Contents
Repurposing Sigma-1 Receptor Ligands for COVID-19 Therapy?
www.ncbi.nlm.nih.gov/pmc/articles/PMC7751758/
2020年11月9日
要旨
特にCOVID-19パンデミックのような新興感染症のアウトブレイクは、患者を治療するというユニークな課題を抱えた医療従事者に直面している。新薬を発見する時間がないため、承認された薬や臨床開発中の薬を再利用することが唯一の解決策である可能性が高い。
コロナウイルス(CoV)の複製は、小胞体(ER)に由来する修飾された膜状コンパートメントで起こり、宿主細胞のERストレスを引き起こし、ウイルスのニーズに宿主細胞機械の適応を促進する経路を活性化する。したがって、ERリモデリングとERストレス応答の調節は、CoVと宿主の相互作用を解明する上で極めて重要であり、新しい治療法である宿主ベースの抗ウイルスアプローチの根拠となる可能性がある。
シグマ-1受容体(Sig-1R)は、リガンドで作動するER膜結合型シャペロンであり、ERストレスの上流モジュレーターとして作用するため、COVID-19患者を治療するための宿主ベースのリパーポージングアプローチの候補となる宿主タンパク質である。Sig-1Rリガンドは、重症急性呼吸器症候群CoV-2(SARS-CoV-2)を含むCoVsに対する抗ウイルス化合物を同定することを目的とした試験管内試験薬物リパーポージョンスクリーンで頻繁に同定されている。Sig-1Rは、適応的宿主細胞ストレス応答の重要なメカニズムを制御し、ウイルス複製の初期段階に関与している。Sig-1Rは、脂質ラフトや耐洗剤性のあるER膜に富み、ここではウイルスのレプリカーゼタンパク質とコロケーションしている。実際、SARS-CoV-2の非構造タンパク質Nsp6はSig-1Rと相互作用している。COVID-19に対するSig-1Rリガンドの活性は、臨床試験で具体的に評価されていない。
このレビューは、COVID-19患者を治療するための宿主ベースの薬物再利用アプローチとしてSig-1Rを標的とする根拠を提供する。非バイアス試験管内試験抗ウイルス薬スクリーニングにおいてSig-1Rリガンドを用いて得られた証拠と、宿主細胞応答に対するSig-1Rの調節効果の根底にある潜在的なメカニズムについて議論している。Sig-1Rを標的とすることは、確立されたウイルス複製を劇的に減少させることは期待されないが、ウイルス誘発性宿主細胞のリプログラミングの初期段階を妨害し、感染の進行を遅らせ、疾患の悪化を防止し、および/または防御免疫応答を成熟させるための時間枠を確保することができるかもしれない。Sig-1Rをベースとした薬剤は、早期介入、予防のみならず、アジュバント治療としても有用であると考えられる。
キーワード
COVID-19, SARS-CoV-2, 抗ウイルス剤, 再利用薬, 薬物再利用, シグマ-1受容体, ERストレス, ウイルス複製
序論
重症急性呼吸器症候群CoV-2(SARS-CoV-2)と名付けられた2019年に新たに出現した新型コロナウイルス(CoV)は、中国の武漢で重症肺炎患者で確認されて以来、高い感染率と関連しており、急速に拡大してパンデミック(COVID-19パンデミック)となっている。残念ながら、ヒトのCOVを治療するワクチンはまだ承認されておらず、新薬の発見と開発には何年もかかると思われる。したがって、承認された薬剤や臨床開発中の薬剤を再利用することは、de novo創薬に比べて時間を短縮し、最終的にはCOVID-19患者により迅速な治療オプションを提供するための実行可能なアプローチとして浮上してきた。
CoVの複製は、構造的にも機能的にも小胞体(ER)と関連しており(Sola et al 2015)CoV感染は、ERストレスのウイルスニーズへの適応を促進するための経路を活性化することがよく知られている。これらは、タンパク質翻訳、ERタンパク質フォールディング能力、オートファジーを含むER関連分解(ERAD)およびアポトーシス細胞死を調節するために宿主細胞のERストレス応答をハイジャックすることを包含する(FungおよびLiu 2014;Fung et al 2014a)。したがって、ERストレス応答の調節は、CoV-宿主相互作用の解明において極めて重要であり、新しい治療アプローチのための根拠を提供するかもしれない。
シグマ-1受容体(Sig-1R)は、ERストレスの上流モジュレーターとして作用する。Sig-1Rは、リガンド操作された膜結合型シャペロンであり、通常は(ミトコンドリア関連ER膜(MAM)と呼ばれるER-ミトコンドリア接触部に存在し、そこでER-ミトコンドリアシグナル伝達およびER-核クロストークを調節している(Hayashi, 2019)。Sig-1Rによるミトコンドリア機能の制御には、バイオエナジェティクスとフリーラジカル発生/酸化ストレスが含まれる。細胞がストレスを受けると、Sig-1RはMAMからER網目網や形質膜に転座し、様々な機能性タンパク質を制御する。その分子シャペロン活性を介して、Sig-1Rは、タンパク質のフォールディング/分解、カルシウム(Ca2+)恒常性、ERストレス応答、オートファジー、および最終的には細胞の生存を調節する(Hayashi and Su, 2007; Su er al 2010; Schrock er al 2013; Vollrath er al 2014; Hayashi, 2019; Delprat er al 2020)。興味深いことに、そのシャペロン活性は、合成Sig-1Rリガンドによって、アゴニスト-アンタゴニスト的に活性化または阻害され得る。
その潜在的な抗ウイルス活性に関しては、SARS-CoV-2や他のCoVに対する抗ウイルス化合物を同定することを目的とした試験管内試験薬物再利用スクリーニングにおいて、Sig-1Rリガンドが頻繁に同定されている。メカニズム的には、Sig-1Rは、ウイルスの複製を促進するためにウイルスによって使用される細胞ストレス経路に関与している(VasalloおよびGastaminza 2015)。したがって、Sig-1Rは、膜質コンパートメント内のウイルスレプリカーゼタンパク質とコロケーションすることが示されており(Friesland et al 2013)非構造(NS)SARS-CoV-2タンパク質Nsp6がSig-1Rと直接相互作用することが最近報告されている(Gordon et al 2020)。Sig-1Rは、げっ歯類(Lever et al 2015)およびヒト(Stone et al 2006)の肺において実質的な密度で発現している。
ここでは、ウイルス感染におけるSig-1Rの役割を支持する薬理学的および遺伝学的データを収集し、CoV全般および特にSARS-CoV-2に焦点を当ててまとめた。Sig-1Rを標的とすることは、ウイルスを標的とした抗ウイルス剤とは異なり、ウイルス感染に重要な宿主細胞の適応細胞メカニズムに対処する、COVID-19患者を治療するための薬物再利用の可能性のあるアプローチであることが明らかになった。
シグマ-1受容体リガンドが抗ウイルス活性を発揮
非コロナウイルスに対する薬理学的知見
抗ウイルス剤としてのSig-1Rリガンドの潜在的な役割についての最初の洞察は、おそらく1984年に発表された(NemerowおよびCopper、1984)。この研究では、トリフルオペラジン、クロルプロマジン、クロルプロマジン、プロクロルプロマジン、プロメタジン、ハロペリドール(非フェノチアジンであるがブチロフェノン)を含む数種類のフェノチアジンが、ヒトヘルペスウイルスであるエプスタインバーウイルス(EBV)によるBリンパ球の感染を阻害することが示された。この頃までには、シグマは、(+)-[3H]SKF10,047が結合するフェンシクリジン、ミューおよびデルタオピオイド受容体とは別の結合部位であると考えられ始めたばかりであった(Su, 1982; Tam, 1983)。また、この頃までに、ハロペリドール、トリフルオペラジン、クロルプロマジンおよびプロメタジンを含む様々な非選択的神経遮断薬がこのシグマ部位に結合することが示された(Su、1982;TamおよびCoock、1984)(表1)が、これはシグマ-1Rが最初にクローン化される12年前のことであった(Hannner et al 1996)。したがって、著者らはシグマのメカニズムには言及せず、これらの薬剤の抗ウイルス効果を、EBV感染の初期段階に関与するカルモジュリン制御細胞内細胞内細胞プロセスへの作用に帰着させた。これらの非選択的Sig-1Rリガンドは、後にコロナウイルスを含む他のウイルスに対して抗ウイルス活性を発揮することが判明した(表1)。
表1 薬効
aIC50 CHIKV、チクングニヤウイルス;HCV、C型肝炎ウイルス;FLUAV、インフルエンザAウイルス;H5N1,鳥インフルエンザA型H5N1ウイルス、その他のサブタイプ;CCHFV、クリミア・コンゴ出血ウイルス。HSV-1,単純ヘルペスウイルス1型;JCV、JC(ジョンカニンガム)ウイルス;デングウイルス、デングウイルス;HCoV-229E、ヒトコロナウイルス株229E;MARV、マールブルグ出血熱ウイルス。MERS-CoV、東呼吸器症候群コロナウイルス;SARS-CoV、ヒトコロナウイルス株OC43(HCoV-OC43)重症急性呼吸器症候群コロナウイルス;EBOV、エボラウイルス;HIV-1,ヒト免疫不全ウイルス1型;SARS-CoV-2,重症急性呼吸器症候群新型コロナウイルス型;EBV、エプスタインバーウイルス
抗ウイルス活性を示すSig-1Rに結合する薬剤は、C型肝炎ウイルス(HCV)感染のさまざまな段階を標的とした阻害剤の発見を目的とした3つの試験管内試験スクリーニング試験で同定された。最初の研究では、国立衛生研究所クリニカルコレクションからの446化合物のセットが、ヒト肝癌細胞Huh-7細胞のHCV感染を試験管内試験で阻害する能力についてアッセイされた。化合物は、標的特異性や作用機序とは無関係に、細胞ベースのアッセイでスクリーニングされた。臨床的に承認された446の低分子アッセイのうち、33の化合物は、低マイクロモル濃度およびサブマイクロモル濃度で細胞毒性を示さず、抗ウイルス活性(Huh-7細胞のHCV感染をビヒクルDMSO対照と比較して85%以上減少させた)を示した。化合物は、HCV感染のいくつかの側面(侵入、複製、および集合を含む)を標的とした。活性な抗ウイルス化合物のいくつかはすでに抗ウイルス活性を有することが知られていたが、それらのほとんどの化合物がHCV感染を阻害する能力は予想外であった。33種類の活性化合物のうち、19種類の化合物(シプロヘプタジン、トレミフェン、フルフェナジン、トリフルオペラジン、CGS 12066B、プロクロルペラジン、ドキセピン、ケトチフェン、アミオダロン、ロフェプラミン、リムカゾール、クロベンプロピット、サルメテロール、アゼラスチン、デスロラタジン、インダトラリン、ハロペリドール、ベンプロペリンおよびカルベジロール)は、Sig-1Rに高〜中等度の親和性で結合していた(表1)。これらの化合物はいずれも非選択的であり、主にSig-1R以外の分子標的に結合するが、85%以上のHCV感染を阻害する活性化合物の60%近くがSig-1Rに対する既知の親和性を有していたことは注目すべきことである。これらの化合物のほとんどはHCVの侵入を阻害し、小胞性口内炎ウイルス(VSV)偽型と比較して選択的な抗HCV活性を示した。別の非バイアス細胞ベースのスクリーニングでは、非HCV用途に処方されている臨床的に承認されている281種類の薬剤の化学ライブラリをアッセイした。12種類の化合物は、細胞バイオマスを有意に減少させることなく、HCV感染を1桁以上減少させた(Mingorance et al 2014)。驚くべきことに、それらのすべて(クロルプロマジン、クロミプラミン、デシプラミン、ペルフェナジン、イミプラミン、ラロキシフェン、タモキシフェン、クロミフェン、ヒドロキシジン、ベンツトロピンおよびフルオキセチン)は、有意な親和性でSig-1Rに結合した(表1)。ヒドロキシジンおよびベンツトロピンは、それらが標的とする複製サイクルのステップを定義するために選択された。両HCV阻害薬は、ウイルス粒子の付着および内部化の下流にあるが、持続的なRNA複製および感染性ウイルス産生が確立される前の段階である感染の初期段階を阻害した。これらの結果から、化合物はHCV RNA複製の初期段階を阻害するという考えが補強された。Sig-1Rの関与についてはこれらの論文では論じられていないが、著者らは、この分子標的に対する親和性がかなりの数の活性化合物によって共有されていることを指摘しており、これによってHCV感染におけるSig-1Rの役割を具体的に扱う研究が喚起された(Friesland et al 2013)。
これらのスクリーニング研究の第3番目の研究では、臨床試験中または治療目的での使用が承認されている化合物1280種が、遺伝子組換え細胞株n4mBidに対するHCV誘発性細胞病理学的効果を緩和する能力についてアッセイされた。彼らは、未処理細胞と比較してn4mBid細胞の生存率を増加させることができる200以上のヒット化合物を発見した。55の主要ヒット化合物のうち、47の化合物はHCVのライフサイクルの1つ以上の側面(侵入、複製、または感染性ウイルスの集合/放出)を40%以上阻害した。興味深いことに、これらのうち19化合物については、Sig-1Rに対する有意な親和性が報告されている。アミオダロン、アミトリプチリン、ベンツトロピン、ブタクラモール、シナリジン、シプロヘプタジン、フルナリジン、フルフェナジン、イフェンプロディル、プロクロルペラジン、ペルフェナジン、ピモジド、プロトリプチリン、キニジン、キパジン-6N、ラロキシフェン、リタンセリン、トリフルプロマジンおよびトリフルペラジンの19種である(表1)。バイペリデンについてもSig-1Rとの相互作用が示唆されており(吉田 et al 2000年)SKF-38393はSig-1Rのアロステリックモジュレーターとして記載されている(Guo et al 2013)。すなわち、HCVに対する細胞保護小分子スクリーンで同定された55のリードのうち21が既知のシグマ-1結合体であった。それらのすべては非選択的であり、典型的にはSig-1R以外の分子標的に対する活性によって知られている。Sig-1Rに対する親和性データは未知であるか、または同定された残りの抗HCV化合物について報告されていないため、Sig-1R媒介メカニズムがそれらの効果に寄与している可能性を排除することはできない。ほとんどの既知のSig-1Rリガンドの保護効果には、侵入および感染性ウイルスの産生、集合、放出の阻害が含まれていたが、それらの中にはHCVの複製を阻害するものもあった。Sig-1Rの潜在的な寄与については議論されなかった。
A549肺上皮細胞における鳥インフルエンザA(FLUAV)H5N1ウイルスによって誘導される細胞死の変化を、RNA干渉(RNAi)スクリーニング法を用いて探索した。これらのスクリーニングでは、ノックダウンすると細胞の生存率が変化する複数の遺伝子が同定され、これらの遺伝子の一部を標的とした薬剤が抗ウイルス活性を有するかどうかがアッセイされた。神経薬イフェンプロジルは、試験管内試験で細胞生存能を増加させ、白血球浸潤および肺損傷を著しく減少させ、最も致死的なインフルエンザウイルス株であるH5N1(Zhang et al 2019)に感染したマウスの生存を改善した。イフェンプロディルの効果は、NMDA受容体の過剰刺激が肺損傷の引き金となり得るので、N-メチル-D-アスパラギン酸(NMDA)受容体におけるその拮抗作用の文脈で議論された。前の研究と著者を共有する別の研究では、FLUAV H5N1ウイルス感染時にA549細胞で異なる発現を示す遺伝子および経路が同定され、いくつかの薬剤が潜在的な治療法としてアッセイされた(Huang et al 2020)。アミトリプチリンは、感染1時間前または感染3時間後にアッセイした場合、H5N1に感染したA549細胞の生存率を48時間増加させ、浸潤細胞数を減少させ、肺損傷を減少させ、肺水腫を改善し、H5N1ウイルス感染マウスの生存率を向上させた。これらの薬剤のインフルエンザA H5N1ウイルス感染に対する効果を媒介するSig-1Rの関与については、イフェンプロディルはSig-1Rに対して高い親和性を示すが、議論されなかった(Hashimoto and London, 1995; Gitto er al)。 複数の受容体およびトランスポーターに結合する非選択的な抗うつ薬であるアミトリプチリンもまた、中等度の親和性でSig-1Rに結合する(Werling et al 2007)。イフェンプロディルおよびアミトリプチリンは、以前にHCV誘発性細胞病理学的効果を阻害することが示されていることに留意されたい(Chockalingam et al 2010)。
フィロウイルスに関しては、エボラウイルス(EBOV)に対する抗ウイルス活性を有する化合物を同定するために、FDA承認薬の系統的な試験管内試験スクリーニングを実施した(Madride et al 2015)。アッセイは、ベロ細胞株において実施した。単一濃度での活性化合物(>50%のウイルス阻害および<30%の細胞毒性)を用量反応アッセイで試験した。承認されたヒトへの投与量、毒性/耐容性および薬物動態データに基づいて、7つの試験管内試験ヒット化合物を選択し、生体内試験での有効性を評価した。その結果,ヒットした7種のうち5種(クロロキン,アミオダロン,プロクロルペラジン,ベンツトロピン,クロミフェン)はSig-1Rに親和性を示した(表1)が,Sig-1Rを介した機序の寄与は論じられていないが,本試験ではSig-1Rに親和性を示した。マウスを用いた生体内試験投与では,アジスロマイシン(100 mg/kg,1日2回,i.p.),クロロキン(90 mg/kg,1日2回,i.p.),アミオダロン(60 mg/kg,1日2回,i.p.)は感染マウスの生存率を向上させたが,この投与レジメンではクロロキンのみが有意な再現性のある有効性を示した。アジスロマイシンとクロロキンもまた、モルモットのEBOV感染モデルで試験されたが、いずれの投与量でも生存率を向上させることはできなかった。別の研究では、同じくFDA承認薬(〜26000種類の薬剤および分子プローブ)を、タイプの種であるザイールEBOVを用いた試験管内試験感染アッセイで試験したところ、複数のメカニズムクラスにまたがる80種類の薬剤について選択的な抗ウイルス活性が見出された(Johansensens et al 2015)。30の活性化合物のセットを優先順位付けした。それらのかなりの数(30種中17種:アステミゾール、ベンツトロピン、ベプリジル、クレマスチン、クロミフェン、クロミプラミン、フルペンチキソール、フルフェナジン、ロメリジン、マプロチリン、ピペラセタジン、プロクロルペラジン、キナクリン、セルトラリン、テルコナゾール、チオリダジン、トレミフェン)は、Sig-1Rに対して親和性を示すことが知られており、それらのほとんどは、他のウイルスを用いた以前の研究で確かに同定されていた(表1)。興味深いことに、マウス EBOV 感染モデルでの結果は、いくつかの薬剤、特にベプリジルとセルトラリンの保護能力を確認したが、これらの薬剤はいずれも驚くべき親和性で Sig-1R を結合する(表1
)。ウイルス侵入アッセイでは、これらの抗ウイルス薬のほとんどがウイルス侵入の後期段階をブロックすることが示された。
最後に、チクングニヤウイルスおよび他のアルファウイルス(セムリキ森林ウイルスおよびシンドビスウイルス)によって誘導される細胞毒性効果の阻害は、クロルプロマジン、ドキセピン、メトジラジン、ペルフェナジン、チエチルペラジン、チオリダジンおよびクロロキン(Pohjala et al 2011)で見出されたが、これらはすべて、他のウイルスに対しても抗ウイルス活性を示す非選択的なSig-1Rリガンドである(表1)。
コロナウイルスに対する薬理学的知見
COVID-19パンデミックの原因ウイルスであるSARS-CoV-2(重症急性呼吸器症候群関連CoV2型)は、ポジティブセンス一本鎖RNA(+ssRNA)CoVの広いファミリーに属している。他のCoVも感冒から中東呼吸器症候群(MERS)のようなより重篤な疾患を引き起こす。229E、NL63,OC43,HKU1,MERS-CoV、オリジナルのSARS-CoVに次いで7番目に人に感染する既知のCoVである(Zhu et al 2020)。系統解析の結果、SARS-CoV-2とSARS-CoVとの間に保存された進化的関係が明らかになった(79.7%のヌクレオチド配列同一性)(Zhou er al)。
本節では、Sig-1Rの関与を支持するデータおよびCoV感染に対するSig-1Rリガンドの治療可能性についてまとめた。
クロロキンおよびヒドロキシクロロキンは、Sig-1Rに結合する(表1)。これらの抗マラリア薬は、異なるウイルスに対して抗ウイルス活性を示している(Sperber et al 1993;Savarino et al 2001;Madrid et al 2013;Ferraris et al 2015;Wang et al 2015)。COVID-19パンデミックの前に、コロナビル科の他のメンバーに対するクロロロキンおよびヒドロキシクロロキンの有効性を支持する証拠もあった。クロロロロキンは、ヒトCoV株OC43(HCoV-OC43)に対して抗ウイルス活性を示すことが記載されている(Keyaerts et al 2009)。HCoV-OC43は、HCoV-229Eとともに、すべての一般的な風邪の10〜30%の原因となっており、感染は主に冬から春先にかけて発生する(Larson et al 1980)。クロロキンはHRT-18細胞におけるHCoV-OC43の複製を阻害し、母親にクロロキン(15mg/kg)を毎日投与した場合、新生児マウスにおけるHCoV-OC43誘発死を防止した。これらのことから、著者らは、クロロキンが将来のHCoVに対する薬剤として考えられる可能性を示唆している(Keyaerts et al 2009)。実際、クロロキンはまた、試験管内試験でSARS-CoVの複製を阻害し(Keyaerts et al 2004年)その後の多くの研究でSARS-CoVに対する抗ウイルス活性が確認されており(de Wilde et al 2014;Dyall et al 2014年)最近ではSARS-CoV-2に対する抗ウイルス活性が確認されている(Jeon et al 2020;Yao et al 2020)。
クロロキンおよびそのヒドロキシアナログは、COVID-19の治療および予防のために最初に提案された最もポピュラーな薬剤であった:NIHサイトに登録されている208件の介入臨床試験では、これらの薬剤を単独または組み合わせて使用した治療が行われている(ClinicalTrials.gov query, 2020)。試験管内試験では、両薬剤はベロ細胞におけるSARS-CoV-2感染を阻害するが、ヒドロキシクロロキン(EC50=0.72μM)の方がクロロキン(EC50=5.47μM)よりも強力である(Yao et al 2020)。この治療法の利点は、このパンデミックの間に調査されてきたが、まだ、これらの薬剤の広範な使用を支持する科学的証拠はない。実際、ヒドロキシクロロキンの効果を評価した最初の臨床研究の結果は、COVID-19患者における本剤の有効性を支持していない(Geleris et al 2020;Mitjà et al 2020;Roustit et al 2020)。しかしながら、予備的研究は、かなりのメディアの関心を喚起し、いくつかの国で安価に製造されているこれらの薬剤の大量かつ無秩序な使用に対する懸念を高めた。一方で、ヒドロキシクロロキンを投与されたCOVID-19患者において、重篤な副作用が報告されている。両抗マラリア薬の副作用は、重篤な網膜症および薬剤の生体蓄積に関連した心疾患を含む、十分に確立されたものである(Palmeira et al 2020)。最近(2020年6月15日)FDAは、入院患者を対象とした大規模無作為化臨床試験から得られた知見に基づき、COVID-19の治療にヒドロキシクロロキンおよびクロロキンを使用する緊急使用承認を取り消した(FDA通信 2020)。これらのアミノキノリンの作用機序は、エンドソームpHを上昇させてリソソソーム酵素を阻害する能力に依存すると考えられている。これは、エンベロープされたウイルスが宿主細胞内に侵入し、その遺伝物質を放出するのを防ぐ(Tripathy et al 2020)。SARS-CoV-2 Sタンパク質のN末端ドメインにおけるガングリオシド結合ドメインへの結合は、ウイルスの脂質ラフトへの付着および侵入のためのACE-2受容体との接触を阻害するためのクロロキンおよびヒドロキシクロロキンのメカニズムとしても示唆されている(Fantini et al 2020)。Sig-1Rに関する唯一の言及は、Sig-1R媒介機構を介したグルタミン酸誘発細胞死に対するクロロキンによる保護を記述した無関係の研究から来ている(Hirata et al 2011)。クロロキンおよびヒドロキシクロロキンの抗ウイルス効果に対するSig-1Rの最終的な貢献は、認識され始めたばかりであるが(Gordon et al 2020;Mirabelli et al 2020年)これらは非選択的Sig-1Rリガンドであり、この分子標的に対する親和性は亜最適である。
非選択的であるが高親和性のシグマ-1リガンドである抗不整脈薬アミオダロンは、Vero細胞におけるSARS-CoVの試験管内試験での拡散を阻害することが報告された(Stadler et al 2008)。アミオダロンは、細胞生存率に影響を与えない濃度で、濃度依存的にウイルス力価を低下させた。SARS-CoVとの直接的な相互作用やウイルス侵入の障害は抗ウイルス活性の説明にはならなかったが、アミオダロンはSARS-CoVのゲノムを細胞質に導入した後、SARS-CoVのライフサイクルを阻害した。カチオン性両親媒性薬物として、アミオダロン(およびその主要代謝物MDEA)は、後期エンドソーム/リソソームに蓄積し、その内腔酸性度を低下させ、ウイルスタンパク質の酸性切断を阻止し、内分泌経路を妨害する(Salata et al 2015)。しかしながら、アミオダロンは、SARS-CoVがそのゲノムを細胞質に送達した場合でも抗ウイルス活性を示し、従って、ポストエンドソームレベルでの付加的なメカニズムが関与している(Stadler et al 2008)。アミオダロンの抗ウイルス活性に対するシグマ-1介在機序の寄与(または寄与していない)については、これらの論文では議論されていない。アミオダロンはまた、遺伝子組換え細胞株n4mBid(Chockalingam et al 2010)Huh-7.5.1細胞におけるHCVの侵入およびアセンブリーステップ(Cheng et al 2013)および様々な培養細胞株におけるEBOV細胞の侵入を阻害することも示された(Gehring et al 2014;Salata et al 2015;Dyall et al 2018)。試験管内試験での有望な結果にもかかわらず、アミオダロンは、モルモットを致死量のEBOVから保護することに失敗した(Dyall et al 2018)。臨床現場では 2014年12月、シエラレオネのフリータウンにあるエボラ治療ユニットの約80人の患者に、1日あたり30mg/kgまでの用量で慈悲療法としてアミオダロンを投与した(ClinicalTrials.gov Identifier NCT02307591,2014)。地域の過去のデータと比較すると、症例死亡率の減少が報告された。残念ながら、この研究は正式な臨床試験ではなく、この結果の統計的有意性は不明である(Turone, 2014; Gupta-Wright et al 2015)。最近、COVID-19関連呼吸不全に罹患した患者が、支持療法とアミオダロンによる適応外の短期治療のみで回復した症例(入院から 2日目から開始し、5日間持続;1日目に15mg/kg/24時間点滴静注、その後400mgを1日2回経口投与)が報告されている(Castaldo et al 2020)。したがって、心室性および上室性不整脈の両方の治療のために広く処方されているアミオダロンは、症状のあるまたは重度のCOVID-19患者を治療するためではなく、SARS-CoV-2感染を予防するための可能な治療法(単独または併用レジメンの一部として)として提案されている(Aimo et al 2020;Sanchis-Gomar et al 2020)。
MERS-CoVに感染した細胞培養物において、348種類のFDA承認薬のセットがスクリーニングされた(de Wilde et al 2014)。4つの化合物(クロロキン、クロルプロマジン、ロペラミド、およびロピナビル)は、低マイクロモル範囲(IC50s 3〜8μM)でMERS-CoVの複製を阻害した。これらの化合物はまた、SARS-CoVおよびHCoV-229Eの複製を阻害した。興味深いことに、クロロキンだけでなく、クロルプロマジンおよびロペラミドもSig-1Rに結合した(表1)。クロロキン、クロルプロマジン、ロペラミドは複製サイクルの初期段階を阻害するのに対し、ロピナビルは複製後の段階を阻害することが、添加時間実験から示唆された。この知見は、Sig-1RがHCV RNA複製の初期段階を調節していることを示す以前の知見と一致している(Friesland et al 2013)。
別の研究では、FDA承認済みまたは高度な臨床開発中の290化合物のライブラリーを用いて、MERS-CoVおよびSARS-CoVに対する抗ウイルス活性をスクリーニングした(Dyall et al 2014)。27の化合物は、MERS-CoVおよびSARS-CoVの両方に対して試験管内試験活性を示した。27の活性化合物のうち、少なくとも19の化合物は、Sig-1Rに有意な親和性で結合した(クロロキン、ヒドロキシクロロキン、メフロキン、アモジアキン、タモキシフェン、トレミフェン、テルコナゾール。シクロヘキシミド、ベンツトロピン、フラスピリレン、チオチキセン、フルフェナジン、プロメタジン、アステミゾール、クロルフェノキサミン、クロルプロマジン、チエチルペラジン、トリフルプロマジン、クロミプラミン) (表1)に示したが、それらの抗ウイルス活性がSig-1Rと関連しているかどうかは議論されなかった。最近、著者らは、この以前のスクリーニングから 20の薬剤に優先順位をつけ、SARS-CoVおよびMERS-CoVを阻害する試験された20の薬剤のうち17の薬剤が、同様のIC50値で、かつ非細胞毒性濃度で、Vero E6細胞に対するSARS-CoV-2の細胞病理学的効果も阻害することを見出した(Weston et al 2020)。全て(アモジアキン、ベンツトロピン、クロロキン、クロルプロマジン、クロミプラミン、フルフェナジン、フルスピリレン、ヒドロキシクロロキン、メフロキン、プロメタジン、タモキシフェン、テルコナゾール、チエチルペラジン、トレミフェン)であるが、2つはSig-1Rに結合することが知られている(表1)。そのうちの2つ、クロロキンとクロルプロマジンは、マウスに適応したSARS-CoVモデルを用いて生体内試験で評価した。薬物治療は、肺でのウイルス複製を阻害しなかったが、マウスを臨床疾患から保護した(Weston et al 2020)。クロロカインだけでなく、抗精神病薬クロルプロマジンのリポジショニングが、COVID-19を治療するために提案されていることに注意されたい(Nobile et al 2020;Plaze et al 2020)。
最近の再配置研究では、SARS-CoVに対する活性により事前に選択された35剤と感染症専門家から推薦された13剤を含む48剤のFDA承認薬を、ベロ細胞におけるSARS-CoV-2に対する抗ウイルス活性についてアッセイした(Jeon et al 2020)。感染細胞は、SARS-CoV-2のウイルスNタンパク質に対する抗体を用いて免疫蛍光により分析した。評価された48の薬剤のうち、24の薬剤は,0.1〜10μMの間のIC50値を有する潜在的な抗SARS-CoV-2活性を示した。クロロカインに加えて、ロペラミド、メフロキンおよびアモジアキンの3つは、Sig-1Rに結合することが知られていた(表1)。それらのすべてが、MERS-CoVおよびSARS-CoVを含む他のCoVに対して有効であることが以前に示されていた(Dyall et al 2014)。
最近の論文では、SARS-CoV-2タンパク質相互作用マップおよび薬理学的データの知見に基づいて、Sig-1Rを標的とすることが強調された(Gordon et al 2020)。薬剤のサブセットをスクリーニングすることにより、Vero-6細胞におけるSARS-CoV-2感染性を効果的に減少させる2つの薬理学的薬剤のセットが同定された:mRNA翻訳の阻害剤およびシグマ-1およびシグマ-2受容体の予測される調節因子。ハロペリドール、PB28,PD-14418およびヒドロキシクロロキンを含む非選択的Sig-1Rリガンド、およびその後クレマスチン、クロペラスチン、プロゲステロンおよびシラメシン(表1)を含む非選択的Sig-1Rリガンドが抗ウイルス効果を発揮することが判明した。ヒドロキシクロロキンはSig-1Rリガンドの中で最も抗ウイルス作用が弱かったが,これはSig-1Rリガンドの分子標的に対する親和性の低さと相関していた。著者らは、シグマ受容体の関与について議論した。彼らは、これらの分子は他の受容体に対しても活性であるが、すべての間で共有されているのはシグマ受容体のみであることを指摘した。例えば、抗精神病薬のハロペリドールはドーパミンD2受容体とヒスタミンH1受容体を阻害し、クレマスチンとクロペラスチンはそれ自体が抗ヒスタミン薬であるが、3つの分子はいずれもシグマ1Rリガンドであり、抗ウイルス活性を発揮する。対照的に、同じくH1受容体とD2受容体を阻害する抗精神病薬オランザピンは、有意なSig-1R活性を有さず、抗ウイルス活性を有さない。著者らはまた、広く使用されている抗鎮咳薬のデキストロメトルファンがプロウイルス活性を示していることを指摘し、その使用には注意が必要であり、COVID-19との関連でさらなる研究が必要であると述べている。デキストロメトルファンだけでなく、カルベタペンタン(Brown et al 2004年)麻薬性鎮痛薬のペンタゾシン(特にその活性(+)-ペンタゾシンエナンチオマー)(Tam and Cook、1984年)およびいくつかの抗うつ薬(Narita et al 1996)は、他のいくつかの市販化合物の中では、プロトタイプのSig-1Rアゴニスト/陽性調節薬と考えられている。したがって、非選択的ではあるが他の潜在的なSig-1Rアゴニストの使用にも注意が必要であろうか。このように、コカインは非選択的なSig-1Rアゴニストであり、コカインへの暴露はSig-1Rを活性化することでHIV感染を促進することが示されている(Roth et al 2005年)。このように、コカインの使用/乱用は危険因子である可能性があるが、私の知る限りでは、コカインのCoV感染への影響は調査されていない。
最後に、最近の出版物では、AIベースの機械学習戦略と結合した定量的高含有形態学的プロファイリングが、SARS-CoV-2に対する有効な単剤を同定するために適用された(Mirabelli et al 2020)。このアッセイは、ウイルスの侵入、伝播、および宿主細胞応答の変調の阻害を含む複数の抗ウイルス作用機序を検出した。ウイルス増殖速度論は、Huh-7細胞における0.2の感染多重度でアッセイされ、感染後48時間でのピークウイルス力価を有する。1,441種類のFDA承認化合物および臨床試験候補化合物のライブラリから,1μM以下の抗ウイルス力を有し,細胞毒性を有しない15種類の用量反応性化合物を同定した。そのうちアミオダロン、ベラパミル、E-52862(S1RA)の3つはSig-1Rに結合することが知られてた(表1)。興味深いことに、E-52862(S1RA)は選択的なSig-1Rアンタゴニストである(Romero et al 2012)。それは、Huh-7細胞(IC50 = 222 nM)およびiPSC由来肺胞上皮2型細胞(iAEC2s)(IC50 = 1 µM)においてSARS-CoV-2に対して強力な活性を示し、限定的な細胞毒性(CC50 > 5000 nM)を示した。E-52862(S1RA)は感染細胞を枯渇させ、宿主調節機構を示唆する細胞変化を誘導した。このことから、S1RAの活性は宿主細胞の機構に依存しており(おそらくHuh-7およびiAEC2s細胞では活性があるが、ウイルス増殖に非常に寛容なVero-6細胞では活性がない)ヒト細胞はこの化合物に対してより反応性が高い可能性があることが示唆された。このこと(および他の実験条件の違い)は、Vero E6細胞株でアッセイしたときにE-52862が活性を示さなかった方法を説明することができる(Gordon et al 2020)。
作用機序
シグマ-1受容体とウイルス侵入
ウイルス侵入の阻害は、非選択的シグマ-1リガンドについて、多くの研究で報告されているが(Chockalingam et al 2010;Gastaminza et al 2010;Cheng et al 2013;Johansen et al 2015;Dyall et al 2018)他の研究では報告されていない(NemerowおよびCooper、1984;Mingorance et al 2014;Stadler et al 2008;de Wilde et al 2014;Gordon et al 2020)。したがって、ウイルス粒子の付着または内部化の防止が、そのような薬剤のSig-1R媒介抗ウイルス効果を占めるかどうかは不明である。
シグマ-1リガンドによるHuh-7ヒト肝細胞へのHCV侵入の阻害は、薬理学的研究で実証されたが(Gastaminza et al 2010)Huh-7細胞におけるシグマ-1RのダウンレギュレーションはHCV侵入に影響を与えなかった(Friesland et al 2013)。このことは、(遺伝子サイレンシングアプローチの場合と同様に)調節性シグマ-1タンパク質の欠乏が、シグマ-1Rで作用するアンタゴニストによって誘発されるウイルス侵入に対する薬理学的抑制効果を模倣していないことを示唆しているかもしれない。したがって、Sig-1R欠損細胞における調節機構の欠如は、Sig-1Rが相互作用している標的タンパク質に対するSig-1Rリガンドが促進する抑制効果と同等のものではないと考えられる。これは、Sig-1Rが物理的なタンパク質-タンパク質相互作用を介して作用するシャペロンの性質に起因する可能性がある(Su et al 2010; Pabba, 2013)。
Sig-1Rは通常、ER、典型的にはMAMに存在するが、細胞がストレスを受けると(ウイルス感染後に予想されるように)Sig-1Rは末梢ERネットワークおよび細胞膜に転座し、様々な細胞表面タンパク質を調節する(Su et al 2010)。このようにして、Sig-1RはER内の重鎖結合免疫グロブリン蛋白質(BiP)、グルコース調節蛋白質78,GRP78,または熱ショック70 kDa蛋白質5,HSPA5としても知られている)と結合する(Hayashi and Su, 2007)。BiPはまた、細胞ストレスに応じてERから細胞表面に転座し、細胞表面上に複数のドメインを露出させ、その基質結合ドメインによるウイルス認識、およびCoVを含むいくつかのウイルスの侵入の促進を含む新しい機能を想定している(Zhang et al 2010)(Chu et al 2018)(図1)。表面付着およびウイルスの侵入を促進するBiPの能力は、MERS-CoVおよびコウモリCoV-HKU9について実証され、SARS-CoV-2について予測されるように、表面S(スパイク)ウイルスタンパク質への結合に依存している可能性が高い(Ibrahim er al)。 Sig-1Rは、ERから細胞膜へのタンパク質トラフィッキングに関与し、BiPと結合し、BiPと同様に、ERストレス時に細胞表面にトラスノケートする(Hayashi, 2019)が、BiPの細胞膜への輸出におけるSig-1Rの関与については、これまで調査されていない。Sig-1Rアンタゴニストは、ERでのSig-1R-BiP解離を阻害する(Hayashi and Su, 2007; Hayashi, 2019)ため、BiPを介したBiPのトラフィッキング、表面発現、そして最終的にはCoVへの付着を阻害する可能性がある。Sig-1Rとは異なり、BiPは非膜結合性のER内腔シャペロンとして記述されている。したがって、Sig-1Rとの相互作用は、BiPの細胞膜への安定化/固定化を可能にする可能性があるが、膜貫通ドメインが同定されており、細胞表面の自律的な再局在化を可能にしている(Zhang et al 2010)。しかし、Sig-1RとBiPとの直接的な相互作用については、特に細胞膜での記述はなかった。同様に、ウイルスの付着/侵入に関与する他の宿主膜タンパク質(例えば、ACE2またはTMPRSS2)との直接的な相互作用や、ウイルス侵入のSig-1R依存性の調節を実証するウイルスエンベロープ構造タンパク質との直接的な相互作用は記述されていない。あるいは、後述するように、Sig-1RはRNA複製および宿主細胞応答の初期段階を制御するが、ウイルスの侵入は制御しないかもしれないが、一方で、Sig-1Rへの結合とは無関係に、多くのSigma-1リガンドが共有する構造的特徴が、ウイルスの侵入を抑制することを説明するかもしれない。
図1 エンドサイトーシスによる重症急性呼吸器症候群CoV-2(SARS-CoV-2)の侵入モデルの提案
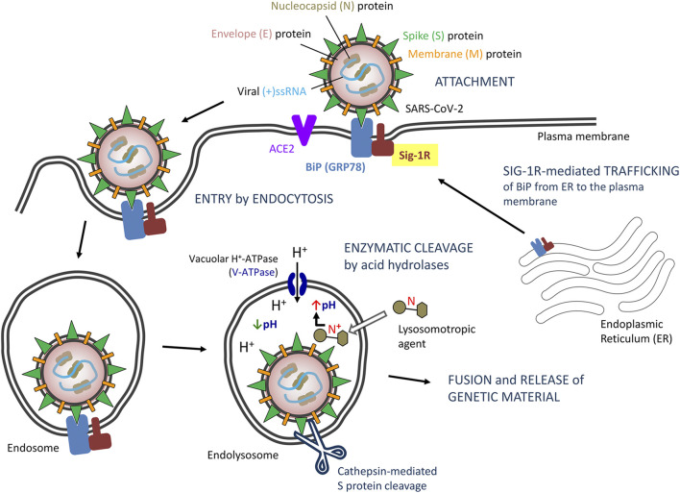
BiP相互作用を介したSig-1受容体(Sig-1R)の役割の可能性。Sig-1Rの役割についての略語および参考文献は本文に記載している。
シグマ-1受容体リガンドはリソゾモトロピック剤として?
SARS-CoV、MERS-CoV、およびSARS-CoV-2を含むCoVの宿主細胞への侵入の基本的なメカニズムは、エンドサイトーシス経路(受容体依存性エンドサイトーシス)である(Glebov, 2020; Yang and Shen, 2020)。SARS-CoV-2のスパイク(S)タンパク質の受容体への結合は、その切断部位を、内膜処理に関与するエンドソーム酸プロテアーゼを含む細胞内プロテアーゼに暴露する(Millet and Whittaker, 2015; Pillay, 2020)。特に、エンドソームカテプシン媒介のSタンパク質切断は、CoVの侵入および感染の開始に重要なステップであると考えられている(Millet and Whittaker, 2015; Glebov, 2020; Wędrowska et al 2020; Yang and Shen, 2020)。
エンドソームおよびエンドソームのリソソームへの成熟は、酸性プロテアーゼに必要とされ、SARS-CoV-2の処理および内部化に重要である、その酸性内部pHによって特徴づけられる(Wędrowska et al 2020)。血漿膜およびリソソーム膜は、弱塩基の結合型に対しては高い伝染性を有するが、塩基のプロトン化型に対しては本質的に不伝染性である(Marceau et al 2012; Homolak and Kodvanj, 2020)。したがって、細胞質で組合わされた弱い塩基は、リソソーム膜を越えてリソソームに入ることができる。リソソームに入ると、リソソームのpHが細胞質のpHよりもかなり低いので、それらは急速にプロトン化され、リソソーム内にロックダウンられるようになる。この結果、薬物のリソソーム内蓄積(イオントラップ)が起こり、ほとんどのリソソーム酵素活性を阻害するのに十分なリソソームpHの上昇(すなわち、リソソソームpHの中和)が起こり、リソソソーム内でプロトン化された薬物は、リソソソーム内にロックダウンられる。リソソソーム内のプロトン化塩基の濃度が十分に高い場合、リソソソーム内に水が浸透圧的に入り、リソソームが膨潤して大きな空胞を形成し(すなわち、リソソソーム空胞化)その結果としてリソソソームの機能が失われる(Aki er al)。
抗ウイルス剤として認められている薬剤の中には、親油性アミン/弱塩基であり、これが蓄積してリソソソームの酸性化を妨げることで、ウイルスの内部化および内部化後の複製部位への輸送を阻害するものがある(Sieczkarski and Whittaker, 2002; Kaufmann and Krise, 2007; Mercer er al)。 このようなリソモトロピズムは、クロロキンおよび他の抗マラリア薬を含む、いくつかの親油性アミンおよびカチオン性両親媒性薬物(Kaufmann and Krise, 2007)によって共有される(Homolak and Kodvanj, 2020)。実際、リソソーム標的化剤は、COVID-19に対する潜在的な治療法と考えられている(HomolakおよびKodvanj 2020)。しかしながら、ウイルス感染症を制御するためのこの作用機序の有効性は、その特異性の低さ、リソソソーム内のpHを低下/回復させ、リソソソームから内包されたアミンを排出するための細胞補償機構(Goldman et al 2009年)およびリソソーム内での実質的な薬物蓄積およびアルカリ化を可能にするための高用量の薬物の必要性によって妨げられているが、これはまた安全性の懸念を提起している。
市販されている様々な薬剤は、リソゾモトロピック剤の一般的な物理化学的特性に適合している。本質的には、ClogP > 2およびpKaが6.5から 11の間にある薬剤は、リソソソームに蓄積することができる(Nadanaciva et al 2011)が、他の物理化学的特徴もリソ運動性に影響を与える(Kaufmann and Krise 2007)。シグマ-1リガンドのファーマコフォアモデル(仮説的アゴニストおよびアンタゴニストの両方)は、疎水性領域に挟まれた正のイオン化可能な基(すなわち、塩基性窒素、通常は二級または三級アミン)を規定しており(Pascual et al 2019)これは潜在的なリゾソモトロピックと一致している(図1)。これは2つの意味合いを持つかもしれない。第一に、リソソーム封鎖は、Sig-1R薬剤がその意図された標的に到達するための障壁を表す可能性があり、その抗ウイルス効果が起こるためにSig-1Rが配置されている他の細胞コンパートメント(例えば、ER、MAMまたは核膜)へのアクセスを減少させるであろう。第二に、リソソソームのトラップは、リソソームの非特異的なSig-1R非依存的な酸性化の欠陥をもたらすことになり、このオフターゲット効果は、このような薬剤にとって付加価値があるかもしれない。しかし、Sig-1Rリガンドのファーマコフォア関連の潜在的なリソソモトロピー性が、抗ウイルス薬のスクリーニングで報告されているいくつかのSig-1Rリガンドの活性を実際に妨げているのか、あるいはどの程度寄与しているのかについては、現時点では不明である。
シグマ-1受容体はウイルスRNA複製の初期段階を制御する
このセクションでは、遺伝子サイレンシングアプローチによって得られたエビデンスについて議論する。薬物再利用試験管内試験スクリーニングにおいて、多数のシグマ-1リガンドが発揮する抗ウイルス活性は、常に気付かれないわけではなかった。以前に記載された薬理学的知見(Gastaminza et al 2010)に続いて、HCV感染においてSig-1Rが果たす役割を調査した。Sig-1R mRNAを標的としたレンチウイルス送達短ヘアピンRNA(shRNA)を用いたRNAiを用いて、Huh-7ヒト肝腫細胞におけるSig-1R発現をダウンレギュレートした(Friesland et al 2013)。4つの異なるshRNAは、無関係なshRNAでトランスフェクトされたコントロール細胞と比較して、異なる大きさでSig-1Rタンパク質サイレンシングを引き起こした。コントロールおよびサイレンス化された細胞を感染性HCVウイルスで接種し、細胞内および細胞外の前駆体感染ウイルスおよび細胞内HCV RNAの産生を測定することにより、感染効率をモニターした。Huh-7細胞におけるSig-1R発現のダウンレギュレーションは、シングルサイクル感染実験におけるHCV RNA蓄積および細胞内および細胞外感染性の低下によって示されるように、HCV感染に対する感受性を比例的に低下させた。すなわち、子孫ウイルス産生は、感染後 24 時間および 48 時間の細胞内 Sig-1R レベルに比例していた。また、その根底にあるメカニズムを探るために実験を行い、Sig-1RのダウンレギュレーションはHCVの侵入に影響を与えず、その発現レベルは、ウイルスRNAゲノムの一次翻訳、持続的HCV RNA複製(定常状態のHCV RNA複製)または粒子の組み立てと分泌に制限を与えないことが明らかになった。しかしながら、シグマ-1発現は、HCV RNA複製を開始するために速度制限的であった。シングルサイクル感染実験におけるSig-1R欠損細胞におけるHCV RNAの蓄積の減少は、一次翻訳の下流にあるHCV RNA複製の確立の欠陥によるものであった。したがって、Sig-1R発現は一次翻訳後早期のRNA複製には速度制限的であるが、持続感染細胞で観察されるように、ウイルス複製機械が確立され、複製が定常状態レベルに達すると、それは無効となる。Friesland et al 2013)による研究におけるもう一つの注目すべき結果は、Huh-7細胞におけるSig-1R発現は、HCV感染に対しては速度制限的であったが、インフルエンザAウイルス(A/WSN/33)またはVSVのようなネガティブセンス一本鎖RNAウイルスへの感染に対しては速度制限的ではなかったことである(Friesland et al 2013)。したがって、HCV(+ssRNAウイルスではあるが、SARS-CoV-2ではない)に感染した培養肝腫細胞(SARS-CoV-2の主要な細胞標的ではない)において宿主Sig-1Rがウイルス複製において果たした役割に関する証拠は、いかなる場合においても、その天然標的細胞におけるSARS-CoV-2に対する証明された機序的相関を示唆するものではない。
全体的に、Sig-1R欠損細胞からのデータは、Sig-1Rが、HCV RNA複製の初期段階を調節するウイルスRNAゲノムのHCV感染、下流への侵入、送達および一次翻訳のためにリクルートされた宿主細胞因子であることを示している(Friesland et al 2013)。これは、Sig-1Rリガンド(ほとんどの研究ではSig-1Rの活性リガンドとして認識されていない)が、ウイルス粒子の付着、内部化および細胞質へのゲノムの送達の後、複製サイクルの初期段階を阻害することが見出された薬理学的知見と一致する(Nemerow and Cooper, 1984; Mingorance et al 2014; Stadler et al 2008; de Wilde et al 2014; Gordon et al 2020)。
シグマ-1レセプターは、ウイルスリプライアーゼ/トランスクリプターゼ複合体の非構造タンパク質とコロケーションし、そして相互作用する
このセクションでは、コロケーションおよびインタラクトームマップ研究から得られたエビデンスについて議論する。Sig-1Rは、HCV複製複合体のNSタンパク質とコロケーションすることが判明した(Friesland et al 2013)。細胞を、ウイルスレプリカーゼの構成要素(NS3,NS4BおよびNS5A)に対して、およびSig-1Rに対して指示された抗体で二重免疫染色のために処理した。模擬感染したHuh-7細胞において、Sig-1Rの免疫蛍光は、ミトコンドリアと並置された細胞質パンクテの優勢な離散性局在、および正常な安静時の非ストレス細胞におけるSig-1Rの特徴的な細胞内分布であるERとコロケーションする拡散性の細胞質パターンを明らかにした(Su et al 2010)。感染中、Sig-1Rの細胞内分布パターンは変化し、感染後48時間で70%以上の細胞が拡散性の核周囲パターンを示した。興味深いことに、Sig-1Rはウイルス感染の初期段階では、ウイルスのNS3,NS4B、NS5Aレプリカーゼコンポーネントと核周囲領域に共局在していた。このことは、Sig-1Rの一部分が元のパターンに戻り、48時間後に観察されたSig-1RのウイルスレプリカーゼNSタンパク質との核内コローカリゼーションは一過性のものであることを示唆している。全体的に、これらの結果は、Sig-1Rがウイルス感染の初期段階でNSタンパク質が蓄積するERの核周囲領域にリクルートされ、HCV RNA複製の開始を調節していることを示唆している。ほとんどのSig-1RとNS3およびNS5Aは、洗浄剤耐性、コレステロールおよびスフィンゴ脂質が豊富な細胞内膜と関連しており、Sig-1RとHCVレプリカーゼの構成要素が類似のER膜環境を標的としており、そこでSig-1Rがプロウイルス機能を発揮する可能性が高いことをさらに示唆していた。特筆すべきことに、このような一過性のシグマ-1の再局在化はERストレス時に記述されており、ストレスに対する細胞応答に寄与することが提案されている(Hayashi and Su, 2007)が、ウイルスが宿主ストレス関連タンパク質を利用して有利な細胞プログラムを展開していることを示唆している。ウイルスの複製および感染細胞の生存の両方を促進するためにHCV感染によって誘導される細胞ストレス経路、ならびにHCV感染におけるSig-1Rのプロウイルス的役割については、これまでにレビューされており(VasalloおよびGastaminza 2015)ここではさらにレビューしない。
最近、SARS-CoV-2タンパク質相互作用マップは、Sig-1Rとの物理的相互作用を啓示した(Gordon et al 2020)。著者らは、29個のSARS-CoV-2タンパク質のうち26個をクローニングし、タグ付けし、発現させ、アフィニティー精製質量分析法を用いてSARS-CoV-2-ヒトタンパク質-タンパク質相互作用を同定した。SARS-CoV-2相互作用タンパク質の約40%は、膜内コンパートメントまたは小胞輸送経路に関連していた。特に、ウイルスNSタンパク質Nsp6はSig-1Rと相互作用することが明らかになった。SARS-CoV-2ゲノムは、14個ものオープンリーディングフレーム(Orf)をコードしている(Masters, 2006; Chan et al 2020; Gordon et al 2020; Wu et al 2020)。ゲノムの3分の2の5′にあるOrf1a/Orf1abは、前駆体ポリタンパク質をコードしており、このポリタンパク質は、レプリカーゼ/転写酵素複合体を形成する16個のNSタンパク質(Nsp1-16)に自己増殖的に処理される。ウイルスゲノムの3’末端には、スパイク(S)エンベロープ(E)メンブレン(M)ヌクレオカプシド(N)構造タンパク質、および付属タンパク質をコードするサブゲノムmRNAから 13個ものOrfsが発現している。ウイルス複製機構は、Nsp3,Nsp4,Nsp6によりER膜に局在していると考えられている。Nsp6は、Nsp3およびNsp4と複合体を形成し、ウイルス複製酵素/転写酵素複合体をER膜に固定する(Oostra et al 2008; AlsaadiおよびJones 2019)。3つのレプリカーゼタンパク質はすべて、ウイルスレプリカーゼ/トランスクリプターゼ複合体のER膜への組み立てに重要な膜貫通配列を含む(Oostra et al 2008)。Nsp6は、7つの疎水性ドメインを含むが、6つの膜貫通ドメインを含み、そのアミノ末端およびカルボキシ末端が細胞質で露出し、C末端の細胞質尾部に保存された疎水性ドメインを含むことが示された(Oostra et al 2008; Baliji et al 2009)。西洋免疫ブロット法により、約 23 および 25 kDa の 2 つの nsp6 生成物が同定されたが、複数の形態の nsp6 が存在する理由は現在のところ不明である (Baliji et al 2009)。レプリカーゼ複合体をER膜に固定する役割に加えて、Nsp6は、二重膜小胞およびオートファゴソーム形成を誘導することが明らかにされている(Cottam er al)。
HCVおよびSARS-CoVを含む陽性鎖RNAウイルスは、ウイルス複製を組み立てるために宿主細胞のER膜を封鎖する。コンボリュート膜、相互に連結された二重膜小胞、および小胞パケットを統合する修飾された核周囲粗ERのネットワークが記載されている(Gosert et al 2002;Knoops et al 2008;Sola et al 2015)。ウイルスレプリカーゼサブユニットは、複雑なER膜に最も豊富に位置し、RNA複製(二本鎖RNA)は二重膜小胞に局在し、小胞パケットは、二重膜小胞の合体から生じ、(出芽た)ウイルス粒子を含む大きな細胞質液胞に発達するように思われた。最終的に、CoVゲノムの複製は、連続的なRNA合成を必要とし(Sola et al 2015)網状小胞ネットワークは、RNA合成に関与するER膜構造を、新しいウイルスの組み立てが起こる部位に接続する構造的および機能的連続体を提供する(Knoops et al 2008)。先行研究によれば、Sig-1Rは、複製の初期段階では必要とされるが、定常状態のHCV RNA複製または感染性粒子の組み立ておよび分泌には必要とされない(Friesland et al 2013)。したがって、ウイルスRNAゲノムの内部化、送達、および一次翻訳は、持続感染において網状網状ネットワーク連続体が完全に発達する前の初期段階で、新たに合成されたウイルスレプリカーゼタンパク質と複合体を形成するSig-1Rのリクルートに先行するであろう。Sig-1RのHCVレプリカーゼタンパク質との初期および一過性のコロケーション(Friesland et al 2013)およびNsp6 SARS-CoV-2レプリカーゼタンパク質とのSig-1Rの相互作用(Gordon et al 2020)は、この仮説を支持している。この相互作用の機能的目的は不明である。即座の仮定は、レプリカーゼ/トランスクリプターゼ複合体のER膜へのアンカーリングが、そのパートナーであるNsp6の提案された役割であるので、Sig-1Rは、ER膜へのウイルス複製機械の挿入を補助するかもしれないということである。しかし、それはまた、機能的なレプリカーゼ/トランスクリプターゼ複合体の多タンパク質アセンブリを支援するために、初期のERリモデリングと網状ネットワークを介したトラフィッキングを促進するために、初期のウイルスの要求に宿主細胞のバイオエネルギーと生合成機械を結合させるために、ER-ミトコンドリアシグナルとER-核のクロストークを調整するために、初期のウイルスのタンパク質の適切な折り畳みまたは膜の配向を可能にするかもしれない。これらの機能はすべて、この常駐型ERシャペロン/足場および動的多能性調節タンパク質が果たす役割と首尾一貫しており、組織間シグナル伝達、バイオエネルギーおよび細胞ストレス応答に関与している(Hayashi and Su, 2007; Su er al 2010; Vollrath er al 2014; Hayashi, 2019; Delprat er al 2020)。
コロナウイルス誘発宿主細胞ストレスにおけるシグマ-1受容体の役割?
培養細胞のCoV感染は、ERストレスを引き起こし、アンフォールドタンパク応答(UPR)ER特異的ストレス応答、およびそれらの下流シグナルを誘導する(Fung and Liu, 2014; Fung er al)。 ERストレスおよびUPRは、特にSARS-CoV-2感染に関与しており(Sureda et al 2020)COVID-19媒介のERストレスを標的とする併用療法が最近提案されている(Banerjee et al 2020)。UPRは、グローバルな翻訳遮断およびERフォールディング能力の増大により、ERの恒常性および細胞生存を回復させることを目的としている。UPRシグナル伝達は、アンフォールドされたタンパク質が3つのERストレストランスデューサー:二本鎖RNA活性化プロテインキナーゼ(PKR)様ERプロテインキナーゼ(PERK)、活性化転写因子-6(ATF6)、またはイノシトール要求酵素(IRE1)を活性化することから始まる。ER内腔シャペロンBiP(GRP78またはHSPA5としても知られている)からの可逆的な解離および他のERコシャペロンとの相互作用は、UPRトランスデューサーの活性化/不活性化ダイナミクスを調節する。BiPは、ミスフォールドされたタンパク質によって活性化されるため、直接的なERストレスセンサーであると考えられている。ストレスを受けていない細胞では、BiPはERストレストランスデューサーのER内腔ドメインに結合し、不活性化状態を維持している(図2)。ERストレスの間、BiPは、アンフォールドおよびミスフォールドされたタンパク質に優先的に結合し、膜貫通型トランスデューサーから解離し、それらの活性化を促進する(Bertolotti et al 2000; Kopp et al 2019)。活性化されると、UPRトランスデューサーは、シグナルをサイトゾルおよび核に伝達し、細胞は、タンパク質合成を低下させ、ERフォールディング能力を増加させることによって応答する。PERK/eIF2α(真核生物開始因子2α)/ATF4経路はタンパク質の翻訳を急速に減衰させ、ATF6およびIRE1α/XBP1(転写因子X-box binding protein-1)カスケードは、適切なフォールディングを促進するERシャペロン遺伝子を転写的にアップレギュレートする(図2)。蓄積されたアンフォールディングされたタンパク質は、正しくリフォールディングされるか、あるいはうまくリフォールディングされずに、ER関連分解複合体(ERAD)ユビキチン-プロテアソーム経路を介して、あるいはオートファジーを介してクリアされる。しかし、長時間のERストレス下では、ホメオスタシスが再確立できず、誤って折り畳まれたタンパク質の蓄積が毒性を帯びると、UPRはアポトーシスによる細胞死を誘発することもある。アポトーシスは、UPR媒介およびCa2+媒介のカスパーゼ活性化経路およびミトコンドリアのリクルートを介して潜在的に誘発される(Kim et al 2006; Fung and Liu, 2014; Karagöz et al 2019)。実際、Ca2+ホメオスタシスは、ERストレスおよびUPRが媒介するアポトーシス誘導において主要な役割を果たしている。ERのCa2+貯蔵量の枯渇は、ERに常駐するCa2+依存性シャペロンおよびタンパク質フォールディングに有害な影響を与え、MAMにおけるERからミトコンドリアへの過剰なCa2+移動(すなわち、ミトコンドリアのCa2+過負荷)は、ミトコンドリアの活性酸素種(ROS)産生/酸化ストレスおよびシトクロムC放出を導く(Carreras-Sureda et al 2018)。最後に、オートファジーはまた、PERK、IRE1,ATF6およびCa2+を含むUPRと共通の上流シグナル伝達を共有する経路によって、ERストレス下で活性化され得る(ERストレス媒介オートファジー)(Song et al 2017)。オートファジーは、分解のためにリソソーム/液胞に送達される二重膜結合構造における細胞質成分の巻き込みによって特徴づけられる。オートファゴソームには、摩耗したタンパク質、タンパク質凝集体、および損傷した小器官が含まれる(Lee et al 2015; Rashid et al 2015; Song et al 2017)。
図2 重症急性呼吸器症候群CoV-2(SARS-CoV-2)が媒介するアンフォールドタンパク応答(UPR)シグナリングの提案モデル
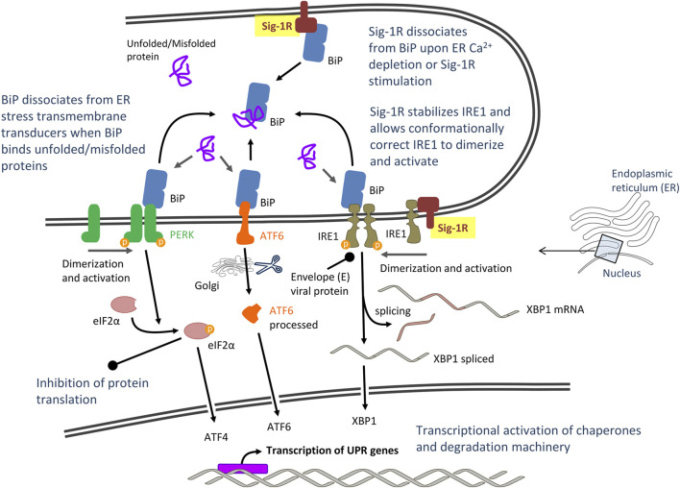
マスターUPR調節因子との相互作用を介したシグマ-1受容体(Sig-1Rs)の潜在的な役割 Sig-1Rの役割に関する略語および参考文献は本文中に記載されている。
ERの折り畳み能力をオーバーロードするタンパク質合成のバースト、ウイルス複製中のER膜の広範な再配置、およびCoVのS(Siu et al 2014)および3aアクセサリー(Minakshi et al 2009)タンパク質などのウイルスタンパク質はERストレスを引き起こすが、ウイルスはUPRシグナリングを管理し、その複製に有利な環境を作り出すためのメカニズムを進化させてきた(FungおよびLiu 2014)。手術的ではあるがハイジャックされたUPRは、選択的なトランスレーショナルリプログラミングおよび転写リプログラミングを行うが、細胞死への感受性は低下しているため、宿主細胞の生存に寄与し、ウイルスの複製を持続させることができるであろう。したがって、CoVはUPRトランスデューサーを活性化するが、いくつかのUPR標的遺伝子の下流への誘導は最小限である。このことは、ウイルスタンパク質の翻訳が増加する一方で、宿主細胞タンパク質の合成を持続的に停止させることに有利である(Bechill et al 2008)。また、SARS-CoVのエンベロープEタンパク質は、UPRのIRE1α/XBP1経路を中和し、アポトーシスを阻害することが示されている(DeDiego er al)。 アポトーシスは感染細胞にとって致命的な宿命であるが、ウイルスの産生および播種を制限することによって宿主を保護することに留意されたい。しかし、すべての証拠が同じ方向性を持っているわけではなく、いくつかの知見は、ERストレス、UPRおよびオートファジー誘導が、細胞宿主のCoVとの闘いにおける生得的な応答であることを支持している。例えば、αCoV感染性胃腸炎ウイルス(TGEV)への感染は、3つのUPR経路(PERK、ATF6およびIRE1)すべてを活性化したが、PERK/eIF2α軸の活性化は、タンパク質翻訳の全体的な減衰を介してTGEVの複製を阻害した(Xue er al)。 PERK経路はまた、プロアポトーシスであるSARS-CoVの3aアクセサリータンパク質を発現する細胞においても活性化された(Minakshi et al 2009)。他の研究では、ウイルス複製に対する正の効果と負の効果が混在していることを指摘している。例えば、IRE1 RNase活性はウイルス複製に好ましくないことが報告されたのに対し、IRE1キナーゼ活性はウイルス複製を増強した(Su et al 2017)。
Sig-1Rはどうか?ERストレス/UPRはPERK/eIF2α/ATF4経路(ATF4はSig-1R遺伝子の5’フランキング領域に結合し、その転写をアップレギュレートする)を介してSig-1Rの発現を誘導する(Mitsuda er al)。 その結果、Sig-1Rのアップレギュレーション、実験的な過剰発現、またはそのリガンド刺激は細胞を保護し、それはほとんどの研究においてERストレスおよびアポトーシスの減少と相関している(Mitsuda et al 2011; Wang er al 2012; Omi er al 2014; Shimazawa er al 2015; Cao er al 2017; Ellis er al 2017; Morihara er al 2018; Zhai er al 2019)であるが、すべてではない(Penas er al 2011; Schrock er al 2013; Alam er al 2017)。二相的な役割も記述されており、Sig-1R介在性の増悪に続いて保護が行われ、それはそれぞれERストレスおよびオートファジー応答のマーカーの増加および減少に伴うものである(Yang et al 2017)。以下の段落では、CoV感染に関連する可能性のあるERストレス応答のいくつかの側面を調節するSig-1Rの役割を支持する証拠をレビューし、議論する。
膜内リモデリング
ERリモデリングは、CoVによって誘導されるERストレス応答の初期の重要な要素である。前述したように、CoVは膜内コンパートメントの恩恵を受け、宿主細胞のER膜の成長およびリモデリングを誘導して網状網状ネットワークを形成する(Knoops et al 2008)。Sig-1Rの枯渇は、ERテザリングの喪失および増殖、ならびにミトコンドリアの異常およびマイトファジーを含むERの異常な形態をもたらし、ERおよびミトコンドリアの構造的および機能的完全性の維持におけるSig-1Rの役割を示唆している(Vollrath et al 2014)。したがって、Sig-1Rの薬理学的ブロックは、ウイルスの複製に必要とされるERのリモデリングを阻害し、ミトコンドリアのエネルギー供給に挑戦する可能性がある。このことは、ERリモデリングおよびウイルスレプリカーゼ複合体のアンカリングが起こるHCV複製の初期段階でSig-1Rが必要とされるという知見(Friesland et al 2013)と一致する。残念ながら、ウイルス感染時のER膜のアーキテクトニクスにおいてSig-1Rが果たす役割は調査されていない。
カルシウムの恒常性
ウイルスは、Ca2+がウイルスの侵入、複製、成熟および放出に不可欠であるため、宿主細胞のCa2+恒常性を乱し、細胞内Ca2+を増加させるメカニズムを進化させてきた(Olivier, 1996; Chen et al 2019)。ERからのCa2+放出または漿膜チャネル/ポンプを介したCa2+進入をブロックすることにより、ウイルス誘発性の異常な細胞質Ca2+増加を阻害することは、ウイルス感染症を制御する戦略として浮上している(Chen et al 2019)。したがって、いくつかのCa2+チャネルブロッカーは、COVID-19で入院した高齢者患者において、死亡率を改善し、挿管および機械的換気のリスクを減少させることが報告されている(Solaimanzadeh 2020)。Sig-1Rは、細胞質膜レベルでのCa2+の侵入(リガンドおよび電圧ゲーテッドCa2+チャネルとの相互作用を介して)およびエンドプラズムストアからのCa2+の動員(イノシトール-1,4,5三リン酸受容体(IP3R)との相互作用を介して)の両方を調節する(Monnnet 2005)。小胞体ストレス(小胞体貯蔵庫からのCa2+枯渇)の下では、Sig-1RはBiPから解離してIP3R3をシャペロンし、小胞体からミトコンドリアへの適切なCa2+シグナル伝達を確保する(Hayashi and Su, 2001; Wu and Bowen, 2008)(図3)。IP3R3を介したミトコンドリアへのCa2+流入の増加は、細胞の生理機能をエネルギー需要に結びつけるための基本的なものであり、ウイルスタンパク質の同化とRNA合成に必要とされる可能性が高いが、ミトコンドリアへのCa2+流入の持続的・過大な増加は、過剰な活性酸素、酸化ストレス、アポトーシスを引き起こす。Sig-1RアゴニストはSig-1RとBiPの解離を引き起こし、Sig-1RとIP3R3の相互作用を可能にし、IP3R3が媒介するミトコンドリアへのCa2+流入を促進するのに対し、Sig-1RアンタゴニストはSig-1RとBiPの相互作用には影響しないが、Sig-1Rアゴニストが媒介する解離を阻害する。Sig-1Rアゴニストについては、ミトコンドリア複合体I活性を増加させ、生理学的シグナルとしてCa2+依存的な方法で適度な活性酸素の増加を誘発するが、複合体IおよびIVの機能不全を減衰させ、病理学的条件下で顕著な抗酸化作用を促進することにより、陰陽作用が記述されている(Goguadze et al 2019)。Sig-1Rアゴニスト(+)-ペンタゾシンでのミトコンドリア膜の処置は、Rac1シグナル伝達を介したBadおよびNADPH依存性の活性酸素の産生のリン酸化を導く(Natsvlishvili et al 2015)。免疫沈降技術は、MAMにおけるSig-1RがRac1,IP3RおよびBcl2と複合体を形成し、Sig-1Rアゴニストは、このIP3R/Sig-1R/Bcl2/Rac1多タンパク質複合体を介して軽度の酸化ストレスを誘導し得ることを明らかにした(Natsvlishvili et al 2015)。まとめると、生体エネルギー結合およびミトコンドリアCa2+オーバーフロー媒介アポトーシスの両方は、MAMにおけるIP3R3を介したCa2+シグナル伝達に依存しており、Sig-1Rによって制御される(Delprat et al 2020)。増加したIP3R1活性を介したERからサイトゾルへのCa2+放出はまた、ERストレスを誘導し、Sig-1Rは同様にIP3R1に結合し、調節する(Kubickova et al 2018)(図3)。これらのメカニズムの微調整制御(強化されたエネルギー供給のために十分であるが、宿主細胞の死を回避するためにあまりにも多くはない)は、効率的なウイルス感染に不可欠である可能性があり、したがって、Sig-1Rの薬理学的調節は、ここでウイルスプログラムを打ち消すための治療の機会を提供していることを示唆している(図3)。
図3 重症急性呼吸器症候群CoV-2(SARS-CoV-2)による宿主細胞カルシウム(Ca2+)のホメオスタシス障害のモデルを提案
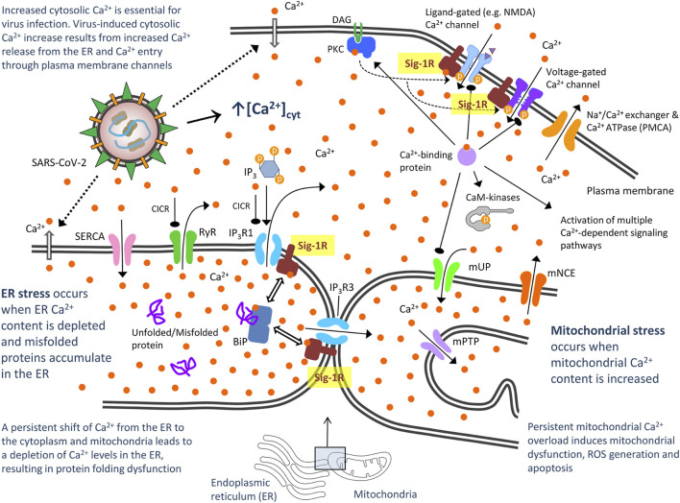
シグマ-1受容体が細胞膜や小胞体のCa2+チャネルとの相互作用を介して果たす役割の可能性 CaMキナーゼ、Ca2+/カルモジュリン依存性プロテインキナーゼ、DAG、ジアシルグリセロール、IP3,イノシトール1,4,5-三リン酸、IP3Rs、イノシトール1,4,5-三リン酸受容体ゲートチャネル、IP3R1はERからサイトゾルへのCa2+放出を、IP3R3はミトコンドリアへのCa2+放出を媒介する。IP3R1はERからサイトゾルへのCa2+放出を、IP3R3はミトコンドリアへのCa2+放出を媒介している。細胞質のCa2+濃度が高いと、IP3R;PKC;プロテインキナーゼC;mNCE;マイトコンドリアのNa+/Ca2+および2H+/Ca2+交換体を阻害することができる。これらは、蓄積されたCa2+をゆっくりとサイトゾルへと排出する;mPTPはミトコンドリア伝染性遷移孔である。一旦、ミトコンドリア内のCa2+がある閾値以上に上昇すると、内膜のこの電圧とCa2+に依存した高導電性チャネルが開き、細胞死メカニズムを活性化する; mUP, mitochondrial uniporter。このチャネルは、二相性の方法でCa2+によってゲートされる。ミトコンドリアへのCa2+取り込みはCa2+/カルモジュリンによって促進されるが、持続的な細胞質Ca2+レベルはユニポーターを不活性化させ、それ以上のCa2+取り込みを妨げる。Ca2+ゲート型Ca2+チャネル(CICR)であり、SERCA、サルコ/小胞体Ca2+ ATPaseである。Ca2+でERを修復する。Sig-1Rの役割については、本文中に参考文献を記載している。
マスターアンフォールドタンパク応答調節因子との相互作用
Sig-1Rは、動的、可逆的、およびCa2+依存的な方法で、ER内腔シャペロンおよびストレスセンサーBiPに結合する(Hayashi and Su, 2007; Ortega-Roldan et al 2013)。BiPは、GRP78とも呼ばれ、ウイルス感染のための重要な宿主因子である。相当量のSARS-CoV Sタンパク質は、感染中にERに蓄積し、BiPおよびUPR選択的経路の直接的な活性化を誘導する(Chan et al 2006)。BiPを標的化することは、ウイルスの生命の多段階を混乱させる可能性があり、CoV感染に対する潜在的な治療アプローチとして最近提案されている(Ha et al 2020)。Sig-1Rは、その嵩高いC末端ルーメンドメインを介してBiPのヌクレオチド結合ドメインに結合する(Ortega-Roldan et al 2013)。ER膜に結合したSig-1Rのルーメン性BiPからの解離は、Ca2+枯渇(ERストレスを示す)または薬理学的Sig-1R刺激によって起こる(図2)。BiPはまた、膜結合型UPRトランスデューサーPERKおよびIRE1のER内腔ドメインに結合し、結合した場合、BiPおよびUPRトランスデューサーの両方が不活性状態のままである。ERストレス中のBiP基質結合ドメインへのミスフォールドされたタンパク質のリクルートは、そのヌクレオチド結合ドメイン内のATPアーゼ活性を刺激し、BiPがPERKおよびIRE1から解離してそれらの活性化およびUPRシグナル伝達カスケードの開始を可能にするADP結合コンフォメーションを採用することを可能にする(Bertolotti et al 2000; Kopp et al 2019年)。BiPとの相互作用を介したUPRのSig-1R調節のための機構的詳細は不明である。Sig-1R-BiPの解離は、UPRを誘導するために(ミスフォールドされたタンパク質およびATP結合とともに)共活性化剤として作用するのか?Sig-1Rはアロステリック誘導剤として作用するのか、あるいはPerKやIRE1と競合してBiPヌクレオチド結合ドメインに結合するのか?Sig-1RはERストレス(Ca2+枯渇)のセンサーであり、UPRの上流制御因子であると考えられている。数多くの細胞アッセイにおいて、Sig-1Rリガンドの抗ウイルス効果を支持しているか?
Sig-1Rは、そのコシャペロンBiPとの相互作用に加えて、ERストレストランスデューサーの1つであるER常駐膜貫通タンパク質IRE1(Mori et al 2013)をシャペロンし、CoVが宿主細胞機械をその要求に適応させ、細胞のアポトーシスに拮抗するために重要である(Fung et al 2014b)。Sig-1Rは、細胞がERストレスを受けているときにIRE1を安定化し、そのような相互作用により、コンフォーマルに正しいIRE1が活性化された形態に二量化することを可能にする(Mori et al 2013)(図2)。IRE1(αアイソフォーム、IRE1α)は、キナーゼ活性に結合したRNase活性を有する。IRE1の活性化のために提案されている異なるモデルがあり、それらのすべては、BiPからの解離、オリゴマー化、およびその細胞質キナーゼドメインの活性化を伴う(Adams et al 2019)。この活性化は、XBP1 mRNAの型破りなスプライシング、および活性転写因子XBP1sのその後の翻訳を可能にする。XBP1sは、タンパク質の恒常性を回復するために、シャペロン、フォルダーゼ、およびERAD経路の構成要素を含むいくつかの標的の発現を促進する(Smith et al 2011)。SARS-CoVのエンベロープEタンパク質は、UPRのIRE1/XBP1経路を打ち消すことが示されており(DeDiego et al 2011)UPRのIRE1/XBP1経路の阻害がCoV感染に重要であることを示唆している。単純ヘルペスウイルス-1の複製について行われた研究では、IRE1ドメインのウイルス複製に対する反対の作用が示され、RNアーゼ活性はウイルス複製に不利であり、キナーゼ活性はウイルス複製を増強することが示された(Su et al 2017)。IRE1のRNase活性は、ウイルスタンパク質の分解につながる可能性のある細胞タンパク質分解経路(ERAD)を活性化し、これはウイルス複製に好ましくない(Su et al 2017)。したがって、ウイルスの複製を促進するために、CoVマウス肝炎ウイルス(MHV)を含む様々なウイルスによる感染において、IRE1のRNase活性が抑制された(Bechill et al 2008)。IRE1のRNase活性は、調節されたIRE1依存性崩壊(RIDD)を介して他の遺伝子を標的とする可能性もある。RIDDは、IRE1が分解される標的転写基質を切断し、mRNA分解を介してERタンパク質負荷を減少させることによりER恒常性の維持に寄与するメカニズムであるが、細胞死につながることも提案されている(Tam et al 2014; Abdullah and Ravanan, 2018)。Sig-1Rは、敗血症および炎症の前臨床モデルにおいて、活性XBP1タンパク質を産生するためにXBP1をコードするmRNAのスプライシングに必要なIRE1エンドヌクレアーゼ(RNAase)活性に関連し、制限する(Rosen et al 2019)。実際に、LPSを負荷したSig-1Rノックアウトマウスは、WTマウスと比較して肝XBP1スプライシングが増加していた。ウイルスがIRE1 RNase活性を損なうメカニズムは不明であるが、Sig-1Rの薬理学的ブロックはIRE1 RNase活性を促進し、その結果IRE1/XBP1依存性の分解経路を増加させる可能性がある。Sig-1Rアンタゴニストリガンドのウイルス複製に対する阻害効果に寄与しているのだろうか。
オートファジー
最後に、CoV感染(SARS-CoV、MERS-CoV、および新しいSARS-CoV-2を含む)は、オートファジーを誘導することが実証されている(Maier and Britton, 2012; Yang and Shen, 2020)。興味深いことに、多様なCoVからのウイルスNsp6の発現は、オートファジーを誘導する(Cottam et al 2011)。MHVおよびSARSのCoVからのウイルス複製タンパク質は、オートファゴソームタンパク質マーカーとコロケーションすることが示されており(Prentice et al 2004a; Prentice et al 2004b)オートファジーは、二重膜小胞の形成およびMHVの複製の両方に関与していることが示唆されている(Prentice et al 2004a)。しかし、オートファゴソームマーカーとSARS-CoVの特定のレプリカーゼサブユニットとのコロケーションは他の研究では観察されておらず(Snijder et al 2006)、オートファジーがウイルスの複製に直接関与していないことを示唆する多くの観察がなされている(Zhao et al 2007)。それどころか、MERS-CoVの増殖がオートファジー過程に抑制効果を及ぼし、オートファジーの増強がMERS-CoVの複製を減少させることが報告されている(Gassen et al 2019)。したがって、オートファジーがウイルスによってその利益のために利用されるのか、あるいは実際にCoV感染に対する保護細胞応答を表すのかは議論の余地がある。オートファゴソームは、ER-ミトコンドリア接触部位に由来し(Hamasaki et al 2013)Sig-1Rは、オートファジーの上流モジュレーターとしてこのMAM交差点で作用する(Schrock et al 2013)。Sig-1Rアゴニストは長時間の治療後にオートファジーを誘発するのに対し、Sig-1RアンタゴニストおよびSig-1Rのノックダウンはオートファゴーム形成を抑制する(Schrock et al 2013)。したがって、機能喪失変異およびSig-1R欠損は、オートファジーの欠損と関連しており、オートファジー空胞の蓄積をもたらす。対照的に、Sig-1Rをヌルバックグラウンドまたはその活性化で再発現すると、オートファジー活性が回復/誘導される(Vollrath et al 2014;MacVicar et al 2015;Christ et al 2019;Yang et al 2019;Christ et al 2020)。Sig-1Rは、一般的な生理学的オートファジー機械のコアコンポーネントではない可能性が高いが、細胞ストレス誘発オートファジーに必要とされるようである(MacVicar et al 2015)。膜貫通型SARS-CoV-2 Nsp6と宿主Sig-1Rタンパク質との既知の相互作用、Nsp6によるオートファジーの誘導、およびオートファジー調節におけるSig-1Rの役割にもかかわらず、Sig-1RがCoV感染の二次的なオートファジー誘導に関与しているかどうか、およびどのように関与しているかは不明である。
