウイルス感染女性のMERS[27]またはCOVID-19症例[22]で観察されたような、サイトカインストームに関連した重篤な転帰の不釣合いな低有病率を、エストロゲンだけでは男性と比較して、生殖機能の低下した女性(閉経後の女性を含む)で説明することはできない。このことは、上述したように、女性の慢性増殖性老化の潜在的に有害な影響に対する追加的かつ既存の防御装置、および重症COVID-19患者の間での女性の低有病率を示唆している。女性のホメオスタシスがどのようにHSRを維持し、慢性炎症性疾患を回避するために細胞老化とSASPに対抗するかは、さらなる焦点を絞った研究のためのオープンな道筋である。
HSRの慢性的な抑制をバイパスし、炎症の解決を再武装する
最近では、老化細胞を優先的に標的とする薬剤(「セノリティクス」)が臨床試験に入っている。セノリティクスは、アポトーシス[113,117]を誘導することにより、老化細胞を選択的に排除し、有害なERストレス-ASP-炎症の悪循環を停止させ、HSRを介して炎症の生理的解決を再確立することを目的としている。老化細胞は機能不全に陥り、若いマウスでも生存率を低下させるが、センolyticsは高齢マウスでも寿命を延ばすことができる[118]。ケルセチンやアジスロマイシンなどのFDA承認の解熱薬の組み合わせは、実際にCOVID-19患者を治療するために提案されている[140]。
発酵細胞における抗アポトーシス経路を標的とする薬物および植物化学物質に加えて、SASPインターロイキン(例えば、IL-1β、IL-6)またはそれらの受容体(例えば、トシリズマブ)に対する抗発酵抗体が、SASPを改善し、低悪性度炎症を決定するNF-κB依存性のプロ炎症経路をブロックすることが示唆されている[117,141]。SASPアンタゴニストによる急性治療はSARSの治療のために示唆されているが[53]、そのような戦略が免疫監視に及ぼす可能性のある結果および望ましくない副作用については、現在まで臨床試験で完全に研究されていない。重要なことに、IL-6は、好中球が媒介するウイルスクリアランスを促進することにより、ウイルス感染に対する予備的な反応を開始する上で重要な役割を果たしている[142]。
一方で、全身温熱療法や発熱域の体温維持だけでも可能であり、生理的な抗原性HSR誘導戦略を伴う。これらのアプローチは、特定の炎症性成分に対する抗体の場合のように、免疫炎症バランスを阻害しないという利点があり、ICU環境や重症患者においても実施可能である[117]。実際に、ヒートショックはマウスマクロファージにおけるNLRP3炎症アソーム活性化とカスパーゼ-1活性の両方を阻害する[143]。HSP70はそれ自体が試験管内試験(in vitro)および生体内試験(in vivo)での腹膜炎においてNLRP3炎症アソーム活性化を阻害するが、HSP70欠損はマウスのNLRP3依存性腹膜炎を悪化させる[144]。急性炎症反応の解決におけるHSRの役割は、重症患者におけるグルタミンの効果と関連している可能性がある[145-147]。したがって、グルタミンは、HSF1 転写活性を増加させるヘキソサミン生化学的経路(HBP)を介して HSR を潜在化させ、したがって、NF-κB 依存性経路をブロックし、敗血症に続く急性呼吸窮迫症候群(ARDS)を予防する [145-147]。このことは、HSP70 と HSR の統合的な役割が関与する肺損傷の軽減や敗血症患者の生存率向上に対するグルタミンの有益な効果がよく知られていることを説明していると考えられる[92,148-150]。
これらのデータに基づいて、SASPに関連した慢性炎症性合併症を持つCOVID-19患者におけるHSRの活発な欠如は、健康とそれに関連した転帰をより危険にさらす傾向があると考えられる。これに関連して、上述のように、発熱域の温度での熱処理は抗発熱性であり、炎症を生理的に解決することができる。COVID-19患者における予後不良の主なハザードの1つは、炎症の解決を妨げる慢性的な低悪性度炎症であるため、熱処理は、これらの慢性疾患患者の組織において著しく低下しているHSF1レベルとは無関係に、抗炎症効果を誘導しうる[48]。制御された熱はNLRP3イン フラマソームを分解し、HuR-SIRT1-HSF1軸を回復させ、炎症の解消を再開させる [143,144]。このことは、慢性的な熱処理が、マウスモデルにおいて動脈硬化性病変の緩和と死亡停止に効果的であることを説明している[151]一方で、卵巣切除ラット[152]やインスリン抵抗性の肥満マウス[94]の代謝状態を改善していることを示している。このため、温熱浴槽療法(37.8~41.0℃)を30分間(37.8~41.0℃)、わずか3週間行うだけで糖尿病患者の血糖値とHbA1c値を低下させることができるという画期的な報告[153]から 20年が経過しているにもかかわらず、温熱療法下の糖尿病患者におけるHSRの全体像を明らかにした臨床試験が実施されていないのは興味深いことである。一部の文化圏では一般的に行われているサウナ入浴の頻度の増加は、心血管疾患患者の致死的転帰や全死因死亡率と逆相関があることを考えると、このことはさらに驚くべきことである[154]。
慢性炎症性疾患における温熱療法の複数の有益な効果に関与するメカニズムは完全には解明されていないが、(サウナまたは温浴により)ヒトの体温を約38~39℃まで上昇させると、一酸化窒素(NO)に基づく内皮機能の改善と慢性的なNOによって誘発されたHSP70発現により抗炎症効果が発揮されるという仮説が立てられている[155]。実際、慢性的な温熱療法による免疫炎症バランスの回復は、免疫抑制的アプローチを必要としない慢性自己免疫疾患の治療においても示唆されている[156]。しかし、ヒトでの長期的な研究が少ないため、各炎症性疾患に適したプロトコルを確立することは困難であり[157]、また、もし副作用の増加が生じた場合には、それをコントロールすることも困難である。
これらの知見は、COVID-19患者への解熱剤処方時の再評価と注意の必要性を強調している。一方、発熱域の体温を受動的に上昇(または維持)させることは、重症患者、特にARDSやSARSを呈する患者では、これまで試みられたことがない可能性である。COVID-19の患者は、炎症が「嵐」効果を引き起こすのを避けるために、抗炎症的アプローチから利益を得るであろう。ステロイドの使用がMERS-CoV-およびSARS-CoV-1感染患者におけるコロナウイルスクリアランスを遅延させていることを考慮に入れると、その瞬間とどのように干渉するかは極めて重要である[158]。したがって、COVID-19パンデミックの最中であっても、受動的温熱療法(または発熱)の効果は、無作為化比較臨床試験(RCT)で調査されるべきである。参加者への害を最小限に抑える傾向のある適応的な試験デザインは、COVID-19の補助的治療法の評価を加速させる可能性がある[159]。
HSRおよびHSR誘導剤の抗ウイルス効果
ウイルスの進化は、細胞生物がタンパク質合成やシャペロン機構の制御をウイルスに奪われないようにするための仕組みと、ウイルスがパリパシーで開発した高度な戦略によって鍛えられた。その意味では、HSRの構成要素もまた、抗ウイルスとしての機能を進化させたのかもしれない。実際、細胞は、細胞またはその内部に到達したウイルスまたはウイルス成分の存在に対する多くの異なる手がかりに応答してHSRを武装させるために、いくつかの独創的なメカニズムを「創造」した。上述したように、ウイルス成分の存在に対する迅速な反応は、UPRに向かって進行するERストレスの引き金となり、それと同時に、抗炎症性HSRを含むタンパク質シャペロン回路を増強する細胞(およびウイルス)タンパク質合成を停止させる。ウイルスは、すべての細胞コンパートメントと同様に、膜レベルで検出することができる。これは、後に HSR を介して抗炎症状態に向かう炎症性応答の NF-κB 活性化で頂点に達する [84]。ウイルス成分は、形質膜(TLR1/2/4/6)またはエンドソーム(TRL3/7/8/9)に存在するToll様受容体(TLR)のレベルで検出され、MyD88-NF-κB経路を介してIFN産生を活性化することができる[160,161]。進化的な理由が何であれ、ウイルス感染はHSRをマウントする炎症を誘導し、その結果、抗炎症性ネガティブフィードバックとして作用する[48]。言い換えれば、発熱を含むHSR誘導因子が抗ウイルス性であるのに対し、ウイルス感染はHSRと並行しているということである[84]。
最も効率的なHSR誘導剤である温熱療法は、一般的な風邪からヒトを保護し、エイズ患者におけるHIV1の転写を阻害するのに有効である[84]。実際、狂犬病ウイルスに感染したマウスは、一般的に20~21℃の環境下で飼育された動物に見られる高い死亡率と罹患率に比べて、35℃の環境下で飼育された場合には長期間生存することが知られている[162]。同様のことが、異なるウイルスタイプの試験管内試験(in vitro)でもHSP70に依存した方法で記述されている[163,164]。さらに、発熱域の高熱は樹状細胞-CD4+ T細胞相互作用回路を活性化し、これはTリンパ球のT中心記憶サブセットを中心としたHSP70依存的な方法で恒常性抗原非依存的な記憶を維持する役割を果たしている[165]。死亡したCOVID-19患者の約75%がリンパ球減少症を呈し[22]、COVID-19患者の80%以上がCD4+およびCD8+リンパ球の両方でT細胞枯渇マーカーを呈していたので、これは絶対に注目すべきことである[166]。
HSRによる免疫学的記憶の浸潤は明らかであるが、回復期の患者がSARS-CoV-2に対する長期的な免疫学的記憶を形成する能力はまだ分かっていないため、COVID-19では特に重要である。この懸念は、回復期患者の血漿中の抗体価が常に高いレベルに維持されているとは限らないという事実によって補強されている[167]。重要なことに、発熱域の温熱療法は、インフルエンザウイルスに感染したヒト単核球におけるマイトジェン刺激の障害を回復させる[168]。実際、ウイルス誘発性炎症と抗炎症性HSRとの間の進化的に保存された平行性は、ウイルスクリアランスと組織保護のために、炎症反応のエスカレートに対する組織保護を調和させる役割を果たしている。
熱効果に関連して、炎症の解決段階で産生されるcyPG(例えば、PGA1、PGA2、PGJ2、15-デオキシ-Δ12,14-PGJ2)は、HIV1を含む、これまでに試験されたいくつかのDNAおよびRNAウイルスに対しても強力な抗ウイルス性を示す[168-172]。これらすべての場合において、抗ウイルス効果はcyPGによって誘発されるHSF1の活性化だけでなく、完全な阻害作用のために誘発されるHSRにも依存していることが明らかになった[173-175]。多くのウイルスが複製に宿主細胞のNF-κB依存性機械を利用するので、HIV1の場合のように、cyPGはNF-κB活性化を直接阻害することによって抗ウイルス性であり[66,174]、またHSP70がNF-κBのp65サブユニットの分解を媒介するので[79]。J型cyPGの直接の前駆体であるPGD2もまた、コロナウイルス誘発NLRP3インフラマソーム活性化の抑制に関与している[111]。それにもかかわらず、cyPGは、SARS-CoV-2感染に対して試験されたことはない。
図1に要約されるように、細胞はHSRの活性化の多くの形態を進化させた。例えば、NF-κBが活性酸素および窒素種(ROS/RNS)の産生を誘発するための炎症性経路は、細胞内グルタチオン(GSH)内容物の枯渇をもたらし、タンパク質チオールの酸化およびアンフォールドタンパク質の量を増加させ、こうしてHSF1およびHSRを活性化する[61,176,177]。HSF1の35位と105位に高度に保存された(化学的に反応性のある)システインが存在することで、HSF1は酸化還元調節を受けやすくなっている[176]。これは、内皮(NOS3遺伝子によってコードされる)および神経(NOS1)NO合成酵素(NOS)を介して、または炎症反応中に誘導性およびNF-κB依存性のiNOS(NOS2)によって、エストロゲンに応答して大量に産生される一酸化窒素(NO)が抗炎症性である理由でもある。
NOがHSRの強力な生理的誘導剤である限り[178]、NOは細胞保護作用および抗炎症作用を示す。例えば、NOは、腫瘍壊死因子α(TNF-α)が肝細胞に対して細胞毒性を有することを抑制する[179]。げっ歯類モデルにおける研究では、NOは試験管内試験(in vitro)および生体内試験(in vivo)の両方でHSP70の急速な蓄積を誘導することが示されている[180]。逆に、覚醒ラットの全身熱処理(41℃、15分)は、さまざまな臓器でNO依存性のHSP70発現を誘導する[181]。さらに、関連するHSP70発現とNO産生は、低血圧を回避するための慢性的な熱処理に対する血圧の適応を媒介していると考えられている[182]。
NOは非常に反応性の高いフリーラジカルであり、炎症性食細胞によって産生される殺菌剤として長い間認識されてきた[183]。細菌および原虫に対するその効果に加えて、NOは抗ウイルス性でもあり、NOおよびINFはウイルス感染に対して相加的な効果を有するが、タイプI-IFNとは独立して自然免疫応答を刺激する[183,184]。NF-κBとIFN調節因子-1は相互作用して、iNOSの発現および産生されるNOの量を増加させる[184]。NOはまた、S-ニトロシル化を介してウイルスプロテアーゼの重要なシステインを共有結合的に修飾することによって抗ウイルス性であり[185]、NO供与体であるS-ニトロソ-N-アセチルペニシラミン(SNAP)は、SARS-CoV-1に対する有効な抗ウイルス性である[186]。繰り返しになるが、COVID-19を含む異なるウイルス感染症におけるNO媒介HSRの役割を評価するヒトにおける現在の臨床報告はない。
ウイルス誘発性ERストレスまたはNOに関連するHSP70は、抗ウイルス性炎症反応に沿ってIFN産生を増強することができ[187]、炎症のHSRに関連した解決の前にウイルスの排除を支援する。興味深いことに、炎症性抗ウイルス反応の過程で十分なNOが産生された後、NOはTh1細胞の活性を抑制し、Th2細胞の活性を増加させる[183]。これは、抗炎症性HSRが起こるのと同時に起こるため、IFNで活性化されたNF-κB依存性の超炎症を回避することができる[79]。さらに、NOは、IL-1β、TNF-αおよびIFN-γを含むプロ炎症性サイトカインによって誘導されるERストレス中の適応性およびアポトーシスUPRシグナリングの間の切り替えを調節する[188]。最後に、HSRを誘導する役割とは別に、NOは血小板に対して抗凝集性であり、これはCOVID-19患者にとって重要であるかもしれないが、それは非生存者の半数がIL-6血漿レベルと直接相関する播種性血管内凝固を含む凝固病[22]を呈するためである。結論として、NOが介在する酸化ストレスおよび血行動態を制御下に維持することができれば、HSR誘導作用および抗血小板作用と組み合わせた強力な抗ウイルス作用は、すでにSARSの治療に採用されているように、COVID-19患者(吸入NO療法および肺血管拡張[190]の適応を有する患者を含む)においても利用できる可能性がある[191]。
細胞外-細胞内HSP70比と免疫炎症状態
HSRに関するもう一つの関連する点を考慮することが重要である。細胞内環境の中でHSRを誘発する代謝的にストレスのかかる状況と同じような状況は、非定型的な分泌機構によってHSP70を含むエクソソームの放出を活性化することができる[192-194]。一旦分泌されると、細胞外HSP70(eHSP70)は、Toll様および他の膜受容体に結合することができ、恒常性を脅かす状態の存在に対するすべての組織へのプロ炎症性サイトカイン様シグナルをもたらす[128,195,196]。細胞外HSPはまた、異なる細胞タイプによって取り込まれ[196]、細胞内空間に入ると、細胞機能、特に中枢神経系における細胞機能に影響を与える真の抗炎症シャペロンとして働く[126,197]。
熱処理に応答してeHSP70を放出する能力は、HSRの完全性の指標であり[198]、中年または健康な老年成人と比較して、高齢の糖尿病患者では障害されている[198]。eHSP70の高レベルは肥満糖尿病患者の血漿中に見られる [89] が、その過剰なeHSP70はインスリン抵抗性および膵β細胞障害と正の相関がある [199]。高レベルのeHSP70の血漿中濃度は高血圧の被験者における動脈硬化性疾患の発症の予測因子であり[200]、細胞外HSP60の増加は冠動脈性心疾患のリスクの増加と関連している[201]。高レベルのeHSP72は敗血症患者の血漿中に見出され、高レベルの血漿酸化プロファイルを伴う場合には致命的な転帰と関連している[202]が、これはおそらく酸化後のeHSP72免疫シグナル伝達特性の損失を伴う[203]。しかしながら、非酸化(ネイティブ)組換えマウスeHSP72の単回投与は敗血症の重症度を減衰させ、マウスの生存率を向上させる。そうでなければ、単球および多形核細胞における細胞内HSP72(iHSP72)発現は抑制され、敗血症の重症患者におけるCD14/HLA-DR発現の低下および死亡率と関連している[205]が、敗血症患者におけるグルタミン投与により抗炎症性HSRは増加するであろう[92,150]。
慢性低悪性度炎症性疾患における抑制されたHSRの基礎的な役割については、HSPシャペロンバランスの崩壊に関連したコンセンサスがある [206] [207]。免疫炎症性のアンバランスは、抗炎症性の細胞内HSP70(iHSP70)およびHSRと比較して、プロ炎症性の細胞外HSP70(eHSP70)の過剰に有利である[128,208-210]。これに関して、異なる臨床状態における細胞外HSP70と細胞内HSP70の比率(別名Heck指数またはHSP70比率のH-index)の比較は、全体的な免疫炎症バランスの指標を提供する[128,211] したがって、H-indexはIL-2対IL-10比率に比例し、その値は超感受性C反応性タンパク質およびHOMA-IR値と直接相関している[89,128]。H-indexは、血漿中のeHSP70および血球中のiHSP70を評価することができれば、測定に採用した技術に関係なく、少量の血液サンプルから得られた結果から計算することができる[48]。併せて、H-indexは、ウイルス感染の進化をモニターし、COVID-19サイトカインストームのように壊滅的になり得る重度の免疫炎症性不均衡を警告するための貴重なツールとなり得る。
構成的に武装したヒートショック応答:コウモリからの教訓
コウモリは、狂犬病、エボラ、SARSコロナウイルス[212,213]、COVID-19病因菌SARS-CoV-2を含む、致死性の高いウイルス性人獣共通感染症の宿主保存場所であることで、最近注目を集めている。このことから、ヒトとそれほど似ていない哺乳類[214,215]が、どのようにして明らかな病気の兆候なしに長期的なウイルス感染をサポートすることができるのかという疑問が生じた。
検出後、ウイルスを破壊するために、ヒト細胞は、主に上皮細胞、線維芽細胞、単球および形質細胞性樹状細胞によって産生される「オンデマンド」タイプのI型IFN(例えば、IFN-α、IFN-β、IFN-ε、IFN-κ、IFN-ω)を採用する[216]。これは、ウイルスの存在を周囲の細胞および免疫細胞に助言する自己分泌シグナルとパラクリンシグナルの両方をデフラグする[81]。IL-12で活性化された細胞傷害性T細胞とTh1リンパ球は、ウイルスに感染した細胞を攻撃するためにII型IFN-γを産生する [161]。ヒトは、サイトカインストームを避けるためにそのような炎症反応の解決を迅速に開始しなければならないと同時に、炎症に基づくウイルスクリアランスを促進するために、細かく調整された免疫炎症バランスを必要とする。
一方、コウモリは、炎症によって誘発された傷害の信号に直面することなくウイルスの量を抑制する抗ウイルス防御の茄多を持っている。コウモリの中には、インターフェロン経路という名前の抗ウイルス免疫応答が永続的にオンになっているものもある[81]。この状態でも、コウモリは過剰なサイトカイン産生に悩まされることはない。IFN応答はJAK-STATおよびNF-κB経路[216]を介してすべてプロ炎症性であるため、持続的で全身性の炎症の維持は、ヒトおよび他の非揮発性哺乳類には耐え難いものであろう。コウモリは、そうでなければ、そのような超警戒的な免疫応答[81]からそれらを保護する抗炎症機能で進化し、全身性炎症性症候群(SIRS)や敗血症の任意のフォームを回避して高揚感のあるIFN媒介炎症を武装することができる(図4)。
図4
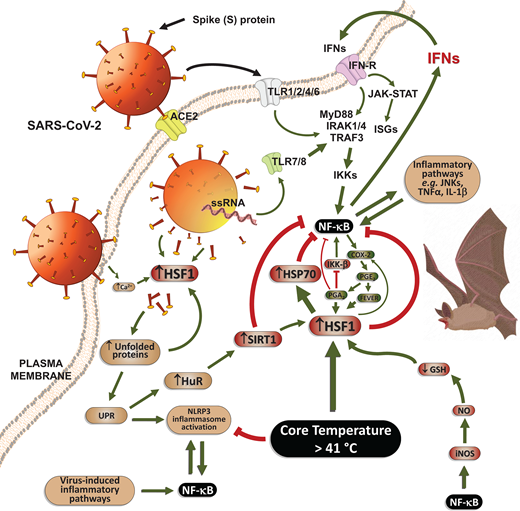
コウモリではプロ炎症性インターフェロン経路と抗炎症性ヒートショック応答(HSR)が構成的に活性化される
ウイルス誘導性インターフェロン(IFN)経路が必要に応じて活性化されるヒトとは異なり、多くのコウモリは、最も致死的なウイルスに対処できるように構成的に活性化されたIFN経路を含む顕著な抗ウイルス性炎症反応を進化させた。同時に、コウモリは、HSRを介して炎症を解決することができるので、これらの動物がサイトカインストームに苦しむことがないように、抗炎症性HSPの構成的に高い発現を提示する。コウモリの固有の抗炎症反応は、HSF1依存性と非依存性の両方の経路を中心としており、コウモリのコア温度が41℃以上に振動することに主に関連している。略語と表記は図1の凡例の通りである。
鳥類[217-219]と同様に、コウモリの翼筋は飛行中に大量の熱を発生し、コウモリのコア温度は41.1-42.1℃の範囲内で振動する[220]。驚くことではないが、コウモリは他の哺乳類と比較して、不釣り合いに高い耐熱性を進化させた。コウモリ(鳥ではない)の細胞は、40℃で数日間培養しても生存し増殖するが、これはHSPに依存した効果である[80]。実際、コウモリ細胞におけるHSP70(HSPA1A遺伝子)の基底発現は、ヒト細胞に見られる発現の少なくとも10倍であり、一方、HSP70の共役型(HSPA8遺伝子)は、ヒト細胞よりもコウモリ細胞の方が200倍近く高い[80]。ER シャペロン GRP78 (HSPA5 遺伝子) の基底発現は、HSF1 依存性および-依存性に関わらず、ヒト細胞よりもコウモリの方が 400 倍以上も多く発現している。さらに、コウモリ細胞では、鳥やマウス、ヒトの細胞に比べて、熱によって誘発されるHSP70sの発現が著しく高くなっている。これらの特徴は、マウスのような同程度の大きさと体表体積比を持つ哺乳類と比較して、コウモリの印象的な保護恒常性、酸化ストレスに対する本質的な抵抗力、および強化された長寿(約34年!)を説明しているかもしれない。
コウモリは、他に類を見ないHSPをベースとした抗炎症性HSRを有しており、ウイルス誘発性炎症反応を常に受けているにもかかわらず、慢性変性疾患やサイトカインストーム[221]を発症することはない(図4)。このような高い抗炎症反応のために、コウモリ免疫細胞はTLRリガンドや無菌刺激に応答してNLRP3インフラマソームの活性化を強力に抑制している[221]。同じことが、MERSコロナウイルスを含む異なるウイルスに対しても起こり、コウモリ細胞のウイルス撃退能力に影響を与えることはない[221]。高い抗炎症応答はまた、HSP70が抗ウイルスIFN-γ産生を増強するという知見も含まれている[187]一方、IFNは2つのレベルでHSP発現を調節する:ヒートショック遺伝子の転写速度を増強することによって、およびHSPをコードするmRNAの安定性を増加させることによって[222]。IFN-αはまた、ウイルス感染細胞においてcyPGによって誘導されたHSP70合成を増強し[223]、IFN-γはエキソソーム経路を介して細胞外空間に向かってHSP70の合成と放出を増加させ、ここでeHSP70はナイーブ樹状細胞に影響を与える[82]。構成的なHSRは、他の動物と比較して少なくとも3つの明確な利点をコウモリに与えている:ウイルス誘発性ERストレス時の迅速なプロテオスタティック応答、INF抗ウイルス活性の強さ、組織損傷を回避するための炎症の迅速な解決です(図4)。
異なる種ではあるが、比較生理学とコウモリの抗ウイルス性および抗炎症性保護経路の類似性は、SARS患者のサイトカインストームを回避したり治療したりする方法について、いくつかの手がかりを与えることができる。これは、一般的な解熱剤の無制御な使用を再考する必要性を強化するものである。さらに、感染症患者、特に慢性炎症性合併症を有する患者における補助的ケアとして、抗炎症性HSRを増幅させるために、患者の体温を発熱域内に維持することについての議論を取り上げている。これらの重要な課題は、さらなる評価に値する。
おわりに
COVID-19の有効な治療法を見出すための多くの努力にもかかわらず、現在進行中の再利用薬剤を用いたRCT[224-226]を含めて、残念ながら、これまでに説得力のある成功した治療法は示されていない。特に、重症化のリスクが高い患者群では、以前の慢性炎症性疾患に関連した抑制されたHSRは、ウイルス感染とより多くの炎症に対応するために、体に負担がかかりすぎる可能性がある。強い炎症反応はSARS-CoV-2を打ち負かすことができる唯一のリソースであるが、悪化した炎症は致命的なサイトカインストームを引き起こす可能性があるという二重の状態が発生している可能性がある。
これらの知見は、次回のRTCでSARS患者のケアと発熱の軽減を再評価するための根拠を提供しているが、図5に描かれているように、多くの未解決の疑問がある。ウイルスに対する自然な反応を利用して、免疫炎症状態のバランスを整えながら寛解に至る患者の割合は実際にはどの程度なのだろうか?抗炎症薬を最初に使用しないようにし、抗ウイルス性炎症の急性期の経過を臨床的にモニターするとどうなるのであろうか?COVID-19が疑われる一般の患者に対しては、解熱剤/NSAIDsの市販薬の使用は控えるべきではないであろうか(図5A)?リスクの高い患者には(いつ)抗炎症治療が必要か?次のRCTでは、支持療法と併用して温熱療法を試験することができるか?
図5
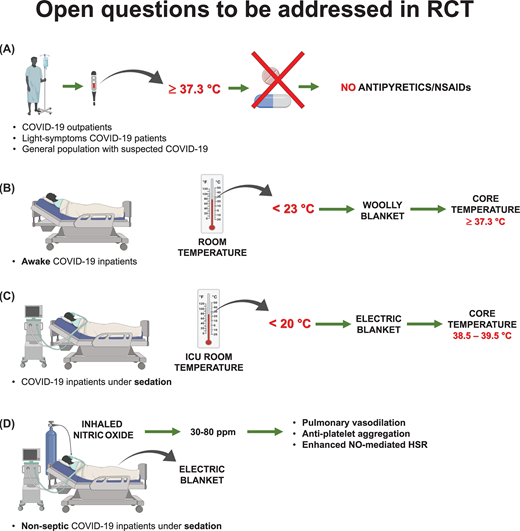
無作為化臨床試験で取り組むべき未解決の問題
(A) 解熱剤/抗炎症剤を最初は避け、抗ウイルス性炎症の急性期の経過を患者が臨床的にモニターできるようにしたらどうなるだろうか?このRCTの同一群を細分化して、COVID-19が疑われる一般集団を対象に、解熱剤/抗炎症剤治療中の患者とそうでない患者を対象にした試験を実施することができる。
(B) 次のRCTで支持療法と併用して温熱療法が試験されるとしたらどうであろうか?
(C)ICUは暖かい場所や熱い場所ではないので、患者の代謝状態を悪化させたり、追加の呼吸努力を必要としたりすることなく、体温をより高い値に維持するために電気毛布の使用(特に鎮静下の患者で)を実施するのか?
(D) NOは抗凝固作用の他に天然の抗ウイルス剤であり、抗炎症性HSRの強力な誘導剤でもあるので、血行動態が不安定でない患者にはNO吸入療法を行うのか?
COVID-19におけるコルチコステロイドの使用はまだ議論のあるところである[142,224]ので、熱の維持または受動的加熱によるかどうかにかかわらず、温熱療法は実行可能な処置として考慮され得る。重要な懸念事項は、ICUが温かくて熱い場所であることはまれであるため、患者の体温が下がらないように注意を払うべきであり、特に入院患者が鎮静される場合には注意が必要であるということである。
患者の代謝状態を悪化させたり、追加の呼吸努力を必要としたりすることなく、体温をより高い値に維持するために電気毛布(図5B-D)を使用することはできるか?
NOは抗凝固剤であるだけでなく、天然の抗ウイルス剤であり、抗炎症性HSRの強力な誘導剤でもあるので、これらの患者(図5D)には、血行動態を不安定にすることなくNO吸入療法を行うことができるであろうか?
さらに、α1アドレナリンがHSP70の発現と放出を強力に誘導することを考慮すると、これらの患者における血管プレッサー治療は、より高いHSRを誘導することができるのであろうか?
適切なRCTはこれらの関連する問題に答えることができる。おそらく患者が解熱剤で炎症を回避しようとするのではなく、高い体温を持つことによって自然に炎症を解決することを可能にすることは、1847年5月末にIgnaz Philipp Semmelweis博士が1847年5月にウィーンで有毒な「subter ungues」の死骸物質を除去するために手洗いをすることを提唱したのと同じくらい単純で積極的な対策であろう[227]。
これはまた、COVID-19の患者が炎症を発症しても、サイトカインストームを起こさずにHSRでそれを解決することができるようにするというゴルディアスの結び目(「箱の外で考える」ことによって解決されるかもしれない難問)が、特に慢性炎症性疾患を持つ患者には解かれようとしていることを示唆しているのかもしれない。
「Quæ medicamenta non sanat, æ ferrum sanat. Quæ ferrum non sanat, æ ignis sanat., æ insanabilia existimare oportet. 」
薬で治らないものはメスで治すことができる。メスで治らないものは熱で治すことができる。熱で治らないものは不治の病と判断されねばならない」
ヒポクラテスの格言