Contents
Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition
2007年12月16日受理
要旨
- ヒトで最も臨床的に重要な膜貫通輸送体の一つであるP糖タンパク質(P-gp/MDR1)は、ABCB1/MDR1遺伝子にコードされている。近年、P-gp/MDR1の構造的特徴が明らかになり、P-gp/MDR1による薬物の結合・輸送に関する生化学的エビデンスの再評価が可能になった。
- P-gp/MDR1は、腫瘍細胞に発現しているだけでなく、ヒトの様々な組織にも存在している。P-gp/MDR1は、腸管上皮細胞、胆管、腎管細胞、胎盤の先端表面、脳や精巣の毛細血管内皮細胞の腔内表面に存在している。
- P-gp/MDR1は、化学療法薬に抵抗性を示したがん細胞に多剤耐性(MDR)の表現型を与える。また、P-gp/MDR1の活性は、様々な薬剤の吸収・排泄に幅広く作用することから、非がん関連の薬物療法においても臨床的に重要な役割を果たしている。
- P-gp/MDR1は、細胞毒性化合物などの有害物質を消化管、胆汁、尿中に排泄する。また、血液脳関門の機能にも関与している。
- P-gp/MDR1の最も興味深い特徴は、その基質が有機陽イオン、炭水化物、アミノ酸、一部の抗生物質などの低分子から、多糖類やタンパク質などの高分子まで、その構造や機能性が大きく異なることである。
- MDR1遺伝子には、かなりの数の一塩基多型が発見されている。これらの一塩基多型は、P-gp/MDR1基質の経口バイオアベイラビリティーの変化、薬剤耐性、ヒト疾患への感受性と関連している。
- 誘導および/または阻害によるP-gp/MDR1活性の変化は、変化した薬物動態および応答を伴う薬物間相互作用を引き起こす可能性がある。
- P-gp/MDR1 の生理機能と薬理学的役割を解明するためには、さらなる研究が必要である。
キーワード
P糖タンパク質(P-gp)薬物動態、血液脳関門、腸管吸収、一塩基多型
はじめに
哺乳類のアデノシン三リン酸(ATP)結合カセット(ABC)トランスポーターのスーパーファミリーは、AからGに指定された7つのファミリーに細分化されている機能的に多様な膜貫通タンパク質の多数を構成している(BorstおよびElferink 2002; Locher 2004; Tian et al 2005; Piddock 2006; Linton 2007; LintonおよびHiggins 2007)。この輸送スーパーファミリーのメンバーは、ATP結合ヒダを取り囲む200アミノ酸の高いアミノ酸類似性を表示する。ABCスーパーファミリーのメンバーであるP糖タンパク質(P-gp)/MDR1は、ABCB1/MDR1遺伝子の転写の結果として発現する。これは、クローン化されたABCトランスポーターの最初のものであり、そのように、P-gpは、化学療法薬に抵抗性を開発した癌細胞に多剤耐性(MDR)表現型を付与する上で重要な役割を果たすことに起因して、ABCトランスポータースーパーファミリーの中で最も特徴づけられたタンパク質である(Romsicki and Sharom 1998; Kwan and Brodie 2005)。P-gp/MDR1は、腎臓、肝臓、腸、副腎、血液脳関門などの多くの分泌細胞の先端膜に発現しており、正常な機能は薬物とその代謝物の排泄を伴う(Thiebaut er al)。 1987; Dean 2002)。このように、P-gp/MDR1は薬物動態に重要な役割を果たしている。
P-gp/MDR1活性は、様々な薬物の吸収および排泄に対する広範な効果のため、非がん関連の薬物療法において臨床的に重要である。P-gp/MDR1の最も興味深い特徴の一つは、その多くの基質が、有機カチオン、炭水化物、アミノ酸、一部の抗生物質などの低分子から、多糖類やタンパク質などの高分子まで、その構造や機能性が大きく異なることである(Wang er al)。
ヒトとマウスのP-gpsの遺伝子メンバーの機能的な類似性は、薬物の吸収、分布およびクリアランスにおけるP-gp/MDR1の重要性を評価する上でのより良い理解を提供するために、トランスジェニックMDRノックアウトマウスを用いた実験を可能にする。MDR1ノックアウトマウス(Mealey 2004)で見られるように、P-gp/MDR1は、多くの薬物の脳内への侵入を防ぐ上で非常に重要であるように思われる。mdr1aおよびmdr1bノックアウトマウスを用いた研究は、体内でのP-gp基質の蓄積の増加を示すP-gpの完全ノックアウトを示している(Mealey 2004)。しかし、mdrノックアウトマウスは、生理的異常を伴わずに健康で繁殖力があることが確認された。本論文では、P-gpの構造、機能、制御、および関連する臨床的意味合いについて、現在の知見をまとめ、更新することを目的としている。P-gp/MDR1の詳細については、Chinn and Kroetz (2007), Higgins (2007), Leschzinerら (2007), Lin (2007), Marchettiら (2007)による最近の優れたレビューを参照されたい。
P-gpのトポロジーと機能的関連性
P-gpのトポロジカルな特徴
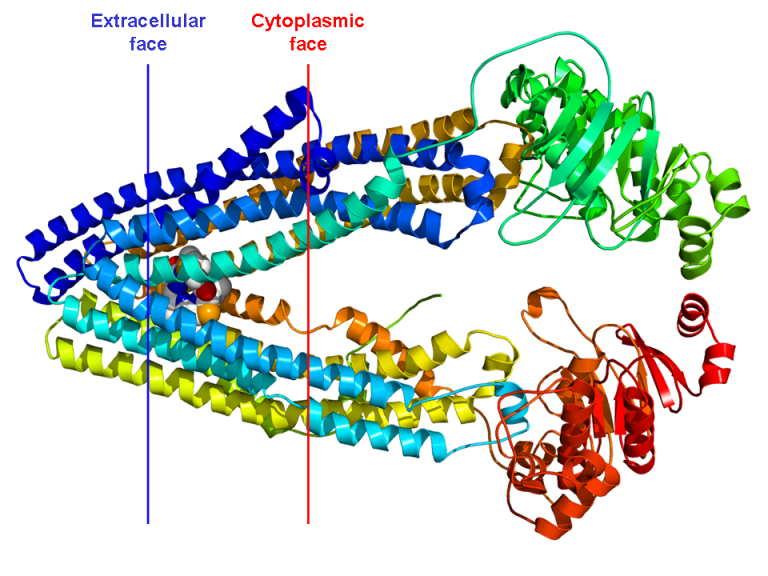
P-gp/MDR1は1276~1280個のアミノ酸からなり、分子量は約170kDaである。一般に受け入れられているP-gp/MDR1のトポロジカル構造のモデルは、分子の各半分がヌクレオチド結合ドメイン(NBD)を含むタンデム二重構造であり、予測された6つの高度に疎水性の膜貫通ドメイン(TMD)を明らかにしている(図1)(Borst and Elferink 2002; Shilling er al 2006)。TMDは、薬物分子が膜を横切る経路を形成していると考えられている。NH2-末端およびCOOH末端、ならびにNBDは、細胞内に配置され、最初の細胞外ループはN-グリコシル化されている。各NBDは、ウォーカーAおよびBモチーフと呼ばれる2つのコアコンセンサスモチーフと、ABCトランスポーターのSシグネチャーから構成される(Walker er al)。 これらのモチーフは、一般的に幅広いATPアーゼに見られ、ヌクレオチドの結合および加水分解に直接関与している。2つの半分子は、高電荷を帯びた「リンカー領域」によって分離されており、プロテインキナーゼCによっていくつかの部位でリン酸化されている(Higgins er al)。 1997)。
図1 P糖タンパク質の提案されたトポロジー。
原文参照
P-gp/MDR1の異なるトポロジカル配向が報告されており、いくつかの研究では、ヌクレオチド結合に伴うP-gpの構造のコンフォーマルな変化が、エピトープアクセシビリティの変化をもたらすことが示されている(Druley er al 2001a, b; Ruth er al 2001)、プロテアーゼ感受性(Julien er al 2000)、薬物結合(Martin er al 2000b)、蛍光および分光学的特性の測定(Sonveaux er al)。 1996, 1999)。このコンフォーマルな変化は、脂質二重層の深さ全体にわたって孔の片側を開き、脂質二重層から中央の孔へのアクセスを可能にする。加水分解サイクルの異なる段階でトラップされたP-gpのための二次元プロジェクションマップはまた、非加水分解性ATPアナログアデニル-イミド二リン酸の結合時およびアデノシン二リン酸(ADP/VI状態)の存在下でのバナデートラッピング後にTMDの細胞外面での実質的なコンフォメーション変化を示唆している(Rosenberg et al 2001)。
P-gpは、トランスポーターの構造分解能のための生物学的材料を得るために、過剰発現、精製、および再構成に成功してきた。この努力の結果、電子顕微鏡を用いた低分解能の構造が得られた(Rosenberg er al 2001, 2003)。初期の研究では、25-Aの電子顕微鏡構造から、その内面にNBDと結合する重要なチャンバーが明らかになった(Rosenberg er al 2001)。別々の触媒状態にトラップされたP-gpのさらなる研究は、タンパク質のTMDの劇的な再配列を明らかにする(Rosenberg et al 2003)。これらの後者の観察は、任意の哺乳類のABCトランスポーターのための唯一の構造データのままである。しかし、基質結合ドメインや輸送過程のダイナミクスの原子レベルでの詳細はまだ不明である。それにもかかわらず、P-gpをはじめとするATP依存性多剤トランスポーターの構造は、現在のところ詳細な構造が明らかにされていない。
多くの研究が、P-gp/MDR1によって媒介される輸送過程の速度論および熱力学を探求してきた(Ramachandra er al)。 1998; Al-Shawi er al 2003; Pleban er al 2005)。アミノ酸残基を置換し、ATPアーゼ活性、基質結合および輸送動態に関して変異体P-gpを特徴付けるために、部位特異的変異誘発が使用されてきた(Ma er al)。 1997; Hafkemeyer er al)。 1998; Loo and Clarke 1999a,b,2000; Song and Melera 2001; Loo er al 2003b, 2004; Tombline er al 2004)。しかし、これらの変異は、P-gpの一次アミノ酸配列全体に存在するため、薬物結合部位の地図を提供することができなかった。
ATP結合ドメイン
両方のNBDは、P-gp/MDR1の適切な機能に不可欠であり、P-gp/MDR1の活性は、ATPの存在に完全に依存している(RomsickiとSharom 1998)。ATP結合ドメインは、しばしば急な濃度勾配(Buxbaum 1999)に対して、膜を介して基質をポンプするためにP-gp/MDR1に必要なエネルギーを提供するためにADPにATPを変換するATPaseとして機能する。
ATPは、全体のP-gpタンパク質の活性を可能にするために両方のNBDに結合する必要がある。しかし、両方の結合したATP分子の加水分解が活性化に必要かどうかは不明である(Kimura er al 2007)。
P-gp/MDR1の主要な構造変化は、ATP加水分解ではなくATP結合時に起こる。それはしばしばATP加水分解が輸送プロセスを駆動すると仮定されているが、データは、P-gp/MDR1への薬物結合親和性の低下は、加水分解ではなく、ATP結合にも起因することを示している(Martin et al 2000b 2001; Rosenberg et al 2001)。したがって、ATP結合は、薬物結合親和性を減少させ、細胞外の環境(中央の水性細孔)に薬物結合部位を露出させる主要な構造変化を駆動するように見える; ATP加水分解は、したがって、単にトランスポーター分子(SaunaおよびAmbudkar 2000)を’リセット’する可能性がある。
P-gpはどのように多くの結合部位を持っているか?
証拠は、P-gp/MDR1が、輸送と調節という2つのカテゴリーに均等に分けられた複数の結合部位を持っていることを示唆している(Martin er al 2000a, b; Shilling er al 2006)。ロダミン123によるHoechst 33342輸送の刺激によって実証されたように、これらの異なる部位は相互作用しているかもしれない(Shapiro and Ling 1997)。P-gp/MDR1のN末端ハーフおよびC末端ハーフの両方が結合部位を含み、これらの2つの部位は、タンパク質全体の構造において単一の領域を生成する可能性がある(Morris er al)。 1994; Loo er al 2003a)。さらに、インドリジンスルホンSR33557および1,4-ジヒドロピリジンのような化合物が、ビンブラスチンの輸送のためのP-gp/MDR1上の結合部位にアロステリックな制御を与えるような、輸送部位とは異なるP-gp/MDR1のアロステリック部位の証拠がある(Martin er al)。 1997)。P-gp/MDR1上の複数の薬物結合部位の存在は、このタンパク質と相互作用することが知られている広範囲の化合物の説明を提供し得る。これまでの研究では、P-gp/MDR1上に2つの主要な基質結合部位、すなわちTMD部位5および6,TMD部位11および12が存在することが決定されている(Wang er al 2003)。
P-gp/MDR1のすべての結合部位は、輸送部位からの作用に影響を与える調節因子とともに、高親和性と低親和性のコンフォメーションを切り替えることができるようである。これは、基質結合および/またはATP加水分解のような刺激によって引き起こされる可能性がある。P-gp/MDR1のコンフォメーション変化は、2H/H交換速度論、タンパク質分解アクセス性、および抗体エピトープ認識の変化を用いて実証されている(Mechetner et al 1997; Sonveaux et al 1999)。
P-gp/MDR1による基質認識、結合、および輸送の分子機構を解明する必要性が強くある。この情報は、P-gp/MDR1による認識をバイパスする新規薬剤の開発を可能にするかもしれない。さらに、P-gp/MDR1に強固に結合し、P-gp/MDR1を不活性化する新しい薬剤が設計され、生成される可能性がある。
P-gp基質の共通の生理化学的性質
P-gp/MDR1 は、様々な物質に対する細胞保護のための流出ポンプとしての性質から、基質は有機陽イオン、炭水化物、アミノ酸、一部の抗生物質などの低分子から多糖類、タンパク質などの高分子まで、その大きさ、構造、機能は多岐にわたっている。P-gp/MDR1は薬剤耐性や薬剤の薬物動態に重要な役割を果たしているため、このタンパク質とその低分子基質との相互作用に必要な分子特性を明らかにするために多くの研究が行われてきた。Seelig (1998)は、ほとんどのP-gp/MDR1基質は、それぞれ2.5 Aと4.6 Aの空間的に固定された2つまたは3つの電子供与体基を有しており、これらの要素の数が多いほど薬物結合の親和性が高くなることを明らかにしている。これに対応して、P-gp/MDR1の膜貫通配列には、基質認識に関与する水素結合性ドナー側鎖を持つアミノ酸の割合が高いことがわかった。さらなる研究では、脂質膜への分割が基質とP-gp/MDR1との相互作用のための速度制限ステップであり、P-gp-基質複合体の解離は、基質とP-gp/MDR1の間に形成される水素結合の数と強度によって決定されることがわかっている(Seelig and Landwojtowicz 2000)。
P-gp/MDR1の基質と非基質を識別することができる複数のファルマコフォアモデルが、文献からの大規模なデータセットから同定されている(Penzotti er al 2002)。基質または非基質としての単層流出の分類に基づく別のより一般的な定量的構造活性関係(QSAR)が確立されている(Ekins er al)。 Cianchetta et al 2005)は、ファーマコフォアP-gp/MDR1 の認識要素は、2 つの疎水性基 16.5 A˚ と 2 つの水素結合受容体基 11.5 A˚ である。P-gp/MDR1の基質と非基質を識別するためのQSARモデルは、53種類の薬剤を用いて計算された分子記述子に基づいて設定されている(Crivori er al 2006)。163化合物のデータセットをP-gp/MDR1基質または非基質として分類するために線形判別モデルが開発された(Cabrera er al 2006)。最近では、パーティクルスウォームアルゴリズムとサポートベクターマシンアプローチを用いて、90%以上の精度でP-gp/MDR1基質を予測するためのより効果的なモデルが設定されている(Huang er al 2007a)。
他の研究では、薬物のP-gp/MDR1への結合能力に寄与する親油性、水素結合能、分子量、表面積などの物理化学的特徴があるかもしれないことが示唆されている(Bain er al)。 1997; Wang er al 2003)。このような化学的特性としては、log-P値が2.92以上の親油性、18原子長以上の分子軸、最高占有軌道(Ehomo)値のエネルギーが高いこと、少なくとも1つの第3級塩基性窒素原子の存在、およびMrが800以下であることが挙げられる。
しかしながら、少数のP-gp/MDR1基質は、サイズが30000ダルトンを超える大きな分子であり、生理学的pHにおいて有機カチオン性である(例えば、ロクロニウムおよびオクトレオチド)(Yamadada et al 1998)。これらの化合物は、一般に、酸素、窒素、および不飽和系内に-軌道を有する基のような電子陰性基の比較的高い濃度を有する。
基質としてのP-gp/MDR1による化合物の認識に必要な分子特性の同定は、所望のP-gp/MDR1相互作用(例えば、流出、阻害、または相互作用なし)に向けてリードの最適化を合理的に指示するのに有用である(Raub 2006)。新規化合物がP-gp/MDR1基質であるかどうかを知ることは、その組織分布(例えば、脳への分布)や消去に関する重要な情報を提供することができ、候補の選択の基準となる可能性がある。ほとんどの抗ヒト免疫不全ウイルス(HIV)薬がP-gp/MDR1基質であることを考えると、P-gp/MDR1による基質としての認識を減らすように化学構造を修飾することは、中枢神経系(CNS)への浸透を改善することにつながり、その結果、より優れた抗ウイルスプロファイルを達成することができる。
P-gp基質とCYP3A4基質のオーバーラップ
CYP3AおよびP-gp/MDR1については、重複する基質特異性および組織分布の広い範囲があり、CYP/P-gpバイ基質として知られている。P-gp/MDR1とCYP3Aタンパク質の両方が、経口投与された薬物のバイオアベイラビリティにおける保護バリアとして相乗的に作用する(Cummins et al 2002)。P-gp/MDR1とCYP3Aの局在は、CYP3A酵素によって代謝される基質の量がP-gp/MDR1によって制御される可能性があることを示している(Cummins er al 2002)。P-gp/MDR1はまた、腸球への受動的吸収後に内腔に戻る薬物の能動的輸送を打ち消すことにも関与している(Cummins et al 2002)。薬物は、内腔と腸球の間を継続的に循環し、CYP3Aが薬物分子に常にアクセスすることを可能にする。これは、最終的には、薬物の濃度の飽和および非吸収の減少をもたらす。したがって、腸内のCYP3Aによって媒介されるP-gp/MDR1のカウンター輸送および代謝の循環は、全身循環に入る分子の濃度を制御することによって、薬物の経口バイオアベイラビリティを低下させる。しかし、CYP3AとP-gp/MDR1のそれぞれの役割を十分に理解することは困難である。
P-gpの阻害剤
特定の薬剤によるP-gp/MDR1の阻害に関する研究のほとんどは、がん細胞における抵抗性の逆転の可能性の調査に関与している。これらのP-gp/MDR1阻害剤の潜在的な治療的用途は、既存の化学療法薬との共同投与(共同投与が他の相互作用を引き起こさないことを条件に)が、P-gp/MDR1によって引き起こされる癌細胞のMDRを否定するのに役立つということである(Dean 2002)。ほとんどのP-gp/MDR1結合阻害剤は、芳香環構造、第三級または第二級アミノ基、高い親油性などの共通の化学的特徴を共有している(Wang er al 2003)が、P-gp/MDR1基質は全体として阻害作用のクラスが異なる。表Iは、いくつかの一般的なP-gp/MDR1阻害剤を示している。表Iに示すいくつかのP-gp/MDR1阻害剤は、化学療法薬耐性を低下させる能力のために、ケモセンシッターとも呼ばれている(Shiraga et al 2001)。臨床現場では、MDRをブロックするための新規な強力なP-gp/MDR1阻害剤を同定することへの関心が高まっている。
P-gp/MDR1の阻害剤がその活性を発揮する主な方法は、P-gp/MDR1に対して非常に高親和性の基質であり、非競合的に結合する(したがって、他の薬剤が結合することを許さない)か、またはATP結合部位でATP加水分解を効率的に阻害するか、またはATP結合に関与するプロテインキナーゼCを阻害することである(Wang et al 2003)。薬剤によって媒介されるP-gp/MDR1の誘導は、プロテインキナーゼCに対して活性を有する非特異的プロテインキナーゼ阻害剤によってブロックされる(ChaudharyおよびRoninsonson 1993)。
P-gpの誘導
P-gpの誘導剤
P-gp/MDR1は、フェニトイン、リトナビル、ネルフィナビル、アムプレナビル、プロベネシド、リファンピシン、およびハーブ抗うつ剤セントジョーンズワートを含む多くの薬剤によって誘導される(表I)。P-gp/MDR1は、多くの化学物質だけでなく、X線照射、紫外線照射、ヒートショックなどの物理的ストレスによっても誘導される(Seelig 1998)。薬物によるP-gp/MDR1誘導は、2つの分野で最も臨床的に関連している。すなわち、薬物吸収および経口バイオアベイラビリティの変化をもたらすP-gp媒介の薬物間相互作用と、化学療法剤に対する癌細胞のMDRの発現である。P-gp/MDR1誘導の程度と臨床的影響は、誘導剤、患者、および共同投与される薬剤に関連する因子に依存する。P-gp/MDR1の過剰発現とその結果としてのMDRは、がん化学療法において大きな問題となっている。
P-gp誘導における核内受容体の役割
ABCトランスポーター遺伝子発現の制御は、多数の核内受容体の関与を伴う(Staudinger er al 2003; Klaassen and Slitt 2005; Urquhart er al 2007)。核内受容体は、ヘテロ二量体として作用する転写因子のファミリーを構成し、プロモーター要素に結合して遺伝子発現を誘導する(Nagy er al 2006)。トランスポーター遺伝子は、膜の回収および再挿入、翻訳および転写を含むいくつかのレベルで制御されている(Trauner and Boyer 2003)。ABCトランスポーターの発現に関連する核内受容体は、肝臓X受容体(LXR)ファルネソイドX受容体(FXR)プレグナンX受容体(PXR)およびペルオキシソーム増殖因子活性化受容体および(PPARおよびPPAR)である(BorstおよびElferink 2002)。多数のキセノバイオティクスによるCYP3A4およびCYP2B6遺伝子の誘導は、PXRの活性化を介して媒介されることがよく知られている(Bertilsson er al)。 1998)。PXRは、ヒトにおけるリファンピシン、フェノバルビタール、およびミフェプリストンを含む多様な数の化合物によって活性化される(Kliewer er al)。1998)。PXRは、げっ歯類Oatp1a4(Staudinger et al 2003)Oatp2(Staudinger et al 2001;KliewerおよびWillson 2002)ヒトMDR1/P-gp(Geick et al 2001)マウスMrp1(Kauffmann et al 2002)Mrp2(Kauffmann et al 2002)およびMrp3(Teng et al 2003)の発現を媒介する。さらに、構成性アンドロステイン受容体(CAR)活性化は、マウス肝臓のMrp2-7 mRNAを誘導し(Maher et al 2005)Mrp4およびスルホトランスフェラーゼ2a1の調節に関与している(Assem et al 2004)。PPARアゴニストであるクロフィブラートは、マウス肝臓における有機陰イオン輸送ペプチド(Oatp1a1,Oatp1a4,およびOatp1b2)のmRNA発現にはほとんど影響を与えないが、Mrp3およびMrp4流出トランスポーターの遺伝子およびタンパク質発現をPPAR依存的に誘導する(Moffit er al)。
ヒトMDR1遺伝子の転写活性の制御は、MDR1プロモーターのコンセンサスシスエレメントに結合するいくつかの転写活性タンパク質に依存する。例えば、Sp1転写因子は、メジャーþ1開始部位の56〜45塩基上流に位置するプロモーター内のGCに富んだ領域に結合する。Sp1と倒立CCAAAT要素(Y-ボックス)の両方が、紫外線照射下でのMDR1活性化に必須である(JinおよびScotto 1998)。追加の転写因子やプロモーター配列も遺伝子の転写に関与している可能性があるが、これはまだ完全には解明されていない。MDR1遺伝子の転写の実際のメカニズムは驚くほど複雑であり、部分的にしか理解されていない。
P-gp/MDR1発現の誘導は、P-gpへの直接的な薬物結合を介して起こるのではなく、むしろ転写レベルでの核内因子によって制御されている(Kuwano er al 2004)。おそらく、MDR1遺伝子の最も重要な核内誘導因子は、Yボックス結合タンパク質1(YB-1)である。YB-1の活性はP-gp/MDR1の発現と強い相関があり、YB-1阻害物質を導入すると、YB-1の発現低下がP-gp/MDR1の発現低下につながることが示された(Kuwano er al 2004)。YB-1は活性化されると細胞質から核に移動し、デオキシリボ核酸(DNA)領域に結合し(Ohga er al)。 YB-1がストレス誘発性タンパク質であることが示唆されている(Kuwano er al 2003)-薬物がMDR1プロモーター領域を誘導するだけでなく、熱、寒さ、紫外線(Chin er al)。 1990)もYB-1活性化を誘導しうることが研究で示されている。
表I. P糖タンパク質の基質、誘導剤および阻害剤
原文参照
YB-1は、細胞増殖、DNA合成、細胞損傷の修復などの機能に関連するタンパク質である(Shibahara er al)。 ヒトの癌の増殖および薬剤耐性の観点からのその重要性は実証されており、化学療法薬の誘導および化学療法によって引き起こされる細胞損傷を介した誘導の両方によって、癌細胞においてYB-1活性が増加することを示す研究(桑野 et al 2003, 2004)が行われている。では、なぜP-gp/MDR1はMDRの観点からこれほどまでに活性が高いのであろうか?それは、ヒトMDR1プロモーター遺伝子の発現とP-gp/MDR1の発現は、P-gp/MDR1によって薬物が輸送されるかどうかにかかわらず、細胞障害性薬物による細胞障害に応答して誘導されるからである(Chaudhary and Roninson 1993)。P-gp/MDR1転写の活性化は、そのストレスが細胞損傷、薬物応答、または他の手段から生じる場合には、細胞における主要なストレス応答機構の一つである(Ledoux er al 2003)。
薬物の薬物動態におけるP-gpの役割
薬物の腸内吸収におけるP-gpと他のトランスポーターの役割の比較
P-gp/MDR1は腸内の上皮細胞に高濃度に存在するため(Dean 2002)経口投与された薬剤の吸収性とバイオアベイラビリティーを変化させる可能性があると考えられている(Mealey 2004)。P-gp/MDR1阻害剤とP-gpの基質であることが知られているP-gp阻害剤との併用投与が血漿中濃度に及ぼす影響を調べるために、多くの研究が行われてきた(Mealey 2004)。ジゴキシン、オピオイド、パクリタキセル、シクロスポリンなどのP-gp基質を経口投与したP-gpノックアウトマウスを用いた研究が行われている(Mealey 2004)。これらの研究では、P-gpノックアウトマウスでは、P-gp基質の血漿中濃度が大幅に上昇することが一貫して示されている。同様に、ヒトの研究では、モルヒネを経口投与する前にP-gp阻害薬を投与した場合、モルヒネを経口投与した場合と比較して、モルヒネの血漿中濃度が2倍に増加することが示されている(Mealey 2004)。この証拠は、腸管上皮細胞における P-gp/MDR1 活性が多くの薬物のバイオアベイラビリティーに大きく影響することを示しており、いくつかの観察結果を指摘している。すなわち、P-gpは、吸収後に上皮細胞から薬物を送り出し、排泄のために腸に戻すという重要な役割を担っているようであること、腸内でのP-gp活性の低下は、薬物のバイオアベイラビリティーの劇的な増加につながる可能性があること、薬物相互作用やMDR1遺伝子の遺伝的変異のような様々な要因によるP-gp活性の増加は、P-gp基質である広範囲の薬物の治療失敗につながる可能性があることである。
基質特異性および体内の位置が類似しているため、CYP3AおよびP-gp/MDR1は、経口薬物吸収を制限する上で相補的な役割を果たす(Wang er al)。 肝臓が薬物のファーストパス代謝の主な部位であるにもかかわらず、CYP3Aを介した代謝は腸の上皮細胞で行われていることが示されている(Canaparo er al)。 多くの経口投与された薬物は、標的部位に到達する前にいくつかの障壁を乗り越えなければならない(Chan er al)。 MRP2およびMRP4は、P-gp/MDR1およびBCRP/MXR(ABCG2)とともに腸球の先端膜/内膜に局在していることが示されており、基質薬物の腸管吸収の主要な障壁を形成していると考えられている(図2)(Chan er al)。2004; Yu er al)。2007)。
薬物の胆道排泄におけるP-gpと他のトランスポーターの役割の比較
肝トランスポーターは、胆汁の形成と外来物質の処分の調節に関与している(Pauli-Magnus and Meier 2006)。肝細胞は、基底側ドメインと先端側ドメインを持つ分極化された形質膜を有しており、血液中の内因性化合物と外因性化合物の胆汁中へのベクトル運動を可能にしている(Alrefai and Gill 2007)。血液に到達した薬物は、その後、肝臓に渡され、そこで代謝されて胆汁排泄の対象となり、多くの場合、MRPや他の重要なABCトランスポーターによって排泄される(Borst and Elferink 2002; Chan er al)。 胆汁成分の管状分泌は、胆汁形成の律速段階を表す。胆汁酸、グルタチオン抱合体、およびキセノバイオティクスは、肝細胞から除去され、ATP依存性の方法でカナル性排出トランスポーターによって胆汁中に濃縮される(Moffit er al 2006)。
図2 腸球におけるトランスポーターのトポロジー
原文参照
P-gp/MDR1/ABCB1は、MRP2/ABCC2,MRP4/ABCC4,BCRP/MXR/ABCG2とともに腸球の先端/内膜に局在しており、多くの基質薬物の腸内吸収に対する物理的障壁を形成していると考えられている(Chan er al 2004)。OATP、OCTおよびPEPT1もまた、この側に位置している。それらの発現レベルは、腸の異なるセグメント間で異なる。一般に、P-gp/MDR1/ABCB1およびBCRP/MXR/ABCG2は小腸で高レベルで発現しており、この分野の多くの人が経口薬物吸収の律速障壁と考えている(Chan er al 2004)。MRP1,MRP3,MRP5,OATPsは腸球の基底側膜に発現している。
肝臓では4つのMRPトランスポーター(MRP2,3,4,6)がかなりの範囲で発現している。肝臓では、MRP2が唯一の管状膜に局在するMRPであり、化学物質の胆汁への排泄に関与している(Marin er al 2005)。また、MRP3とMRP4は基底側膜に局在し、肝細胞から血液中への化学物質の排出に関与している(Marin er al 2005)。MRP6も基底側膜に局在していると考えられているが、このトランスポーターの高親和性基質はこれまでに同定されていない。MRPは代謝物の肝内排泄に重要な役割を果たしており、肝臓でのMRP発現の調節は薬物の体内動態を変化させる可能性がある(Marin er al)。
有機陽イオンと陰イオンの両方が、基質特異性が重複する有機陽イオン輸送体(OCT)とOATPのグループによって肝細胞に取り込まれる(Chandra and Brouwer 2004)。既知のOATPはいずれも非共役ビリルビンを輸入しない(Chandra and Brouwer 2004)。有機陰イオン(ビリルビンおよびグルタチオンを含む)は、通常、マイクロソームで共有結合によって修飾された後、肝細胞を横切って胆汁中に輸送される。これらの結合体はMRP2によって胆汁中に分泌される(Ballatori er al 2005)。これらのトランスポーターは肝細胞の基底側膜に発現しており、基質特異性がかなり重複している。MRP1は非共役ビリルビンと共役ビリルビンの両方を輸出するが、MRP3,MRP4,OSTは共役胆汁酸塩を優先的に輸出する(Ballatori er al 2005)。これらの受容体は、正常肝臓では低レベルの発現を示すが、胆汁性胆汁症では高発現である(Alrefai and Gill 2007; Geier er al)。
腎薬物排泄におけるP-gpと他のトランスポーターの役割の比較
P-gp/MDR1,MRP2/ABCC2およびMRP4/ABCC4は主に腎上皮細胞の先端膜(腔内膜)に局在しているが、MRP1/ABCC1およびMRP6/ABCC6は基底側膜に発現していることが示されている(Kim er al)。 1996; Evers er al)。 1998; Inui er al 2000; Matsuzaki er al 2005; Van de Water er al 2005; Yu er al 2007)。MRP2およびMRP4の基質は、トランスポーター機能を欠いた動物の腎クリアランスに変化を与えることが示されている(Van de Water er al 2005)。これらのトランスポーターは、腎尿細管細胞の細胞質から尿中に化合物を輸出する。したがって、これらのトランスポーターの基質は、糸球体濾過で予想されるよりも高い腎排泄を有することが予想される。抗HIV薬であるテノホビルは、MRP2とMRP4によって近位尿細管細胞から積極的に排泄される(Ray er al)。 これらのトランスポーターの詳細な薬物動態学的役割を理解するためには、さらなる研究が必要である。
さらに、OATP、OCT(OCT1-3)およびOAT(OAT1, 3, 4)トランスポーターファミリーのメンバーが近位尿細管細胞の基底側膜に存在することが確認されている(Terada and Inui 2004; Van de Water et al 2005; Launay-Vacher et al 2006; Sekine et al 2006)。OAT3は、プラバスタチンの腎排泄に関与していることが示されている(Nishizato er al)。 OCTの基質は、OCT1およびOCT2を欠損したマウスにおいて、腎クリアランスを大幅に減少させ、血漿中濃度を増加させることが示されている(Jonker et al 2003)。一方、2つのペプチドトランスポーターであるPEPT1およびPEPT2は、近位尿細管細胞の管腔膜上に存在し、ブラシ境界膜を越えて-ラクタム系抗生物質などのペプチド様薬物の尿細管再吸収に関与することが示された(Ganapathy er al)。 この再吸収過程は、糸球体濾過で予想されるよりも低い腎クリアランスをもたらす。さらに、この取り込み過程では、近位尿細管細胞の細胞質中の薬物濃度が上昇し、腎臓での毒性効果につながる可能性がある。抗生物質セファロリジンの腎毒性はOAT3の機能と関連している(Jung er al 2002; Deguchi er al 2004)。
テノフォビルによるMRP2阻害は、テノフォビルとジダノシンの相互作用に寄与していると考えられる。これら、2種類の抗レトロウイルス薬を併用すると、ジダノシン濃度-時間曲線下面積(AUC)が44~60%増加する(Kearney er al)。 これは、テノフォビルによるヒト有機アニオントランスポーター1による近位尿細管細胞へのジダノシンの活性取り込み阻害(Kearney et al 2004)またはジダノシンの分解に関与する酵素であるプリンヌクレオシドホスホリラーゼの阻害(Ray et al 2004, 2006)によって起こると考えられる。しかし、MRP2阻害剤であるジダノシンもMRP2基質であると仮定すると、ジダノシンAUCの増加は、管状筆界膜や他の組織におけるMRP2媒介の流出を阻害することによっても達成され得る。ラミブジン、アバカビル、ネビラピンで維持されていたHIV関連多中心キャッスルマン病患者のMRP基質ビンブラスチンの生命を脅かす毒性(好中球減少症)には、いくつかのMRPの阻害も寄与している可能性がある(Kotb et al 2006)。別のHIV関連ホジキン病患者も、ABVD(ドキソルビシン、ブレオマイシン、ビンブラスチン、ダカルバジン)化学療法とロピナビル・リトナビルをベースとした抗レトロウイルス療法で治療を受けた際に、生命を脅かす好中球減少を経験した(Makinson er al)。 ビンブラスチンとロピナビル-リトナビル相互作用は、化学療法投与前後のロピナビル-リトナビル中断で管理され、6回の薬剤サイクル後に完全寛解と免疫・ウイルス学的成功を得た。
血液脳関門におけるP-gpと他のトランスポーターの役割の比較
血液脳関門は、脳内皮細胞を接続するタイトジャンクションによって形成され、したがって、循環血液から細胞傍および細胞間の経路を介して脳への化合物の侵入を制限する(Banks 1999; Loscher and Potschka 2005; Couture er al 2006; Dallas er al 2006; Girardin 2006; De Boer and Gaillard 2007)。血液脳関門は、タイトジャンクションおよびP-gp、BCRP、およびMRP1,2,4,および5などの多数のABCトランスポーターの存在により、特に解剖学的およびトランスポーター関門として機能する(図3)(BorstおよびElferink 2002; De Boer er al 2003; HawkinsおよびDavis 2005; LoscherおよびPotschka 2005; Couture er al 2006; Dallas er al 2006)。このように、血液-脳関門は、潜在的に有害な内因性および外因性物質から脳を保護することにより、脳の恒常性維持に寄与している(Terasaki and Ohtsuki 2005)。脳毛細血管内皮細胞の先端膜・腔内膜に局在するP-gp/MDR1とBCRP/MXRが、薬物の脳内浸透の主要な障壁となっていることはよく知られている(Terasaki and Ohtsuki 2005)。
機能研究では、ヒトMRP2/ABCC2が血液脳関門における役割を担っていることが明らかになっている(Jedlitschky er al 2006)。また、MRP1/ABCC1は、外来物質(ソマトスタチン類似物質など)から脳組織を保護することにも関与している(Mercier er al)。 MRP1は脈絡膜上皮細胞の基底側膜に局在しており、薬物や毒性物質の頭頚髄液への浸透を阻止している(Yang er al 2007)。脳微小血管内皮細胞と腸管上皮細胞におけるMRP2,MXR/BCRP、MDR1の局在が類似していることから、これらのトランスポーターが腸管ブラシ境界膜と血液脳関門で外来物質に対する生理的障壁として機能していることが示唆される。MRP4とMRP5は、血液脳関門を形成する脳毛細血管内皮細胞に位置している(Kusch-Poddar er al 2005)。MRP1,MRP4,MRP5は脳毛細血管内皮細胞の内腔側に明確に局在している(Kusch-Poddar er al 2005)。
図3 血液脳関門におけるトランスポーター
原文参照
脳毛細血管内皮細胞の先端側/内腔側膜に局在するP-gp/MDR1(ABCB1)およびBCRPタンパク質が、薬物の脳内浸透の主要な障壁となっていることはよく知られている(De Boer er al)。 MRP1,MRP2,MRP4,およびMRP5は、血液脳関門の脳毛細血管内皮細胞の内腔側(先端側)に明確に局在している。これらのトランスポーターはアストロサイトやミクログリアにも発現している。
脳の研究が進歩しているにもかかわらず、中枢神経系の疾患は、大部分の薬剤が血液脳関門を通過しないため、治療が非常に困難なままであり、また、これらの疾患の病因は不明である。血液脳関門は、中枢神経系に作用する薬剤の98%以上の送達をブロックしている(Taylor 2002; Girardin 2006)。脳への十分な浸透性は、中枢神経系に作用する薬剤のリード化合物を設計するための必須条件である。中枢神経系の副作用を制限するために、脳への浸透性もまた、非中枢神経系作用薬の開発にとって重要である。したがって、両薬剤の血液脳関門伝染性は、臨床使用に先立って評価することが有用である。P-gp/MDR1および他のABCトランスポーターは、薬物の脳への浸透を制限し、その結果、多数の薬物の有効性および中枢神経毒性を調節することができる(Begley 2004; Deeley er al 2006; Girardin 2006)。
膨大な数の薬剤がP-gp/MDR1の基質であるため(例えば、癌や抗ウイルス剤)脳腫瘍やHIVなどの特定の中枢神経系疾患に対して治療レベルの薬剤濃度を達成することは非常に困難である。中枢神経系疾患を治療するためには、治療レベルの薬物が脳に到達することが必要であり、特に高齢化が進み、脳卒中、アルツハイマー病、パーキンソン病などの神経変性疾患の罹患率が高まる中、血液脳関門がもたらす薬物送達の課題はやむを得ないものである。脳研究の進歩にもかかわらず、中枢神経系疾患の治療は非常に困難なままである。その理由は、大部分の薬剤が血液脳関門を通過しないことと、基本的な生物学の理解不足と適切な標的に到達できないためである(Girardin 2006)。薬物の脳内への限られたアクセスを回避するために、リポソーム、ナノ粒子、ペプチドベクター戦略、P-gp/MDR1モジュレーター、内皮タイトジャンクションのモジュレーター、または浸透圧修飾などの薬物送達システムを含む様々なアプローチが研究されてきた(Siegal and Zylber-Katz 2002)。
血液脳関門伝染性は、アルツハイマー病、後天性免疫不全症候群(AIDS)髄膜炎、および精神疾患などの様々な疾患状態の下で変化する(Begley 2004; Deeley er al 2006; Girardin 2006)。これらの疾患状態は、電気化学的およびpH勾配の変化を介して、あるいは生理的バリアの炎症によって、血液脳関門の伝染性を直接的または間接的に変化させ、薬物が拡散しやすくする。これは、治療用の濃度でも起こりうるが、中枢神経系に過剰に蓄積し、細胞内濃度が有毒になる可能性がある。酸化ストレスは、脳毛細血管内皮細胞におけるP-gp/MDR1の発現を変化させる可能性がある(De Boer er al 2003)。さらに、疾患は、血液脳関門におけるP-gp/MDR1および他の活性な細胞間輸送システムをアップまたはダウンレギュレートし、最終的には中枢神経系の障害および細胞死につながる可能性がある。したがって、これらの疾患を治療するために開発された薬剤は、このような方法で直接または間接的に血液脳関門伝染性に影響を与えることができる。
P-gpを介した薬物間相互作用
患者において、薬物間相互作用は非常に頻繁にリスクとなり、患者に対する軽度の不快感のような単純な副作用、または極端な場合には生命を脅かす毒性をもたらす可能性がある。
薬物間相互作用におけるP-gp/MDR1の重要性がますます明らかにされており、このトランスポーターと相互作用する薬物(基質として、阻害剤として、または誘導剤として)の共同投与は、共同投与された薬物の薬物動態および薬力学に影響を与える薬物間相互作用をもたらす可能性があることが一般的に認められている。2006年9月に米国食品医薬品局(USFDA)により、薬物トランスポーターを媒介とする薬物相互作用研究に関する新しいガイダンスの草案が発表された(http://www.fda.gov/Cder/drug/drugInteractions/default.htm;Huang et al 2007bも参照のこと)。様々なトランスポーターの中で、P-gp/MDR1は臨床的意義が最もよく理解されており、医薬品開発の際に評価するのに適していると考えられる。USFDAガイドラインは次のように強調している。
適切な試験管内試験プローブを使用し、相互作用する薬剤を慎重に選択して初期の生体内試験を行うことで、開発プロセスの初期段階で薬物間相互作用の可能性を研究し、観察された相互作用を必要に応じてプロセスの後半で評価するための更なる研究を行うことができる。
このような初期の研究は、一般集団、特定の集団、個人における用量、濃度、反応関係についての情報を提供することができ、薬物間相互作用の結果を解釈する上で有用である。より大規模な臨床試験では、初期の試験で予測された相互作用の確認や発見が可能となり、また、相互作用の可能性に応じて行われる用量調整やその他の処方の変更が、望ましくない薬物間相互作用を回避するのに十分であることを確認することができる。
USFDAガイドラインでは、CYP及びトランスポーターを媒介とする薬物間相互作用を研究するための 戦略も取り上げられている(Huang er al)。 治験薬がP-gp/MDR1の阻害剤/誘導剤である可能性を検討する際には、ジゴキシンやキニジンやロペラミドのようなP-gp/MDR1の他の既知の基質を 選択することが適切であろう。治験薬の輸送が阻害又は誘導される可能性があるかどうか(P-gpの基質として)を評価する際には、リトナビル、シクロスポリン、ベラパミル等のP-gpの阻害剤や、リファンピン等の誘導剤を検討すべきである。薬物がCYP3A基質でもある場合には、リトナビルのようなP-gp/MDR1およびCYP3Aの両方の強力な阻害剤を用いて阻害を研究すべきである(Huang er al)。2007b)。
Caco-2細胞、MDCK-MDR1細胞およびLLC-PK1 MDR1細胞を用いた双方向輸送アッセイ、腫瘍細胞、cDNAトランスフェクトされた細胞、およびP-gp/MDR1のcRNAを注入した卵母細胞を用いた取り込みおよび流出アッセイなど、いくつかの試験管内試験アッセイモデルがある。および再構成P-gp/MDR1を発現する様々な組織または細胞からの膜小胞を用いたATPアーゼアッセイは、P-gp/MDR1媒介の薬物間相互作用の研究のために一般的に使用される(Huang er al 2007b; Lin 2007)。) 双方向輸送アッセイは、他の方法よりも直接的な方法で薬物の流出を測定するため、P-gp/MDR1の基質および阻害剤を同定するための決定的なアッセイと考えられている。ATPase activity assayとuptake/efflux assayは化合物を迅速にスクリーニングすることができるが、P-gp/MDR1の基質と阻害剤を区別するために設計されたものではない。さらに、文献データは、ATPアーゼ活性アッセイおよび蛍光指示薬アッセイの両方が、比較的低い伝染性を有するP-gp/MDR1基質の同定に失敗することが多いことを示唆している。双方向輸送アッセイは、P-gp/MDR1基質としての高伝染性化合物の同定に失敗する可能性があるが、高伝染性化合物の同定に失敗しても、この状況では、P-gp/MDR1は、これらの化合物が膜を横断するための重要な障壁ではない可能性が高いため、懸念されないであろう。
P-gp/MDR1は、多数の構造的にも薬理学的にも多様な基質薬物の薬物動態に影響を与えることが報告されており、共同投与された薬物または食物、およびハーブ成分によるP-gp/MDR1の阻害または誘導は、予期しない治療結果につながる薬物動態相互作用をもたらす可能性がある(Marcettti et al 2007)。上で議論されているように、多くの薬剤は、アップレギュレーションまたはダウンレギュレーションを介してP-gp/MDR1の発現および活性を変化させることができる。これは、P-gp/MDR1への直接結合、ATPアーゼ活性の阻害(例えば、プロテインキナーゼC阻害剤)または核内受容体発現の変化など、多くの異なるメカニズムを介して起こり得る。P-gp/MDR1が特定の薬剤(必ずしもP-gp/MDR1基質である必要はない)によって過剰または不足発現されると、別のP-gp/MDR1基質の薬物動態および吸収が実質的に変化することがある。
ハーブレメディによって引き起こされる薬物間相互作用の領域は、患者がすべてのハーブレメディが安全で「自然なもの」であると信じているため、過小評価されることが多いが、大多数の医療従事者は一般的なハーブレメディについての深い知識を持っていない(Skalli er al 2007; Zhou er al 2007b)。最も著名で一般的に服用されているハーブレメディの一つがセントジョンズワートである。セントジョンズワートは、CYP3A4とP-gp/MDR1の誘導剤としての活性のために広範囲に研究されている。シクロスポリン、タクロリムス、イリノテカンなどの他の様々な薬剤との併用により、複数の薬物間相互作用を引き起こすことが何度も報告されている(Zhou er al 2004; Madabushi er al 2006)。通常の状況では、P-gp/MDR1とCYP3A4が一緒になって、多くの経口吸収された薬剤に対して非常に効率的なバリアを構成している。CYP3A4とP-gp/MDR1に対する誘導作用により、セントジョンズワートは多くの薬物の定常血漿中濃度を低下させることが示されている。セントジョンズワートは、CYP3A4酵素の量を誘導して薬物代謝を増加させ、P-gp/MDR1の発現を増加させ、薬物排出機能を強化することにより、これらの潜在的に深刻な相互作用を引き起こす(Hennessy er al 2002)。この結果、CYP3A4およびP-gp/MDR1の発現において、経口吸収のために既に非常に効率的な障壁となっているものの有効性が増加する。
しかしながら、P-gp/MDR1は多くの臨床薬物間相互作用において重要な役割を果たしていると考えられているが、P-gp/MDR1と他のトランスポーターおよび薬物代謝酵素(特にCYP3A4)との薬物処分における相互作用が主な原因で、P-gp媒介薬物相互作用を定義することは困難である。P-gp/MDR1,CYP3A4,MRP1/他のトランスポーターの基質特異性が重複していることや、ほとんどの薬物トランスポーターに対して選択的な阻害剤が存在しないことから、薬物間相互作用におけるP-gp/MDR1の寄与を判断することは困難である。
多剤耐性(MDR)における潜在的な治療標的としてのP-gp
P-gp媒介MDR
1970年代に同定されたP-gp媒介性または古典的MDRは、よく特徴づけられた実験現象である(Biedler and Riehm 1970)。腫瘍細胞におけるP-gpの過剰発現は、MDRを引き起こし、その結果、広範囲の細胞傷害性薬剤に対する癌化学療法が失敗する。古典的MDRは、以下のような特徴を有する(Gottesman er al 2002):
- 化学的に無関係な一連の薬剤間の交差抵抗性
- がん細胞における薬物蓄積の減少。
- P-gpの発現の増加。
- 様々な化合物による表現型の逆転。
P-gpを介したMDRに最も頻繁に関与する薬剤は、真菌または植物由来のもので、アントラサイクリン系薬剤(主にダウノルビシンやドキソルビシンなど)やVincaアルカロイドなどがある。これらのグループ内の薬物とは別に、多数の他の非関連性の化合物が、P-gp媒介のMDRを誘導することができる(例えば、エピポドフィロトキシン、アクチノマイシンD、コルヒチン、タキサン類、およびアントラセンジオン誘導体)(Gottesman et al 2002)。これらの薬剤はすべて疎水性であり、ほとんどが弱塩基性である。
新規で強力なMDRリベラーの開発
MDRを回避するために、P-gp/MDR1の薬理学的に有効な阻害剤の同定と発見に多くの努力が注がれてきた。P-gpを介したMDRを克服するために、確立された薬理学的薬剤と新規化合物の両方からなる数世代の阻害剤が開発されてきた。エリスロマイシン、タモキシフェン、ケトコナゾール、シクロスポリンなどの第一世代のP-gp/MDR1阻害剤は知られているが、いずれも非常に強力な阻害剤ではない(Bartra er al)。 したがって、腫瘍に課された複数の薬剤耐性を逆転させる程度にP-gp/MDR1を阻害する能力を有するためには、一般的な毒性が高すぎる。例えば、エリスロマイシンは、治療レベルでCYP3A4を阻害し、QT間隔の延長およびスティーブンス・ジョンソン症候群を引き起こし得る(Katoh er al)。 癌治療を改善するためには、これらのトランスポーターを関与させ、回避し、または利用するための新規で効果的な戦略が必要とされている。
トランスポーター媒介の流出を回避する新規抗がん剤の設計は、MDRを回避するための可能性のあるアプローチである。エポチロンは、P-gp/MDR1に認識されないパクリタキセルのような作用機序を持つ新規の微小管標的薬である(Szakacs et al 2006)。エフラックスの対象となるほとんどの抗がん剤は、現在のところ化学療法レジメンには代えがたいものであるため、抗悪性腫瘍活性を維持しながら化学的に輸送されやすいように改変することが、魅力的な解決策となるだろう。このような改変はしばしば薬物のバイオアベイラビリティまたは有効性を低下させるが、いくつかの新規な薬剤がこのアプローチを用いて開発されている(Perego er al 2001)。また、製剤を改良して流入速度を高めることにより、薬物の細胞内濃度を上昇させることができる。ポリエチレングリコールコートリポソーム中のドキソルビシンのカプセル化は、従来のドキソルビシンよりも安全であり、時にはより効果的であるかもしれない(Vail et al 2004)。ABCトランスポーター、特にP-gp/MDR1,BCRP、およびMRPの過剰発現は、試験管内試験および生体内試験の両方でMDRの原因として一貫して示唆されている(Loscher and Potschka 2005)。
阻害剤はトランスポーターにアロステリックに結合し、基質または阻害剤の濃度に関係なく一般的に不可逆的である、P-gp/MDR1に対する新規の非競合的な阻害剤の開発に関心が寄せられている。例えば、SR33557は、腫瘍のMDRを逆転させるために開発された新しいP-gp/MDR1阻害剤である(Martin er al)。 1997)。阻害剤とトランスポーターの間には共有結合が形成され、基質と結合することも、膜を通過するために必要な構造変化を受けることもできない程度にトランスポーターを修飾する。トランスポーターが永久に機能しなくなると、エンドサイトーシスによって細胞膜から除去され、リゾチームによって分解される。トランスポーターの機能を再開させるためには、新しいトランスポーターを合成するか、細胞膜に移動させる必要がある。
P-gpの薬理遺伝学
P-gpの一塩基多型(SNP)について
MDR1遺伝子は7番染色体の長腕に位置し、コアプロモーター領域と29のエクソンから構成され、全長209kbである。MDR1は多型であり、様々な民族集団において多くの変異が記録されている(Chinn and Kroetz 2007)。白人を対象とした初期の研究では、15の多型が同定されたが、そのうち12の多型はタンパク質の配列を変化させなかった(Hoffmeyer er al 2000)。これらのサイレント変異は、エクソン境界に近いイントロンや、アミノ酸を変化させないウォブル位置に位置している。3つの多型は、エクソン2,5,および11においてタンパク質の変化をもたらした。エクソン2にはAsn-21をAspに変化させる多型が含まれており、エクソン5の変異はPhe-103をLeuに変化させる。現在のところ、これら、2つのアミノ酸変化の重要性を示す研究は非常に少なく、これらの変異の発生率は一般集団で12.9〜18.1%である。G1199A多型はエクソン11に位置しており、これはSer-400をAsnに変化させる。RNアーゼ保護解析において、遺伝子の残基26777および2995において、2つの追加の遺伝的多型部位が同定された(Mickley et al 1998)。MDR1遺伝子の残基26777において、GからTへの変化は、アミノ酸をAlaからSerへと変化させた。 同様に、2995における一塩基対のミスマッチ(GからA)は、アミノ酸をAlaからThrへと変化させた。A3320C SNPは、対立遺伝子および遺伝子型分布のために461人のドイツ白人のスクリーニングで明らかにされた(Cascorbi et al 2001)。
MDR1 SNPの対立遺伝子頻度は、異なる民族間で大きく異なる。MDR1遺伝子の残基2677および3435に位置するSNPは、他のSNPよりもはるかに頻度が高い(Hoffmeyer er al)。 アフリカ系アメリカ人(10%)は他の集団に比べてG2677T対立遺伝子の頻度が比較的低いが、白人(42-46%)メキシコ系アメリカ人(40%)およびアジア系アメリカ人(45%)はこの変異対立遺伝子を保有する可能性が高い(Chinn and Kroetz 2007)。全体的に、対応する野生型対立遺伝子の存在は変異型対立遺伝子の存在よりも頻度が高い。対照的に、世界中の人々は、野生型よりもC3435Tヘテロ接合体またはホモ接合体である可能性がはるかに高い(Sheffeler et al 2001)。黒人およびアフリカ系アメリカ人は、MDR1 3435変異型が優勢な数少ない集団の一つである。一般に、MDR1のSNPは、研究されたすべての集団で見られる(Cavaco et al 2003;Kaya et al 2005;Pechandova et al 2006)。特定の突然変異対立遺伝子を持つ確率は、ある集団ではより高い。
MDR1遺伝子型がP-gp機能および薬物動態に及ぼす影響
心不全に一般的に使用される薬剤であるジゴキシンは、P-gp基質である(Bartnicka er al)。 ジゴキシンは治療域が狭いため,過剰な副作用を伴わずに治療効果を得るためには,適切な血漿中濃度を維持することが重要である。健康な日本人114名を対象とした研究では、C3435T対立遺伝子を持つ被験者では、単回経口投与後のジゴキシンの血中濃度が低いことが明らかになっている(Sakaeda er al)。 MDR1のSNPがP-gp/MDR1基質の経口バイオアベイラビリティーを低下させることが報告されている。しかしながら、相反するデータも報告されている。Kim et al 2001)は、G2677T/A遺伝子型と、既知のP-gp基質であるフェキソフェナジン(TT<GT<GG)のクリアランスとの間に有意な逆関係があることを報告している。しかし、別の研究では、C3435TおよびG2677G/A遺伝子多型とフェキソフェナジン薬物動態との間には何の関係も示されていない(Drescher et al 2002)。C3435Tホモ接合のTT被験者はCT被験者よりも20%高いジゴキシン血漿中濃度を示し、CC被験者は48時間ジゴキシン尿中回収率が高い傾向がある(Verstuyft et al 2003)。相反する結果は、MDR1遺伝子型と薬物曝露量の減少との関連性の妥当性を疑問視するものである。C3435T変異はアミノ酸の変化をもたらさないため、このSNPが正常なP-gpの発現および機能を阻害する可能性は低いと考えられる。一方、MDR1遺伝子の転写は非常に複雑であり、まだ完全には解明されていない。さらに、上述の2つの研究では、被験者へのジゴキシンの複数回投与が関与していた。したがって、これらのデータは、ジゴキシンの代謝および排泄に影響を与える因子の影響を受けている可能性が高い。薬物排泄におけるP-gp/MDR1の役割、およびC3435Tホモ接合体の被験者が他の遺伝子型よりも低いジゴキシン尿中回収率を有することの証明のために、変異型P-gp/MDR1の関与を排除しないことは合理的である。
シクロスポリンは、移植後の臓器拒絶反応を防ぐために使用されるもう一つのP-gp/MDR1基質である(Hsiao er al 2006)。腎移植患者を巻き込んだ研究では、術後3日目にシクロスポリンAの吸収が、C/T3435およびT/T3435のグループ化された集団よりも野生型の方が有意に高いことが分かった(Foote et al 2007)。移植後1ヶ月の時点では、MDR1 SNPsとシクロスポリン曝露との間に相関は見られなかった。これらの結果は、日本のジゴキシン試験で見られた基質経口吸収の低下と一致している(Morita er al)。 しかし、MDR1 SNPs遺伝子型によって生じる薬物の腸内吸収のこの変化は、永久的ではなく、短命である(Foote er al 2007)。別の研究の観察は、同様の所見を示唆した(Anglicheau et al 2004)。C1236T対立遺伝子関連のより高い用量調整ピーク薬物濃度およびシクロスポリンAの濃度-時間曲線値の下での用量調整面積は4時間のみ持続した(Anglicheau et al 2004)。
MDR1の遺伝子変異は、P-gp/MDR1基質の経口吸収および腎排泄を減少させ、単回の経口投与では血漿中濃度が低くなるが、多回投与では高濃度になることがわかっている(Hoffmeyer er al 2000)。これらの効果の持続時間が短いため、このような可変性のある薬物曝露に関する知識は、臨床の現場では役に立たないかもしれない。
MDR1多型と標的細胞濃度、薬物反応および薬剤耐性
適切な治療薬の十分に高い血漿中濃度は、常により良好で迅速な応答に等しいとは限らず、疾患組織で発現したP-gp/MDR1は、薬剤の細胞内濃度に有意な影響を与え得る(Crouthamel et al 2006)。MDR1 SNPは、P-gp/MDR1基質の標的細胞内濃度に同様の影響を与え、薬剤耐性につながると考えられている。
P-gp/MDR1は、末梢血単球中のCD56þ、CD8þ、CD4þ、CD19þ、その他のサブ集団に発現している(Klimecki et al 1994)。CD4þ細胞はHIVの主要な標的であるため、そのP-gp/MDR1活性は多くのHIVプロテアーゼ阻害剤の細胞内濃度に影響を与える可能性がある(Woodahl er al)。 スイスの研究では、MDR1遺伝子型3435TTの患者は、CTやCC遺伝子型の患者に比べてCD4þ細胞数の有意な増加とウイルス感染の著しい改善を経験した(Fellay er al)。 他の長期的な研究は実施されていないが、後者の証拠は、HIV感染の拡大を遅らせ、治療成績を改善する上でMDR1多型が果たす役割を浮き彫りにしている。さらに、MDR1 SNPはHIV患者のAIDS発症率を低下させる可能性がある(Kedmi er al)。 現時点では、HIV患者管理におけるMDR1 SNPの重要性を支持するためには、より多くのエビデンスが必要である。
試験管内試験での研究では、G1199Aを発現する組換え上皮細胞は、対応する野生型を発現する細胞よりもビンブラスチンやビンクリスチンに対してより耐性があるようであることが明らかにされている(Woodahl er al 2004)。ドキソルビシンに対しても同様の耐性が両遺伝子型で認められた。さらに、蛍光基質であるロダミン-123の蓄積量は、G1199Aホモ接合体では野生型を発現する細胞に比べて約4.75倍であった。この現象は、P-gp/MDR1の排出能力と経上皮伝染性の変化によって説明できる。さらに重要なことに、変異体細胞はすべてのP-gp/MDR1基質に対する抵抗性の増加を示さないので、G11999Aが薬物標的におけるP-gp/MDR1発現のレベルを変化させないことは十分に可能である。別の試験管内試験研究では、G1199A遺伝子型が5種類のHIVプロテアーゼ阻害剤の経上皮伝染率を増加させることが確認された(Woodahl et al 2005)。パクリタキセルとカルボプラチンで治療された卵巣癌患者群において、ヘテロ接合型患者の平均無増悪生存期間は、野生型患者が19ヶ月であったのに対し、2ヶ月のみであった(Green er al)。 これは、試験管内試験で発見されたG1199A遺伝子変異に対する高い抵抗性と一致している(Woodahl et al 2005)。おそらく、G1199A遺伝子多型は、翻訳されたP-gp/MDR1が薬剤の流出と経上皮伝染性を増加させ、標的組織内での治療薬濃度の低下を引き起こすのではないかと考えられる。G1199Aの変異対立遺伝子は、その対立遺伝子頻度が比較的低いため、その関連性は疑問視されているが、白人ではアフリカ系アメリカ人と比較して少なくとも2倍の頻度で存在する(Kroetz et al 2003)。しかし、抗がん剤治療は、白人患者に適した場合はどこでも、ドキソルビシンのような影響の少ない基質を選択することにより、最適化される可能性がある。一方、この遺伝的変異によって影響を受ける薬物基質を同定するためには、より多くの研究が必要である。最近、MDR1の1199ヌクレオチド位置にある新規のG-T変異が発見され、白血病患者において2.3%の対立遺伝子頻度を示した(Crouthamel et al 2006)。この研究は、G1199A SNPがドキソルビシン、パクリタキセル、ビンブラスチン、ビンクリスチンに対する抵抗性の増加と関連していることを改めて検証した。興味深いことに、G1199T SNPは、トポテカンを除くこれらの薬剤の耐性を野生型の4分の1まで低下させた。これらの研究結果から、G1199T変異型はMDR1依存性の化学抵抗性が低いのに対し、G1199A多型はより高い抵抗性を示している可能性がある。全体的に、MDR1エクソン11遺伝子型解析は、化学療法の用量を最適化し、薬剤耐性の発生率を低下させることで、より良い臨床転帰を得るのに役立つ可能性がある。
これまでのところ、C1236T多型に関する研究はほとんど発表されていない。比較研究では、C1236T遺伝子型とリンパ増殖性疾患の治療における薬剤耐性との有意な関連は認められていない(Goreva er al 2004)。その結果、C1236T多型は、標的細胞の薬物濃度およびP-gp/MDR1機能にほとんど変化がないため、臨床では関連性がない。
漢民族のてんかん患者746人と対照者179人を対象とした研究では、薬剤抵抗性てんかん患者は薬剤反応性てんかん患者に比べてC3435Tホモ接合型を保有している可能性が高いことが示された(Kwan er al)。 この関連性は、白人を対象とした研究では認められなかった(Tan er al 2004; Sills er al 2005; Sills er al 2005)。これらの研究は、異なる民族集団における抗てんかん薬反応におけるこの多型の役割の複雑さを浮き彫りにしている。別の研究では、これらの知見に挑戦し、薬剤耐性てんかん患者はC3435Tホモ接合体よりも野生型MDR1 3435である可能性が高いことを示唆した(Siddiqui et al 2003)。矛盾した矛盾した証拠は、てんかん制御におけるC3435T SNPの役割をさらに複雑にしている。それにもかかわらず、てんかんは臨床的にいくつかの主要なタイプに分類されており、それに対応する薬物療法は他のタイプの発作ではしばしば禁忌であることに注意することが重要である。さらに、化学的および構造的に多様な抗てんかん薬のすべてがP-gp/MDR1基質であるわけではない(Sills et al 2002)。残念ながら、これらの研究のいずれも、被験者のてんかんの臨床的性質を文書化したものではなかった。したがって、C3435T SNP遺伝子型とてんかん患者における薬剤耐性の増加との関連は、今のところ決定的なものではない。
C3435T多型に関連する矛盾した所見は、タクロリムスを用いた研究からも報告されている。トルコの腎移植患者において、1ヵ月目と6ヵ月目にC3435T TT遺伝子型を有する患者では、タクロリムスの1日投与量が有意に低かった(Akbas et al 2006)。興味深いことに、移植後6ヶ月と12ヶ月の時点で、野生型の患者ではTT遺伝子型とCT遺伝子型に比べて、用量調整されたタクロリムスのトラフ濃度が有意に低くなっていた(Akbas er al)。 しかし、日本人の腎移植患者では、C3435T多型はタクロリムスの薬物動態パラメータに影響を及ぼさないことが明らかになった(Tada er al)。 このことは、薬物療法の治療成績におけるこの多型の役割をさらに疑問視している。小児心臓移植患者を対象に実施された試験では、プレドニゾン離乳の早さとC3435TおよびG2677T遺伝子型との間に有意な関連性が認められている(Zheng er al)。 さらに重要なことは、後者の研究では、小児心臓移植患者においてC3435T遺伝子型とG2677T遺伝子型の関連性が認められたことである。C3435T遺伝子型とG2677T遺伝子型の間には密接な関連があるため、上記のC3435T SNPに関連した陽性の結果は、G2677T遺伝子型の発現が見落とされたことによるものである可能性がある。
対照試験の結果は、この後者の考え方に一致する。この研究では、117人の統合失調症患者におけるMDR1多型の対立遺伝子頻度とオランザピンに対する治療反応を比較した(Bozina et al 2006)。G2677T遺伝子型とより良い治療反応との間に有意な関連が認められた。対照的に、C3435T多型では有意な関連は認められなかった。さらに、G2677TおよびG2677Aのジゴキシン、ジダノシンまたはフェキソフェナジンによる飽和動態パラメータは、類似のタンパク質発現レベルにもかかわらず、野生型とはかなり異なることが見出された(Schaefer et al 2006)。野生型と比較して、G2677T遺伝子型およびG2677A遺伝子型のビンクリスチンの最大輸送率は、それぞれ50%および3倍に増加した。このことは、MDR1 2677 SNPの薬物動態学的影響が、C3435T SNP研究のためのいくつかの偽陽性結果を発生させるという考えをさらに補強している。別の研究では、ベラパミル、ジゴキシン、ビンブラスチン、およびシクロスポリンAを試験管内試験で細胞間輸送活性および細胞内蓄積について調べた(Morita er al)。 5種類の多型MDR1(2677G/3435T、2677A/3435C、2677A/3435T、2677T/3435C、2677T/3435T)を発現する細胞と、対応する野生型を発現する細胞との間には、有意差は認められなかった。その結果、相反するエビデンスの存在により、MDR1 SNPに関連する薬剤耐性のメカニズムは依然として不明である。
SNPのタイプに応じて、MDR1遺伝子変異は、P-gp/MDR1基質の標的細胞濃度に影響を与え、治療抵抗性を増加または減少させることができる(Rund et al 1999)。これらの遺伝子変異は、医療専門家が投与量、臨床モニタリング、および治療法の選択を最適化するための貴重なツールを提供する。MDR1遺伝子の2677残基および3435残基における遺伝的変異の正確な役割は、相反する証拠があるため不明のままである。
MDR1 多型と疾患感受性
P-gp/MDR1 の機能の変化は、様々な組織や臓器の潜在的に毒性のある有害物質への曝露を増加させることから、遺伝的に障害のある患者の P-gp/MDR1 活性が疾患感受性を増加させているのではないかと疑われてきた(Ho er al)。 潰瘍性大腸炎患者249名、クローン病患者179名、健常対照260名を対象に遺伝子型解析を行った(Ho er al)。 共通のハプロタイプと潰瘍性大腸炎との間には、クローン病ではなく、非常に有意な関連が観察された。さらに、P-gp/MDR1の下部腸内発現に関連するC3435T多型を有するヒト患者は、潰瘍性大腸炎患者の中で過剰に発現している。今回の報告は、潰瘍性大腸炎のリスクを決定する際のMDR1遺伝子の寄与を支持する説得力のある証拠を提供するものであるが、クローン病のリスクを決定するものではない。血液脳関門のP-gp/MDR1は毒素を排除することで脳を保護しているので、突然変異によるP-gp/MDR1の機能不全は、いくつかの神経疾患の発症に寄与する可能性がある。実際、3435 TT遺伝子型は、早期発症パーキンソン病群で最も高く、後期発症群では2番目に高く、対照群では最も低かった(Drozdzik er al)。2003)。また、早期発症パーキンソン病患者では野生型に比べてC3435T対立遺伝子の発現頻度が高いことも報告されている(Furuno er al)。 C3435T対立遺伝子はP-gp/MDR1の発現低下と関連しており、パーキンソン病は環境毒素の脳内蓄積に起因すると考えられていることから、MDR1多型遺伝子型は排出能力の低下により、神経細胞をより高濃度のP-gp/MDR1基質に曝露している可能性が高いと考えられている(Furuno er al)。
MDR1多型は、潰瘍性大腸炎、パーキンソン病、およびおそらく他のヒト疾患への感受性の増加と関連している。この知識は、医療専門家が特定の疾患の可能性を低減するための早期かつ適切な介入を行うのに役立つ。しかし、食生活、生活習慣、人種、環境などの他の要因も病気の罹患率や進行に大きな影響を与えることを認識することが重要である。
結論と今後の方向性
今回紹介した情報から、P-gp/MDR1を深く理解することの重要性が明らかになった。P-gp/MDR1は、外来生物の排泄やがん細胞の抵抗性に大きく関与するタンパク質であるため、そのメカニズム、構造、機能を十分に理解することは、多くの基質、誘導剤、阻害剤が知られているため、多くの異なる分野での薬物療法の理解を深めることにつながると考えられる。現在、薬学の分野では、P-gp/MDR1の知識と薬物投与レジメンに影響を与える可能性についての知識は、非常に過小評価されている。医療従事者がP-gp/MDR1の阻害誘導を引き起こす可能性のある薬剤を十分に認識していることは稀であり、したがって、知識が不足しているだけで、臨床現場での薬物間相互作用の可能性が高まっている。しかし、がん治療におけるP-gp/MDR1の役割は、ますます認識されつつある。MDRにおけるP-gp/MDR1の役割がますます広く認識されるようになるにつれ、P-gp/MDR1の発現が増加している腫瘍を有する患者において化学療法の奏効性を改善するための研究が開発されている。P-gp/MDR1は、まず、MDRの発症における役割と、その発現に寄与する様々な因子を有していることから、新しい化学療法薬の開発に理想的な候補である。化学療法の一環としてP-gp/MDR1阻害剤を併用することは、P-gp/MDR1を阻害しながらも広範囲の副作用を引き起こさない理想的な薬剤を見つけることを目指して、広く研究が進められているが、P-gp/MDR1は全身で広範囲に活性を発揮するため、難しい課題となっている。P-gp/MDR1はYB-1によって活性化されたストレス誘発タンパク質であると考えられており、この分野にも創薬の可能性がある。ストレス誘発性のYB-1の転写を低下させる薬剤は、腫瘍細胞におけるP-gp/MDR1の発現レベルを低下させる可能性がある。将来的には、より効果的な化学増感剤を含むがん治療のための新しい薬物療法レジメンの開発が期待されており、より効果的ながん治療を可能にし、がんに対する薬物療法のより良い結果が得られるようになることが期待されている。
MDR1遺伝子については、様々な民族間でかなりの数のSNPが発見されている。MDR1 SNPは、P-gp/MDR1基質の経口バイオアベイラビリティを一時的に低下させることが見出されているが、定常状態ではより高い薬物曝露が起こる。それらはまた、変異の特定のタイプに応じて、薬剤耐性の増加または減少に関連している。MDR1多型はまた、いくつかのヒト疾患に対する感受性の増加と関連している可能性がある。最後に、MDR1多型は、P-gp関連の薬物間相互作用によって引き起こされる薬物動態効果を増強したり、相殺したりする可能性があるが、この分野ではさらなる研究が必要とされている。また、P-gp/MDR1の発現や活性に及ぼす年齢、性別、人種、SNPの相互作用、そしてそれがP-gp/MDR1基質薬の薬理学、治療効果、毒性や副作用にどのように影響するかは、P-gp/MDR1基質薬の臨床的使用に大きく影響する分野である。このような知識ベースが開発されれば、治療効果の予測が容易になり、個々の患者さんに合わせた薬物療法が可能になる可能性がある。
上記の知識は、薬物投与量の最適化、臨床モニタリング、治療の嗜好性に影響を与えることができる。医療専門家は、高額な疾患の発症を防ぐために適切な介入を行うことができる。これは、患者と医療システムに財政的に利益をもたらすことができる。P-gp関連の薬物間相互作用の結果をより正確に予測することができ、臨床現場での不必要な毒性や合併症を回避することができる。将来的には、P-gp/MDR1の活性を操作し、治療成績を最適化するための新薬が開発されることが期待されている。
P-gp/MDR1,BCRP、およびMRPを含む輸送タンパク質が経口バイオアベイラビリティを低下させ、組織分布を変化させる能力は、薬物設計に明らかな意味を持つ。実際、薬物の体内動態や安全性に影響を与えるトランスポーターの同定は、創薬プログラムの新たな課題となっている。第一に、薬物が薬理学的障壁を自由に越えることができるのか、それともP-gp/MDR1のようなABCトランスポーターによって薬物の通過が制限されているのか、第二に、薬物がABCトランスポーターを阻害することによって他の化合物の通過に影響を与えることができるのかを知ることが不可欠である。その結果、医薬品候補の輸送感受性の評価は新規治療薬開発の重要なステップとなっており、製薬業界では創薬プロセスにおいてP-gp/MDR1の感受性評価を日常的に採用している。
