Contents
Neurodegenerative Diseases: Potential Effect of Glutathione
要旨
神経変性疾患は、中枢神経系や末梢神経系の神経細胞の機能が徐々に低下し、最終的には神経細胞が死滅することを特徴としている。酸化ストレスは、神経変性過程に関与する重要なメディエーターであり、様々な形態の細胞死を誘発する重要なイベントである可能性があるという大きな証拠がある。
ここでは、酸化ストレスに起因する神経細胞の損失は、抗酸化分子であるグルタチオン(GSH)の急激な減少によって引き起こされるのではないかという仮説を検討した。
グルタチオンの生理的なレベルの低下や、神経細胞内のグルタチオン関連酵素の活性の変化は、神経変性疾患の発症や進行に関与していることが示唆されている。GSHは神経系における細胞の酸化還元恒常性の維持に重要な役割を果たし、様々な酸化的障害から神経細胞を保護している。GSH の枯渇は酸化ストレスを増大させ、タンパク質の凝集を増加させ、異なる神経細胞集団において細胞死を引き起こす可能性がある。パーキンソン病やアルツハイマー病では、酸化ストレスと神経細胞のGSH低下が大きな影響を与えていることが明らかになっている。
1. 序論
この30~40年、世界的な平均寿命の伸びに伴い、特に高齢者を中心とした脳の神経変性疾患が社会的な負担となってきている。欧州連合(EU)の共同プログラム-神経変性疾患研究(JPND)では、2030年には欧州人口の4分の1(25%)が65歳以上となり、現在の16%から大幅に増加するとしている[1]。そこで、研究者たちは、加齢に伴う神経変性疾患(アルツハイマー病)の研究に特に力を入れている。アルツハイマー病やパーキンソン病などのアルツハイマー病は、可逆性や有効な治療法がないことから注目されている[2]。神経変性疾患(神経変性疾患)は、神経細胞の損傷や神経細胞の喪失が徐々に進行し、運動機能や認知機能が低下することで知られている。治療は、病気の生理を研究し、病気の進行を抑えるメカニズムを解明することよりも、症状を軽減することが主な治療法となる[3]。神経変性疾患の媒介となる病態はまだ十分には解明されていないが、神経変性疾患患者の脳内では酸化ストレス(酸化ストレス)の上昇が観察されており、活性酸素種(ROS)が重要なイベントであることが多くのエビデンスから示されている[4]。ここ数十年の幅広い研究で、加齢に伴う神経変性の発症は、抗酸化力の低下と酸化的損傷の増加によるものであることが示されている。累積的な活性酸素は、ミトコンドリアの機能障害に加えて、脂質、タンパク質、DNAなどの生体分子の損傷を引き起こす可能性がある[5]。神経変性疾患 のパンデミックの増加と患者の生活の質を大きく阻害することから、神経細胞におけるグルタチオンレベルの向上など、効果的かつ新規の治療法の開発が必要とされている。グルタチオン(GSH)は主要な抗酸化物質であり、そのレベルは神経変性疾患と同様に加齢に伴って低下することがわかっている。現在、科学者たちは、これらの疾患におけるグルタチオンの枯渇の役割を完全に理解し、それを利用してGSHをベースとした治療法を開発しようとしている。グルタチオンは、活性酸素への曝露によるリスクから細胞を保護・防御する役割を担っており、特に脳ではその役割が明らかになっていることから、GSH のホメオスタシスは必須の細胞成分である。このように、GSHの恒常性の障害やGSH依存性酵素の不活性化は、活性酸素に対する主要な防御バリアの破壊につながり、その結果、細胞は酸化ストレスによる損傷に対してより脆弱になる[6]。
本章では、一般的なアルツハイマー病の開始と発症に対する酸化ストレスの刺激効果の観点から、酸化ストレスに焦点を当てた。また、グルタチオンが神経細胞を酸化ストレスによる損傷から守り、その枯渇に起因する細胞内変化がアルツハイマー病を悪化させることの重要性を示した。
2. 酸化ストレスと神経変性疾患
2.1 脳内の活性酸素
人間の脳は体重のわずか2%を占めているにもかかわらず、その酸素要求量は体の酸素消費量の20%と推定されている。脳は、ミトコンドリアで消費された酸素の4%がスーパーオキシドスーパーオキサイドイオン(O2–)に変換される活性酸素種(活性酸素種)の最も発生しやすい臓器に分類され、特に強力な酸化剤として、またラジカル反応の開始剤として非常に高い反応性を持っている。主にスーパーオキシドによって開始される3つのラジカル反応がある “図1” (i) スーパーオキシドジスムターゼ(SOD)の影響下で、スーパーオキシドは過酸化水素(H2O2)に変換され、その後、GSHペルオキシダーゼ(GPx)またはカタラーゼによって水と分子状酸素に変換される。(ii) H2O2はまた、フェントン反応を介して脳内で高濃度に見られる鉄と反応してヒドロキシルラジカル(OH.)を形成し、脂質過酸化を誘発することができる。(iii) スーパーオキシドはまた、酵素ニューロンNO合成酵素(nNOS)によって脳内で大量に生成される一酸化窒素と相互作用する。この反応はフェントンの100万倍の速さで、ヒドロキシルラジカルの1万倍も遠くに広がる有毒な酸化物質ペルオキシナイトライト(ONOO-)を生成する。ONOO-の有害な影響は、高分子(DNA、タンパク質、脂質)の酸化、アミノ酸の硝酸化、ミトコンドリア酵素の不活性化など様々で、ミトコンドリアの機能不全を引き起こす。現在では、酸化された内因性高分子の副産物である特定のマーカーを検出することが可能になっている。例えば、4-ヒドロキシル2,3-ネナール(HNE)は不飽和脂質酸化のマーカーであり、タンパク質付加体の不可逆的な形成やGPx活性の阻害など多くの細胞毒性効果を持ち、H2O2の上昇に寄与している[7]。
図1.活性酸素の発生と活性酸素/RNSの消去におけるグルタチオンの意味合い
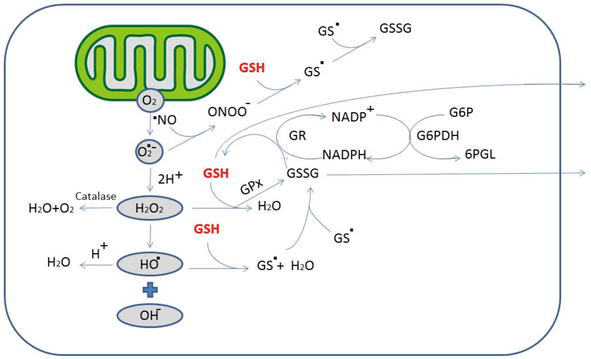
ミトコンドリアの呼吸の結果として、O2からスーパーオキシド(O2-)が生成される。この後者は、スーパーオキシドジスムターゼ(SOD)によって過酸化水素(H2O2)に変換され得る。ヒドロキシルラジカル(-OH)およびヒドロキシルアニオン(OH-)のような多数の他の活性酸素は、H2O2から生成され得る。ヒドロキシルラジカルおよび一酸化窒素またはペルオキシナイトライトは、GSSGを形成するGSHと直接相互作用してもよい。GSHはGPxによって触媒されるH2O2や他の過酸化物の還元のための電子供与体として機能し、その結果、GSSGに変換される。
2.2 神経変性における酸化ストレスの根底にある役割
多くの証拠が、ニューロンの特徴的な特性のために、ニューロンが活性酸素に対して特別な感受性を持っていることを示している。高エネルギー要求、高酸素消費、高レベルの鉄、多価不飽和脂肪酸、そして特に抗酸化保護機能が低いという特徴があるからである。神経細胞の防御酵素系は、SOD、カタラーゼ、GPx活性が他の臓器に比べて低く、弱い。また、必須の抗酸化成分であるグルタチオンは脳内に低濃度で存在している。これらの知見は、神経変性疾患における活性酸素の関与を示唆している[4, 8, 9]。
活性酸素は、神経変性疾患の病態を理解するために行われた研究の重要な軸の一つであった。核酸、タンパク質、脂質などの高分子の酸化誘導、ミトコンドリア機能不全、グリア細胞の活性化、アミロイドβ沈着、アポトーシス、プロテアソーム機能不全など、様々なメカニズムを通じて、神経変性疾患の病態に酸化ストレスが強く関与していることが多くの研究で確認されている[3, 5, 10, 11]。これらの神経変性のメカニズムは、多くの有害な細胞経路に関与していることが全身的なレビューで明らかになった。これらの経路が複雑に絡み合うことで、疾患発症に最も大きな影響を与えることが観察されている[12]、アポトーシス、サイトカイン産生や炎症反応、プロテアソーム機能不全などが観察されている。現在、神経変性疾患(神経変性疾患)の病態形成に及ぼす酸化ストレスの影響や、神経変性疾患の有望な治療法として抗酸化物質の有効性に注目が集まっている。
3. グルタチオンの恒常性と神経変性疾患
グルタチオン(GSH)は、正常な脳機能において重要な意味を持つチオール含有トリペプチドである。GSHはグルタミン酸、システイン、グリシンから形成される。グルタミン酸のγ-カルボキシル基は、N末端のグルタミン酸残基とシステイン残基を連結しており、特異的なペプチド結合である。この特異的なペプチド結合は、細胞内ペプチダーゼによる開裂からGSHを保護し、その加水分解を防ぎ、GSHを細胞内で適度に安定にする。さらに、GSH構造中のC末端グリシン残基の存在は、細胞内γ-グルタミルシクロトランスフェラーゼによるGSHの開裂を防ぐ。システイン残基は、GSHの活性を担うチオール基を提供するため、GSHの有効な機能性成分である。また、システイン残基はGSHの酸化形態では分子間ジペプチド結合を形成している。主要な酸化形態であるグルタチオンジスルフィド(GSSG)は、酸化され、分子間ジスルフィド結合によって接続されているGSHの2つの残基を含む。
3.1 GSHの含有量と合成
GSHは、血液(15μM)と脳脊髄液(脳脊髄液)(5μM)と比較して高濃度(2-3mM)で脳に存在している[13,14]。いくつかの証拠は、GSHが血液脳関門(BBB)を越えて非常に貧弱にそのまま輸送されることを実証している。しかし、血液は脳GSHの主要な供給源ではない可能性が高い。これは、熱心な脳システムがあることを示している[15]その合成in situを保証する。
一般的に、脳内のGSHの恒常性を維持するためには、少なくとも以下の2つのメカニズムが考えられる。
(i)脳内のGSHのターンオーバー中にグルタチオン成分(システイン部位)が回収されリサイクルされる可能性があり、
(ii)脳内グルタチオン合成のための前駆体(システイン、システイン含有分子)が血液脳関門を越えて輸送される可能性がある[13]。
システインは神経細胞のGSH合成の律速基質である[15, 16]。対照的に、グルタミンまたはグリシンの利用可能性は、神経細胞のグルタチオン合成を制限しない[13]。したがって、システイン単独では、神経細胞のGSH合成に不可欠なアミノ酸である[17]。システインの神経細胞への取り込みは、ナトリウム依存系、主に興奮性アミノ酸トランスポーター(EAAT)によって媒介される[18]。EAATは、中枢神経系における細胞外グルタミン酸の除去に重要な機能を持っている[19]。EAATは、興奮性アミノ酸、例えばグルタミン酸およびアスパラギン酸だけでなく、システイン、特にEAAT3,グルタミン酸に匹敵する速度でシステインを輸送することができるEAAC1としても知られているEAAC1も輸送することができる[19]。
シスチンは、ジスルフィド[20]連結を持つ2つのシステインの酸化形態であり、遊離システインの他のソースであり、脳細胞のいくつかのタイプのGSH合成のための基質として採用されている。シスチン部位は、(i)γ-グルタミルシスチンまたは(ii)シスチニルビスグリシンとして脳内に輸送され、脳内のGSHの可能性の起源である[21]。システインはアストロサイトにおけるグルタチオンレベルの維持に特に重要であるが[22]、神経細胞がグルタチオンを取り込むことができないため、神経細胞のGSH合成には重要ではない。したがって、ニューロンはシステインを利用することができず、むしろグルタチオン合成のためにシステインの利用可能性に依存しているため、システインまたはシステイン前駆体の含有量がニューロンにおけるグルタチオンレベルを決定する[20]。システインに加えて、ニューロンは、グルタチオンの前駆体としてCysGly、γGluCys、およびN-アセチルシステイン(NAC)などのシステイン供与体を利用することができる。メチオニンの存在は、神経細胞のグルタチオンレベルを上昇させない[23]。メチオニンは肝臓におけるシステインの主要な前駆体であり、GSH合成に必要なシステインの50%を供給する。しかし、脳内のシステインの生産におけるその役割は無視できるものであり、したがって、神経細胞のGSH合成は、メチオニン[16]の供給に関連していない。グルタチオンの外因性前駆体の中では、ジペプチドであるCysGlyが最も重要である。CysGlyはマイクロモル濃度でニューロンによって効率的に利用されている[24]。
アストロサイトは、ニューロンに比べて高レベルのGSHを貯蔵・合成している[13, 25, 26]。これは、ニューロンがGSHを直接取り込むことができないためである。また、ニューロンはGSH合成にシスチンではなくシステインを利用するのに対し、アストロサイトは両方を利用する[27, 28]。以上のことから、ニューロンは主にアストロサイトに依存して、ニューロンのGSH合成に必要なシステインを供給していることがわかる。アストロサイトによって放出されたGSHは、γ-グルタミルトランスペプチダーゼ(γGT)[29]による開裂プロセスを経て、γ-グルタミル部位と神経細胞のグルタチオンの必須前駆体であるジペプチドCysGlyを産生する。ジペプチドCysGlyは、アストロサイトのために記述されているように、ペプチドトランスポーターを介して神経細胞に取り込まれる可能性がある[30]。ジペプチドCysGlyは、ニューロンに入ると、ニューロン・エクトペプチダーゼによって加水分解され、システインとグリシン[20]を提供し、その後、グルタチオン合成のための前駆体として取り込まれる。グルタチオンは、ATP [13,20]に依存する2つの連続した酵素ステップによって合成される。最初のステップは、グルタミン酸とジペプチドを形成するためにシステインとの間の最初の反応を媒介するγ-グルタミルシステイン合成酵素(GCL)を含む、γ-グルタミルシステイン(γGluCys)は、順番にGSHを生成するためにグリシンと結合する。十分な量のグルタチオンが合成されると、GCLが阻害されるというフィードバックが起こる[31]。逆にGSHが枯渇すると、短期的にはGCL活性が上昇し、結果的にGSH合成が増加する。
3.2 酸化ストレスに対するGSH活性
成体の哺乳類の脳はエネルギー需要が大きく、ほとんどブドウ糖の代謝に頼っている。ブドウ糖のほとんどは、エネルギー要求を満たすために二酸化炭素に完全に酸化される。ブドウ糖を酸化するこの非常に高い能力は、脳が驚くべき速度で活性酸素を産生する可能性があることを示している。この活性酸素産生の増加は、カタラーゼなどの防御機構のレベルの低さと、脳内の脂質含有量の高さとを組み合わせている。これらはすべて、脳が酸化ストレスに対して特に脆弱である可能性を示している。
GSHは、高レベルの活性酸素を減少させ、脳内の酸化的損傷を最小化する上で主導的な役割を果たしている(図1)。この重要性は、細胞内GSHの増加がこの損傷を改善する一方で、酸化ストレスはGSHの枯渇によって悪化することを示すいくつかの研究によって確立されている[32]。GSHはスーパーオキシド[33]、NO[34]、ヒドロキシルラジカル[35]、ONOO-[36]と直接相互作用し、脳の酸化ストレスを防御する重要な成分である。GSHのスーパーオキシド消去能力は、NACやシステインよりも高い[37]。さらに、GSHはこれらのラジカルに対する酵素的防御が不可能であるため、原理的にヒドロキシルラジカル捕捉剤であると考えられている。一方、GSHは酵素触媒による酸化還元サイクルに参加している。グルタチオンの酸化還元反応において最も重要な酵素は、毒性のあるH2O2(または脂質過酸化物、ROOOH)をH2O(またはROOH)に還元する際の主要な役割のために、グルタチオンペルオキシダーゼ(GPx)である。GSHは、GPxによって触媒されるH2O2または他の過酸化物の還元のための電子供与体として機能し、その結果、GSSGに変換される[21]。グルタチオン酸化還元サイクルは、グルタチオン還元酵素(GR)によって完了する。このGSHの酸化還元サイクルは、サイトゾルとミトコンドリアで行われ、GSHはサイトゾルで合成された後、活性酸素の主要な細胞内供給源であるミトコンドリア[38]でコンパートメント化される[39]。また、カタラーゼはH2O2をH2Oに還元するが、脂質過酸化物を無害化することはできず、ほとんどの組織のミトコンドリアには存在しない。これらの理由から、GPxは細胞呼吸中に絶えず発生するH2O2からミトコンドリアを保護する上で特に重要である[40, 41]。ミトコンドリアは、全細胞GSHの5〜15%を含んでいる[42]。このミトコンドリアのGSHプールの維持は、酸化ストレスに対する神経細胞の感受性の主な決定因子である高親和性GSH取り込み系[43]の作用によって行われる[44]。脳のミトコンドリアのこのプールの枯渇は、不可逆的な損傷[45]と死につながるH2O2の毒性効果に対してそれらをより脆弱になる。ミトコンドリアが酸化ストレスの障害から保護されていない場合、小器官は、ミトコンドリア膜の崩壊(ΔΨ)とマトリックスのコロイド浸透圧膨潤[46]に関連付けられているミトコンドリア透過性遷移(mPT)の誘導で最高潮に達するプロセスを介して不可逆的に損傷を受けるようになる。また、GSHは4-ヒドロキシヘキセナール(過酸化脂質)を含む脳ミトコンドリアのmPTを誘導する多くの薬剤を無害化する[39]。これらの知見は、GSHが脳などのミトコンドリアの完全性を維持する上で高い意義を持っていることを示している。さらに、GSHはグルタチオンS-トランスフェラーゼ(GST)の基質となり、GSHに依存した脂質過酸化物の還元を触媒する。上記に加えて、還元されたグルタチオンと別の防御ラインであるビタミンEとの間には、潜在的な相乗的関係がある。このビタミンは、脂質過酸化を抑制するために細胞膜に取り込まれる抗酸化物質としてよく知られている[47]。脂質は、活性酸素を鎮め、それによって、α-トコフェロキシルラジカルに変換するα-トコフェロール(ビタミンE)によって活性酸素から保護されている。この後者は、GSHによって非酵素的にα-トコフェロールに再還元される[48]。この反応と GPx および GST によって触媒される反応はペルオキシダーゼ活性を持ち、生体膜中の多価不飽和脂肪酸に対する H2O2 の損傷効果(脂質過酸化)に対する脳の保護バリアを形成する [49]。
多くの研究で、過酸化水素の脳に対する特異的な毒性が実証されている[42, 43, 50]。この過酸化物は、その毒性に特に敏感な神経細胞においてアポトーシスを誘導する[51]。それにもかかわらず、ニューロンはH2O2を無害化することができるが、明らかにこの能力は、彼らがH2O2の神経毒性効果[45, 46, 52]の調節で考えられる役割を果たしているアストロサイトでより大きくなる。H2O2に対する神経細胞の防御システムは主にグルタチオンの酸化還元サイクルに基づいている。GSHのこの役割は、H2O2がニューロンに適用されたときのGSHの急速な酸化によって明確に示されている[53]。細胞内GSHの枯渇はミトコンドリアの損傷を強制し,細胞死を引き起こす.アポトーシスは、酸化経路を介したフリーラジカルの誘導を介して媒介されると仮定されている。このように、GSH の枯渇とアポトーシスとの直接的な因果関係は、神経細胞において証明されている [54]。また、GSH 枯渇は細胞死の進行の初期段階の特徴であることが明らかになっている[55]。
3.3 神経変性疾患におけるグルタチオンの意義
活性酸素と抗酸化防御システムの間のバランスの破壊が、パーキンソン病(PD)やアルツハイマー病などの多くの一般的な神経変性疾患の開始または進行を誘発する主なマニピュレーターであることが以前に強調されてきた。これらの疾患のそれぞれは、主に酸化ストレスを含む多くの因子に依存している。しかし、酸化ストレスと神経変性との因果関係については、一般的な神経変性疾患におけるGSH系抗酸化ネットワークの制御異常に焦点を当てているため、本編では割愛させていただく。パーキンソン病、アルツハイマー病 [6]。
3.3.1 パーキンソン病(PD)
パーキンソン病の主な病理学的特徴は、黒質パーコンパクトと呼ばれる脳の領域にあるドーパミン作動性ニューロンの喪失であり、ドーパミン作動性ニューロンの細胞内集合体であるレビー小体が存在し、これらのニューロンの死に寄与している可能性が高い。黒質黒質のニューロンはドーパミンを産生するが、これは神経伝達物質(化学伝達物質)であり、黒質から脳の他の部位に信号を伝達する。これらの脳の他の部分は、総称して「大脳基底核」と呼ばれている。黒質質のニューロンと大脳基底核のニューロンの間のコミュニケーションは、スムーズで目的意識のある動きを生み出する。黒質のニューロンが大量に損傷を受けると、ドーパミンの喪失により大脳基底核の正常な機能が妨げられ、パーキンソン病の運動症状である振戦、硬直、平衡感覚障害、自発的運動の喪失を引き起こす [56]。
ドーパミン作動性SN細胞は通常、細胞質のドーパミン(DA)からDA-o-キノンへの自己酸化[57]または酵素による酸化[58]により生成された黒色のニューロメラニンで色素沈着し、これが重合する。通常、このプロセスは、ドーパミン作動性SN細胞をレンダリングするH2O2の生産を伴っているが、おそらく酸化ストレスに対して特に敏感である。DA酸化の高い基底レベルを持つドーパミン作動性SNニューロンは、色素沈着が激しく、パーキンソン病の変性に対して特に脆弱であることが報告されている[59]。
パーキンソン病SNの初期段階で起こる最も顕著な特徴的な変化は、ニグラルGSHの大規模な喪失である[60]。このGSHの喪失は、生合成酵素の活性変化とは無関係であり、GSSGレベルの上昇を伴わない[61]。GSHの急激な低下は、γ-GTの活性が上昇し、細胞からのGSHとGSSGの両方の除去が増加したことに起因していることが示唆されている[61]。GSH枯渇はパーキンソン病に特徴的なものであり、他の大脳基底核の神経変性疾患には見られないことは興味深い[62]。
GSH枯渇は、色素性SN細胞の細胞質に間接的にエンドトキシンを形成し、パーキンソン病におけるこれらの神経細胞の変性に寄与している “図2” 前述のように、パーキンソン病患者のSNではγ-GTの活性が有意に上昇している[61]。この酵素は、遊離システインのドーパミン作動性SNニューロンへの転座と、これらの細胞からのGSHの放出に関与している。このように、システインの主要な貯蔵形態である神経GSHの重大な損失は、細胞内にますます輸送される遊離システインを、一連の連続した反応によって酸化性ドーパミンと結合し、DHBTs(ジヒドロベンゾチアジン)を形成する可能性が高くなる[63]。これらの化合物は致死的であり、深遠な神経行動応答、特にミトコンドリア複合体Iの不可逆的な阻害を引き起こすDHBT-1を呼び起こす[64]。
図2 パーキンソン病におけるSNドーパミン神経細胞におけるGSH枯渇の影響
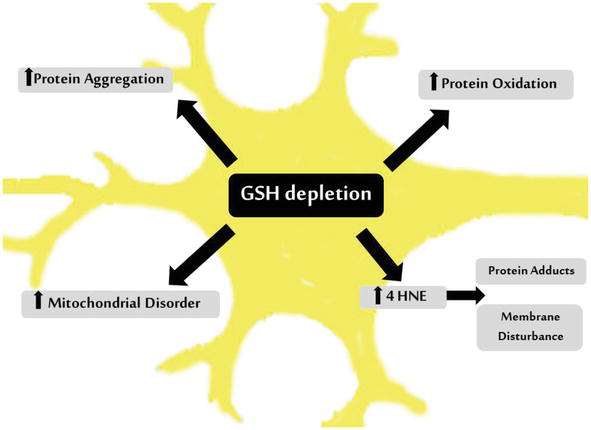
GSHの劇的な損失は、レビー小体を形成するタンパク質の凝集、複合体I活性の阻害に起因するミトコンドリア機能不全、タンパク質の酸化および脂質過酸化副産物4HNEの劇症的な効果を含む酸化的損傷と関連している。
SNにおけるレビー小体の存在もまた、主に神経変性に関与するパーキンソン病の特徴的な特徴である[65]。細胞内の複数のタンパク質は、ユビキチン(Ub)-プロテアソーム経路(UPP)によって分解される。この経路では、熱ショックタンパク質(HSP)の一族に属するUbタンパク質がチオール基と共有結合し、誤って折り畳まれたタンパク質や損傷を受けたタンパク質をプロテアーゼ26S複合体に移動させて分解に寄与している[65]。UPPに寄与する3つの酵素がある:E1(Ub活性化酵素)とE2s(Ubキャリア)はUbを共役に準備するが、このプロセスの主要な酵素はE3(Ub-タンパク質リガーゼ)であり、活性化されたユビキチンを分解されるタンパク質基質に移動させる[66]。UPPの構成要素の欠損またはその活性の欠如は、α-シヌクレインタンパク質の蓄積とそれに続く凝集をもたらし、レビー小体の形成につながる。ドーパミン作動性SNニューロンにおけるGSHの枯渇は、E1活性の低下とそれに続くUPPの障害につながる[67]。この知見は、GSHが酸化ストレス時に酵素の活性部位が酸化されるのを防ぎ、Ub-プロテアソーム経路での機能を維持していることを示唆している。
さらに、パーキンソン病SNでは、酸化ストレスの増加に伴いGSHが早期に消失すると、酸化されたタンパク質が増加することが明らかになった。パーキンソン病の初期段階では、ドーパミン作動性ニューロンにおける損傷タンパク質の蓄積・凝集による障害を防ぐために、HSPタンパク質が高レベルで発現している。疾患が進行すると、これらの防御はタンパク質の凝集体の蓄積を制御できなくなる [68]。
酸化ストレスはまた、ミトコンドリアを標的とし、その機能のすべてを妨害する。ミトコンドリア障害は、パーキンソン病の病態に関連する神経変性を媒介するメカニズムにおいて重要な位置を占めている[69]。グルタチオンはミトコンドリアにおけるヒドロペルオキシドの解毒における主成分であるため、脳内でのグルタチオンの枯渇は、活性酸素の増加を介してミトコンドリア障害を促進すると考えられている。ミトコンドリアは、そのすべての機能を妨害する可能性のある酸化ストレスに対する脆弱性によって知られている。ミトコンドリアのヒドロペルオキシドの解毒の主成分として機能することにより、GSHはミトコンドリアに影響を与える酸化的な障害を減少させる可能性がある。したがって、脳内のGSHの枯渇は、活性酸素の増加を介してミトコンドリアの傷害を促進すると考えられている[70]。
シナプスのミトコンドリアでは、酸化的リン酸化の制御における主要な役割は、25%の阻害で、エネルギー代謝が妨害され、ATP合成が大幅に影響を受ける複合体Iに起因している。同様の効果を発現させるためには[71]、80%までの複合体IIIおよびIVの阻害が必要である。SNにおける減少した複合体Iの活性は、パーキンソン病の脳におけるかなりの生化学的特性として知られている[72]。証拠は、神経GSHレベルの初期枯渇がミトコンドリアの複合体I活性阻害とそれに続くミトコンドリア機能不全に直接つながる可能性があることを示唆しており、最終的にパーキンソン病に関連するドーパミン作動性細胞死を誘導する。コンプレックスIは酸化ストレス時のミトコンドリア酵素に最も深刻な影響を与える[73]。脳内のGSH利用可能性の低下による酸化ストレスは、ミトコンドリア複合体Iの活性阻害の主な原因であると考えられている。このような複合体Iの酸化ストレスに対する感受性は、タンパク質のチオール(SH)基の酸化と、この複合体の中にアクセス可能な酸化感受性の高い鉄-硫黄中心が存在することによって説明されていると考えられる[74]。
GSHは、タンパク質のSH基を還元状態に保ち、酸化を防ぐことで、チオール依存性タンパク質の活性を制御していることが知られている[75]。GSHは酸化されたチオール基と共役してタンパク質-SS-Gを形成し、その後、GR、チオレドキシン、またはタンパク質ジスルフィドイソメラーゼによってタンパク質とGSHに再還元される。さらに、ドーパミン細胞に存在するGSHは、ドーパミンの酸化によって生じたキノンと結合し、タンパク質中のSH基との反応を阻害する[76]。
最後に、酸化的障害の間、副産物としてアルデヒドが形成される; これらのタイプの最も一般的なものは4HNEである。この後者は、その流動性の変化を引き起こす膜に組み込むことができる[77]。さらに、4HNEは、それらを不活性にするNa/K ATPasのような重要なタンパク質との付加体を形成することができる。GSHは、GSTを介してそれを共役することによって4HNEのレベルを減らすのに役立つかもしれない。また、GSH の欠損は、4HNE 付加体の高レベル化につながると考えられている。
3.3.2 アルツハイマー病(AD)
アルツハイマー病は最も一般的な加齢性神経変性疾患であり、高齢者に影響を与える進行性の認知症として知られている。この疾患は、アミロイドβ(アミロイドβ)プラークと神経原線維性タングル(NFT)の沈着によって病理学的に特徴づけられる[14]。アルツハイマー病脳の細胞外空間におけるアミロイドβペプチドを主成分とするアミロイドプラークの存在は、疾患の主な特徴である。アミロイドβレベル、特に最も神経毒性の高いペプチドであるアミロイドβ42の過剰は、アルツハイマー病の家族性形態の出現を引き起こす。このアミロイドβ42の増加は、可溶性オリゴマーの形成をもたらし、シナプス機能の恒久的な変化を引き起こす。それと並行して、アミロイドβ42は凝集して大部分がβシートに富んだフィブリルを形成し、局所的な炎症反応(ミクログリア症やアストロサイトーシス)を亢進させる。シナプス棘の喪失および神経性ジストロフィーも観察される。時間の経過とともに、これらの事象は、酸化ストレス、イオン(例:カルシウム)のホメオスタシスの変化を含む生化学的変化をもたらす [79]。アミロイドプラークは、アルツハイマー病の進行につながるシグナル伝達経路の引き金となる決定因子である。最近の証拠は、アミロイドβプラークが脳内および初代ニューロン培養においてニューロンのアポトーシスを誘導することを示唆しており、このアミロイドβによって誘導されるニューロン死がアルツハイマー病患者に見られる認知機能の低下の一端を担っている可能性がある。さらに、凝集したアミロイドβは細胞内のp38マイトジェン活性化プロテインキナーゼ(MAPK)を活性化し、タンパク質タウの過リン酸化とニューロン内部での神経原線維のもつれ(NFT)の形成を引き起こし、微小管を不安定にしてニューロンの機能喪失を引き起こす。
可溶性アミロイドβオリゴマーはEAAC1が介在するシステイン取り込みを阻害し、培養ヒト神経細胞においてGSHの損失を引き起こすことが実証されている[81]。このことは、海馬の錐体ニューロンにおけるEAAC1の異常蓄積を示すアルツハイマー病患者の剖検脳[82]や、疾患の進行に伴ってGSH/GSSG比が低下しているアルツハイマー病患者の脳[83]からも裏付けられている。
以上のことから、アルツハイマー病におけるEAAC1機能障害という概念を強調することができる。
酸化ストレスはアルツハイマー病の主要な病因因子と考えられている。酸化ストレスにはGSHの枯渇が大きく関与していることから、本疾患の発症・進展に関与していると考えられている。NMR分光法を用いた最近の臨床研究では、健常者と比較してアルツハイマー病患者ではGSHレベルが枯渇していることが示された[84]。この所見は臨床的に深い意味を持つ可能性がある。さらに、アルツハイマー病患者の血液サンプルの分析では、年齢と性別をマッチさせた対照者と比較して、赤血球中のGSH濃度の低下が示された[85]。これはアルツハイマー病の前臨床段階である軽度認知障害(MCI)でも観察されている。MCI患者では、健康な年齢の対照群と比較して、海馬のGSH/GSSG比やGST活性が低下していることが明らかになった[86]。これらの結果によると、GSH代謝の障害はアルツハイマー病の発症に先行していることが示唆される。GPx-1遺伝子とGST遺伝子の遺伝子多型は、アルツハイマー病の正の危険因子として同定された[87]。これは、アルツハイマー病におけるGPxおよびGST活性の低下の理由となり得る[88]。
以前に述べたように、活性酸素形成は、アミロイドβ凝集によって誘導され、海馬ニューロンにおけるHNEの生成を含む多くの酸化的損傷および代謝障害を引き起こし、その結果、そのような障害の毒性を媒介する可能性がある[89]。いくつかの研究では、年齢をマッチさせた対照群と比較して、アルツハイマー病患者の脳内脂質過酸化の増加が示されている[90]。脂質過酸化の結果として、二次的な生理活性アルデヒドであるHNEは、アルツハイマー病後期の被験者のいくつかの脳領域で高レベルで産生される [89]。アルツハイマー病の進行におけるHNEの重要な役割は、多くの知見によって支持されている。したがって、MCI患者の脳内では、タンパク質と結合したHNEのレベルが増加していることが観察された[91]。アルツハイマー病では、ATP合成酵素、グルタミン合成酵素、DRP-2,MnSODなど、多くのタンパク質が有意にHNE修飾されていることが明らかになった。これらのタンパク質は、細胞内シグナル伝達、エネルギー代謝、解毒を含む多くの細胞機能に加えて、脳細胞の構造機能の調節に大きな意味を持っている。GSHは、培養神経細胞において、アミロイドβやHNEによって誘発される酸化損傷を抑制することが明らかになった。このことから、GSHの枯渇はアミロイドβやHNEによって刺激された酸化的障害を悪化させ、病気の発症を加速させることが示唆された。
4. 結論
GSH は、過去数十年にわたり脳内で集中的に研究されてきた興味深いテーマである。このような研究の目的は、神経変性疾患の進行を防ぐための細胞内GSHの役割を理解することだけでなく、神経変性疾患に関連した細胞機能障害のメカニズムを明らかにすることにある。GSH 枯渇は、GSH ホメオスタシスの乱れや GSH 関連酵素の修飾など、様々な原因によって引き起こされる 神経変性疾患 の一般的な特徴である。GSHおよびGSH依存性酵素の調節障害に起因する複数の細胞障害は、ミトコンドリアの機能障害、酸化的損傷の増加、細胞内シグナル伝達経路の障害、タンパク質凝集、そして最終的には細胞死に寄与している。
神経変性事象におけるグルタチオン依存性反応のネットワーク障害の関与をより正確に明らかにし、これらの事象を予防または制限するための新たな方法を見つけるためには、さらなる研究が必要であることに留意することが重要である。また、神経変性疾患患者に対するより効果的なアプローチ療法を提案することも重要である。
