

Contents
Unified theory of Alzheimer’s disease (UTAD): implications for prevention and curative therapy

www.ncbi.nlm.nih.gov/pmc/articles/PMC4947325/
著者:Michael Nehls
2016年7月15日オンライン公開
要旨
本レビューの目的は、アルツハイマー病の統一理論(UTAD)を提案することである。それは人間の進化の歴史から発せられる3つの概念に基づいている。
(1)後天的な知識の世代を超えた転送による生殖における進化的優位性に起因する人間の長寿を説明する祖母仮説(おばあさん仮説)。その結果、老年期のメンタルヘルスは人間の遺伝的プログラムの既定の経路であって、アルツハイマー病の発症ではないと主張されている。
(2) したがって、神経細胞の若返り(ニューロンの若返り)や成体海馬の神経新生(成人海馬神経新生)のように、老齢になっても効率的に機能するメカニズムは、生存に重要な個人的な経験を記憶するために必要な生涯の能力を提供している。多数の実験的および疫学的研究からの累積証拠は、生産的な成人海馬神経新生を損なう行動的および環境的危険因子が、エピソード記憶のパフォーマンスの低下と心理的回復力の低下につながることを示している。これは、新規性の回避、視床下部-下垂体-副腎(HPA)軸の調節障害、コルチゾールの過分泌につながり、シナプト毒性アミロイドβの蓄積とオリゴマー化、慢性神経炎症、ニューロンインスリン抵抗性などのアルツハイマー病の主要な発症機序を牽引している。
(3) 生物学的成長過程の基本的な要件を定義した最小値の法則(長期増強)を成人海馬神経新生に適用することで、生活習慣の違いによる成人海馬神経新生の障害やHPA軸の調節障害がなぜ、どのようにしてアルツハイマー病のプロセスを開始するのか、また、毒素やApoE4などの環境的・遺伝的危険因子がそれぞれ、このような不自然な条件下でどのように疾患の促進因子に変化するのかを説明している。その結果、UTADは精神衰弱の予防のための合理的な戦略と、アルツハイマー病の原因治療のためのシステム生物学的アプローチを提供し、病気のプロセスの十分な早期に全身的な介入を開始する場合には、治療可能な可能性さえある。
したがって、UTADの有効性を検証するためのテストとして、早期アルツハイマー病患者に対する個別化された全身生物学的治療が提案され、本レビューで概説されている。
キーワード
統一理論アルツハイマー病(UTAD)、祖母仮説(おばあさん仮説)、神経細胞の若返り(ニューロンの若返り)、成人海馬神経新生(成人海馬神経新生)、最小値の法則(長期増強)、治療的アルツハイマー病治療、原因性アルツハイマー病予防
背景
アルツハイマー病は、海馬のエピソード記憶能力の障害に続いて、認知能力や社会性の低下が進行するのが特徴である。アルツハイマー病は認知機能低下の主な原因であり、治療薬が開発されていないため、世界的に研究が盛んに行われている。疫学的、生化学的、分子的、遺伝学的、動物学的研究は、複雑な病気のプロセスに異なる入口を提供し、それがアルツハイマー病の病因についての異なる理論につながった。
主な原因および主要な危険因子としての年齢から始まり、アルツハイマー病は、高リン酸化および調節障害されたタウ[1]、中毒仮説[2-4]、慢性感染症[5-7]、マイクロバイオーム組成[8]を含むオリゴマーアミロイドβ(アミロイドβ)カスケード仮説によって説明されている。神経細胞のインスリン抵抗性 [9、 10]、血液脳関門(BBB)の物理的・機能的破壊 [11、 12]、複数の原因による慢性神経炎症 [13]、神経細胞の若返り障害(ニューロンの若返り) [14]、シナプス障害 [15]、その他多数の説がある。
これらの理論はすべて、多かれ少なかれ、加齢そのものが主な病因であるという信念に深く組み込まれている。実際には、老化自体がアルツハイマー病の主な原因であるという考えは非常に深く私たちの思考に染み付いているとアルツハイマー病についての任意の科学論文のほぼすべての導入に表示される強制的な文は、めったに挑戦されていない。しかし、私が主張するように、 “年齢は主な原因-ドグマ “に挑戦する深刻な議論の数があるだけでなく、包括的な原因としての老化はまた、それ故にこの衰弱性疾患の病因と病態を説明する、よく知られている環境や行動の危険因子の長いリストを含むすべての重要な知見を組み込んでいる “アルツハイマー病の統一理論”(UTAD)の開発を妨げているだけでなく、実際、UTADの欠如は、効果的な予防策や治療法の開発を試行錯誤に制限し続けている。このような特異的なメカニズムに焦点を当てた治療的介入は失敗し続けている[16]。さらに、アルツハイマー病の完全な理論よりも多かれ少なかれ任意に選択された危険因子の修正にレジメンの基礎を置く予防試験も、これまでのところ全体的な成功には限界があった [17、 18]。対照的に、提案されたUTADは、アルツハイマー病の主要な原因としての年齢自体の概念を克服し、アルツハイマー病の病因と病態の包括的な説明を提供する。また、アルツハイマー病を高確率で予防するために必要な個人の人生を変える介入を数多く提案することができる。さらに、UTADは、このレビューの最後に概説されるように、治療レジメンのための論理的な枠組みを提供するかもしれない。
私は提案された理論を「UTAD」と呼んでいることを指摘したいと思うが、それは現在知られているすべての主要な行動、環境、または遺伝的危険因子が個別に、または組み合わせでどのようにアルツハイマー病プロセスを開始または加速させるかの全身的な神経生物学的枠組みを提示しているため、すべての理論ではない場合はほとんどの理論に適用される一定の注意事項にもかかわらず、私は指摘したいと思う。私は、私の検索で可能な限り包括的かつ網羅的にしようとしたが、例えば、”アルツハイマー病の危険因子”、”成人海馬神経新生を阻害する因子 “や国立生物工学情報センターのPubMedデータベース内の “神経炎症 “のようなキーワードを使用して、いくつかのマイナーな危険因子が見落とされている可能性があるが、それは彼らがUTADを検証したり、改ざんするかどうかを見ておく必要がある。リスク因子についても同様であり、今日すでに作用しているがまだ特定されていないものや、将来の個人のライフスタイルの選択や文化の発展に由来するものもある。逆に、いくつかの危険因子のカテゴリー(例えば、環境毒素や化学物質)では、包括的なリスト(例えば、UTADが提案するアルツハイマー病プロセスの重要なメカニズムに悪影響を及ぼす現在知られているすべての化学物質のリスト)は、その理解を深めることにはならず、したがって、このレビューの範囲を超えてしまうので、私は意図的に例だけを列挙した。UTADの主要概念が受け入れられれば、UTADによればアルツハイマー病の中心にある神経生物学的メカニズムを阻害するすべての危険因子が認識され、より効率的に調査できるようになることを願っている。最後になるが、UTADの文脈での行動的危険因子は、自由意志や行動の自由についての議論につながるかもしれないが、これもまた、このレビューの範囲を超えてしまう。
加齢は必要だが、アルツハイマー病の原因ではない
2-デオキシ-2-[フッ素-18]フルオロ-D-グルコースを用いたポジトロン断層撮影による海馬/側頭葉のインスリン抵抗性の測定(F歯状回-PET/CT)は、アミロイド-PET診断[19]に加えて、アルツハイマー病の最初の臨床症状が現れる数十年前から予測値を持つ、アルツハイマー病の非常に特異的で感度の高いバイオマーカーとなっている[20]。アルツハイマー病の発症には明らかに時間が必要であり、したがって、アルツハイマー病発症のリスクは年齢とともに増加すると結論づけることができる。しかし、相関関係は先験的に因果関係とは一致しない。この場合、発症に時間を必要とする病気のために、年齢は単に前提条件かもしれないが、必ずしも原因ではない。もし本当に年齢が病気の原因だとしたら、アルツハイマー病は人間の老化の自然な結果であるだけでなく、アルツハイマー病との戦いは人間の本性との戦いであり、勝つことは非常に難しい。
しかし、幸いなことに、多くの証拠がこの説明に反論している(例えば[21、 22]参照)。特に人間の生活史の観点から見ると、もし年齢そのものが本当に主な危険因子であったならば、なぜ前世紀の初め頃にはアルツハイマー病は本質的に知られていなかったのであろうか?最近の推計によると、アメリカだけでも年間約36000人の新規症例が発生しており、この病気は非常に一般的なものとなっている[3]。しかし、19世紀後半に出版された神経学の教科書には、アルツハイマー病に似た病態についての言及すらなく[23]、1906年にアロイス・アルツハイマーが初めてこの病態についての報告書を発表した際には、特異な脳疾患として記述しており[24]、以前から知られていなかったことを示唆しているそして、32年後の1938年にはまだ、アルツハイマー病脳病理の特徴としてアミロイド斑と神経原線維のもつれについてのアルツハイマーの記述は、病理学の包括的な教科書には載っていないであった[25]。当時、アルツハイマー病の結果としての死亡は非常にまれな出来事であったと提案されるかもしれない。
アルツハイマー病の有病率は前世紀半ばには日本と同じような稀なものだったのかもしれない。最近の研究では、20世紀後半のアルツハイマー病有病率の7倍(!)の増加は年齢ではなく、むしろ特定の生活習慣因子が説明しているという重要な証拠が提供されている[26]。この劇的な増加は、主に1961年から 1985年の間に行われた伝統的な日本人の食事/ライフスタイルから西洋のものへの変化と強く関連していた。特に、アルコール摂取量の大幅な増加に加えて、肉類と動物性食品の消費量がそれぞれ7倍と4倍に増加した。動物性食品と肉は、過剰な鉄(以下で詳述するように、特にApoE4キャリアのリスクを高める)、高度な糖化最終生成物(AGEs)やアラキドン酸などの化合物を含むため、アルツハイマー病のリスクを高めることが知られている(これも以下で詳述する)脳内の酸化ストレスや炎症を増加させることが示されている(これも以下で詳述する)。食生活の変化は、日本の65歳以上の人々のアルツハイマー病有病率の劇的な増加と並行して、1985年の低1%から 2008年には約7%、約20年のラグタイムで上昇した。この有病率の傾向は、平均寿命の変化や遺伝的なものでは説明できない。このような比較的短い間隔は、喫煙による肺がんなどの他の行動性疾患ではむしろ知られている[27]。
この画期的な研究の著者によると、日本におけるアルツハイマー病有病率はピークに達している可能性があり、他の高度工業化社会と同様に、これらのアルツハイマー病の原因となる行動因子は1985年以降わずかに変化しているだけなので、それ以上増加することはないだろう。その結果、彼らは研究の一つのレビュアーが警告し、 “日本人が伝統的な日本の食生活に戻らない限り、日本のアルツハイマー病率は減少する可能性は低い” [28]低率から高率へのアルツハイマー病発症率の同様の増加は、新興国でも観察される。例えば、インドの農村部における年齢別アルツハイマー病発症率は、米国よりも約4倍低いことが示された[29]が、その上昇はむしろライフスタイルに強く影響を与える経済成長率と平行している[30]。同様に、米国に住むアフリカ系アメリカ人の間でのアルツハイマー病の有病率は、彼らの祖国の年齢にマッチしたアフリカ人と比較すると数倍高い[31、 32]。アメリカに移住してアメリカ人の生活様式を採用した日本人は、アルツハイマー病リスクを高めた [33]。したがって、民族的な出自ではなく、むしろ採用された現代的なライフスタイルがアルツハイマー病リスクに影響を与えている[34]。つまり、個人の生活史がアルツハイマー病リスクを形成する上で決定的な役割を果たしているのかもしれない。

Framingham Heart Studyの再解析によると、30年間で認知症の発症率はほぼ半減した[35]。認知症の5年間の年齢および性差調整済み累積ハザード率は、第1期(1970年代後半および1980年代前半)では100人当たり3.6人、第2期(1980年代後半および1990年代前半)では100人当たり2.8人、第3期(1990年代後半および2000年代前半)では100人当たり2.2人、第4期(2000年代後半および2010年代前半)では100人当たり2.0人であった。第1期の発生率と比較して、第2期では22%、第3期では38%、第4期では44%減少している。繰り返しになるが、これらのデータは、年齢自体がアルツハイマー病の主な原因であることと矛盾している。研究の著者によると、この減少の主な要因は特定されていないが、この肯定的な傾向は、少なくとも高校の卒業証書を持っている人の間でのみ(!)観察されたことに注意することが重要である。低学歴者では、アルツハイマー病リスクは40年間で66%上昇していた。これは、教育および/または所得、したがって社会経済的要因が重要な役割を果たす可能性があることを示している。しかし、社会経済的地位が低いと寿命が伸びるのではなく、むしろ短くなるので、年齢を除外することは確かに可能である[36]。同様の減少を報告した別の研究 [37] の著者によると、降圧剤、脂質低下剤、抗糖尿病薬の使用量が増加したこと、また、戦時中のようなストレスの少ないライフイベントを経験したことが、これらの肯定的な傾向に寄与した可能性がある。これらを総合すると、人生の選択や人生経験がアルツハイマー病の病因論的意義を持っているように思われる。
動物モデルに基づいた研究の結果もまた、加齢がアルツハイマー病の因果関係にあることに反論している。実験動物は、西洋社会の生活に非常に似ている面もあるが、いわゆる標準的な住宅条件の下で飼育されている。この “西洋型のライフスタイル “は、彼らは不自然な標準的な12時間の暗/光のリズムに拘束されているので、社会活動を欠いている、と比較して、動物は慢性的な睡眠不足に苦しんでいる。さらに、不自然な一日おきの給餌パターンはまた、実験研究で動物に日常的に適用され、特にアルツハイマー病研究では、実験結果の誤解につながる。例えば、身体活動や断続的な絶食(間欠断食)が自然であるような野生の生息地で生活している動物ではなく、定住しているケージに入れられた動物が、自由に摂食されていることが、細胞や分子レベルまでのすべてのデータを相関させ、解釈するための基準となっている。対照的に、より自然なハウジングは、環境エンリッチメント(EE)と呼ばれ、より大きな囲いの中での社会活動と環境の複雑さ(例えば、操作可能な物体の存在、登山や運動のための構造物、採食の機会、隠れる場所や営巣場所)を含み、したがって、より自然な状況は、実験条件とみなされている[38]。しかし、環境エンリッチメントの下での条件は、重要な点で私たち自身の前近代的な生活様式を模倣しており、それゆえに私たちの遺伝的プログラムも適応される一般的な条件となっているので、その逆であるべきである。したがって、標準的な住宅条件は、身体的・精神的な幸福のための重要な要因が排除され、老化とアルツハイマー病の結果が研究されている実験条件とみなされるべきである。この視点の変更は、アルツハイマー病を主に欠乏性疾患、すなわち、生物の行動要件とその実際の生活条件との間の矛盾の結果として見なすのに役立つだろう。
参考記事

提案されたUTADに沿って、環境エンリッチメントの中で維持された古いマウスであっても、以下に詳細に概説されるように、アルツハイマー病に関して重要な側面である成体海馬神経新生(成人海馬神経新生)[39]の堅牢な5倍の増加を示している。さらに、彼らは不安や抑うつのような行動の減少を示し [40]、標準的な住宅条件で維持されている動物と比較して、記憶力の改善を示している [41]。したがって、一度我々は進化の観点から、老化やアルツハイマー病の動物モデルを見始めると、身体運動、間欠断食、社会活動や必須栄養素の実験的欠陥(現在の標準的な住宅条件の下で)は、アルツハイマー病の明白な原因となるだろう(下記参照)。例えば、間欠断食の1年間であっても、アルツハイマー病のトリプルトランスジェニックマウスモデル(APPswe、PS1M146V、およびタウP301L変異[42]を発現している)で認知機能の低下を防止し、老化ではなく、むしろ給餌パターンは、メモリ[43]に長期的な効果を持っているという証拠を提供している。以下にさらに多くの例を挙げるが、これらの例を組み合わせることで、1つの重要な結果を得ることができる。老化とアルツハイマー病に関する動物研究(疫学研究と同様に)から得られたほとんどの結果は、進化の観点から再解釈する必要がある。「西洋式」の住宅の下での「正常な」老化は自然なものではなく、同じことがヒトのアルツハイマー病にも当てはまるかもしれない。
ライフスタイルの選択が開始され、遺伝的素因は、アルツハイマー病の発症を加速させる
進化は主に有利な遺伝的変異体の選択によって駆動する。前世紀の先進国でのアルツハイマー病の発生率の劇的な傾向と新興国での同様の上昇は、今日ではしたがって、簡単に遺伝的影響によって説明することはできない。それは、アルツハイマー病が2型糖尿病、肥満、高血圧、動脈硬化症、アルツハイマー病のすべてのよく知られているリスクファクター[44]のよく知られている増加を平行して同様のライフスタイルの変化に従うことがより可能性が高いである。
特定のライフスタイルの選択と アルツハイマー病 リスクと推定遺伝的素因との相互作用を示す間の相互作用を説明するために役立つ別のかなり粗雑な例は、米国から来ている。一般的な人口と比較して、ナショナルフットボールリーグ(NFL)の選手は、大うつ病を開発するリスクが3倍に増加している[45]と彼らのキャリアの間に3つ以上の脳震盪を発生させた人は、無症候性軽度認知障害(MCI)[46]のための5倍の高いリスクを持っていた。NFL研究の著者によると、外傷性脳損傷はアルツハイマー病や他の形態の認知症の原因となる危険因子である。アルツハイマー病診断時の平均年齢は53.8歳であり、これらのケースでは、アルツハイマー病リスクを高めるのは年齢ではないことを示している。他にもいくつかの研究でこれらの知見が確認されている(レビューは[47]を参照してほしい)。一般的にタックルまたはタックルされる前にかなりの運動量を構築する特にスピード選手では、一般的な米国の人口と比較して アルツハイマー病 と筋萎縮性側索硬化症から生じる有意な 6 倍以上の死亡率を示した。興味深いことに、ApoE4キャリア(アポリポタンパク質多型の特定の遺伝子型のバリアントのすなわちキャリア)は、ApoE4は、ApoE2とApoE3とは対照的に、特にBBBがプロ炎症性の侮辱に脆弱になるという実験的観察と一致している、脳震盪を媒介とするアルツハイマー病リスクにより敏感である。この変化は、複数の血液由来の神経毒性タンパク質の神経細胞への取り込みを増加させるとともに、微小血管脳血流障害をもたらす。ApoE4を発現する動物モデルでは、これらの血管障害は神経細胞の機能障害に先行し、神経変性変化を引き起こす可能性がある[48]。
ApoE4多型は散発性アルツハイマー病の最も一般的な遺伝的危険因子として知られており、白人の15%がキャリアである。ApoE4はアルツハイマー病の発症に必要でも十分でもないが、ApoE4対立遺伝子の1つまたは2つのコピーを持つことは、それぞれ約3〜12倍の遅発性アルツハイマー病リスクを増加させる[49]。興味深いことに、ApoE4はヒト固有の対立遺伝子であり、最も先祖代々のものであり、進化におけるその出現はヒトの寿命の劇的な増加を示す(レビューは[50]を参照のこと)。対照的に、ヒトのApoE3対立遺伝子はアルツハイマー病リスクに関しては中立的であるようであり、その突然変異は人類の歴史の中でずっと後に出現した;その頻度は、現在白人の75%がキャリアであると人類の進化の間に増加した。なぜApoE4対立遺伝子は完全に健康に有益なApoE3またはApoE2対立遺伝子と自然淘汰によって置き換えられていない、それさえもアルツハイマー病リスクを低下させるように見える?いくつかの可能な説明がある。例えば、高齢になると健康に有害な対立遺伝子は、拮抗的な多元性によって、若い個体に何らかの利益を与えるので、集団の中で存続する可能性があると仮定されている[51]。
ApoEは、エストロゲンとプロゲステロンの産生のためのコレステロール前駆体の主要な供給者である。ある最近の研究によると、少なくとも1つのApoE4対立遺伝子を持つ女性は、ApoE2またはApoE3対立遺伝子のみを持つ女性よりも平均黄体プロゲステロンのレベルが有意に高い。したがって、ApoE4は受胎可能性、ひいては生殖能力において有利である可能性がある [52]。別の研究では、ApoE2やApoE3と比較した場合、ApoE4は若く健康なヒトにおいて良好なエピソード記憶と記憶に関連した神経資源の経済的利用に関連することが明らかになったが[53]、他の研究ではそのような利点を示す証拠は得られなかった[54]。
ある人にとってのApoE4の利点が何であれ、我々は、アルツハイマー病に関して任意の遺伝的変異の欠点に関するすべての私たちの観察は、劇的に最近の歴史の中で彼らのライフスタイルを変更したヒト集団(またはケージに入れられた動物)で観察されたことを認識する必要がある。我々は単にApoE4が中立であろうか、あるいは現在の行動と環境の欠乏が省略されている場合、高齢者の認知健康のための利益を提供する可能性があるかどうかを知らない。実際、特に認知に関するApoE4多型の有害な効果は、修正可能な危険因子に強く依存する可能性があることが顕著である(レビューについては[50]を参照)。例えば、いくつかの研究では、身体活動への参加は、主にApoE4キャリアのアルツハイマー病リスクを減少させるようである[55]、”おばあちゃん仮説”(下記参照)に沿って、長い人間の寿命の進化は、おそらく身体的および認知的健康の両方の高いレベルを維持するために高齢者を必要としたと主張する。上記で議論したNFLの例のように、最近のコホート研究では、ApoE4は私たちが不健康なライフスタイルの選択肢に対してより脆弱になるという証拠を提供した。ある研究では、高コレステロール血症は主にApoE4キャリアの間で認知機能障害と関連していた[56]。したがって、上記で概説されているように、受胎可能性の利点は、コレステロール値の上昇につながる変更された行動時に不利になる可能性がある。以下に詳述する別の研究では、健康的な地中海式食生活(地中海式ダイエット)に準拠したApoE4被験者は、他の遺伝子型[57]と比較したときに脳の健康に関する最大の利益を持っていた。この傾向は、特にApoE4対立遺伝子のキャリアは、アルツハイマー病リスクが2分の1に低下したのに対し、非ApoE4キャリアはそのような利点がなかったのに対し、魚の栄養摂取から利益を得ているという知見によって確認された[58]。さらに、最近の研究では、脳脊髄液(脳脊髄液)中のフェリチンレベルが認知的に正常(すなわち、年齢とともに平均的に精神的に低下する)、MCIおよびアルツハイマー病被験者の認知パフォーマンスと負の関係があり、MCIからアルツハイマー病への転換の速度を予測したという証拠を提供した[59]。興味深いことに、脳脊髄液-フェリチンレベルの上昇は、ApoE4遺伝子型と強く関連していることがわかった。進化の観点から、ApoE4キャリアは、彼らの脳が低い栄養鉄の可用性の状況で十分な鉄を提供されたように利点を持っていたかもしれない。今日の食事は通常、大量の鉄を豊富に含む動物性食品(現代の日本社会のために上記のように概説されている)ので、鉄代謝におけるApoE4の潜在的な生理学的利点は、活性酸素種(ROS)の生産の強化を促進し、悪影響を及ぼすアルツハイマー病の進行に影響を与える脳の鉄の上昇につながる、反撃するかもしれない。
したがって、散発性アルツハイマー病の最も一般的な遺伝的危険因子であるApoE4は、我々の進化の最も長い部分で優勢な条件の下で脳の健康に重要であるかもしれないことが考えられる[50]。そのため、今では不健康なライフスタイルの選択(職業的に後天的な頭部外傷、コレステロール値や鉄分摂取量の増加につながるライフスタイルを好む、または低運動量など)の下でのみ危険因子とみなされるかもしれないApoE4対立遺伝子は、したがって、アルツハイマー病の原因ではなく、むしろそのような(不健康な)条件の下で促進剤として作用する可能性がある。実際、各ApoE4対立遺伝子は用量に関連したアルツハイマー病の早期発症に影響を与える[60]。状況は、いわゆる肥満の対立遺伝子に似ているかもしれないが、これは単に、実際に間欠断食と採食に成功した後のごちそうとの間を交互に行き来する生活条件の下で、エネルギー保存に特に効率的であることによって、先祖の体力を実際に向上させた遺伝的変異をキャリアに与えるものである。
現在では、最小限の身体的支出で安定したエネルギー供給が可能な生活条件の下では、同じ遺伝子が「病気の遺伝子」とみなされている。「肥満遺伝子」と特にApoE4の両方のキャリアのための良いニュースは、彼らが健康的なライフスタイルへの復帰から最も恩恵を受けるだろうということである。一緒に考えて、アルツハイマー病の病因は、身体運動、健康的な食品の摂取、断続的な断食と社会的活動のよく知られた欠損によって引き起こされる他の現在の「文化が媒介する病気」とは異なっていないように見える。アルツハイマー病のパンデミックは、単純に、高度に不活動であり、過剰な消費と職業に特化した不活動なライフスタイルが発達したことで、延長された家族の絆やリラックスする時間を大きく排除していることに起因しているのかもしれない。アルツハイマー病 を予防し、治療するためには、真の原因となる危険因子がどのように相互作用するかのより良い理解が必要かもしれない。しかし、それ以上に、私たちはよりオープンマインドになる必要がある。私たちは、アルツハイマー病を自然な(遺伝的な)病気や老化の原因となる結果として捉えることをやめ、アルツハイマー病は環境要因や行動的欠陥(すなわち、よく知られている危険因子)によって引き起こされることを受け入れ始めるべきである。その結果、アルツハイマー病は予防可能であるべきであり、病気の初期段階では、我々は(再)私たちの人間の特定の進化の生活史から得られる私たちの脳のすべての本質的な要件を満たすライフスタイルを想定していることを条件に、さらに治るかもしれない。
人間の長寿の進化
我々は進化の産物である。したがって、ADのような人間の病気を説明する場合、その病因と病態を完全に理解しようとするならば、進化論に基づかなければならない。
熱力学の第二法則は、あらゆる情報の自然な不安定性を記述している。したがって、生命は主要な障害を克服し、遺伝情報を保存する方法を見つけなければならなかった。自然の解決策は、遺伝物質を繰り返し複製することであった(情報内容を失わないためには、お気に入りのデータストアを頻繁にコピーする必要があるのと似ている)。複製プロセス後の品質管理メカニズムは、DNA複製プロセスを効率的に継続する能力を持つ複製を選別することである。このメカニズムにより、刻々と変化する環境に適応し、まったく異なる生殖戦略を進化させることができた。一方の細菌は、少なくとも数個の遺伝子コピーの生存確率を高めるために、大量生産と大量遺伝的変異に取り組んでいる。一方ヒトは、種として存続するためには、比較的少数の子孫を残すだけでよい。その大きな脳は、複雑な社会的協力関係を築き、獲得した知識を利用して子孫の生存確率を高めることを可能にするからである。前近代のカナダとフィンランドにおける大規模かつ完全な多世代人口統計記録によると、祖母が早く亡くなった家族と比較した場合、生後10年を経過するごとに、少なくとも2人以上の孫が生殖年齢に達していた[61]。したがって、祖母が60歳を超えれば超えるほど、つまり70歳、80歳、90歳(つまり、年齢が精神的な衰えを引き起こすとされる年齢)になればなるほど、孫の数を増やし、彼らが成人まで生存する可能性を高めることができ、長寿は世代を超えた世代性によって作用する選択形質となったのである。心理社会的な意味での世代性とは、次の世代を確立し導くことへの人間の関心を指す。
この特殊な進化戦略は人類史の初期に発達し、今日では祖母仮説として知られている [62] 。祖母仮説は、遺伝的に最も近いいとこであるチンパンジーが閉経を迎えてから数年で死んでしまうのと比較して、私たちの並外れた長寿の進化的起源を説明している [63] 。狩猟採集社会は平均寿命が低いので、長寿はむしろ現代の現象であるという祖母仮説に対する反論は、否定することができる。実際、現代生活から隔離された現存する狩猟採集社会では、成人に達した人の約3分の2が70歳まで生存しており、80歳の老人に遭遇しても例外ではないかもしれない[64]。狩猟採集民の生殖年齢は15歳から45歳の間であったため、祖母は、自分の最後の子供が約15歳の出産年齢に達したときに、初めて自分の子供の養育から独立する。従って、独占的な孫育て、つまり母親業から独立した年月は、60歳前後から始まり、直近の子供の最初の孫が成人するまで、少なくともあと15年は続かなければならなかった。興味深いことに、祖母仮説はシャチの拡大家族の観察によって最近裏付けられた[65]。人間と同じように、メスのオルカは閉経後も数十年生きることができ、その繁殖戦略も生涯にわたって獲得した知識の活用に依存している。特に、閉経後の最高齢のメスはサケの採餌場で群れを率い、特にサケの資源が少ないときに狩りを組織する。

というのも、生殖戦略としての長寿の進化には、獲得した人生経験を親族に与えるために、脳が特に高年齢でよく機能することが必要だからである。さらに、祖母仮説が予言するように、人間の発達の長い前近代的な領域で活動していた条件に近く、遺伝的プログラムが絶妙に適応している条件下では、人間の脳は経験的知識を獲得する能力を生涯維持する [66] 。もうひとつは、オートファジーと細胞内構造と細胞小器官の再生のメカニズムによる、すでに存在するニューロンの生涯にわたる維持、すなわちニューロンの若返り(NRJ)である。NRJ[67]と成人海馬神経新生[68]の両機構は、高年齢でも効率的に機能しているように見え、これは祖母仮説(後述)と一致しているが、特定の環境的な合図を必要とし、私たちのライフスタイルの選択に依存している。
生涯にわたる神経細胞の若返り(NRJ)
ミトコンドリアは、エネルギー産生、代謝、カルシウムシグナル伝達、多くの生合成経路など、広範な中核的細胞機能において重要な役割を果たすことにより、細胞の生体エネルギー恒常性を維持している。[69]。また、代謝シグナルやストレスシグナルの伝達、活性酸素種(ROS)などのフリーラジカルの産生などのプロセスにおいても、様々な機能を発揮している。もともとは「必要悪」、すなわち不完全な酸化的代謝の副産物として想定されていた活性酸素は、現在では細胞生理学において不可欠なシグナル伝達機能を持つことが認識されている。[70]。ミトコンドリアが損傷した場合のみ、過剰に蓄積した活性酸素が炎症反応を引き起こし、成人海馬神経新生を阻害するだけでなく、ミトコンドリア宿主に細胞死プログラムを実行させる。したがって、ミトコンドリア機能の慢性的な障害は、多くの神経変性疾患の病因を悪化させる。この種の酸化ストレスに対する複雑な防御機構は、臨機応変に適応されており、内因性に生成された活性酸素封鎖分子や酵素(α-リポ酸、スーパーオキシドジスムターゼ、グルタチオンなど)、および多くの生物活性栄養素(植物由来のビタミンやポリフェノールなど)から構成されている。
加えて、ニューロンのようにターンオーバーが低いか全くない細胞は、NRJのプロセスを通じて細胞内の若さを維持することで、個体の高年齢までしか生存できない。[72]。細胞の生体エネルギー恒常性維持の中心は、細胞のエネルギー需要がエネルギー供給によって満たされるように、健全なミトコンドリア集団を維持することである。[71]。この目的のために、損傷した高分子とミトコンドリアを含む細胞小器官の交換は、活動的で高度に制御されたプロセスとして進化し、[73]、その開始には行動的な合図が必要である。従って、老化の促進 [74]、記憶力の低下、AD [75]は、これらの手がかりの欠如によって引き起こされる可能性がある。逆に、機能しない高分子や損傷したミトコンドリアの蓄積は、何年にもわたって損傷の速度が修復やターンオーバーの速度を上回ると、細胞機能に影響を及ぼし、症状が現れる。[76]。したがって、オートファジーは老化、炎症過程、細胞死を遅らせるために重要である。
オートファジーのマスターレギュレーター/阻害因子は、哺乳類ラパマイシン標的キナーゼ(mTOR)である。[78]。この細胞内キナーゼは、細胞外成長因子の刺激、栄養の利用可能性、エネルギーの供給に関する情報を統合する重要なシグナル伝達ノードとして機能している。[79]。真菌の代謝産物であるラパマイシンがmTORを阻害することが偶然発見され、mTORの代名詞となっただけでなく、mTORの機能を解明する主要な分子ツールとなった。ラパマイシンを投与すると、mTORを阻害することでオートファジーが活性化され、[80]、ケージに入れられたマウスの老化と認知機能の低下を遅らせることがわかった。[81]。逆に、ケージ飼育(標準飼育)により、重要な行動的手がかり(例えば、身体活動や間欠的絶食など、後述)が排除され、mTORの活性が不自然に高くなり、ミトコンドリア形成のマスターコントローラーであるペルオキシソーム増殖剤活性化受容体γコアクチベーター1α(PGC-1α)[82]の活性が低くなる可能性がある(後述)[83]。したがって、ADのマウスモデルで観察される老化と認知機能の低下は、自然条件下での老化プロセスを反映しておらず、人為的なものと考えられる。
参考記事
mTORを不活性化する行動的な合図としては、脳内の機能不全に陥った小器官や高分子のオートファジーを促進することが示された運動 [85]の他に、基本的にすべての真核生物において老化を遅らせ寿命を延ばすことがよく知られている慢性カロリー制限(CCR)がある。[86]。しかし、CCRは生理学的な手がかりなのだろうか、それともむしろ、進化的な観点から見てより自然な食事パターン、すなわち間欠的絶食をある程度シミュレートした実験研究の産物なのだろうか(下記参照)。しかし、断続的絶食はCCRよりも実験的に手間がかかるため、あまり研究されていない。とはいえ、オートファジー、特にマイトファジーは、酵母からハエ、ミミズ、魚、げっ歯類、さらにはアカゲザルに至るまで、調査された基本的に全ての種において、mTORの阻害を通じてCCRによって活性化されることが判明しており、[87]、それによってmTOR主導型の老化が減速される。[88]。CCRは寿命を延ばすだけでなく、中枢神経系を神経変性疾患から保護する。一方、カロリーの過剰摂取は明らかに脳の老化促進と関連しており、オートファジーの抑制により神経変性疾患のリスクが高まる。[89]。
とはいえ、断続的絶食はCCRと比較して、mTORによる老化促進や認知機能低下をより強固かつ安定的に抑制することが示された[90]。これは、断続的絶食中の代謝状態の主なホルミシス様シグナル分子であるケトン体のアセト酢酸(AcAc)とD-β-ヒドロキシ酪酸が、CCRよりも絶食中に効率よく生成されるという事実によって説明される[91]。これら2つの呼吸性燃料は、様々な生理学的 [92] または病理学的状態 [93]において、動員された脂肪酸から肝臓で内因的に大量に(最大150g/日)産生される。ヒトでは、D-β-ヒドロキシ酪酸の基礎血清レベルは低マイクロモル域であるが、12~16時間の絶食後には数百マイクロモルまで上昇する。[92]。重要なことは、血中グルコースとインスリンが低い場合 [94]、脳が必要とするエネルギーの最大60%がケトン体から得られ、グルコースを主燃料として置き換えることができることである。[95]。長時間の持久的運動後にも、D-β-ヒドロキシ酪酸が1~2ミリモルまで上昇する。[96]。生理的に適切なケトン体産生量の増加は、一晩絶食することですでに達成されており、朝絶食を解除する前に身体を動かすと、さらにケトン体産生量が増加する可能性がある。[97]。これは、胃が空っぽの状態で狩猟や採集に出かけた私たちの祖先が直面した状況を模倣している可能性が高い。
長鎖飽和脂肪酸も中鎖飽和脂肪酸もBBBを通過することができないため、ケトン体への変換によってのみ、エネルギーを必要とする脳が最大のエネルギー貯蔵庫である脂肪組織にアクセスすることができる。実際、ケトン体の生成によって、絶食中のグルコース要求量が減少し、糖新生タンパク質貯蔵量が維持されるため、生存能力が大幅に向上するのである[98]。興味深いことに、ここでも祖母仮説と一致するが、高齢者は断続的絶食中に少なくとも若年成人と同程度の効率でケトン体を生成し、[99]、ケトン食に対する代謝反応も加齢の影響を受けないようである。[100]。
上述したように、断続的絶食がCCRよりも優れているという観察結果は、進化の観点からも非常に理にかなっている。なぜなら、慢性的な飢餓ではなく、むしろ採食に成功した後の絶食と高カロリー食の摂取の間を周期的に変化させることが、古代の正常であったからである。重要なことは、系統的に保存されている遺伝プログラムが、断続的絶食(断続的絶食)から生じる代謝変化を、細胞内再生の開始の行動的合図として利用していることを、最近の証拠が示唆していることである。[101]。細胞の若さを維持するためには、CCRによって飢餓状態にする必要はない。ケトン体産生を誘導するのに十分な時間(例えば一晩で12時間)の絶食期と、身体の総エネルギー需要を満たすことができる食事期を交互に繰り返すだけで十分なのである。これとは対照的に、現在の常態は恒常的な摂食パターンであり、その結果、mTOR活性が恒常的に高くなり(PGC-1αレベルも低くなり、後述)、細胞の若返りが抑制される。一方、長時間の運動はmTOR活性を低下させ [102]、おそらくケトン体産生を増加させる。
これらを総合すると、体を動かすことと断続的絶食によってmTORを抑制することは、健康を維持するための自然な方法である。しかし、その代わりに、健康的なライフスタイルを錠剤にすることができるのではないかという期待も常にある: ラパマイシンは、ヒトでは臓器移植における拒絶反応の予防に使用される免疫抑制剤として承認されており、一方、ADにおける有効性を検証するための臨床試験が進行中である(総説は[103]を参照)。しかし、断続的絶食、栄養豊富な食事、定期的な有酸素運動と睡眠には、mTORを阻害する以上の有益な機能があることを考えると、生活習慣を変えることなく薬物で長期的な健康が得られるかどうかは疑問である。例えば、間欠的絶食(断続的絶食)や運動がもたらすよく知られた恩恵的効果の多くは、PGC-1αのアップレギュレーションによって媒介される。しかし、PGC-1αの活性化はまた、例えば活性酸素解毒酵素の発現を誘導することによって、神経炎症 [66]を軽減する。[104]。したがって、老化の2つの原因である慢性的なミトコンドリア機能障害と神経炎症(後述)は、PGC-1αのレベルの低下によって説明することができ[105]、運動不足、断続的絶食、またはその両方、あるいは以下で詳細に議論される他の多くの行動的欠陥のいずれかに由来する可能性がある。従って、行動上の欠乏を一つの薬物で補うことができるかどうかは疑問である。

まとめると、行動によって抑制された細胞の若りは、すべての臓器系に影響を及ぼし、加齢による虚弱につながる。しかし、身体的な衰え、特に認知機能の衰えの主な原因として、虚弱や老化そのものを責める罠にはまってはならない。[106]。「なぜなら、それは現在の行動とその結果(例えば、加齢に伴う認知能力の平均的低下)の統計的平均値を反映しているに過ぎないからである。実際、私たちは不健康な老化にもかかわらず寿命を延ばしている。しかし、この傾向を逆転させる能力は、祖先の祖母とともに進化した遺伝子プログラムのおかげで、私たち自身の手にある。
アミロイドβ(Aβ)の生理的役割
海馬複合体(HC)、すなわち原皮質海馬と嗅内皮質は、個人的な人生経験を記憶するための中心的な器官である。[108]。人生の一瞬一瞬は唯一無二であり、語彙のように繰り返すことはできないため、脳のこの古い部分は、重要な情報を瞬時に学習する能力を維持してきた。この能力は、採集や狩猟のときのような新しい地形での空間的ナビゲーションのための前提条件であり、個人的な交流や語りから社会的な学習をするときにも必要であり、長期的に自分の思考を記憶するときにも重要である。生存と繁殖にとって重要である可能性を示す指標として、HCは感情的な効果によって記憶する経験を選択する。(残念ながら、語彙がすぐに記憶できるほどエキサイティングであることはほとんどない)。こうして中脳は、何が起こったか(内容情報)、どこで起こったか(空間情報)、いつ起こったか(時間情報)という、人生のエピソードの相互に関連する少なくとも3つの側面を記憶する。HCの記憶容量には限りがあるため、日々獲得される内容情報は一時的にしか記憶されない。重要な(感情的にエキサイティングな)ライフイベントを生涯にわたって記憶するために、内容情報は夜間睡眠の初期の徐波睡眠(SWS)期に大脳新皮質に統合される。[109]。夜間睡眠の後半では、新しい記憶は急速眼球運動(REM)相の間に以前の経験と架橋される。時空間情報、すなわち記憶された出来事の文脈上の「いつ、どこで」は、海馬の歯状回(歯状回)ネットワークで長期的に維持され、新皮質の記憶痕跡の検索(再活性化)の指標として使用される(下記参照)。
海馬の高速学習は、グルタミン酸を主要な神経伝達物質とするシナプス長期増強(LTP)のメカニズムに依存している。グルタミン酸は特定のシナプス結合の強さ(重み)を変化させ、それによって新しい海馬の記憶痕跡を作り出す。神経細胞の活動が増加すると、同じシナプスでβ-アミロイド(Aβ)の産生と放出が同時に増加し、Aβは学習記憶の形成に重要な役割を果たす[111]。Aβは主に40個のアミノ酸を持つ単量体(Aβ40)で分泌され、わずかに凝集する傾向がある。逆に、Aβ42は生産量が少なく、オリゴマー、プロトフィブリル、フィブリルを形成しやすい。これらの凝集体は、ADの脳斑に含まれるAβの主な形態である。[112]。この観察と、凝集型Aβ42がある種のAD変異によってより大量に産生されるという事実 [113] から、病理学的作用はオリゴマーに、Aβの生理学的特性はほとんどモノマーにのみ帰属することになった。これはアミロイドカスケード仮説の基礎でもある。
しかしながら、低濃度のAβであってもある程度のオリゴマー化は起こりうるため、Aβのオリゴマー状態が様々であることから、ペプチドの本来の状態や、どのような状態で多機能を発揮するのかを解明することは困難である。[114]。いずれにせよ、正常な健康な脳における可溶性Aβの濃度はピコモルの範囲と推定されており、その種類はモノマーから高次のオリゴマーまで様々である。[116]。Aβの重要な生理的機能の一つは、グルタミン酸放出確率の調節因子として働くことであり、[117]、Aβ産生の活動依存的調節がシナプス可塑性の負のフィードバック調節因子として働く [118]。低濃度のピコモル濃度では、AβはLTPを増加させるが、高濃度のナノモル濃度では、Aβは増強の減少につながることが示されている。[119]。これは、Aβが用量に応じて、新しい記憶の迅速な形成をサポートするアゴニストとして作用するか、新しい記憶の痕跡が、記憶されるべき後続の出来事によって上書きされるのを防ぐアンタゴニストとして作用するという、提唱されている作用様式と一致している[120]。
参考記事

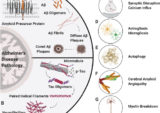
この負のフィードバックループが損なわれると、Aβが過剰に産生されるか、あるいはほとんど消去されなくなる。その結果、プラークが形成されなくても、より高濃度の可溶性オリゴマーAβが形成され、シナプス毒性作用を発揮するようになる。[121]。このことは、早期ADの臨床症状を有し、Aβ沈着のスキャンが陰性である、いわゆる非アミロイド病態が疑われる症候性患者の割合が相対的に高いことを説明するかもしれない。[122]。可溶性Aβオリゴマーの分布が、プロトタイプのアミロイド斑よりもADの認知機能低下とよく相関することが明らかになり、[123]、以前のアミロイドカスケード仮説はオリゴマーカスケード仮説に取って代わられた。[124]。
多量のオリゴマーAβの蓄積は神経炎症を誘発し、酸化的損傷を引き起こし、グリコーゲン合成酵素キナーゼ-3β(GSK-3β)の活性化を含む複数のシグナル伝達事象に悪影響を及ぼす。例えば、GSK-3βの活性化は成人海馬神経新生を阻害し、神経炎症とアポトーシスを促進する(総説は[126]を参照)。さらに、GSK-3βのアップレギュレーションは、アミロイド前駆体タンパク質(APP)とタウのリン酸化につながる。これらは、それぞれアルツハイマー病(AD)の特徴であるAβプラークと神経原線維のもつれにつながる病理学的プロセスに関連している。[127]。GSK-3βの活性は、微小管の安定化因子であるタウを、高リン酸化された非機能性状態(p-タウ)に移行させる。[128]。タウ病態は進行性で、タウの機能喪失 [129]と、病的なp-タウ凝集体による毒性機能の獲得 [130]の両方を介して、罹患したニューロンに有害な影響を及ぼす。それゆえ、高濃度のオリゴマーAβだけでなく、むしろ機能不全のp-タウがAD進行の重要なドライバーとみなされるかもしれない。このことは、p-タウの沈着が病期とより密接に相関するという事実によって裏付けられている[131]。
参考記事
しかし、Aβとp-tauのどちらがAD発症の結果を引き起こすのだろうか?この疑問は長い間議論されてきた。例えば、アミロイドカスケード仮説の支持者は、Aβプラーク形成の原因因子として主に加齢を非難し、それゆえ薬理学的努力の焦点はアミロイドプラークの除去に向けられた。皮肉なことに、不溶性プラークではなく、前述のように、むしろ過剰な可溶性オリゴマーが有害であることが最近明らかになった。[132]。しかし、新たに提唱されたオリゴマーカスケード仮説から発展した現在の薬理学的戦略によってAβオリゴマーを除去しても、有害であることが判明するかもしれない。ADの主な原因が無視されたままであるだけでなく(下記参照)、Aβはシナプス可塑性に必要であるからである。例えば、LTPと記憶における健全な生理学的機能は、分泌されたAβ単量体(長さも濃度も異なる)と複数のオリゴマー状態との間の微妙なバランスに依存しているようである(総説は[133]を参照)。この注意点は、ADの実験動物モデルにおいて、ヒトの治療研究にも用いられているAβに対する様々な抗体によるAβ除去が、神経細胞機能障害の修復に効果がないばかりか、有害な皮質過活動を引き起こした理由を説明するかもしれない。[134]。この予期せぬ所見は、ヒトを対象とした研究で免疫療法による認知機能改善が見られないことの細胞学的説明の可能性を与えるものであると結論されている。[135, 136]。言い換えれば、Aβに対する唯一の攻撃は神経細胞の恒常性を阻害し、学習と記憶のさらなる低下を引き起こす。
従って、現在の重要な治療戦略やADの少なくとも病気の進行を遅らせる戦略は、患者の状態を悪化させる可能性さえある。Puzzoらが最近述べたように [133]: Aβをもっぱら「悪い」タンパク質としてとらえるあまり、この病気の他の重要な側面に目を向けることができなくなっている。著者らはさらに、「Aβは乳児の脳ではすでに神経細胞内に存在し、脳の可塑性が高い8歳まではさらに増加し、そのときには神経細胞の約半数がAβ免疫陽性である」と指摘している。成人期には、Aβはニューロンの大部分に存在するが、高齢者では20%減少する。「さらに、Aβの脳内沈着には、Aβシードのみが重要であり、モデル動物の年齢は関係ない。しかし、年齢でないとすれば、他にどのような要因がAβシードの形成につながるのだろうか?」この重大な疑問に対する答えのみが、AD予防戦略や真に因果関係のあるAD治療への道を開くことになる。しかし、Aβ濃度の上昇は必然的なものでも、老化の自然な結果でもないことは、ますます明らかになってきている。むしろ、他の要因が関与しており、興味深いことに、そのほとんどは様々な方法で修正可能である。
睡眠中の記憶成長
睡眠の重要な機能の一つは、新しい海馬の記憶の痕跡を大脳新皮質に定着させて長期保存し、既存の知識体系に統合することによって生涯の経験を得ることである。シナプスのホメオスタシス仮説によると、これはSWS中の記憶内容の反復によって達成されるが、選択的な長期抑圧を含むシナプス重みの差次的な再正常化によっても達成される。これに対応して、次の覚醒期に新しい記憶の海馬での符号化を可能にするためには、前日の情報収集期に蓄積したAβレベルを低下させ、 LTPの潜在能力を完全に再活性化させる必要がある。それゆえ、Aβクリアランスは睡眠のもう一つの能動的機能として発達した可能性があり、間質空間の拡大によって達成される。その結果、間質液と脳脊髄液の対流交換が著しく増加し、BBBを越えてAβが排出される。[138]。記憶機能に関するこの回復作用は、より大量のシナプト毒性Aβの濃度依存的凝集のリスクも低下させる。したがって、マウスやヒトの睡眠不足が、大量のオリゴマー凝集が起こる臨界閾値以上にAβ濃度を上昇させることは、重要な発見である。[140, 141]。
参考記事
睡眠のもう一つの重要な機能は、成人海馬神経新生のための時間的空間を提供することである。睡眠中、高濃度では成人海馬神経新生を抑制するコルチゾールは低下するが、成人海馬神経新生を促進するインスリン様成長因子1(IGF-1)、成長ホルモン(GH)、メラトニン、BDNFは上昇する。したがって、長時間の睡眠不足は成人海馬神経新生にとって有害であり、[142]、それによって海馬の記憶能力とパフォーマンス(下記参照)を高める可能性のある明確なコード(指標)の数が減少する。[143]。同様に、成体で生まれたニューロンを訓練後に切除すると、以前に獲得した記憶が破壊されることが示された。[144]。歯状回の新生顆粒細胞は、時空間的な文脈を記憶することで、以前と類似しているが新しい経験を区別し、それによって新しい記憶と以前の記憶との干渉を軽減する上で特に重要である。[145, 146]。さらに、海馬記憶指標理論(HMIT)によって概説されたように、記憶する、すなわち、それぞれの事象に固有に新皮質に分布した記憶痕跡にアクセスし、それを検索し、文脈的な経験を再構築するためには、記憶された事象の海馬の時空間情報が必要である。[147]。HMITによれば、海馬-皮質系による遠隔記憶の統合には、海馬のインデックスの維持が必要である。したがって、我々は成人海馬神経新生によって生成された新しいニューロンによって記憶された時空間的文脈によって、人生のエピソードを記憶している。[144]。したがって、遠隔記憶は、成体で生まれた新しい歯状回ニューロンが生涯にわたって作られることによって、最もよく維持される。[149]。時空間記憶容量の拡大は、それによって自伝的記憶を継続的に拡大するための前提条件にもなる。[150]。このことは、成人海馬神経新生が障害されると、エピソード新皮質エングラムにリンクするインデックスの海馬アーカイブが記憶容量を使い果たすだけでなく、恐怖の過剰般化や持続的な心的外傷後ストレスにつながる、以前の経験と新しい経験の間の弁別エラー(干渉)を引き起こす理由を説明する[151]。最近、早期ADのトランスジェニックマウスモデルにおいて、海馬の記憶エングラム(index)細胞を光遺伝学的に直接活性化すると、自然想起の手がかりを用いた長期記憶テストでは健忘であるにもかかわらず、記憶が想起されることが示された。興味深いことに、歯状回エングラム細胞の穿通路シナプスにおけるLTPを光遺伝学的に誘導すると、ケージに入れた動物の「年齢に依存した」スパイン密度と長期記憶の両方が回復する。このことは、例えば環境エンリッチメントのADモデルにおいて、なぜ社会的活動が記憶の低下を防ぐのか、したがって、年齢ではなく、むしろ不自然な生活習慣がこれらのモデルにおいてADを引き起こすのかを説明するものである。
拡大する指標アーカイブにおいて、新しい経験と以前の経験との干渉が減少することは、成人海馬神経新生の重要性と、このプロセスがいかに持続性を促進するか [154]、また文脈記憶の精度を高めるか [155]を包括的に説明する。効率的に記憶するために、成体で生まれた海馬の脳細胞はLTP、すなわち学習に対してより低い閾値を示し、その一方でLTPは樹状突起スパイン密度をアップレギュレートし、最大の変化は成熟の初期段階、つまり既存の回路とシナプスを形成し始める時期に起こる。[156]。このようなメカニズムにより、成熟しつつある若年成体生まれの歯状回細胞は、新しく経験した出来事によって、特に情報量の多いニューロンへと変化している。興味深いことに、成人海馬神経新生を刺激するのに必要な条件、例えば自発的な運動は、海馬のスパイン密度を増加させ、シナプス形成の増加とシナプス除去の減少、さらに成熟ニューロンの生存率の増加をもたらす。[157]。この観察に続いて、成人海馬神経新生の主要な機能は、新しいニューロンそれ自体の産生だけでなく、比較的若いニューロンのシナプス特性を持つ新しいニューロンの産生を含むという仮説を示唆する研究者もいる。[158]。このメカニズムにより、歯状回では1年に細胞のほぼ2%が入れ替わるため、成人海馬神経新生は、新しい経験を得るために重要なこの脳領域を、高齢になっても若く保つ可能性がある。しかし、成人海馬神経新生の開始と新しい歯状回ニューロンの成熟と統合のための行動的合図を与えれば、UTADに沿ったものになる(下記参照)。
成人海馬神経新生は気分を制御し、抗うつ作用を持つ
ADにおいて、脊髄周囲皮質(PRH)と脳内側部皮質(LEC)が最初に侵される皮質領域のひとつであることは興味深い。[160, 161]。両領域は、穿通路投射を介して、新生歯状回ニューロンに対して最も重要な内容情報(すなわち「何が起こっているか」)を提供していることが判明した(総説は[162]を参照)。さらに、海馬のCA3領域からの逆投射信号は、内側嗅内皮質(MEC)からの直接信号を受け、新生歯状回ニューロンに対して必要な時空間情報(すなわち「いつ、どこで、何が起こっているか」)を提供する。[163]。両方の入力は、事象やパターンの分離に必要であるようだ。[164]。海馬の変性がADの最も顕著で初期の特徴のひとつであり、[165]、穿通路が損傷を受ける最初の構造のひとつである。[166] ことや、海馬内白質の病変負荷が進行性MCIと強く関連している。[167] ことから、慢性的に障害された成人海馬神経新生がAD発症において病因学的に重要な役割を果たしているのではないかという仮説が支持される。この仮説は、行動上の欠陥や環境毒素がADHを障害し、ADのリスクを増加させることが知られているという証拠によってさらに支持されている(下記参照)。したがって、非生産的な成人海馬神経新生は、病理組織学的にだけでなく、機能的にもADの病因に関係している可能性がある。したがって、この関連性と生産的な成人海馬神経新生の要件を理解することは、ADの新たな理解と、予防や治療介入のための原因究明戦略の開発に役立つかもしれない。
成人海馬神経新生によって生成された新しいニューロンは、空間ナビゲーション、エピソード学習、記憶検索に必要である。[168]だけでなく、直接的に [169] または間接的に [170, 171] HPA軸を制御することによって、気分、特に心理的回復力(ストレスに対する抵抗力)を制御する。成人海馬神経新生が障害されると、中程度のストレスの後にコルチコステロンのレベルがベースラインまで回復するのが遅くなり、デキサメタゾンによるHPA軸の抑制が弱くなることから、実験的に新しい成体生まれの歯状回-ニューロンを欠損させた高齢マウスでは、HPA軸のフィードバックが損なわれていることがわかる。選択的セロトニン再取り込み阻害薬(SSRI)の一種であるフルオキセチンのような抗うつ薬が抗うつ効果を発揮するには、無傷の海馬神経発生ニッチと生産的な成人海馬神経新生が必要であることが示されたことで、心理的回復力の調節に活性成人海馬神経新生が決定的に関与していることがさらに証明された。[172]。この抗うつ機序はげっ歯類で同定され、ヒト以外の霊長類でも確認された。[173]。同様に、最近のヒトの研究では、抗うつ薬治療による海馬体積の有意な増加が報告されている。[174]。逆に、成人海馬神経新生の障害は不安(新奇探索行動の抑制)、大うつ病 [175]、慢性的なコルチゾールの上昇を引き起こし、これら3つはすべて早期ADの特徴的な警告サインであり、潜在的な原因となる危険因子である。[176, 177]。興味深いことに、HPA軸の調節異常は、少なくともADのMCI段階から起こり、疾患の進行を促進することが示された。[178]。さらに、フラミンガム心臓研究のデータを再解析したところ、老年期のうつ病は認知機能の低下に先立って発症し、認知症およびADの有意な危険因子であることが判明した。[179]。
参考記事
慢性的に乱れた成人海馬神経新生によって引き起こされるHPA軸の調節障害がADにつながるかもしれないというもう一つのヒントは、早期AD患者が一貫して基礎血漿コルチゾール値の上昇を示し、[180]、低用量DEX抑制に対する感受性も低下しているという知見である。[181]。このため、コルチゾール測定は信頼性の高いADバイオマーカーとして示唆されている。[182]。磁気共鳴画像法(MRI)研究から、ADにおける最も早い発症事象のひとつが非効率的な成人海馬神経新生によって引き起こされるかもしれないというさらなる証拠が得られた:Aβ測定よりも海馬の萎縮がMCIからADへの進行時間を予測したのである。[183]。
HPA軸の調節異常がADにおける記憶力低下の重要な原因因子であるかもしれないという仮説に対するさらなる支持は、最近のプロスペクティブなワシントン/ハミルトンハイツ・インウッド・コロンビア・エイジング・プロジェクトから得られており、その結果、晩年の抑うつ症状は記憶力低下に先行するが、その逆はないことが明らかになった。[184]。加えて、年齢、性別、学歴、血管疾患を含む疾病負担とは無関係に、ベースライン時に認知機能に病的な変化がなかった人においても、うつ病のスコアが高いほど認知機能が急速に低下することが予測された。この観察は、うつ病の併存が、神経原線維もつれ負荷の増加 [185]や認知機能低下の速さ [186]など、AD病態の程度や進行と関連することを示した以前の研究の証拠と一致している。
慢性的なHPA軸の調節障害はAD病態を促進する
急性ストレスは、感覚入力の増加を促進する生理学的変化を即座に引き起こす。生命の危機に瀕した状況で感覚を鋭敏にすることは生存にとって重要であるが、こうした変化は、過剰な興奮性グルタミン酸放出による情報過多や海馬神経毒性の危険性をはらんでいる。[187, 188]。したがって、ストレスによって誘発されるHPA軸のアップレギュレーションが、グルタミン酸放出の調節因子として作用する海馬Aβ [189]の産生を同時に増加させることは、有用な適応であるように思われる。さらに、Aβ単量体はホスファチジルイノシトール-3-キナーゼ経路を活性化し、アポトーシスを抑制して興奮毒性死からニューロンを保護する。さらに、単量体Aβは、エネルギー代謝が高い状況において、酸化ストレスからニューロンを保護する。[192, 193]。
HPA軸の活性化は、どのようにしてシナプスAβ放出を増加させるのだろうか?最近明らかになったメカニズムのひとつは、ストレス応答神経ペプチドであるコルチコトロピン放出因子(CRF)によるアミロ生成γセクレターゼ活性のアップレギュレーションである[194]。さらに、グルココルチコイドを実験的に投与すると、過剰なAβを分解することが知られているインスリン分解酵素の産生が低下するため、高齢の非ヒト霊長類において海馬のAβ濃度が上昇することが判明した。[195]。さらなる試験管内試験および生体内試験の実験により、グルココルチコイド投与もまた、アミロイド前駆体タンパク質(APP)とAβ切断酵素の定常状態レベルを上昇させることにより、Aβ産生を亢進させるという証拠が得られた[196]。興味深いことに、これらの実験ではグルココルチコイドがタウの蓄積を増大させることも示されており、このホルモンが慢性的に活性化すると神経原線維のもつれの発生を促進する可能性があることを示している。著者らは、AD患者にみられる悪名高い高濃度のグルココルチコイドは、単に疾患プロセスの結果ではなく、むしろADの発症と進行に中心的な役割を果たしている可能性があると結論づけた。この考えを支持するものとして、生涯大うつ病患者におけるコルチゾールの慢性的な分泌過多は、MCIやADの症状がなくても、脳内にAβの著しい蓄積をもたらす。[197]。
まとめると、急性のストレス状況で海馬のAβの放出が亢進するのは、有用な神経保護メカニズムであるように思われるが、慢性的なストレスや成人海馬神経新生の障害による慢性的なHPA軸の調節障害が、心理的回復力の低下(ストレス抵抗力の弱化)や慢性的なコルチゾールの過剰分泌につながる場合、この亢進が問題となる可能性がある。したがって、私たちの生体はどのようにして生涯にわたって生産的な成人海馬神経新生と機能的なHPA軸の調節を維持するのかという疑問が生じる。
生産的な成人海馬神経新生と最小限の法則(LOM)の主な要件
動物、特にヒトにおいて成人海馬神経新生が発見されて以来、多くの研究により、成人海馬神経新生は高度に制御された現象であり、局所因子(神経原性ニッチ)、サイトカイン、成長因子、および多くのホルモンの制御下にあることが明らかになった。[198, 199]。餌を蓄える鳥類 [200]や歌を学習する鳥類 [201]の研究から、機能的に海馬に似た脳構造が季節ごとに成長する現象や、成人海馬神経新生と行動(例えば餌を蓄えたり歌を学習したり)が動的に相互に関連していることが明らかになった。[202]。この種の研究は哺乳類にも拡張され、例えばカンガルーネズミ(Dipodomys)の海馬の大きさは、その自然な空間利用パターンに密接に依存していることが示された。[203]。また、シロアシネズミ(Peromyscus leucopus)のような光周性の生物は、環境の日長を監視して、生理や行動において季節に適した適応を行うが、その前に成人海馬神経新生による脳体積の調整が行われる。[204]。脳の大きさはエネルギー消費量に対応するため、成人海馬神経新生によるこのような環境と行動の緊密な制御は、進化の観点から理にかなっている。したがって海馬の大きさは、記憶容量に対する個体の重要な必要性に応じて、大きくなったり小さくなったりする。私たちのアイデンティティと祖母仮説の原理は、生涯記憶し続ける能力に依存しているため、ヒトの成人海馬神経新生は季節的な成長だけでなく、むしろ生涯成長し続ける性質を持っている。

カンガルーネズミ

シロアシネズミ
生涯にわたって生産的な成人海馬神経新生を維持するための環境的、行動的な手がかりや重要な要件は何だろうか?人類はその歴史の大部分を狩猟採集民として生き延びてきたのだから、生産的な成人海馬神経新生の複雑な制御は、人類が何十万年も生き延びてきた原則的条件に強く適応していたと考えるのが妥当である。進化の観点から見ると、部族の生存は、狩猟や採集が成功するまで、身体活動と飢餓の条件下で採集地を記憶することに依存しており、社会的絆(集団での狩猟)と年長者からの学習(例えば狩猟戦略)が決定的に重要であった。したがって、社会的・身体的活動、日々新たな挑戦があり、それがストレスとなり、必須栄養素を豊富に含む新鮮で変化に富んだ食物の摂取によって中断される断続的絶食が、成人海馬神経新生(下記参照)のポジティブな調節因子であることは驚くにはあたらない。したがって、生産的な神経新生に必要な基本的条件は、前近代の生活様式によって自然に備わっていたのである。これとは対照的に、以下に詳述するように、運動不足、孤独感(実際に感じられるものであれ、感じられるものであれ)、慢性的な苦痛、あるいは不健康な西洋式食事(WeDi)と恒常的な食事パターン(ad libitum)との組み合わせは、成人海馬神経新生の負の調節因子であり、上に概説したような有害な結果をもたらす。
神経幹細胞の増殖から始まり、前駆細胞の拡大、成長、成熟(分枝形成とシナプス形成)を経て、最終的に既存の歯状回ネットワークに統合されるまでには、約3カ月、もしかしたらそれ以上かかるかもしれない[198, 205]。増殖そのものが厳密に制御されており、活性化と抑制シグナルからの除去の両方が必要であるという事実に加え、新生前駆細胞のうち、成熟した統合型歯状回ニューロンには一桁の割合しかならず、ほとんどの細胞は早期に死滅する。[206, 207]。成人海馬神経新生は各段階において、特定の環境的手がかり(通常はホルモンを通じて伝達される)だけでなく、特定の脳の構成要素(必須多価不飽和脂肪酸、コレステロールなど)、社会的インプット(記憶に残る新しい人生経験)、エネルギーを必要とする。これらの要因はそれぞれ個々に成人海馬神経新生を制限する可能性があるため、「最小の法則」が適用される。この「法則」によって、ADをどのように効果的に予防・治療できるのか、また、なぜ単剤治療による介入が失敗し続けるのかを理解することができるからである。
生産的成長は制限因子によって制限されるという原則は、1828年にカール・シュプレンゲルによって農業科学で初めて定式化され、[208]、1840年にユストゥス・フォン・リービッヒによって最小の法則(LOM)として拡張・普及された(シュプレンゲル・リービッヒLOMの参考文献と歴史については [209]を参照)。LOMは、成長は利用可能な資源の総量によってではなく、最も希少な資源によって制御されると述べている。これは、成人海馬神経新生を成長プロセスであるとみなすならば、非常に重要な概念である。LOMは、成長または成熟に不可欠な因子の欠乏は、定義上(不可欠である以上)、他の因子で補うことができないことを説明している。したがって、新しい歯状回ニューロンの増殖、成長、成熟を制限するような、それぞれの行動的、環境的合図や他の必須成長因子の欠乏は、必然的に成人海馬神経新生の障害につながり、コルチゾールの過剰分泌、うつ病、AD発症リスクの増大という結果をもたらす。
疫学的研究により、ADや大うつ病の危険因子が数多く同定されているが、それらの多くはLOMによれば成人海馬神経新生の制限因子であり必須因子である。海馬の成長のダイナミックスには、海馬が縮小する可能性も含まれているため、欠落している各要因は生産的な成人海馬神経新生を妨げるだけでなく、海馬サイズの縮小やうつ病やADのリスク上昇にもつながる。ADのような潜在的な多因子疾患にLOMを適用した場合、個々の生活習慣が通常いくつかの欠損につながるため、介入試験は、そのような欠損(すなわち危険因子)の1つ、あるいは限られた数だけを恣意的に排除しようとするものであるが、弱い効果しか得られない。以下は、生産的な成人海馬神経新生に必要な条件を列挙したもので、その多くは経済的に先進的な社会では通常不足しているものである(図1とand22も参照):
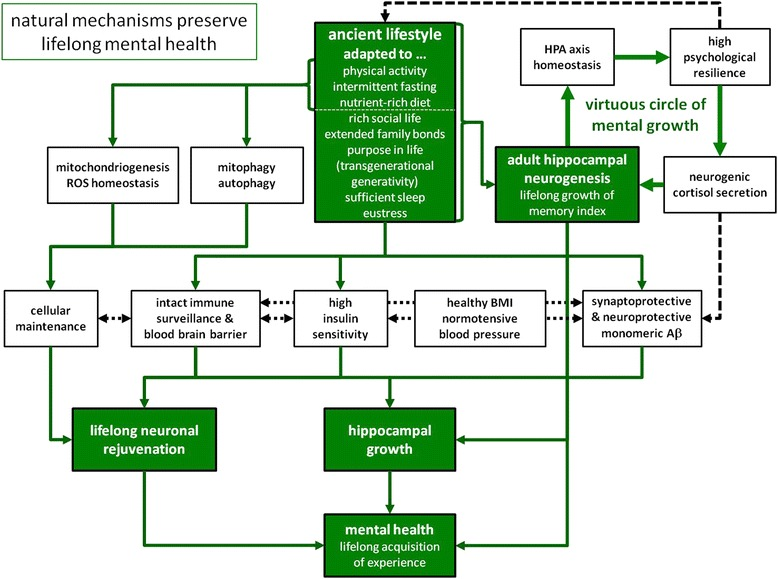
図1
「自然のメカニズムが生涯にわたって心の健康を保つわれわれの遺伝子プログラムは、人類の生活史の大部分を支配してきた生活様式によく適応している。古代の生活様式は、広範かつ日常的な身体活動、絶食と栄養豊富な食事の交互の段階、十分な睡眠とストレス解消によって特徴づけられていた。さらに、生存は拡大した家族の絆に決定的に依存していたため、豊かな社会生活とともにあった。例外的に長い閉経後の期間は、祖母仮説によれば、人類の長寿の現在の主な説明となる、世代を超えた世代交代という目的を果たした。したがって、このような自然条件下では、すべての主要な生理学的システム(免疫、心臓血管機能、エネルギー代謝など)が積極的に相互作用し、それによって、生涯にわたって知識を習得し、高齢になるまで精神的健康を維持することを可能にするために、神経細胞の若返りや成人海馬の神経新生などの主要なメカニズムを支えている。詳細は本文を参照のこと」
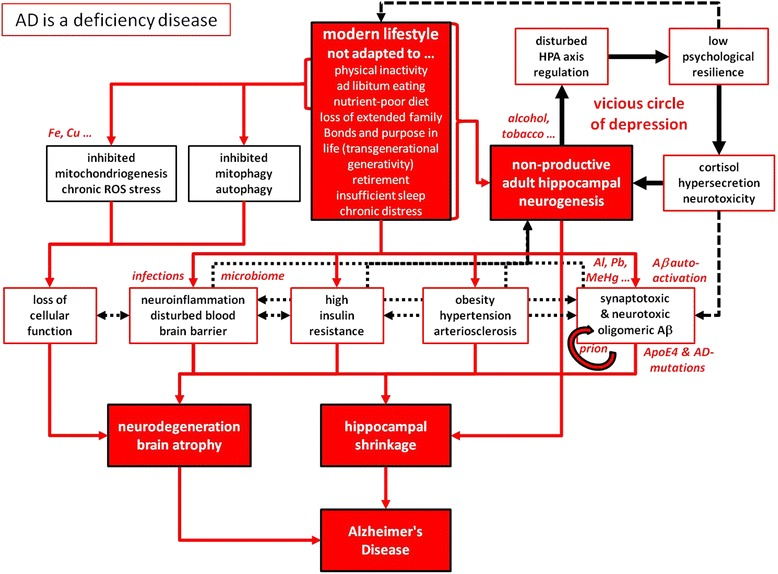
図2
「ADは欠乏症である」私たちの遺伝子プログラムは、運動不足、栄養の乏しい食事の自由食パターン、高度に競争的な労働環境の要求から発せられる慢性的な苦痛、しばしば家族の絆の喪失などを個々に組み合わせることを意味する、現代のライフスタイルを定義する急速かつごく最近の変化に適応していない。さらに、このような状況下では、定年退職という概念は、人類の文化史の中で後期に発明されたものであり、進化の観点から見た晩年期の主な目的を打ち消すものである。精神的健康にとって必要不可欠な要素の欠乏は、定義上、遺伝的プログラムによって補うことはできない。その結果、最小の法則によって定義されるように、個々の欠損は神経細胞の若返りを妨げ、特に生産的な成人海馬神経新生の妨げとなる。うつ病の神経細胞相関として、HPA軸の調節障害とコルチゾールの過剰分泌、その他の病態生理学的結果(神経炎症と血液脳関門の破壊、インスリン抵抗性、高血圧、動脈硬化)は、このような生活習慣由来の欠損から生じ、神経毒性Aβの蓄積、特に海馬の縮小、一般的な脳の萎縮につながり、それゆえADの特徴としてよく知られている。このような行動不全の状況下では、環境毒素、慢性感染症、遺伝的素因がADの進行を加速させる。異なる病理学的プロセス間の相互作用は、ADプロセスを暴走現象にする多数の悪循環を活性化させるが、これはシステム生物学的アプローチによってのみ阻止され、図1に描かれた状況に逆転させることができる。
適度だが長時間の身体活動は、加齢したヒトにおいても脳容積を増加させる。[210]。このことは、進化の観点から見ても非常に理にかなっている: 前近代社会では、身体活動は生存のための必須条件であり、活動する身体からのホルモン信号は、体力だけでなく、自伝的記憶能力も高めるメカニズムとして使われている(下記参照)。ある意味、身体を動かすことは、新しい経験が期待できるというシグナルを脳に送ることになる: 歩けば歩くほど、より多くの経験をすることになり、海馬の記憶指数が大きくなる。今日、私たちは走るか走らないかの選択を迫られ、ほとんどの人が努力を惜しむテクノロジーを取り入れ、座りがちなライフスタイルを送っている。運動不足の増加は、体力、日常的な家事や社会的交流の能力、運動能力、認知能力を低下させ、生活の質全体に多大な影響を及ぼす。
よく管理された無作為化研究では、認知症のない120人の高齢者を有酸素運動群(n=60、平均年齢67,6歳)とストレッチ対照群(n=60、平均年齢65,5歳)に無作為に割り付けた。[211]。MRIで測定した海馬のベースライン容積に、両群に差はなかった。健康であることに加え、プログラムに参加するための前提条件は、座りがちなライフスタイルであることで、介入前の過去6カ月間、毎日30分以下の身体活動であったと定義された。最初の6週間のトレーニングの後、有酸素ウォーキンググループは、最大心拍数の60~75を目標心拍数ゾーンとして毎日40分のウォーキングを行い、ストレッチとトーニングのコントロールグループは、ダンベルやレジスタンスバンドを使ってエクササイズを行い、ヨガのシークエンスを含むバランス改善のトレーニングを行った。1年間の介入後、有酸素運動群では海馬体積が約2%増加し、認知症でなくとも高齢者では海馬体積が毎年1~2%減少するのが普通であることを打ち消した。[212]。このような容積の減少は、上に概説したように、認知障害を発症するリスクを増加させる。ストレッチング対照群では、この加齢に伴う体積減少のパターンに当てはまり、1年間のインターバルで1.4%の体積減少が見られた。長期にわたる軽度の有酸素運動が成人海馬神経新生の増強に優れていることは、げっ歯類でも示されている[213]。実際、短時間の激しい運動でも成人海馬神経新生にプラスの効果はなかった。[214]。
興味深いことに、ウォーキング群における個々の有酸素性体力の向上は、海馬体積の増加と強い相関があった。成長は主に歯状回を含む前部で起こった。前部海馬の細胞は、言語記憶能力だけでなく空間記憶能力の獲得を仲介しており、[215]、後部海馬に比べて加齢に伴う萎縮を起こしやすい。[216]ため、これは重要な発見である。したがって、有酸素運動による海馬体積の増加は、成人後期においても記憶機能を増強することを示している。

身体活動の変化は、健康全般や寿命にも重要な影響を及ぼす。祖母仮説と同様に、70歳男性の不自然な座りっぱなし行動は、90歳までの生存確率を54%から44%に低下させることが示された。[217]。運動不足は多くのいわゆる。「生活習慣病」を引き起こすが、それらは慢性的に不健康な生活習慣が原因ではなく、加齢が原因であるかのように、長寿病という不適切な名前で呼ばれることもある。[218]。BDNF、神経成長因子(NGF)、グリア細胞由来向神経性因子(GDNF)などの向神経性因子の発現レベルの低下は、加齢とADに強く関与している。[219]。しかし、年齢との相関は因果関係とは一致しない。むしろ、これらのホルモン因子はすべて運動によってプラスに調節されるため、脳の老化促進はむしろ高齢者の座りがちな生活習慣によって引き起こされることが説明され、[222]、したがって老化自体とはあまり関係がないように思われる。
この因果関係の方向性、すなわち生活習慣から長年にわたる脳の「変性」への方向性は、動物実験で検証された。ある実験では、座りっぱなしのADトリプルトランスジェニックマウスにおけるGDNFのダウンレギュレーションが、自発的な運動によって回復し、シナプス機能が改善した。[223]。この特定のADモデル三重トランスジェニックモデル(3×Tg-AD)は、PS1M146V(プレセニリン1変異体)、APPSwe(β-アミロイド前駆体タンパク質変異体)、tauP301L(p-tau変異体)の導入遺伝子を保有し、アミロイドプラークとp-tauもつれ病態の両方がシナプス機能に及ぼす潜在的なAD治療薬の影響を評価するために特別に開発された[42]。別の実験では、NGFは運動によっても発現が上昇し、成人海馬神経新生を刺激することが示された。[224]。さらに、BDNFは成体で生まれた歯状回ニューロンの分化と成熟を促進し、[225]、運動によって海馬から放出されることが示された。[226]。
上記の有酸素歩行介入研究では、運動群における海馬体積の増加は血清BDNF濃度の増加とも関連しており、少なくともラットとブタでは、海馬のBDNF濃度と並行していることが示された。[227]。神経成長因子ファミリーの一種であるBDNFは、AD患者の海馬と側頭皮質で減少していることが判明しているため、運動によって海馬のBDNF分泌が誘導されるという観察は重要な知見である[228]。ヒトを対象とした別の介入研究では、海馬体積の減少が、記憶能力の低下と有意かつ直接的に関連しており、中強度の運動によって打ち消すことができるというさらなる証拠が得られた。[229]。興味深いことに、運動によって誘発されるエリスロポエチン(EPO)と血管内皮増殖因子(VEGF)の両方が、それぞれ全身または局所的な酸素濃度の低下に反応して放出され、成人海馬神経新生を増強する。VEGFは運動誘発性成人海馬神経新生に必要であることさえ示された。[230, 231]。EPOは、可塑性、シナプス結合性、記憶関連ニューロンネットワークの活性を調節することにより、海馬依存性の記憶を増強することがわかった。[232]。また、成人海馬神経新生を活性化し、[233, 234]、BDNFの発現を増加させることで海馬ニューロンを保護する。[235]。
慢性疾患、特にADの予防における身体活動の重要性は、BDNFが海馬ニューロンにおけるPGC-1α依存性のミトコンドリア生合成とシナプスの形成と維持の促進の両方を刺激する能力を持っていることでも示されている。[236]。運動によるPGC-1αの活性化もまた、ADを引き起こす神経炎症を抑制する上で重要な役割を果たしている。[237]。運動が脳の健康にとって重要であることは、身体活動が成人海馬神経新生を刺激し、記憶機能を改善することを確実にするホルモンシグナル伝達経路がますます特定されつつあるという事実によって、さらに立証されている。上記のBDNF、NGF、GDNF、EPO、VEGFに加えて、身体運動は、ジヒドロテストステロン [238]を介して成人海馬神経新生を刺激することが発見され、驚くことではないが、ジヒドロテストステロンは、身体活動のよく知られた抗うつ効果を媒介する。[239]。アンドロゲン活性とADの間のリンクの直接的な支持は、前立腺がん治療の一環としてのアンドロゲン遮断がAD発症リスクを2倍にするという証拠を提供した最近の研究から得られた。[240]。
GH [241]とIGF-1 [242]もまた、有酸素運動の反復に反応し、成人海馬神経新生を刺激し、神経保護作用を持つ。線維芽細胞増殖因子2(FGF-2)もまた、運動によって海馬で直接誘導され、[243]、ADのマウスモデルにおいて海馬機能を回復させることが示された。[244]。FGF-2はADの潜在的な治療法として議論されているが、[245]、LOMが予測するように、単一の治療手段がADの治癒に成功する確率は低い。さらに、セロトニンの中枢放出は身体活動依存性成人海馬神経新生に必要であり、そこでもセロトニンは、若年成体だけでなく老齢マウスにおいても、直接的かつ鋭敏な調節的役割を果たしている。[246]。この知見から、運動誘発性成人海馬神経新生を理解することは、うつ病や加齢に伴う認知機能低下の予防だけでなく、治療にも役立つ可能性があるという結論に至った。同様に、脂肪細胞から分泌されるアディポネクチンは、運動誘発性成人海馬神経新生を媒介し、[247]、抗うつ薬として作用するようである。[248]。興味深いことに、いくつかのホルモンやサイトカインは、運動する筋肉自体から直接分泌されるため、内分泌器官とみなすことができる。[82]。成人海馬神経新生を促進するホルモンであるイリシン [249]とメテオリン様ホルモン [250]は、活動筋のみから分泌されるようである。
最後になるが、健康や記憶に対する身体活動の効果は、適度なレベルのコルチゾール [251]によっても媒介される。身体運動によるプラスの効果が中強度レベルでピークに達し、それによって成人海馬神経新生が活性化されるのに対し、極端な身体運動による過剰なコルチゾールレベルは成人海馬神経新生を抑制する。[252]。極度の身体運動にしばしば伴う心理的ストレス以外の機序の1つは、緊張した筋肉からIL-6のような炎症性サイトカインの放出が増加し、下垂体からの副腎皮質刺激ホルモン(ACTH)分泌が活性化され、その結果、高レベルのコルチゾール分泌が誘導されることであろう。[253]。このコルチゾールの過剰分泌は、神経炎症を悪化させることがわかった。[254]。対照的に、定期的で適度な運動は、全身の炎症を抑制する。[255]。同様に、適度な身体活動は抗酸化防御機構を高めるが、激しいレベルの身体活動は抗酸化予備能を枯渇させる。[256]。したがって、極端なレベルの身体運動は神経学的な悪影響につながるため、避けるべきである。[257]。したがって、心身の健康を維持するためには、適度な強度で頻繁に運動するバランスの取れた生活スタイルが理想的であり、一方、運動不足(過剰な運動も同様)は可能な限り避ける必要がある。
上記のような有害な因果関係の連鎖を引き起こす成人海馬神経新生 [258]の活性化が不十分であることに加え、運動不足は過剰なAβのクリアランスを低下させるため、AD発症リスクを直接的に高める。座りがちな生活習慣は、BBBに存在するAβの主要な輸送タンパク質である低密度リポタンパク質受容体関連タンパク質1(LRP1) [259]をダウンレギュレートすることが判明している。運動不足はまた、主要なAβ分解酵素の一つであるネプリライシンの活性化を阻害する。[260]。さらに、座りっぱなしでいると、持続的な不毛な神経炎症 [237]、インスリン抵抗性、糖尿病、内臓肥満につながる。最近の研究によると、これらの状態はすべてAD発症のリスクを高める。
治療を考える上で重要なことは、ADと診断された場合であっても、身体を動かすことは有効であるということである。最近のパイロット研究では、AD患者を対象とした在宅身体活動介入プログラムが、12週間後および24週間後に臨床症状および機能的能力の発達、ならびに家族介護者の負担に及ぼす影響について調査された。[262]。坐位の)対照群では日常生活能力の低下がみられたが、(身体活動の)介入群の患者は認知的に安定していた。実行機能と言語能力を分析した結果、介入群では意味語流暢性にかなりのプラスの効果が認められた。実際、身体活動的な患者は介入中に改善したが、対照群では悪化が続いた。その結果、介護者の負担は介入群では安定していたが、対照群では悪化した。
LOMが物語っているように、ADを効果的に予防または治療するには、身体運動だけでは不十分である。それにもかかわらず、ほとんどの研究ではLOMが無視され、さらにいくつかの研究では介入群の日常的な運動量が非常に少なかったため、研究者はAD予防のための運動を軽んじた。[263]。残念なことに、この種の研究は科学的標準(1つの変数だけを変更する)であるため、誤解を招く結果を招き、その普及は健康的なライフスタイルの重要性に関する一般の理解を損なう可能性がある。しかし、身体を動かすことは、満たされるべき自然な(そして進化の観点からは明白な)要件のひとつに過ぎない。もうひとつは、必須栄養素の安定供給である。
成人海馬神経新生への栄養効果
(成人の)神経新生とシナプス形成に不可欠な構成要素であり(総説は[264]を参照)、NRJにとっても重要な栄養素は数多くある。例えば、n-3系多価不飽和脂肪酸(PUFA)、フラボノイド、抗酸化物質が豊富なベリー類、赤ブドウやその他の果物に含まれるポリフェノールであるレスベラトロールは、すべて成人海馬神経新生を刺激することが示されている。[265]。加えて、これらの物質は酸化ストレスを軽減し、炎症プロセスをダウンレギュレートする(総説は[266]を参照);また、強力な抗アミロイド生成特性を持つものさえある。[267, 268]。
参考記事
対照的に、赤身肉や加工肉、動物性脂肪、精製された穀物、菓子類を多く摂取する典型的なWeDiは、必須栄養素が少なく、動物実験でシミュレーションしたところ、BDNFなどのニューロトロフィンの脳内濃度が著しく低下し、神経細胞の可塑性や学習が阻害された。[269]。このような現代的な食生活は、活性酸素レベルの上昇と神経炎症も引き起こし、成人海馬神経新生を阻害し、慢性的なコルチゾールの過剰分泌によってHPA軸を調節できなくなる。[270]。高度糖化最終生成物(AGEs)がADに関与しているという証拠もかなりある。[271]。砂糖、果糖、コーンシロップ、および一般的にグリセミック負荷の高い飲料を含む食品は、断続的な高血糖エピソードを引き起こす。これはAGEsの内因性生成の一因となる。加えて、食肉製品、特に工業的畜産による食肉は、大量の外因性AGEsを供給する(総説は[272]を参照)。AGE(加齢ではない)誘発性の神経炎症過程は、インターロイキン-1(IL-1) [273]、インターロイキン-6(IL-6) [274]、腫瘍壊死因子α(TNF-α) [275]などの炎症性サイトカインによって媒介され、神経変性過程を悪化させ、さらに生産的成人海馬神経新生を減少させることが示された。[276]。
AGEsによる慢性的な神経炎症の問題をさらに深刻にしているのは、オメガ6系(n-6系PUFAs)の摂取量が絶対量だけでなく、典型的なWeDiでは一般的に摂取量が少ないn-3系PUFAsとの相対量でも多いことである。[277]。一般的に、n-6系PUFAであるアラキドン酸(AA)に由来するケモカインは炎症促進作用があるが、例えばドコサヘキサエン酸(DHA)のようなn-3系PUFAに由来するケモカインは抗炎症作用がある。[278]。炎症促進作用と抗炎症作用は、それぞれ創傷の修復と治癒に不可欠であるが、両方のタイプのPUFAをバランスよく摂取する必要がある。しかし、工業的に生産される食品の進歩に伴い、n-6系PUFAとn-3系PUFAの比率は、近代以前の食事では1対1程度であったのが、現在のWeDiでは20対1までと、健康的な(そして「自然な」)比率へと劇的に変化している。[279]。このようになった理由の一つは、n-6系PUFA、すなわちリノール酸を多く含む植物油、例えばひまわり油(最大70%含有)やコーン油(最大60%含有)が広く使用されるようになったことである。これらの多価不飽和油の使用は、血清コレステロール低下作用があるとして食品業界によって宣伝されたが、ランダム化比較試験から得られた利用可能な証拠に基づく最近の研究では、食事中の飽和脂肪をリノール酸に置き換えると、血清コレステロールが30mg/dL(0.78mmol/L)減少するごとに、冠動脈性心疾患またはすべての原因による死亡リスクが22%増加することが示されている[280]。健康的な代替品としては、エクストラバージンオリーブオイル [281-283]、有機栽培されたキャノーラ油 [284]、亜麻仁油 [285]などを適度に使用することで、n-6系PUFAとn-3系PUFAの比率をより健康に保つことができる。

現代の食生活でn-6系PUFAが増加しているもう一つの理由は、肉類と高脂肪乳製品の大量消費にある。これらの製品が集約的な畜産に由来するという事実は、消費者の健康への悪影響を悪化させる。なぜなら、これらの動物の不自然な「生き方」が、製品中のn-6 PUFA対n-3 PUFA比に悪影響を及ぼすからである[286, 287]。その理由は、肥育のための餌、運動不足、養殖動物のストレスかもしれない。最後になるが、植物由来のn-3系PUFAからDHA(またはもう一つの生理学的に重要なn-3系PUFA産物であるエイコサペンタエン酸(EPA)への変換は、ヒトでは非常に非効率的な代謝プロセスであるため、WeDiにおける魚(動物性DHAとEPAの主要供給源)の消費量が少ないと、これら2つのn-3系PUFAの供給が絶対的に不足することになる。さらに、n-6系PUFAの摂取量が多いため、植物由来のn-3系PUFAからDHAとEPAへの代謝変換率がすでに低くなっていることがさらに損なわれている。[288]。その結果、n-6系PUFAとn-3系PUFAの深刻なアンバランスが生じ、早期老化、神経炎症プロセス、うつ病、ADを引き起こす。[289, 290]。
高濃度のDHAは脂質の過酸化を抑制し、[291]、EPAとともに脳の炎症と認知機能障害を軽減する。[292]。さらに、多くの異なる神経保護細胞メディエーターが、DHA、EPA、そしてもう一つの重要なn-3 PUFAであるドコサペンタエン酸(DPA)に由来している(総説は[293]を参照)。例えば、DHA由来のニューロプロテクチンD1(NPD1)は、細胞の完全性を恒常的に維持するためのシグナル伝達を誘導し、アポトーシス性および炎症性のシグナル伝達経路を不活性化する能力により、神経保護作用を示す。[294, 295]。さらに、NPD1は抗アミロイド活性が証明されたメディエーターであり、[296]、これはもともとDHAに起因するものであった。[297]。DHAはBDNF [298]とGDNF [299]をポジティブに制御することが示され、それによって成人海馬神経新生とシナプスの成長を高めるために相乗的に作用する。さらに、AAやDHAのようなPUFAは、すべての神経細胞組織にとって不可欠な構成要素であり、脳膜脂質のおよそ30%を占めている。[300]。これらを総合すると、DHAが欠乏した食事は神経新生を制限し、その供給は健全な脳の発達とその後の人生における神経可塑性の維持に不可欠である。[301]。このような観察を裏付ける最近の知見として、WeDiに典型的な、栄養密度の高い食品の摂取量が少ないことと、不健康な食品の摂取量が多いことは、それぞれ独立して、ヒトの左海馬体積の減少と関連していることが判明した。[302]。対照的に、n-3系PUFAであるDHAとEPAを混合したサプリメントを1日あたり約1g摂取すると、軽度の記憶障害を有する高齢者においてエピソード記憶の結果が有意に改善した。[303]。
参考記事
少なくとも、19世紀に始まったいわゆる「近代的な生活様式」が出現する以前の数十万年にわたる人類の進化において、人類はn-3系PUFAとn-6系PUFAを十分かつほぼ同量含む食事に慣れていたと考えるのが妥当であろう(総説は[297]を参照)。必須DHA(およびEPA)の安定的かつ継続的な供給源となる海洋食への変化が、刃物石器技術やその他の文化的進歩に代表されるように、人類の文化的発展の進歩とともに起こったように見えるのは、単なる偶然ではないかもしれない。[304]。実際、海洋食物網への安定したアクセスが、人類の進化の最終段階において重要な役割を果たしたに違いないという仮説も、化石の証拠から揺るぎない支持を得ている。[305]。これにより、植物由来のn-3 PUFAをDHAやEPAに効率的に変換する能力がなくても、ヒトは大きな脳を進化させることができたのかもしれない。したがって、ヒトは、DHAだけでなくEPAの有害な不足を引き起こす食習慣のごく最近の急激な変化には全く適応していない。
必須n-3系PUFAの不足が神経炎症と成人海馬神経新生の乱れを引き起こしている一方で(そのため、EPAとDHAを含むサプリメントが原発性うつ病に好影響を及ぼすことが示された。[306])、魚介類の摂取が認知症とADのリスクを低下させることを示す研究結果も現在までにかなり収束している。[307]。
しかし、すべての研究者がこれに同意しているわけではなく、患者の食事に魚を加えることによるAD予防の大きなブレークスルーは達成されていないため(試験のリストは[289]を参照)、より良い成功率を期待して、より長期的なヒト試験を主張する研究者が多い。[308]。とはいえ、どのコホートにおいても、DHAの治療(またはその他の成人海馬神経新生を損ねるような欠乏症)の恩恵を受ける可能性があるのは、実際にDHA(またはその他のそれぞれの欠乏症)が欠乏しており、上記のLOMによれば、生産的な成人海馬神経新生とNRJのためのその他の必須要件に欠乏症がない参加者だけであることに注意すべきである。したがって、たとえ介入試験を長く行ったとしても、付随する欠損をすべて是正しない限り、成功率は上がらないかもしれない。いずれにせよ、発表されたほとんどの症例において、DHA単独ではAD(または大うつ病)の予防や治療に効果がなかったからといって、十分なDHAの摂取が不可欠ではないと結論づけるべきでない。
しかし、一つの重要な疑問が残る: DHA(およびEPA)の安定的な世界供給はどこからなされるべきなのか?想定される世界的な消費ニーズを満たすために、世界の漁業の持続可能性に対する懸念が高まっている。天然魚や有機養殖魚の選択的消費に代わる経済的・生態学的選択肢は、有機栽培された微細藻類由来のDHAとEPAを豊富に含む食用油の供給を増やすことである[309]。そうすれば、魚に含まれるメチル水銀(MeHg)汚染の問題も回避できる(下記参照)。
地中海食(MeDi)

標準的なWeDiに代わる健康的な食事法はMeDiである。[310]。地中海沿岸諸国にはそれぞれ独自の料理があるため、MeDiと呼ばれる食事は人為的なものであることを認識しておく必要がある。とはいえ、この食事の主な側面には、オリーブ油、豆類、精製されていない穀類、果物、ナッツ類、野菜の消費量が比例して多いこと、魚の消費量が中程度から多いこと、乳製品の消費量が抑えられていること、赤ワインの消費量が控えめであること、魚以外の肉や魚以外の肉製品の消費量が少ないことなどが含まれる。[311]。最近の研究によると、MeDiを実践している高齢者は、WeDiを実践している高齢者と比較して、脳の萎縮が少なく、5年間の加齢と同様の効果があるという。この研究の著者が指摘しているように、特に魚の摂取量が多く、肉の摂取量が少ないことが、脳の構造に対するMeDiの利点に寄与した2つの重要な食品要素である可能性がある。別の研究では、このような食事介入がAD予防に一役買っているという証拠が示された。ここでは、MeDiの遵守度が低い認知障害のない人は、遵守度が高い人に比べて、臨床AD患者と同じ脳領域、すなわち、嗅内皮質、眼窩前頭皮質、下頭頂小葉、下側頭皮質および中側頭皮質、後帯状皮質で皮質の菲薄化がみられた[57]。興味深いことに、MeDiのアドヒアランスが高いApoE4被験者が、他のすべてのサブグループの中で最も高い効果を示した。これは、不健康な生活習慣のもとでは、ApoE4が主にAD過程を促進するという前述の役割と一致している。同様の予防効果が伝統的な日本食にも認められ(総説は[313]を参照)、このことは、上述したように、昔の日本におけるAD有病率の低さを少なくとも部分的には説明できるかもしれない。
重要なことは、MeDiの利点は、慢性疾患を予防する他の食事療法と同様に、その製品だけに起因するものではないということである。食品の加工技術や調理法にも強く影響されるのである。力学的および疫学的証拠から、食品加工が固有の植物化学物質の質に影響を与え、それがひいては炎症に関連する慢性疾患に対する食品の保護特性に影響を与えるという説得力のある証拠が得られている(総説は[272]を参照)。
参考記事
ビタミン
WeDiは一般的に重要な栄養素が少なく、ビタミンや必須微量元素の摂取量の減少につながる。LOMに従い、ビタミンA [314]、ビタミンB群 [315]、ビタミンB1 [316,317]、ビタミンB3 [318,319]、ビタミンB6、ビタミンB9、ビタミンB12(下記参照)、ビタミンC(包括的な総説は[320]を参照)、ビタミンD(下記および[321]を参照)、トコフェロールE [322]など、個々の欠乏は生産的成人海馬神経新生を阻害し、ADのリスクを高めることが示された。それぞれの欠乏症が成人海馬神経新生に与える影響や、その結果としてのうつ病やADのリスクについて詳述することは本総説の範囲を超えるため、ここでは2つの例のみを紹介する。とはいえ、AD予防(および治療)においては、それぞれの欠損に対処する必要がある。
例1
ビタミンB6、B9、B12のいずれかが栄養不足になると、L-メチオニンおよびL-システインアミノ酸代謝の中間産物であるホモシステインの蓄積が増加する。ホモシステインの血中濃度の上昇は、ADにおける認知機能低下の予測因子である。[323]。ホモシステインは、神経細胞の一酸化窒素合成酵素の活性化とフリーラジカルの形成を介して酸化ストレスを誘発し、[324]、神経細胞死を著しく増加させる。[325]。さらに、ホモシステインの上昇は成人海馬神経新生に有害であることが示され、[326]、細胞増殖に必要ないくつかのシグナル伝達経路を阻害することにより、神経細胞前駆体の増殖を阻害する。[327]。死後の神経病理学的所見とMRI所見に基づく最近の10年間の研究では、ホモシステインの上昇がAβの蓄積と脳の萎縮に関係していることが確認された。ホモシステインが最も高い四分位群では、内側側頭萎縮のオッズ比が3.78、脳室周囲白質増多のオッズ比が4.69であった。[328]。すべての関連は、一般的な血管危険因子を含むいくつかの潜在的交絡因子から独立していたため、ビタミンB6、B9、B12のいずれかの欠乏は、ホモシステイン上昇によるADの独立した危険因子とみなされるべきである。
しかし、ビタミンB群の補充によってホモシステイン値が低下すると、MCIにおける脳萎縮の加速速度が有意に遅くなる(プラセボ群では年間1.08%であったのが、積極的治療群では0.76%に低下した)[329]という事実にもかかわらず、LOMによれば、他の本質的な危険因子も除去されない限り、ADの完全な予防も治癒も期待できない。図1に概略を示したように、すべての必須因子を提供する予防・治療スキームのみが成功する可能性がある。したがって、潜在的な微量栄養素の欠乏を管理することは、AD予防・治療戦略の重要な一側面でしかない(総説は[313, 330]を参照)。
例2
ビタミンDの欠乏は、骨密度を低下させ、多くの一般的ながん。[331]のリスクを増加させるだけでなく、高齢者だけでなく若年者においても認知障害を引き起こすことが知られている。[332]。ビタミンD欠乏症の全体的な有病率は広範囲に及んでいる。例えば、最近の研究 [333]によると、米国では平均約41.6%で、黒人(82.1%)が最も高く、次いでヒスパニック系(69.2%)である。ビタミンDの欠乏は成人海馬神経新生の異常を引き起こす。[334]。最近のメタアナリシス解析の結果は、ビタミンDの低濃度がうつ病と関連するという仮説(成人海馬神経新生が生産的でない場合のセンチネル疾患としてとらえる)と一致していたが、例えば、ビタミンDの補充がうつ病の予防や治療に本当に有用かどうか [335]、認知機能低下の予防や治療に有用かどうか [336]を調べるために、より多くのランダム化比較試験を行うことが常に求められている。ビタミンDの欠乏が特定の神経成長因子やそれぞれのシグナル伝達経路をどのように調節不全に陥らせるかをよりよく理解することが科学的に強く求められていることは理解できるが、[337]、必須ビタミンが欠乏すると何がうまくいかなくなるのか、より科学的な理解を待っても、今まさにそのような欠乏に苦しみ、深刻な病気を発症しやすい人々の助けにはならない。さらに、繰り返しになるが、このような介入は、ベースライン時に十分な血清ビタミンDを有している試験対象者を助けるものでもない。[338]。第二に、上述したように、生産的な成人海馬神経新生に必要な、欠乏している他のすべての必須因子が同時に提供された場合にのみ、ビタミンD補給の恩恵を十分に受けることができる。このようなことはめったに行われないので、AD予防に関して弱い結果や否定的な結果が出たからといって、問題の欠損がADと無関係ではなく、是正されるべきだという誤った結論に至ってはならない。
実際、最近の研究結果から、ビタミンDの欠乏は全死因性認知症およびADのリスクを大幅に増加させることと強く関連していることが確認されている。[339]。逆に、食事からのビタミンD摂取量が多いほど、ADのリスクが有意に低下することが示された。[340]。さらに、最近の研究では、血清25-ヒドロキシビタミンD濃度が70nmol/Lの場合、心血管疾患死亡リスクが最も低く、ビタミンD濃度が不十分な場合(50nmol/L以上)と比較して、脳萎縮が2倍以上抑制されることが示された。私見では、これらの結果を総合すると、プライマリ・ケア医だけでなく政策立案者にも、すべての必須栄養素(ビタミンDもその一つ)の欠乏のスクリーニングを全国的な予防プログラムの一環として行うよう、早急に行動を起こすことが必要である。
参考記事
微量元素
ADの予防と治療における同じ論理が、例えばセレン、亜鉛、リチウムのような必須微量元素の欠乏にも当てはまる。
セレン依存性酵素(セレン酵素)は、体全体、特に神経系における酸化的損傷を予防し、回復させるのに必要だからである。[344]。血清セレンの最適範囲は85μg/L前後で、若年成人における抑うつ症状のリスク低下と関連していた。[345]。興味深いことに、一次神経幹細胞(NSCs)の分化を誘導すると、ミトコンドリア総数と活性酸素産生全体が直ちに増加することが最近示され、日常的な成体神経新生の結果として酸化ストレスが生成されることが示唆された。[346]。この代謝適応プロセスは、あらゆる種類の幹細胞系に共通しているようである。[347]。したがって、セレンが不足すると、成人海馬神経新生による活性酸素産生の増加に対する緩衝能が制限され、その結果、成人海馬神経新生が非生産的になり、うつ病やADのリスクが高まる可能性がある。
亜鉛は成人海馬神経新生に必要なもう一つの主要微量元素である。[348, 349]。大うつ病患者において、血清中の亜鉛濃度が低いことは、免疫系のマーカーの活性化の増加と相関しており、成人海馬神経新生の障害につながっていた。さらに、予備的な臨床研究では、抗うつ療法における亜鉛補給の有益性が実証された。前臨床研究では、亜鉛の抗うつ活性がネズミのうつ病モデルの大半で観察され、うつ様症状の誘発、神経新生と神経細胞生存の低下、あるいは学習・記憶能力の障害における亜鉛欠乏の原因的役割が明らかにされた(総説は[350]を参照)。加えて、亜鉛はアミロイド前駆体タンパク質(APP)の酵素的非アミロイド原性プロセシング経路とAβの酵素的分解に不可欠である。[351]。
リチウムはまだ一般的には微量元素として認識されていないが、いくつかの証拠から有力な候補として挙げられている。[352]。例えば、リチウムへの低用量長期暴露は、抗老化能力を発揮し、動物モデルにおいて死亡率を明確に低下させる。[353]。ヒトでは、疫学的研究により、他の精神疾患の中でも、飲料水中のリチウム濃度と気分、うつ病、自殺率との間に逆相関があることが示されている。[354]。慢性リチウム治療を受けた高齢の双極性障害患者(認知症のリスクが高い)とリチウム治療を受けなかった双極性障害患者を比較した研究では、治療群の有病率は年齢を比較可能な一般集団と同等であったのに対し、リチウム治療を受けなかった患者では認知症の発生率が6倍、すなわちそれぞれ5%対33%であったことが示された[356]。別の研究では、リチウムを投与すると、無投薬群と比較して両半球の海馬の容積が増加することが示され、この効果は平均約4週間の短期間の投与でも明らかであった。[357]。重要なことは、標準的な治療量だけでなく微量でもリチウムを摂取すると、認知症、自殺、その他の行動学的転帰のリスクが低下することであり、これらの病理学的プロセスの主要な調節因子に対する薬理学的干渉が示唆されている。[358]。つまり、リチウムは重要な細胞シグナル伝達経路を自然に制御しており、食事中のリチウムの不足は、それゆえ疾患リスクの増加を引き起こす可能性がある。
参考記事

リチウムが2つのキナーゼGSK-3αとGSK-3βの活性を負に調節することが示されており、低用量リチウム治療の効果の相対的特異性と感受性の両方を説明できるかもしれない(下記参照)[359]。オリゴマーAβによるGSK-3βの活性化は、神経炎症、タウのリン酸化、成人海馬神経新生の障害 [360]を促進することから、リチウムによるGSK-3の阻害は、生体内試験でタウオパチーと神経変性を抑制する。[361]。同様に、リチウム投与はADモデルマウスにおいて成人海馬神経新生、神経病理、認知機能を改善することが示された。[362]。さらに、生後2カ月からリチウムを投与した「ADマウス」は、非投与のトランスジェニックマウスと比較して、老人斑の数が減少し、大脳皮質と海馬における神経細胞の減少がみられず、BDNFレベルが上昇した[363]。このような効果を得るためには、双極性障害における標準的な高用量治療の約1ミリリットルのリチウムを投与すれば十分であった。したがって、この研究の著者らは、アルツハイマー病の予防と治療における(事実上副作用のない)微量リチウムの使用を支持するデータであると考えている。
実際、リチウムの長期投与は、無作為化比較試験において、無記銘MCIに対する疾患修飾作用の予備的証拠をすでに示しており、そこでは、リチウム投与群ではMCIからADへの転換が少なかった。[364]. リチウム投与は、高リン酸化タウの脳脊髄液濃度の有意な低下と、アルツハイマー病評価尺度(Alzheimer’s Disease Assessment Scale)の認知下位尺度および注意課題の成績向上と関連していた。ADがさらに進行した段階では、300μgのリチウムを1日1回投与するマイクロドーズ治療が、15カ月間の完全評価期間中にアルツハイマー病患者を安定させた。[365]。例えば、治療群ではミニメンタルステート検査(MMSE)の成績の低下は認められなかったが、対照群では同期間中に低い得点が観察され、有意差は3カ月後に生じ、徐々に増加した。
重要なことは、リチウムは効率的な成人海馬神経新生に必要であり、AD特異的な病理学的プロセスを阻害する以外に、細胞を若返らせるオートファジーにも影響を与えることである。リチウムはイノシトールモノホスファターゼ(IMPase)の活性を阻害し、ミオ-1, 4, 5-トリリン酸(IP3)の減少をもたらすことがわかった。このIP3活性の低下により、mTORとは無関係にオートファジーが誘導される。[366]。この文脈で、塩化リチウムが線虫Caenorhabditis elegansの寿命を延ばし、[367]、おそらくミトコンドリアの若返りによって、[368]、リチウムが進化的に高度に保存されたメカニズムでその効果を発揮することを示唆していることに注意することは重要である。日本における大規模な疫学調査では、リチウム濃度が同程度に低い飲料水の摂取が、全死因死亡率と逆相関していることが判明している。[353]。したがって、リチウムの不足は、加齢や虚弱(死亡率を増加させる)とオートファジーの障害、そしてADと成人海馬神経新生の障害とを結びつけており、この神経変性疾患におけるもう一つの重要かつ修正可能な危険因子を示しているのかもしれない[369]。地域によっては、地元の水道水を飲んだり、市販のミネラル豊富な湧水を飲んだりすることで、微量のリチウムを摂取することができる。したがって、このような水を1日にコップ1~2杯飲むことによるリチウムの微量摂取は、潜在的な治療価値があるだけでなく(下記参照)、重要な新規微量元素の摂取不足を減らすだけで、安全な予防策にもなる。

脳の活性化、成人海馬神経新生、神経細胞の若返りにおける断続的絶食の重要性
狩猟採集は危険な事業である。それゆえ、安全な生活圏を離れて野生に行くことは、胃袋が満たされている限り、私たちの祖先は確実に避けてきた。その結果、空腹時でも脳と身体がうまく機能するようなメカニズムが進化したに違いない。特に海馬のエピソード記憶機能は重要で、どこに食べ物があり、どこに捕食者が潜んでいるかをよく覚えておくことが、生存、ひいては繁殖成功の鍵だったからだ。断続的絶食と海馬の成長をリンクさせることは、我々の祖先が頼りにしていたメカニズムである(下記参照)。しかし、ヒト科動物の進化の歴史の大部分において、食物を獲得する必要性が日々の大きな課題であったのに対し、現代社会の人々にとっては、常に食物が供給されることが当たり前になった。[66]。その結果、実験動物にも日常的に自由食パターンが適用されるようになった。例えば、標準的な飼育環境で自由食を与えているトランスジェニックADモデルは、AD様症状を発症し、高カロリー食は記憶障害を加速させることさえある。[370]。対照的に、このようなマウスをより自然な断続的絶食食で飼育すると、野生型マウスだけでなく、認知機能や脳構造も改善した。[371]。ヒトのAPPswe、PS1M146V、tauP301L変異を発現するADのトリプルトランスジェニックマウスモデルでも、断続的絶食を1年間続けると認知機能の低下が抑制され、[42]、加齢そのものではなく、むしろ長期の摂食パターンが記憶に長期的な影響を及ぼすという証拠が得られた。[43]。興味深いことに、断続的絶食のポジティブな効果は、Aβやタウの病理学的変化とは無関係であった。これは、断続的絶食が、例えばタンパク質シャペロンや抗酸化酵素の活性化、神経炎症の抑制 [372-374]、あるいはNRJの活性化 [375]など、別のメカニズムによって脳細胞を保護し、それによって再生、認知能力の向上、健康寿命の延長を促進することを示唆している。[376]。さらに、「空腹ホルモン」であるアシル-グレリンは、歯状回ニューロンにおける神経原性転写因子Egr-1のレベルを増加させることによって、神経原性シグナル伝達経路を活性化することが発見された。[377]。断続的絶食はまた、海馬においてFGF2とBDNFの発現を誘導し、[378]、新しく生成されたニューロンの生存を促進し、成人海馬神経新生の増強に寄与する。[379]。BDNFはまた、興奮毒性、エネルギー欠乏、酸化ストレスから歯状回ニューロンを保護する。[380, 381]。
ADのもう一つの特徴は、側頭葉における神経細胞のインスリン抵抗性の増大である。インスリン抵抗性の海馬神経細胞がグルコースを取り込めないことも、ADの進行の原動力である。[382]。動物実験における。「標準的な飼育環境」と疫学研究で観察された西洋的ライフスタイルの「正常さ」のために、神経細胞のインスリン抵抗性の有害な状態は、加齢の主な結果とみなされるADと同様であり、座りがちなライフスタイル、肥満、WeDi、高コルチゾール分泌、産生増加(成人海馬神経新生障害)と除去障害による有毒なAβレベルの蓄積の結果とみなされることはまれである。逆に、実験的に断続的絶食のような食事パターンに変更し、日常的な身体活動を増やすと、インスリン感受性、脂肪酸の動員、ケトン体の産生が増加する(これはADにおける海馬ニューロンのエネルギー飢餓を回避する)。重要なことは、遺伝的プログラムを利用したこのようなライフスタイルの適応は、AD予防に重要であるばかりでなく、多くの慢性疾患の予防・治療プログラムにも不可欠な部分であるということである。[383]。
興味深いことに、ケトン体D-β-ヒドロキシ酪酸は、断続的絶食や運動中に肝臓から脳などの末梢組織へエネルギーを運ぶだけでなく、D-β-ヒドロキシ酪酸は重要なシグナル伝達代謝物でもあることが判明した。[91]。例えば、D-β-ヒドロキシ酪酸はGPR109Aとしても知られる脂肪細胞受容体HCAR2(ヒドロキシカルボン酸受容体2)を介して作用し、ナイアシン(ビタミンB3)が成人海馬神経新生を誘導することで知られるアディポネクチン[318]の分泌を刺激する[247]。D-β-ヒドロキシ酪酸はまた、HCAR2を活性化することで、それ自身の産生を負に制御し、それによってケトアシドーシスを予防し、脂肪貯蔵の効率的な利用を促進する。さらに、D-β-ヒドロキシ酪酸は、マウスやヒトの交感神経節に最も多く発現しているGPR41としても知られるFFAR3(遊離脂肪酸受容体3)に拮抗することにより、交感神経系(絶食時のエネルギー節約につながる)を抑制する[384]。
さらにD-β-ヒドロキシ酪酸は、ヒストン脱アセチル化酵素(HDAC)を阻害することで、エピジェネティックな再プログラミングを介して、例えばオートファジーやストレス応答経路のような、細胞内再生や細胞生存をサポートする遺伝子の発現を制御し、神経保護や長寿につながる。これらの遺伝子プログラムは高度に保存されている。例えば、D-β-ヒドロキシ酪酸はHDACを阻害することで、保存されたストレス応答経路の活性化、耐熱性の向上、グルコース毒性の低下、ADに関してはオリゴマーAβの毒性の遅延など、多くのメカニズムによって線虫の寿命を延ばす。[385]。同様に、D-β-ヒドロキシ酪酸は海馬培養を低酸素症やグルタミン酸興奮毒性 [386]、Aβ毒性から保護する(総説は[387, 388]を参照)。さらに、D-β-ヒドロキシ酪酸は神経炎症 [389]に対抗し、神経細胞のインスリン抵抗性の機序としてインスリンの糖化(インスリン-AGEs)から保護し、抗脂質過酸化作用によってミクログリアの生存率を高める。[390]。D-β-ヒドロキシ酪酸はまた、オートファジーを刺激し、培養皮質ニューロンにおけるグルコース欠乏によって誘発される神経変性を予防する。[391]。さらに、低酸素誘導性因子-1α(低酸素誘導性血管新生に関連する主要成分であり、神経保護反応の制御因子)のD-β-ヒドロキシ酪酸誘導性アップレギュレーションは、脳卒中モデルにおける神経細胞損失の減少につながる[392]。また、上述したように、広範な運動や断続的絶食によってケトジェネシスを増加させると、PGC-1αが活性化され、mTORが抑制され、その結果、少なくとも神経細胞レベルでは長寿となる。
この文脈で、断続的絶食の健康上の利点は、例えばCCRのような全体的なカロリー摂取量の減少を必要としないことに注意することが重要である。例えば、1日8時間しか食事を与えないマウス(すなわち、毎日16時間断続的絶食を行うマウス)は、全体的なカロリー摂取量を変えることなく、高脂肪食誘発性肥満に抵抗性がある。[393]。ケトーシスが1日単位で長ければ長いほど、健康上の結果は良好であり、朝食は1日のうちで最も重要な食事であるという広く信じられている信念に疑問を投げかけるものでさえある。[394]。実際、肥満やADのような合併症のリスクを減少させるのに十分なケト新生を可能にするのに十分な食間の間隔がある限り、朝食を抜くことは遅めの食事を除くのと同じくらい代謝的に有益であると仮定することは合理的である。[395]。
まとめ
まとめると、内因性シグナル伝達代謝産物D-β-ヒドロキシ酪酸は、神経細胞のインスリン抵抗性の条件下で脳に燃料を供給するだけでなく、遺伝的・エピジェネティックなプログラムを有利に変化させることによって、記憶機能を改善する。これは、進化の過程で培われた長期的な脳の健康の原則が、今日でも有効であることを示している。[396]。断続的絶食や長時間の運動に加えて、食後のインスリン分泌につながるグリセミック指数の高い食品の摂取を控えることで、脳を保護するケトジェネシスが日中も活性化する。さらに、食事中にケトジェネシスを増加させる自然な可能性さえ存在する。
絶食を伴わないケトジェニック脳内燃料補給
中鎖脂肪酸を60%まで含むバージンココナッツオイルのような中鎖脂肪酸(中鎖脂肪酸)を摂取すると、ケトン体の産生が上昇する。摂取された長鎖脂肪酸とは対照的に、中鎖脂肪酸は水溶性であるため、門脈を経由して肝臓に直接運ばれ、そこで効率的にケトン体に変換される。[399]。そして実際に、中鎖脂肪酸の急性投与は、AD患者の記憶能力をいくらか改善するようであり、[400, 401]、記憶改善の程度は、生成されたD-β-ヒドロキシ酪酸の血漿レベルと正の相関があることが判明した。しかし、その効果は、病期があまり進行していない場合にのみ観察された。[402]。さらに、これらの研究すべてにおいて、全体的な受益効果の弱さに加えて、ApoE4保有者ではより顕著ではなかった。これらの観察結果はいずれも、D-β-ヒドロキシ酪酸レベルを増加させるために中鎖脂肪酸を薬剤のように使用しても、断続的絶食や長時間の身体活動によって生じる可能性のある健康上の利益をすべて完全に提供できるわけではないことを示しているのかもしれない。したがって、海馬の代謝、成人海馬神経新生、神経細胞の若返りにケトジェネシスが重要であるとしても、単にケトジェニック中鎖脂肪酸を食事に加えるだけでは、ADの予防や治療には不十分である。それにもかかわらず、研究者の中には、ケトジェニックなエネルギー代謝の活性化だけで、ADの完全な統一理論を基づかせようとする傾向がある。[403]。
様々な脳領域における遺伝子発現に対するエネルギー制限と身体運動の効果を比較すると、これら2つの異なる環境因子は、脳細胞の可塑性に対して重複する効果を持っているが、同時に異なる効果も持っているという考え方が支持されている。[66]: ランニング運動は主に神経幹細胞の増殖を促進し、二次的に新しく生成されたニューロンの生存率を高めるが、 断続的絶食は基礎幹細胞の増殖を支援することに加え、何よりもまず、 新しく生成されたニューロンの生存率を高める。[379]。この点から、運動とエネルギー制限が成人海馬神経新生 [404]と神経可塑性に相加的な効果を示すことは明らか: 走るホイール運動と毎日のエネルギー制限の両方が、走るだけのマウスやエネルギー制限だけのマウスと比較して、海馬歯状回ニューロンの樹状突起スパイン密度を増加させた。[405]。進化の観点からすると、これらの知見は、断続的絶食と採食によって脳の神経回路が構造的・機能的に改善され、生存に有利になるという考え方と一致している。このような生理学的効果は、ヒトへの介入研究が一つの側面(ココナッツオイルや中鎖脂肪酸だけを提供したり、ライフスタイルの選択として断続的絶食だけを提唱したりするような)にしか焦点を当てない場合に、明確な効果を示せない理由を説明するものである。また、ほとんどの人が日常的に大量の単糖類と不健康な脂肪を摂取しているWeDiが、なぜ慢性的な病気になるのかも説明できる。このような条件下では、多くの人々が栄養価の高い食品を奪われるだけでなく、効率的で健康を保護するケトジェネシスもほとんど起こらない。[406, 407]。
これらを総合すると、断続的絶食と身体運動の重要性に加えて、食事からの中鎖脂肪酸摂取は、非絶食状態におけるケトン体の魅力的な(間接的な)供給源となりうる。中鎖脂肪酸の健康的な供給源としてのココナッツオイルの摂取は、神経細胞のインスリン抵抗性、神経炎症、Aβ-毒性に対してさらなるプラスの効果をもたらし、HDLを増加させることによってLDL/HDL-コレステロール指数を改善し、ADの他のいくつかの主要な進行因子を改善する。[408]。したがって、ヴァージンココナッツオイルの使用は、炒め物や焼き物用の多価不飽和油やバターの安全で健康的な代替品として、またADの予防と治療のための包括的戦略の重要な一部として、強く推奨される。
体組成と内臓脂肪
一晩で12時間以上の断続的絶食、栄養豊富で低血糖の食事、定期的な運動を含むライフスタイルは、ほぼ必然的に、痩せ型で筋肉質という、採食の祖先に近い体組成になる。残念なことに、肥満と「加齢に伴う」筋肉の減少が健康上の大きな問題となっている。脂肪細胞は内分泌機能を持つアディポカイン(筋肉細胞のミオカインのようなもの)を産生し、脳を含む本質的にすべての臓器の生理機能に影響を与えるため、この傾向は脳機能にも悪影響を及ぼす。[410]。内臓脂肪の不足と増加は、いずれもこれらのアディポカインの不健康な調節につながる。特に、内臓脂肪型肥満の蔓延は、慢性炎症 [411]、インスリン抵抗性 [412]、高血圧 [413]、高コレステロール血症 [414]、動脈硬化 [415]、そして最終的にはメタボリックシンドローム(多かれ少なかれ、上記で引用したADリスク因子のすべてからなる状態)の発症など、ADのいくつかの疾患リスクの上昇を引き起こす。[416-418]。
ある種のアディポカインの調節異常は、提唱されているADの中心的メカニズム、すなわち成人海馬神経新生にも直接的な影響を及ぼす。例えば、レプチンは食物の摂取とエネルギー代謝を視床下部で調節することでよく知られているアディポカインである。しかし、レプチンは成人海馬神経新生、軸索成長、シナプス形成、樹状突起形態、ニューロン興奮性、神経保護、さらにはAβレベルの調節にも顕著な影響を及ぼす。[419]。こうした多面的な作用により、レプチン投与はADのトランスジェニックマウスモデルにおいて、脳の病理を軽減し、記憶を改善することが示された。[420]。逆に、肥満によるレプチンの過剰発現は、レプチン抵抗性を引き起こし、それゆえ成人海馬神経新生とうつ病を障害する。[421]。肥満と糖尿病のマウスモデルでは、レプチン抵抗性がADに関連したタウ病態を促進することが示された。[422]。逆に、成人海馬神経新生の顕著な活性化因子である神経保護作用のあるアディポネクチンは、中枢性肥満によって発現が低下する。[423]。
これらを総合すると、アディポカインは主要な脳機能に直接的、間接的に影響を及ぼすため、中年期の中心性肥満がADの原因的危険因子の大きなリストに加わることになる[424]および[425]。したがって、ADの原因となる予防戦略は、身体活動、少なくとも12時間のオーバーナイト断続的絶食、健康的な食事(MeDiなど)を奨励することにより、内臓脂肪蓄積を減少させることを目指すべきである。
社会参加と生きがい
脳は、生存と繁殖の成功の可能性を高めるために環境的な課題を克服するという1つの中心的な機能を持っている。特に成人海馬神経新生は、必要不可欠な人生経験を生涯にわたって収集することを可能にするので重要である。しかし、認知能力は認知的挑戦によってのみ向上する。新しい経験がなければ、生まれたばかりの歯状回-ニューロンは既存の歯状回-ネットワークと相互接続しない。シナプスが機能しなければ、ニューロンはアポトーシスを起こす。[426]。したがって、生産的な成人海馬神経新生が起こるためには、人間の脳は高年齢になっても記憶に残る活動を必要とし続ける。人間のように生存が社会的支援に依存する存在にとって、社会的活動は重要性が高く、したがって感情的影響も大きいため、記憶に残る最高の経験も生まれる。つまり、精神的健康は社会的相互作用に依存しているのである。老後の引退は人類の長い人生の歴史の一部ではなかったし、祖父母が家族のサポートにあまり関わらない(あるいは関わらない)ような緊密な家族の絆の崩壊につながる現代の個人主義も、ますます頻繁になっている。生きがい尺度のスコアが低い人は、スコアが高い人に比べてADのリスクが2.4倍高いことが判明している。[427]。
参考記事
ヒトを対象とした研究における相関関係は、先験的に因果関係とは一致せず、時には逆の因果関係を反映することさえあるため、動物モデルは、異なる因果関係の可能性を区別するために重要であり、因果関係のある相関関係がある場合には、その根底にある分子メカニズムを同定するのにも役立つ。動物実験では、複雑で困難な環境の経験が脳の可塑性を高め、構造と機能の両方を改善することが示された。環境エンリッチメント(EE)の効果は、神経病理学的条件下で特に重要: 複雑な(自然)環境での経験は、家族性ADに関連するトランスジーンを保有するトランスジェニックマウスの成人海馬神経新生障害を回復させる。[428]。海馬細胞の生産的増殖は、EE後に有意に改善した。さらに、EEは海馬のLTPを有意に増強した。さらに、成人海馬神経新生の増強は、環境的に豊かになったマウスの海馬と大脳皮質におけるp-tauとオリゴマーAβのレベルの有意な減少を伴っていた。ADの別の動物モデルでは、社会的相互作用がBDNF依存性の成人海馬神経新生を増加させることにより、記憶障害を回復させた。[429]。また、長期間の環境エンリッチメントは、シナプト毒性AβオリゴマーによるLTP阻害を防ぐことさえわかった。[153]。これらの実験から、環境調節がAD脳の障害された表現型を救うことができるかもしれず、社会的・認知的挑戦を増やすことによって脳の可塑性を誘導することが(初期のADと診断された患者にしばしば起こるような減少ではなく)、貴重な治療・予防手段になるかもしれないと結論づけることができる。実際、ヒトを対象とした研究では、米国の代表的な高齢者サンプルにおいて、ベースラインの社会的統合度が高いほど記憶力の低下が遅いことが予測された。[430]。最も統合度の低い患者の記憶力は、最も統合度の高い患者と比較して2倍の速度で低下した。さらに、これらの研究群では、認知機能の低下が社会的相互作用の低下を引き起こすという逆の因果関係を示す。証拠はなかった。
一方、系統的な認知トレーニングやそれぞれのコンピュータを用いたプログラムは、認知機能の長期的な改善や日常生活動作の困難さの軽減をもたらすが、これらのような活動は社会的(感情的)学習をシミュレートするものではないため、成人海馬神経新生の改善、すなわち新生歯状回ニューロンの生存率の改善にはつながらない可能性がある。コンピュータを利用したメンタルトレーニングは、訓練されたスキルには利点があるが、[431]、認知トレーニングが認知症の発症率の低下と関連することが見出されなかったのは当然である。[432]。進化的な観点から見ると、生産的な成人海馬神経新生に基づいて、高年齢まで精神的に健康であり続ける人間の能力は、他の活動を必要とする。特に、祖母仮説に即して言えば、他者(あるいは少なくとも親族関係)のためになるような関心や行動を示すことによって、生成的であること、また生成的であると感じることは、中年期以降の非常に重要な発達目標である。そこで最近の研究では、エクスペリエンス・コープス(EC)と名付けられた世代間市民参加プログラムに参加することが、高齢者の生成性に対する自己認識に有益かどうかを検討した(上述のように、生成性とは心理社会的な意味であり、次世代を確立し導くことへの関心を指す)。介入群に無作為に割り付けられた参加者は、ボルチモアの公立学校システム内にボランティアとして2年間配置された。この結果は、世代を超えた市民参加プログラムへの参加が、高齢期における世代性の自己認知を肯定的に変化させるだけでなく、脳の萎縮を防ぐという、史上初の大規模な実験的実証となった[433]。特に、ECプログラムの対照群の男性では海馬の減少が2年間続いたのに対し、社会的活動群の男性では脳の容積が0.7~1.6%増加した。[434]。これらの知見は、目的をもった社会的活動は、高齢者によくみられる認知症になりやすい部位の脳容積の減少を止めるだけでなく、逆に減少させることさえあることを示している。
対照的に、本人が意図しない、あるいは強制された社会的孤立はストレスとなり、成人海馬神経新生を混乱させることが、最近マーモセットサルで示された[435]。さらに、生後早期の心的外傷は、HPA軸の調節不全を引き起こし、後年、不安様行動などの有害な結果をもたらし、成人海馬神経新生を障害し続ける。動物モデルでは、海馬ニューロンのコルチコステロイド受容体(CR)のエピジェネティックなダウンレギュレーションにより、循環コルチコステロイドレベルの感知が阻害され、長期的なHPA軸の調節不全につながる可能性が示された。[436]。同じ原理がヒトでも働くという証拠がある: GR遺伝子のプロモーター領域におけるDNAメチル化レベルは、小児期または若年期に経験したストレスフルなライフイベント(両親の離婚、重病など)の数と累積的に正の相関があることが示された。[437]。コルチゾールの感知が低下すると、HPA軸の負のフィードバック調節が妨げられるため、大うつ病やADを発症するリスクが高まる。例えば、小児期の虐待はHPAストレス反応を変化させ、自殺のリスクを増加させるが、これはニューロン特異的GRプロモーターのエピジェネティックな差異と密接に関連していることが判明した。[438]。これに対応して、職場での慢性的な問題や慢性的な障害性疾患、離婚、近親者の死など、慢性的に苦痛を伴う生活は、海馬の大きさと逆相関することが示され、これはうつ病患者の認知障害と関連していた。[439]。最近行われた38年間の縦断的集団研究によると、中年女性におけるこのような心理社会的ストレス因子は、その後の人生におけるADのリスクを増加させるようである。[440]。しかし、ストレスフルな出来事を経験したすべての人がうつ病になったり、ADを発症したりするわけではない。実際、研究データのより詳細な追跡分析で明らかになったように、心理的回復力があり、そのような苦痛な出来事に対処している人は、後年ADを発症しにくい。研究者らは、中年期における「神経質」(不安、恐怖、不機嫌、心配、妬み、不満、嫉妬、孤独を特徴とする)の程度が高いほど、長年の苦痛やAD認知症リスクの増加と関連することを発見した。特に、「神経質」の得点が高く、外向性の得点が低いパーソナリティは、AD認知症のリスクが最も高いことを示した。[441]。「神経質」と「内向性」の両特徴は、HPA軸の調節障害と関連している。[442, 443]。この集団研究の著者らは、中年期の「神経質」はAD認知症リスクの増加と関連しており、苦痛がこの関連を媒介すると結論づけている。したがって、ストレスフルな出来事ではなく、それらのストレス要因に対する反応が長期的に精神的健康に影響を及ぼすのである。どちらの場合も、生産的な成人海馬神経新生を再活性化することによって心理的回復力を高めることが第一目標でなければならない。
必要とされるライフスタイルの変化は、認知機能低下のリスクを軽減するが、少なくとも最初のうちは、一般的にストレスの多いものとみなされる。しかし、それらはまた、やりがいのある経験(より多くの身体的・精神的フィットネス、より多くの社会的活動)をもたらし、それによって歯状回の前駆細胞をストレスホルモンの上昇による悪影響から緩衝する。[254]。特にオキシトシンは、コルチゾール濃度が高いストレス状況下でも成人海馬神経新生の強力な活性化因子であり、[444]、ストレス因子に対する行動的・生理的反応を正しく調節することが判明している。[445]。ヒトでは、オキシトシンは社会的報酬の条件下で海馬で放出される。[446]。オキシトシン以外にも、特に海馬の神経原性「ニッチ」におけるニューロペプチドY(NPY)の活性が、ストレス因子に対する行動の回復力と関連しており、[447]、成人海馬神経新生に対する特定のストレス因子/報酬体験のプラスの効果を媒介する。[448]。興味深いことに、心的外傷後ストレスの動物モデルにおいて、NPY-ergic系がEEによるストレス対処反応と不安軽減の主要なメディエーターであることが最近判明した。[449]。ヒト以外の霊長類の成体では、ストレスコーピングが成人海馬神経新生を刺激することから [450]、本総説で概説されているように、成人海馬神経新生促進行動と組み合わせてストレスコーピングを促進するようにデザインされた心理療法は、ヒトにおいても同様の有益な効果をもたらすはずである。[451]。特に、大うつ病やADを発症する高リスク群に属する被験者は、このような介入から利益を得るはずである。結果として、ヨガやマインドフルネスに基づくストレス軽減プログラム [452]や、過去のストレスフルな人生経験を克服して心理的回復力を高める心理療法は、ADの予防・治療戦略の一部となるべきである。[453]。
睡眠の重要性
睡眠不足は海馬の学習と記憶に害を及ぼす。上で概説したように、成人海馬神経新生は睡眠不足の結果に特に敏感である。[454]。特に、慢性不眠症では海馬亜野の萎縮が観察され、睡眠の断片化は成人海馬神経新生の減少だけでなく、アンモニア角(CA)亜野の神経細胞喪失を引き起こし、慢性的な睡眠障害のある患者は特に認知障害を起こしやすくなる。[455]。
睡眠障害がADリスクを増大させる様々なメカニズムに加えて [456]、途切れることのない睡眠不足は、脳の健康に対するメラトニンの働きを妨げる。メラトニンは、松果体から日没時に分泌され、深い眠りを組織化する。メラトニンは神経炎症を強力に抑制することがわかった。[457]。さらに、新しく発生した歯状回ニューロンの樹状突起の成熟を促進し、その生存をサポートする。[458]。このような神経原性ホルモン作用は、GHやBDNFのような睡眠中にアップレギュレートされるホルモンメディエーターによって支えられている。さらに、生産的な成人海馬神経新生にとって重要なことだが、深い睡眠中は、IL-1、IL-6、TNF-αのような抗神経原性免疫メディエーターの発現だけでなく、コルチゾールの分泌も減少している。重要なことは、覚醒時に上昇するAβ濃度が睡眠中に低下することである。このAβの日周パターンが睡眠不足によって損なわれると、有毒なAβの蓄積とオリゴマー化が起こる。[459]。興味深いことに、ADの前臨床段階においてさえもAβの沈着は睡眠の質の低下を引き起こし、[460]、全身的な治療アプローチにおいて対処すべき悪循環をもたらす。
最近、BBBの破壊は、それ自体が老化の結果であることが示唆された(神経炎症、2型糖尿病、ADと同様)。その原因は、海馬におけるオリゴマーAβの蓄積であることが判明した。[11, 461]。それゆえ、BBBの機能不全はADの原因であり、結果でもあると考えられている。[462]。しかし、議論されているように、自然な状態、特に十分なレム睡眠がある状態では、脳はこのような老化の影響と考えられるものから保護されている。実際、一般的に睡眠を制限すると [463]、特にレム睡眠を制限すると、BBBの伝染性が増加することがわかったが、短時間の深い睡眠でも、BBB機能の深刻な変化を迅速かつ効果的に回復させることがわかった。[464]。これらの観察結果は、レム睡眠がBBBの物理的なバリア特性を制御しているだけでなく、Aβ蓄積もBBB伝染性も加齢の必然的な結果ではない可能性を示唆している。
参考記事
この文脈では、睡眠障害は外傷性脳損傷の最も一般的な結果でもあり、プロのNFプレーヤーの脳震盪後のADリスク上昇を説明できるかもしれない(上述)。睡眠が頻繁に中断される状態が慢性化すると、その原因が何であれ、細胞修復の低下と神経炎症、BBB機能の低下、神経可塑性の変化、神経変性、成人海馬神経新生と海馬容積の減少、海馬の可塑性と機能の低下を引き起こす。[142, 465]。したがって、睡眠不足と日中の眠気の増加は、人口統計学的要因や臨床的要因とは無関係に、認知症の危険因子である。[466]。
これらを総合すると、睡眠の質を改善することは、認知機能低下の予防や治療を目的とした治療法において重要な要素である。正常な睡眠サイクルを取り戻すための治療法としては、睡眠衛生の改善(www.ncbi.nlm.nih.gov/pubmedhealth/PMH0072504)、マインドフルネストレーニング [467]、認知行動療法(特に精神的外傷体験を克服する必要がある場合)などが考えられる。メラトニン [468]、類似体 [469]、またはその前駆体であるトリプトファン(セロトニンの前駆体でもあり、ビタミンB3のプロビタミンでもある)などの薬物療法は、行動上の原因が特定され除去されるまでの急性の状況において役立つ可能性がある。
参考記事
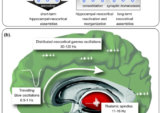

生涯を通じた脳の健康の複雑なメカニズム
祖母仮説から予測されるように、ヒトの繁殖戦略(と遺伝的選択)は、次の世代の生存確率を高める経験を生涯にわたって獲得することに依存している。図1に描かれているように、また前文で概説したような進化論的観点から見ると、ヒトの遺伝子プログラムは前近代的なライフスタイルに絶妙に適応し、経験豊かな高齢者の世代を超えた世代交代に必要な重要な条件、すなわち高年齢までの精神的健康を提供するように最適化されているように見える。ビタミン、必須脂肪酸、微量元素を毎日摂取すること、十分な身体活動、断続的絶食、高年齢まで目的を持った豊かな社会生活、そして再生のための十分な時間(睡眠)が、人類の進化の大部分において、環境と行動の「正常性」を特徴づけていた可能性が高い。例えば、十分なビタミンCが常に食事に含まれていたため、私たちの祖先は、エネルギーコストのかかるビタミンC産生装置をコードする遺伝子を維持する必要がなかった。これらすべての異種因子が常に利用可能であることに依存する重要な生理機能のひとつが成人海馬神経新生である。しかしLOMによれば、成人海馬神経新生は他の生物学的成長過程と同様、すべての必須要件が満たされた場合にのみ生産性を発揮する。成体で生まれた歯状回-ニューロンは、新規性を登録し、それを以前の経験と比較することによって、HPA軸の活動を調節する。したがって、生産的な成人海馬神経新生は、新しい挑戦(新規性追求)への関心を高め、心理的回復力を高めることにつながるかもしれない。これにより、神経細胞の成熟と脳の長期的な健康の前提条件である、新しい経験をする可能性が高まる(総説は[470]を参照)。図1に示すようなこの自然なプロセスを、私は「精神成長の好循環」と呼んでいる。生産的な成人海馬神経新生に必要な行動因子や環境因子のほとんどは、例えばインスリン感受性、血圧や微小循環、免疫機能やBBB機能、細胞の若返りなど、生体の他の重要な生理機能にも影響を与え、それによって間接的に高年齢までの神経細胞の生存に影響を与える。これらの行動因子はすべて、Aβのホメオスタシスを最適化する。覚醒時にAβが産生され、シナプスと神経保護濃度が確保され、その後、海馬の学習によって新しい経験を効率的に獲得するために、睡眠中にAβが分解される。これらの複雑な環境的、社会的、行動的、生理的、分子的プロセスを総合すると、私たちの遺伝的プログラムの指導の下で、生涯にわたって心の健康を維持する必要があるようだ。
ADは欠乏によって引き起こされ、環境毒素によって加速される
これとは対照的に、図2(ADにつながるすべての主要な危険因子と欠乏を含む)に示すように、また各個人の生活スキームにもよるが、1つまたはいくつかの質的欠乏がさまざまな量で組み合わさって、成人海馬神経新生による新しい歯状回ニューロンの効率的な生成、成熟、統合を制限している。上述したように、また図2に描かれているように、成人海馬神経新生が効果的でないと記憶容量が制限され、「抑うつの悪循環」が始まる。提案されたモデルは、もともと海馬の縮小とうつ病の間の生理学的関連を説明するためにサポルスキーによって提案された3つの相反するモデルを調和させるものであり、[471]、慢性的または重度の心理社会的ストレス以外のうつ病の多くの異なる原因に対して論理的根拠を提供するものである。直接的な原因としては、前述の生産的成人海馬神経新生に不可欠な要件の欠乏(行動障害)や、環境毒素による生産的成人海馬神経新生の妨害が挙げられるが、これについては後述する。間接的な原因とは、これらの欠乏(または毒素)の行動上の結果であり、BBB機能を阻害し、神経細胞のインスリン抵抗性、肥満、高血圧、動脈硬化、血液供給の減少を引き起こし、行動上の欠乏を悪化させる。不健康な生活習慣があれば十分であるため、加齢そのものが原因とは考えられない。とはいえ、成人海馬神経新生を阻害しAβの蓄積を増加させる神経炎症過程は、ほとんどの場合、加齢の結果とみなされている。[472]。しかし、神経炎症は、しばしば主張されるように(例えば、[473]を参照)、本当に加齢それ自体が原因なのだろうか、それともむしろ、修正可能な生活習慣要因(および不自然な飼育条件下での動物実験の誤った解釈)の結果なのだろうか?最近の研究結果は後者を示唆しており、神経炎症は若年AD患者と比較して高齢AD患者でははるかに少ないことが判明している[474]。ADの原動力である慢性炎症が本当に加齢によって誘発されたものであれば、逆の結果が予想されるはずである。神経炎症はむしろ多因性であり、生産的成人海馬神経新生を妨害することが判明したのと同じ行動障害や環境因子が寄与している(総説は[475]を参照)。上述した栄養因子に加えて、これらの修正可能な因子は、慢性的なストレス [476]、栄養不足の結果として脳内でAβオリゴマー化の種として働く可能性のあるアミロイド産生微生物(総説は [477]を参照)、座りがちなライフスタイル [478]、睡眠の質の低下 [479]、内臓肥満 [480]、歯科衛生状態の悪化(総説は [481]を参照)、または慢性感染症の他の原因 [482, 483]など多岐にわたる。うつ病とADは、成人海馬神経新生が障害されているだけでなく、神経炎症も潜在的な原因として共通しているため、Berkらは、UTADにも関連する質問をしている。彼らの答えはこうだ: 修正可能な危険因子 [484] である。
図2にさらに描かれているように、MCIやAD初期のバイオマーカーであるHPAの活性化、つまりコルチゾールの過剰分泌 [178] は、Aβの過剰産生につながる。睡眠不足や身体的・社会的活動不足は、BBBを介したAβクリアランスを阻害する。[259, 485]。産生の増加とクリアランスの減少の両方が、Aβの蓄積と神経毒性オリゴマーへの形成につながる。さらに、細胞外マトリックス(ECM)のエクトプロテインキナーゼとホスファターゼ [486]が、Aβの凝集と毒性を増強することが判明した。[487]。この新たな分野が、現在の行動学的欠陥や環境毒素とどのように関連しているかはまだ明らかではないが、ECM活性の生理機能が、他のすべてのプロセスと同様に高度に制御されていないとすれば、驚くべきことである。
図2の丸い矢印で示したように、有毒なAβは少なくとも2つのメカニズムによって自身の生成を促進する。1)老人斑や細胞内神経原線維もつれの原因物質であるAβやタウタンパク質は、「プリオノイド」とも呼ばれ、タンパク質がプリオンのような性質を示すことを示している[488]。2) オリゴマーAβは、GSK-3βの活性化を通じてAPPのアミロゲン化を促進し、タウの有害なリン酸化も引き起こす。[126]。これらの神経変性促進過程はすべて相互に作用し、海馬の縮小と皮質の菲薄化を促進する。AD発症の初期段階では、さらに悪循環が進行するため、原因と結果を区別することはしばしば困難である。とはいえ、いったん悪循環が始まると、さまざまなレベルで有害なプロセスを加速させる傾向がある。図2には、うつ病の粘性円(連続した黒い矢印)の他に、いくつかの他の円(破線の黒い矢印)が含まれている:
- 上述したように、睡眠障害はさまざまなメカニズムによって毒性Aβの蓄積を増加させる。しかし、毒性Aβは睡眠も障害する。[490]。少なくともADの初期段階においては、睡眠障害は記憶障害の重要な原因かもしれない。
- 動脈硬化とADはともに、同様の行動障害と関連している可能性があり、このことが併存率の高さを説明しているかもしれない。[491]。さらに、動脈硬化性低灌流は、アミロ化促進性APPプロセッシングの活性化とAβクリアランスの減少を引き起こし、脳内のAβ蓄積を増加させるが、[492]、Aβは血管壁に蓄積する傾向があり、酸化ストレス、炎症、内皮機能障害を引き起こし、動脈硬化プロセスを促進する。[493]。
- 毒性AβはBBBの機能を乱し、この重要なバリアの破壊を、例えば細菌やウイルス感染を脳に侵入させ、疾患の進行を促進させることによって、結果的に、また病因の連鎖の中間的なリンクとしている。[462]。
- 毒性Aβと慢性的に上昇したコルチゾール濃度の両方が、海馬(側頭葉)のインスリン抵抗性を増強する。[496]。神経細胞のインスリン抵抗性は成人海馬神経新生の障害を悪化させ、「うつ病の悪循環」によって、神経細胞のインスリン感受性を回復させるために必要な、身体的・社会的活動への参加や栄養豊富な食事の摂取といった生活習慣の改善に対する患者の能力を低下させる。
- Aβによって活性化されたGSK-3βは、Aβを増加させるだけでなく、タウをリン酸化し、タウはプリオンのように振る舞うことが示された。[497]。神経細胞の損傷とプリオンの凝集体は神経炎症を悪化させ、その結果神経変性が促進される。[498]。高リン酸化タウもまた酸化ストレスを引き起こし、それがタウの高リン酸化を引き起こす。[129]。
AD治療を成功させるためには、自己維持と疾患促進の悪循環をすべて断ち切る必要があることは明らかである。これをどのように達成できるかは、以下の治療法の提案で概説する。このような計画には、成人海馬神経新生を阻害し、疾患プロセスを楽しませ、悪化させる可能性のあるあらゆる種類の環境毒素を除去することが含まれなければならない:
- タバコの煙はラットの成人海馬神経新生を減少させ、グリア形成を促進する。[499]。そのため、ヘビースモーカー(1日2箱)はADリスクが2.6倍上昇する。[500]。ニコチンは海馬ニューロンの神経新生と可塑性を損なうためである。[501]。
- 1日2杯以上のアルコール摂取は、ADを約2~3年、多量の喫煙(1日20本)と合わせると約4~6年早める。また、不健康な生活習慣を選択した結果(上記で詳述したとおり)を悪化させるApoE4対立遺伝子を持つ患者では、ADの発症が平均10年早まる。[502]。高濃度のアルコール摂取は、霊長類モデルにおいて成人海馬神経新生を効率的に阻害し、その効果はアルコール摂取を中止しても2カ月間持続することが示された。[503]。アルコールによる成人海馬神経新生の持続的減少は、非アポトーシス的メカニズムによる神経変性の増加と並行していた。興味深いことに、過去には禁酒も危険因子と考えられていたが、[504]、この所見は確認されていない。[505]。したがって、少量の飲酒は統計学的に有害ではないかもしれないが、AD発症リスクを減少させるものではない可能性が高い。
- トランス脂肪酸(TFA)は、加工食品(揚げ物やファストフードの多く)または全脂肪乳製品や反芻動物の肉製品に由来するもので、ADに関与している。[506]。両者とも、低比重リポ蛋白(LDL)コレステロールを等しく効率よく増加させ [507]、健康に悪影響を及ぼすことが示されている。[508]。TFAは心血管系疾患による死亡リスクを高め [509]、高齢者の認知機能低下率を増加させるが、これも摂取源に関係ない。[510, 511]。健康な被験者を対象とした疫学研究では、TFAがインスリン抵抗性、血圧を上昇させ、慢性炎症をも引き起こすことが示された。さらに、TFAはn-3系PUFAのDHAへの変換を阻害し、脳への蓄積を制限する。[512]。DHAの欠乏は、BDNFレベルの低下 [513]、生産的成人海馬神経新生の阻害 [514]、神経炎症 [308]を引き起こすことが示されている。最近のin-vitro研究では、TFAがアミロイド前駆体タンパク質(APP)のアミロイド形成プロセシングを増加させ、Aβの過剰産生をもたらすという説得力のある証拠が示された[515]。さらに、TFAはAβのオリゴマー化と凝集を促進することが示された。まとめると、TFAの多量摂取は、供給源に関係なく、多くの経路からADのリスクを増大させ、病気の早期発症も引き起こす可能性がある。
- ニトロソアミンは、脂質過酸化、Aβやp-tauの増加、神経炎症、神経細胞のインスリン抵抗性などを特徴とする神経変性だけでなく、運動機能や空間学習の障害を引き起こす。[516]。ニトロソアミンはタバコの煙に含まれている。また、亜硝酸塩漬けの塩で保存された加工肉やチーズなど、多くの製品に含まれる亜硝酸塩や第二級アミンからも、かなりのレベルのニトロソアミンが生成される。これも(大量生産された)肉やチーズ製品を避ける理由のひとつである。
- ビスフェノールA(BPA)は不要なホルモン活性を引き起こし、成人海馬神経新生、空間学習、記憶を損なうことが示されている[517]。BPAは、水筒のような一般的な消費財の多くに含まれている。BPAを含むエポキシ樹脂は、水道管の配管や多くの食品・飲料缶の内側のコーティングに使用されている。BPAフリー製品を宣伝するために)代替品として使用される他のビスフェノール類(B-Z)への暴露も、同様の有害な影響を示す[518]。したがって、BPAフリー製品は必ずしも安全ではなく、消費者向け商品からすべてのビスフェノール類を除去することを支持する[519]。
- 農薬: ラウンドアップ(グリホサート系除草剤)は最近、海馬における脂質過酸化、グルタミン酸興奮毒性、酸化的損傷を増加させることが示された。[520]。広く使用されているピレスロイド系殺虫剤デルタメトリンは、海馬でアポトーシスシグナルを誘導し、成人海馬神経新生を障害する。[521]。カーバメート系農薬であるカルボフランにも同様の作用が観察された。[522]。慣行農法で広く使用されている化学物質がこのような神経毒性を示すことから、ADの予防と治療には有機農法で生産された農産物を使用する必要がある。[4]。
- アルミニウムはADの進行を促進する作用があるかもしれない。[523]。アルミニウムはプロアポトーシスシグナルを増強し、海馬のBDNFを減少させ、動物モデルの空間学習に悪影響を及ぼす。実際、脳細胞のあらゆる生化学的機能がアルミニウムの影響を受けているようである(総説は[2])。さらに、アルミニウムはAβのオリゴマー化において架橋剤として重要な役割を果たしている可能性がある。[524]。これらを総合すると、ADの予防と治療においては、アルミニウムへの曝露を避けるべきである。
- メチル水銀(MeHg)は、セレンの酵素結合部位でセレンと競合し、全身、特に神経系の酸化的損傷を予防し、回復させるのに必要なセレン酵素を、極めて特異的かつ不可逆的に阻害する。セレン酵素の阻害は、MeHg毒性に伴う病理学的影響の近因であると考えられている。[525]。食事からのセレン摂取量は、メチル水銀(MeHg)毒性に対する脆弱性と逆相関している。このことは、MeHgを含む食品を母親が摂取した場合、Seがモル比で過剰になり、子どもの予後が悪くなる一方で、MeHgを含むがセレンが豊富な海産魚を食べると、子どものIQが向上する理由を説明している。[526]。同様に、MeHgの摂取は海馬の発達や成人海馬神経新生を障害し、[527]、認知発達の遅れ [528]やAD [529]と関連している。とはいえ、最近の研究では、水銀濃度が上昇しているにもかかわらず、魚介類の摂取はADの神経病理を軽減することが示された。[58]。この議論の余地のある結果は、DHAとセレンの両方が魚介類に高濃度で含まれているという事実によって説明されるかもしれない。興味深いことに、この研究では、ApoE4キャリアの場合、魚介類を多く摂取することが最も有益であり、逆にApoE4キャリアの場合、DHAやセレンの摂取が不足すると害が大きくなる。これらを総合すると、MeHgの少ない食事が勧められるが、少なくともそれと同じくらい重要なのは、セレンとn-3系PUFA(DHAとEPA)の濃度を十分に高く保つ食事である。
- 鉄は多くの代謝プロセスに必須であるが、制御されないまま放置されると、活性酸素発生の強力な触媒として関与する。鉄は、この遷移金属の主要な細胞内貯蔵物質であるフェリチンと複合体を形成する。[530]。上述のように、脳脊髄液(CSF)中のフェリチン濃度の増加は、認知能力およびMCIからADへの転換の予測速度と負の相関がある。ApoE4は(ApoE2やApoE3とは対照的に)脳への鉄の取り込みを促進するため、[59]、鉄分の多い動物性食品を大量に摂取する保因者は、特に脳鉄の上昇、活性酸素産生の亢進、AD進行の影響を受けやすい。これが、動物性食品の少ない食事がADリスクを低下させ、ペスコ・ベジタリアンが他の一般的な食事と比較して死亡率が最も低い理由の一つかもしれない[531]。上記のCSF-フェリチン研究の著者によれば、MCIでCSF-フェリチン値が高い人は、デフェリプロン [532]のようなキレート剤を服用することで、ADへの移行を3年ほど遅らせることができる。副作用の少ない別の選択肢として、α-リポ酸による治療がある。[533]。α-リポ酸は、鉄過剰症から生じる酸化ストレスを逆転させるのに非常に有効であることが示されている。[534]。α-リポ酸にはAD治療に有用な特性が多数あるため、α-リポ酸は以下に提案する治療スキームの一部となる。
- 銅は鉄と同様、人間の成長と発達に不可欠な元素である。そして同様に、遊離銅につながる過剰摂取は、シナプスタンパク質の発現や海馬のシグナル伝達経路に特異的な変化をもたらし、酸化ストレスや神経細胞のアポトーシスを引き起こすことで、神経毒性や空間記憶障害の一因となる[535]。飲料水に含まれる無機銅は直接吸収され、血清中の遊離銅プールを上昇させるため、ADの進行の原因となる。[536]。遊離銅濃度が高い場合、3つの治療法が勧められる:(1)銅で汚染された水やサプリメントを避ける、(2)亜鉛濃度を上げるとアルツハイマー病患者の血清遊離銅が有意に減少することが示されたため、亜鉛濃度を正常に戻す。[3]、(3)遊離銅を低下させるα-リポ酸による治療 [537]。
- 薬物療法: 胃酸抑制剤の使用は、ADの危険因子であるビタミンB12欠乏症 [538]の存在と有意に関連していた。一方、これらの薬剤はマウスの脳内のAβレベルを増加させ、ヒトの認知症リスクの増加と関連することが明らかになった。[539]。認知症発症の増加は、三環系抗うつ薬のドキセピン、抗ヒスタミン薬のクロルフェニラミンやジフェンヒドラミン、膀胱抑制薬のオキシブチニンなどの抗コリン薬の長期使用(3年以上)と関連している。[540]。症例対照研究では、ベンゾジアゼピン系薬剤(睡眠と不安のコントロールに使用)の使用とADのリスクとの関連が示された。[541]。最近の研究では、WeDiを投与したラットの記憶と海馬の構造が、ベンゾジアゼピン系のミダゾラムによる短期治療に対して特に脆弱であるという証拠が示された。[542]。繰り返しになるが、一般的に使用されている多くの薬剤のような人工的な化学物質が、UTADによって概説された複雑な病態メカニズムを阻害し、ADリスクを増大させる可能性が常に存在することを示すには、この短い数の例で十分であろう。
AD予防戦略
UTADは、認知機能の低下とアルツハイマー病(他の文化的背景を持つ病気と同様)が、修正可能な危険因子に大きく依存していることを理解するための枠組みを提供した。図3の上部は、行動障害がADリスクの増加につながる可能性のある主要な領域を模式的に描いている。緑色の線で示したように、さまざまな行動が成人海馬神経新生や長期的な脳の健康に必要なその他の主要な生理機能に影響を与える。しかし、LOMが予測したように、どれかひとつが、欠けている別のものを完全に置き換えたり、補ったりすることはできない(ただし、成人海馬神経新生の刺激に関しては、身体的なものと激しい社会的なもの、すなわちオキシトシンを誘発する活動の間には、機能的な重複があるかもしれない)。身体活動 [263]やn-3系PUFAの補充 [543]のような一側面にのみ焦点を当てた介入試験が、ADリスクを最小限しか低下させないか、全く低下させないのは、生物学的過程における各因子の必須性によって説明できる。
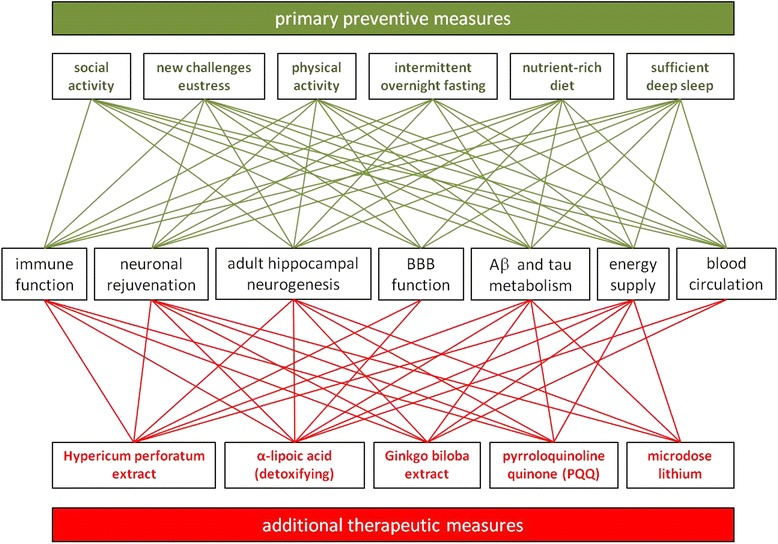
図3
「ADの予防と治療のためのシステム生物学的プログラム」生涯にわたる精神的健康の維持という遺伝子プログラムの「関心」をサポートするために、ADを予防するための主要な対策は、豊かな社会生活、ユーストレスと記憶に残る経験を提供する日々の新しい挑戦、身体活動と十分な深い睡眠、そして十分な量の必須栄養素で構成されている。緑色の線で示したように(そして本文で概説したように)、これらの対策はそれぞれ、精神的健康の維持に必要な主要な生理学的システムやプロセスに、直接的または間接的に良い影響を与える。ADと診断された場合、これらのシステムは損なわれるだけでなく、図2に概略を示し、本文で詳述したように、暴走現象につながる。したがって、予防措置の実施と生活習慣に起因する欠陥の是正に加え、提案する治療的介入は、ADのプロセスを推進し加速させる様々な悪循環を中断させることが示された活性成分の全身的な組み合わせで構成される。それぞれは、特定の主要な病態生理学的プロセスを阻害する能力によって選択された。しかし、赤い線で示したように、また本文で詳しく説明したように、提案された活性成分はすべて、ADプロセスによって障害されたさまざまな機能を再活性化するのに役立つ。
高齢者のライフスタイルを変える最も包括的な実験のひとつに、フィンランド認知障害予防介入研究(FINGER)がある。[17]。介入群の参加者は食事指導と認知トレーニングを受け、社会的・身体的活動プログラムに取り組んだのに対し、対照群は一般的な健康アドバイスのみを受けた。この研究の著者によると、包括的なアウトカム測定により、全体的な認知と日常活動に関連性の高い認知領域(例えば、実行機能、処理速度、複雑な記憶課題)の両方に対する有益な効果が示唆された。とはいえ、提案されたUTADによれば、FINGERでさえ、長期的な脳の健康にとって重要ないくつかの要件を考慮していない。FINGERの詳細(http://www.alzheimersprevention.org/downloadables/FINGER-study-report-by-ARPF.pdf)によると、例えば、ストレス軽減や睡眠の質向上のための具体的なプログラムはなく、断続的絶食やケトジェネシスの有益性に関する指示もなく、潜在的なビタミン欠乏のモニタリング(したがって是正)もない(例えば、ホモシステインの測定はない)。最近の研究結果によると、推奨されている1日400~800単位のビタミンDでさえ、AD予防には十分でない可能性が高い。[339]。さらに、必須微量元素の潜在的欠乏のモニタリングと補正は実施されなかった。
参考記事

しかし、UTADによれば、認知機能低下を予防するための効率的なプログラムは、個別的かつ包括的である必要がある。そのためには、標準的な健康診断に加え、ビタミン、必須微量元素の完全測定、さらに上記の環境毒素の潜在的蓄積の分析が必要である。有害金属の血中濃度が高い場合は、介入措置としてα-リポ酸治療が勧められる。生活習慣を変えてもなお不足が残る場合(例えば、土壌中のセレン量が少ないためにヨーロッパ諸国で生産される食品中のセレン濃度が低すぎる場合、あるいは、単に紫外線が皮膚に十分に届かない北の方や冬に住んでいる場合 [544]、あるいは、水道水中の濃度が低いためにリチウムの摂取が不十分な場合など)には、特定の栄養補助食品が予防戦略の重要な一部となる。
ADの原因療法
ADの病態は進行性であり、治療介入はできるだけ早期に開始すべきである。ADは、ステージ1または自覚的認知機能低下(SCD)(詳細な分類については、[545]を参照)から、ステージ2またはMCI [546]、ステージ3または軽度/早期AD、ステージ4または中等度AD、ステージ5または後期/重度ADへと段階的に進行する。
UTADによる原因療法は、上記のAD予防スキームと同じように、すべての原因因子を取り除くことを目的としている。成人海馬神経新生の行動障害のために、患者は明らかなうつ病を呈するか、少なくともコルチゾール分泌過多を示す。したがって、コルチゾールの状態を分析し、治療中も定期的にモニターする必要がある。健康的なライフスタイルへの復帰を指導することに加え、治療レジメンは、病気のプロセスを維持し、さらに加速させる上記の悪循環を中断させることを目的としている。この治療法が確立されれば、少なくとも第1期から第3期の患者はエピソード記憶能力を完全に回復する可能性がある。というのも、ADのこれらの初期段階までは、エピソード記憶障害が優勢であり、患者はまだADに起因する認知機能低下に苦しんでいないからである。この仮定は、デール・ブレデセンが最近行った研究の結果と一致している。そこでは、ステージ1(3人)、ステージ2(2人)、ステージ3(3人)の患者10人のうち8人が、UTAD(下記参照)の結果と同様の治療スキームを遵守した後、エピソード記憶を回復した。ステージ4の患者1人は状態を維持し、ステージ5の患者1人は治療不可能であった。これらの結果はどちらかというと症例ベースであるため、より大規模な試験による確認が必要である。しかし、個々の欠損の除去に続く成功は、この論文で提案されたUTADと、その結果としての予防と治療に対する意味合いと完全に一致している。一方、認知機能とエピソード記憶機能における同様の改善は、本論文で概説した治療計画を実施し始めた個人診療所(ドイツ、フライブルク、W. Karner & B. Karnerによる私信)の最初の3人のステージ3のアルツハイマー病患者においても観察されている。
治療プログラムの完了には約6カ月かかると推定している。この計算はいくつかの仮定に基づいている: 第一に、不健康な生活習慣が非常に包括的で深く根付いている場合は特にそうであるが、生活習慣を変えるには数週間かかる。第二に、成人海馬神経新生は一度再活性化されると、新しい成体で生まれた歯状回ニューロンを完全に統合するのに数ヶ月を要する。さらに、その期間は、同様に非生産的な成人海馬神経新生を標的とする必要がある大うつ病の治療と大差ない。さらに、ブレデセンのパイロット研究では、ほとんどの患者が約4~6カ月後に有意な改善と認知機能低下の逆転を示したと報告されている。現在、ドイツのフライブルグでも同様の報告がなされている(W. Karner & B. Karnerによる私信)。
生産的な成人海馬神経新生を再始動させるだけでなく、提案されている治療法は、神経細胞の若返りプログラムを再活性化し、図2に示されているようなADの悪循環を中断させることを目的としている。さらに、長引く感染症やあからさまな感染症は治療する必要があり、これには歯科治療や、ADプロセスの加速を抑えるためにマイクロバイオーム組成を改善するためのプロバイオティクスによる治療が含まれる(上記参照)。上記で概説した個々の予防法(図3の上部)に加えて、以下の治療法が提案されている(図3の下部):
オトギリソウエキス(HPE)
UTADが提唱しているように、成人海馬神経新生の障害によるコルチゾール分泌過多は、ADにつながる因果の連鎖の主要なリンクである。コルチゾール分泌過多は、病気の進行を促進するだけでなく、患者の治療に対するコンプライアンスを低下させ、特に生活習慣の改善に対する積極性やアドヒアランスを低下させる。このうつ病のような苦境を克服する一つの方法は、SSRIクラスのような標準的な抗うつ薬による治療である。例えば、ある研究では、慢性的に投与されたSSRIタイプの薬剤であるフルオキセチンの抗不安・抗うつ活性には、若いニューロンの成熟と機能的特性が必要であることが示され、その結果、よく知られている治療効果の発現遅延が説明された。[549]。別の研究では、SSRIタイプの抗うつ薬が、トランスジェニックADマウスや健常人のAβ産生を有意に減少させた。[550]。海馬の神経新生がゆっくりと進行するのとは対照的に、Aβ産生を制御するメカニズムははるかに速いことが判明した。どうやら、セロトニン受容体の刺激は細胞外シグナル制御キナーゼ(ERK)シグナル伝達カスケードを素早く活性化し、それがセロトニン依存性のAβ抑制に必要であることが示された。[551]。SSRI型抗うつ薬によるERKシグナルの活性化は、非アミル化αセクレターゼ活性を増加させ [552]、γセクレターゼ活性を低下させ [553]、両者ともAβレベルを低下させる。
SSRIタイプの抗うつ薬に代わるものとして、セイヨウオトギリソウ(HPE)の抽出物がある。ドイツのミュンヘンにある補完医療研究センター(Centre for Complementary Medicine Research)の報告では、29件の臨床試験の大規模なメタアナリシスに基づいて、次のような結論に達している。[554]: 利用可能な証拠は、含まれる臨床試験で試験されたHPEが、a)大うつ病患者においてプラセボよりも優れていること、b)標準的な抗うつ薬と同様の効果があること、c)標準的な抗うつ薬よりも副作用が少ないことを示唆している。一方、2015年に発表されたドイツの単極性うつ病の患者ケアガイドラインS3では、中等度大うつ病の治療において、HPE900mgのエビデンスに基づく効果は、主要なSSRIであるシタロプラム(20mg/日)と治療上同等であると評価されている(http://www.leitlinien.de/mdb/downloads/nvl/depression/depression-2aufl-vers3-lang.pdf)。HPEは優れた忍容性を特徴とする。同様に、別のメタアナリシスでは、HPEはSSRIと有効性で差はないが、大うつ病性障害の管理において有利であり、有害事象が少ないため患者のコンプライアンスが高いと結論づけている。[555]。
HPEは、多くの抗AD特異的作用を有する:(1)HPEは、慢性コルチコステロン治療による不安/抑うつ様状態を効率的に逆転させる[554]、(2)慢性コルチコステロン治療下のマウス海馬における前駆細胞の増殖低下を相殺する、(3)コルチコステロン誘発の長期的な海馬幹細胞増殖抑制を減少させる、(4)およびコルチコステロンにより減少したスパイン密度を改善する(5)。HPEの生物学的に強力な成分の一つであるハイパーフォリンの合成誘導体であるテトラヒドロハイパーフォリン(THH)(6)は、野生型マウスとADモデルの両方で成人海馬神経新生を活性化する[556]。(7)HPEの投与は、脳内Aβ42レベルを有意に低下させ(8)、新皮質ニューロンを毒性Aβから救い、(9)認知機能を正常レベルに回復させ、(10)試験管内試験および生体内試験でミクログリアを活性化する[557]。機序的には、可溶性Aβ42種の減少は、BBBに位置するATP結合カセット(ABC)トランスポーターのスーパーファミリーのメンバーであるABCC1トランスポーターの活性の増加の結果であることが示され、ABCC1トランスポーターは血流へのAβ排泄に基本的な役割を果たすことが判明した。同様に、HPE投与は、Aβ排出に関与するもう1つのABCトランスポーターであるABCB1を活性化することにより、二重トランスジェニックADモデルにおけるAβ蓄積を減少させた。[558]。(11)さらに、THHはAPPのアミロジェニック処理を阻害することがわかった[559]。この作用機序により、(12) THHはマウスADモデルにおいてタウのリン酸化とシナプス毒性を防いだ。[560]。(13)そして最後になるが、HPEを投与したマウスは、白色脂肪組織におけるアディポネクチンのレベルが上昇し、インスリン感作作用を示した[561]。
これらを総合すると、図3(赤線)に示すように、HPEはADに特異的な複数のプロセスや悪循環に対して作用する。とはいえ、HPEは一般的に安全であり、米国では処方箋なしで入手可能であるが、HPEは解毒作用をもつ肝酵素CYP 3A4を有意に誘導することが知られているため、経験豊富な医師の指導のもとでのみ服用すべきである[562]。したがって、HPEを1日当たり約1gの標準量で治療的に投与すると、臨床効果が減弱し、市販されている医薬品の少なくとも50%を占めるすべてのCYP 3A4基質に対する必要量が増加する可能性がある。
α-リポ酸
α-リポ酸は酵素的補酵素であり、好気性ミトコンドリアのエネルギー産生に必須である。[563]。すべての動物細胞におけるデノボ合成は、中間代謝におけるその役割に必要なすべてのα-リポ酸を供給するように見えるが、α-リポ酸はまた、効率的に食事から吸収することができる。自然界には(R)-(+)-エナンチオマーのみが存在し、その生物学的活性において優れている[564]。化学合成における副産物としての(S)-(-)-エナンチオマーや、多くの市販のα-リポ酸製品は、例えばインスリン抵抗性を低下させるα-リポ酸の能力のような、天然型の重要な生物学的活性を阻害する可能性さえある[565]。したがって、α-リポ酸についてさらに論じる場合、私は(R)-(+)-エナンチオマーを参照する。
α-リポ酸は、適量であれば安全であり、ADや関連する認知症の治療において高い関心を集めている(総説は[566]を参照):
- (1)α-リポ酸とその還元型であるジヒドロリポ酸(DHLA)は、ヒトの生体内で容易に形成され、上記のようにADのプロセスを促進する可能性のある酸化還元活性遷移金属をキレートするのに非常に効率的である。α-リポ酸によって体内に蓄積された毒素が除去されることで、ヒトの生物学への悪影響が軽減され、生理機能が回復する可能性がある(総説は[567]を参照)。重要なことは、α-リポ酸も、DHLAもトリカブトから鉄を、スーパーオキシドジスムターゼから銅を除去することはできず、α-リポ酸 補給は金属枯渇を引き起こすことなく、酸化還元活性遷移金属の不安定なプールのみを調節することを示唆している[568]。
- (2)α-リポ酸とDHLAは、ラジカルを容易に消去し、活性酸素と脂質過酸化生成物を消去し、過酸化水素とヒドロキシラジカルの生成を抑制することができるため、理想的な抗酸化物質である。例えば、α-リポ酸は酸化ダメージを減少させ、抗酸化防御を高め、ミトコンドリア機能を改善することで、グルタミン酸やオキシダントによる細胞死を減少させることが示された[569]。
- (3) α-リポ酸は、還元型グルタチオンを増加させるなど、他の抗酸化物質を再生する。
- (4) α-リポ酸はまた、グルタチオン合成酵素やその他の抗酸化保護酵素を誘導する。このようなミトコンドリア保護作用は、炎症によって誘発される成人海馬神経新生の障害を減衰させる[570]。
- (5) α-リポ酸は炎症プロセスをダウンレギュレートし、フリーラジカルと細胞毒性サイトカインの放出を抑制する。上述したように、
- (6) α-リポ酸はインスリン受容体経路を感作し[571]、高齢動物におけるグルコース取り込みを増加させる[572]。
- (7)α-リポ酸はPGC-1αの発現を増加させ、低下した脳血流を活性化し、それによって局所虚血から保護する[573]。ミトコンドリアの生合成を刺激することは、α-リポ酸が加齢やADにおける認知機能障害を改善するための重要なメカニズムの一つである[574]。PGC-1αを活性化することにより、α-リポ酸の効果はオルガネラに限定されるものではなく、加齢に伴う脳エネルギー代謝の障害を回復させるミトコンドリア-細胞質-核連絡の機能的かつ効果的な調整に存在する[572]。このことは、脳卒中モデルにおけるα-リポ酸治療が、神経回復と機能回復を促進することが最近示された理由を説明するかもしれない[575]。
- (8) α-リポ酸は、オリゴマーAβ、過剰な鉄、その他の神経毒の有害な影響からニューロンを保護する[576]。
- (9)α-リポ酸はまた、BBBを安定化させることが示された[577]。
- 最後になるが、(10) α-リポ酸は、AD脳で減少していることが知られているアセチルコリン(ACh)産生を増加させる。[578]。
これは、最近の研究で、α7含有ニコチン受容体(α7-nAChR)を介した内因性AChシグナルが、海馬の成体で生まれた歯状回ニューロンの成熟と統合を促進することが解明されたため、特に興味深い。これは成人海馬神経新生におけるACH/α7-nAChR-システムの深い役割を示しており、逆にα7-nAChRが失われると、歯状回に加わるニューロンが減少し、加わったニューロンが統合される程度が低下するため、海馬の回路と機能が時間とともに徐々に損なわれることを示している。[579]。実際、運動による海馬でのACh放出の増加は、成人海馬神経新生に対する運動の刺激効果に寄与しているかもしれない。[580]。したがって、生理学的および/または薬理学的なコリン作動性刺激は、成人海馬神経新生をサポートすることによって、高齢の動物でも認知機能低下を改善する可能性が高い。コリンアセチルトランスフェラーゼの活性化 [581]により、α-リポ酸は脳のACHレベルを上昇させ、成人海馬神経新生をサポートする。Holmquistら [566]によって要約されている: α-リポ酸は、ACh合成経路の刺激を介してコリン作動性認知機能障害の症状を改善するための主要な根拠を提供する。
α-リポ酸の作用様式は、ドネペジル [582]のようなアセチルコリンエステラーゼ阻害剤(AChEI)とは大きく異なっており、分解を阻害することによってACHを上昇させるという事実に注意すべきである。このことは、α-リポ酸の望ましくない副作用プロファイルが比較にならないほど低いことを説明するかもしれない。複数の付加的な機能性(すなわち、抗炎症作用、抗酸化作用、カルボニル消去作用、金属キレート作用、プロエネルギー作用、神経保護作用)により、α-リポ酸による治療はADの進行を遅らせることが期待される。これはMCIにおけるAChEIとは対照的であり、ランダム化比較試験のシステマティックレビューによると、AChEIはADや認知症の発症を遅らせることとは無関係であった。さらに、安全性プロファイルから、AChEIに関連するリスクは無視できないことが示された。[583]。2015年に実施された別の、より最近のメタアナリシスは、対照群と比較した場合、AChEIによるMCIの治療は利益をもたらさないという証拠を提供した。行動療法を用いた場合、対照群と比較してわずかな認知的利益が観察され、これは現在の使用を正当化するために用いられるかもしれない。しかし、著者らは、このわずかな有益性の臨床的意義は不明であり、現在のエビデンスはMCIの治療にAChEIを使用することを支持するものではないと結論づけている[584]。
α-リポ酸を1日600mg投与すると、軽度から中等度のアルツハイマー病患者、特に2型糖尿病を合併している患者においてMMSEスコアが改善した[585]。48カ月にわたる研究では、1日600mgのα-リポ酸が軽度認知症患者の認知機能を安定させ、病気の進行は極めて緩やかであった(MMSE:-0.6ポイント/)[586]。また、中等度の認知症患者においても、2年目の進行率は、未治療患者やAChEI投与患者について報告されたデータよりも劇的に低かった。したがって、彼らの研究は二重盲検、プラセボ対照、無作為化ではなかったにもかかわらず、α-リポ酸による治療はADに対する有望な「神経保護」治療の選択肢になりうると結論している。重要なことは、これらの研究はすべて上述の欠乏条件下で行われたものであることを再認識することである。本総説で概説した予防策を含む治療スキームに組み込めば、単に認知機能低下を安定化させる以上の効果が得られるかもしれない。
α-リポ酸は多くの国で市販の栄養補助食品として入手可能であるが、ほとんどの製剤には2つの可能なエナンチオマー(R)-(+)-リポ酸と(S)-(-)-リポ酸がラセミ混合物として含まれている。α-リポ酸はインスリンを感作するため、経験豊富な医師の指導の下でのみ(R)-(+)-リポ酸を投与すべきである。この点で、α-リポ酸のような金属キレート剤が、消化管からのビタミンB12の取り込み機構を阻害する可能性があることに注意することが重要である。[587]。このことはα-リポ酸では直接示されていないが、ビタミンB12が欠乏するとα-リポ酸の効果が相殺される可能性があるため、長期治療中はビタミンB12をモニターすべきである。同様に、高用量のα-リポ酸はビオチン欠乏症を引き起こす可能性があり、α-リポ酸治療中の患者はビオチンを補給する必要がある[588]。とはいえ、1日量600mgのα-リポ酸は、特に包括的な治療計画の下で適用される場合、AD治療のための安全なマルチモーダル薬物である。
イチョウ葉エキス
上記で概説したように、微小循環の障害はADの原因であり結果であり、これら2つの疾患プロセスの間に悪循環を形成している可能性がある。したがって、
- (1) 微小循環を増加させ、
- (2) 高齢患者においてもラジカル消去能を有意に改善するイチョウ葉エキスの能力は、ADの治療において非常に有用である。[589]。これらの効果は、多数の強力な薬理学的活性成分に起因すると考えられる。例えば、標準化されたEGb-761には、フラボノイド配糖体(例えばケルセチン、ケンフェロール、イソラムネチンなど)が約24%、テルペノイド(うちギンコライドA、B、C、Jが3.1%、ビロバライドが2.9%)が6%、有機酸が5~10%含まれている(総説は[590]を参照)。ギンコライドの各成分は、ADの病理学的メカニズムに対してそれぞれ異なる作用を持っている(総説は [591]を参照):
- (3) 上記の2つの効果に加え、EGb-761は、Aβ42誘発性細胞アポトーシス [592]、
- (4) Aβ毒性 [593]、
- (5) さらにはAβ産生 [594]を減少させることができる;
- (6) イチョウ葉エキスは、タウ代謝を積極的に調節する。[595]、
- (7) 酸化ストレスを軽減し、ミトコンドリア呼吸を改善する。特にフラボノイド、ビロバライド、ギンコライドの一部(BとJ)は、高い神経保護能を示す(総説は[596]を参照)。
- (8) 食事誘発性肥満ラットにおいて、イチョウ葉エキスはインスリンシグナル、脂質異常症、体脂肪率を改善し、それゆえ機能不全に陥ったエネルギー代謝をサポートする。[597];
- (9) EGb-761は、閉経後のモデルとして卵巣摘出ラットを用いると、血小板と海馬の両方で、シトクロムcオキシダーゼ活性、ミトコンドリアのアデノシン-5′-三リン酸、グルタチオン含量の低下から保護し、閉経後の女性における中枢神経変性に対する保護剤としての役割の可能性を示唆した[598];
- (10) イチョウ葉エキスを大腸炎モデルに投与すると、神経炎症に関与する炎症性サイトカインであるTNF-αとIL-6のアップレギュレーションが用量依存的に抑制された。[599];
- (11) 最後になるが、イチョウ葉エキス投与は高齢マウスにおいても成人海馬神経新生に有益な役割を示し、コントロールと比較して三次樹状突起が発達した新生歯状回ニューロンが有意に増加した。[600]。
最近行われた2つのメタアナリシスでは、EGb-761を240mg/日、半年以上投与することで、認知機能障害や認知症における認知、機能、行動、全体的な変化の低下が安定するか、遅くなると結論付けられている[601, 602]。その結果、2016年1月にADの専門家グループがドイツの認知症患者ケアガイドラインS3を発表し、そこで初めて軽度から中等度のアルツハイマー病患者の治療にイチョウ葉(特にEGb-761)の使用が提案された(http://www.dgn.org/images/red_leitlinien/LL_2016/PDFs_Download/038013_LL_Demenzen_2016.pdf)。しかし、イチョウ葉エキスとCYP3A4で代謝される医薬品を併用する際には、EGbがCYP3A4遺伝子の発現を誘導するため、注意が必要である。[603]。
これらを総合すると、ADに関連するEGbの複数の作用機序、AD発症の初期段階ですでに活性化している悪循環を遮断する可能性、そして、EGb-761のようなEGbは、複数の薬剤を服用している高齢の患者であっても、1日240mgまでの用量で一般的に忍容性が非常に高いという事実が、図3に概略を示したような全身的な治療スキームにおいて有用であることを示している。
ピロロキノリンキノン(PQQ)
PQQは、細菌の酸化還元補酵素として最初に発見された。[604]。PQQは強力な植物成長因子でもあるため、動物の食物連鎖にも含まれている。PQQは、様々な生物学的機能に関与し、明らかな生存上の利益(例えば、新生児の成長や生殖能力の向上)をもたらすと報告されている[605]。また、PQQの補給は、認知機能、免疫機能、抗酸化機能、心筋梗塞や神経性虚血からの保護にも効果がある(総説は[606]を参照)。例えば、化学的に定義された食餌によってPQQを奪われたマウスは、結合組織代謝の機能的欠陥を示し、成長が遅れ、不妊である。[607]。PQQ欠乏動物はまた、細胞のミトコンドリア含量が低いことを示し、これはわずか200-300μg/kgのPQQを添加すると逆転する[608]。PQQは、その栄養学的重要性、固有のフリーラジカル消去および酸化還元モジュレーター特性、哺乳類組織における補酵素機能[609]から、潜在的な新しいビタミンB群として提案された[610]。PQQは脳虚血モデルで神経保護作用を示したことから[611]、神経変性疾患の治療薬として薬理学的に注目された。驚くべきことに、PQQは、パーキンソン病につながる一連の現象の原因であるα-シヌクレインの神経毒性オリゴマー化を阻害することが見出された。[612]。PQQの抗線維形成機能は、すぐにマウスプリオンタンパク質やAβ-42にも拡張された[613]。さらに、PQQはAβ誘発神経毒性から神経細胞を保護することが示された。[614]。
ADのような疾患はミトコンドリア機能障害と関連しているため、PQQの補充は、cAMP応答エレメント結合タンパク質(CREB)とPGC-1αの両方の活性化を介して、ミトコンドリア形成を効率的に刺激することから、さらに有益である可能性がある。[615]。提案されているAD治療スキームの一環として、PQQの補給は、ミトコンドリアの生合成とNRJの活性化において、上記で概説したAD予防策、すなわち断続的絶食と身体活動を支援することになるであろう。実際、食事性PQQは、ヒト被験者において炎症およびミトコンドリア関連代謝の指標を変化させることが示された。[616]。さらに、小規模の臨床試験では、1日20mgのPQQを8週間摂取することで、活力、疲労、緊張-不安、抑うつ、怒り-敵意、錯乱の指標が有意に改善し、生活の質、食欲、睡眠、強迫観念、痛みの指標も改善することが示された。[617]。二重盲検プラセボ対照比較試験において、PQQ含有食品を12週間摂取した中高年者の精神状態は、注意力、識別・処理能力などの高次脳機能の一部を向上させた。[618]。特に微量リチウムとの併用により、PQQはADにおいて有用な治療効果を発揮する可能性がある(下記参照)。
参考記事

リチウム
図3に模式的に示し、上述したように、少量のリチウムを毎日摂取することは、脳の健康を維持するための重要な予防法である。ADにおけるいくつかの自己維持シグナル伝達経路におけるリチウムの干渉のため、微量リチウムの日常摂取(リチウム含有飲料水で少なくとも300μg)は、治療的意義も高い: リチウムは有害なAβとp-tauの生成を減少させ、ミトコンドリアの若返り、ひいてはエネルギー代謝にプラスの影響を与える。さらに、成人海馬神経新生の生産性にも重要であるようだ。興味深いことに、本研究における治療法の提案と一致して、LazzaraとKimは最近、神経変性疾患におけるリチウムの多重作用と治療の可能性についてコメントした。[369]。彼らは、高用量リチウムのみの治療は、一貫性のない有効性と潜在的な副作用のため、神経変性疾患に対する治療選択肢としては適切でないかもしれないと指摘している。しかし、低用量リチウムと他の潜在的または既存の治療化合物との併用は、神経変性疾患の症状や疾患の進行を軽減する有望なアプローチである可能性がある。
実際、低用量リチウムとPQQの補充が強力な相乗効果を示す証拠が、マウス二重トランスジェニックADモデルで最近示された。その結果、比較的低用量のリチウムとPQQの併用は、リチウム単独と比較して、障害された学習と記憶の回復、海馬LTPの促進、大脳Aβ沈着とp-tauレベルの低下において、より強力な効果を示した。著者らによると、この研究は、微量リチウムとPQQの相乗効果により、複数の疾患原因メカニズムを標的とした新規AD治療戦略の有効性を実証した。[619]。
同様に、UTADによれば、健康的な食生活の一環として、セレンや亜鉛などの必須微量元素、必須n-3系PUFAsであるDHAやEPA、ビタミンなどを毎日十分に摂取することが治療計画の一部である。とはいえ、治療の初期段階では、潜在的な欠損をすべて評価し、食生活の変更によって欠損の発生がなくなるまで、サプリメントによって直ちに治療する必要がある。
まとめと考察
世界中で5万人以上のAD研究者が、増え続ける科学的データを構築し続けており、その一つ一つが未解決のパズルの新たなピースとなっている。パズルを分析的に分割しても、全体像には近づかないようだ。豊富なデータと矛盾する仮説の中から道筋を見つけるために、私は基本的な仮定をした: つまり、進化論は正しいのだから、相反する解釈を調和させるためのガイドであると同時に、確固たる土台として機能するはずだということである。そして実際に、ADを現代の伝染病と解釈することは、この観点から理にかなったものとなり、ADの行動学的原因、既知の危険因子、ADを克服する方法の理解を論理的に説明するUTADにつながった。とはいえ、”Alzheimer “という検索語は、米国国立生物工学情報センター(NCBI)で8万件をはるかに超える論文を検索した。そのため、ここで提案する行動欠損症としてのUTADとの整合性を一つ一つチェックすることは不可能であった。私が研究した一つ一つについて論じることさえ不可能であった。しかし、矛盾を見つけるたびに、それは研究計画や結果の(誤った)解釈によって説明された。非常に多くの場合、これらは相関性=因果性、あるいは正常性=自然性という誤った思い込みの結果であった(例えば、不自然な条件下で飼育された動物の老化は、自然な老化やADの自然な経過を反映しているとする)
UTADは、人間の類まれな長寿をもたらした進化論的論理と、なぜ我々の遺伝的プログラムがADや他の心を蝕む病気のような病気から我々を守るべきなのかを説明している。このプログラムは、他のプログラムと同様に、それが調整された条件下で最もよく機能する。近代化は遺伝プログラムが適応するよりも早く私たちの生活様式を変化させたため、また生産性の高い成人海馬神経新生は多くの文化が生んだ欠陥に特に弱いため、UTADの重要な部分であるLOMは、ADが多数の異なる個人の欠陥によって引き起こされる理由を簡単に説明してくれる。さらに、UTADは、現代社会におけるAD(大うつ病と同様)の流行や、経済成長率と並行して新興国におけるADの発症率や有病率が着実に上昇していることを理解するのに役立つ。しかし、ADの介入試験がしばしば失敗するか、限られた成功しか示さない理由も説明できる。介入群における個々の疾患の原因となる欠損をすべて修正するのではなく、限られた修正手段を用いて万能の解決策を提供するのである。介入群の中で、この(そしてこの!)特定の欠損に苦しむ数少ない個人だけが、欠損の矯正の恩恵を受けるからである。認知症との闘いに勝ちたいのであれば、UTADによれば、すべての(!)個々の危険因子(欠陥)を積極的に是正することのみが、成功する戦略であることが判明するかもしれない。
2006年、ブリントンとワンは、加齢に伴う成人海馬神経新生の低下の原因はまだ完全には解明されていないが、顆粒下帯(SGZ)の微小環境におけるFGF-2、IGF-1、VEGFのような成長因子の損失が、神経発生能の低下仮説の主要な一因であると論じている。[620]。彼らが知らなかったのは、2013年に発表されたブレイクスルー研究 [150]の結果に従い、高年齢であっても神経原性の可能性が残っているということである。高年齢まで神経原性潜在能力が持続していることの証明は、AD後期の死後脳の分析によってさえ示されている。[621]。興味深いことに、AD脳で観察されたこのアップレギュレーションは、疾患に関連した成人海馬神経新生の喪失がADにおける認知機能障害の重要な原因であるという仮定を覆す証拠であると主張する者もいる。[622]。したがって、この総説で提案されているように、成人海馬神経新生による行動障害を取り除くことによって成人海馬神経新生を再活性化することは、治療の選択肢にはならないかもしれない。しかし、これは妥当な反論だろうか?AD患者や動物モデルでは、増加、減少、変化なし、病期に依存した変化の両方が報告されているが、[623]、ADでは完全に成熟した機能的ニューロンの生成が損なわれているというのが、この分野の一般的なコンセンサスである(総説は [624]を参照)。実際、p-tauを介したAβシグナルは、ADにおいて異所性神経細胞周期の再エントリーを引き起こし、異常で非生産的な神経新生によって海馬の神経変性を引き起こす。[625]。その結果、神経前駆細胞(NPC)の増殖が増加しても、移動性の神経芽細胞の数も分化したニューロンの数も増加しないことが示された。[626]。このような病原性の影響はすべて、成人海馬神経新生を阻害する神経炎症性微小環境がADに蔓延していることと、生産的な成人海馬神経新生に必要な行動刺激や必須栄養素を海馬から奪い続ける変わらない生活習慣に起因している可能性が高い。例えば、ADにおけるNPCの非生産的な増殖は、トランスフォーミング成長因子β1(TGFβ1)のような、特に疾患の後期に作用する免疫調節因子の複合作用によっても引き起こされる可能性がある。
生理的な条件下でのTGFβ1シグナルは、主にNPCの増殖をアップレギュレートし、同時に神経発生幹細胞(NSC)を静止期に維持しながら、その機能分化を支援することによって新しく生成されたニューロンの生存を促進することによって、潜在的なAβ毒性からニューロンを保護し、[627]、成人海馬神経新生を増強する。[628]。対照的に、慢性的な神経炎症(上述したような複数の行動不全によって引き起こされる)のような病的条件下では、慢性的に上昇したTGFβ1レベルが疾患の進行に寄与する。[630, 631]。例えば、TGFβへの慢性的な曝露は、活性化した幹細胞の細胞周期のG1期を長くし、それによって神経前駆細胞の細胞周期の進行を損なう。[632]。同時に、高コルチゾールとAβの毒性レベル、そして炎症に起因する他の多くの病態作用が、歯状回ネットワークにおける成体生まれのニューロンの成熟と統合に害を及ぼす。
成人海馬神経新生に大きな治療的可能性があるという仮説は新しいものではなく、NSCを移植すればいつかADが治るかもしれないという期待を抱かせた。[68, 633]。このような楽観的な見方から、ADの動物モデルにおける認知機能低下に対する幹細胞移植の効果を調べる研究が盛んに行われた。[634]。ある実験では、増殖した幹細胞をマウスの海馬に移植すると、アポトーシス細胞死や酸化ストレスを抑制することによって、Aβが誘発する神経毒性や認知機能低下が改善されるという、予期せぬ証拠さえ得られた。[635]。これは確かに心強いニュースではあったが、一つの根本的な疑問が残った: 少なくともADの初期段階であれば、海馬の脳室下帯で再活性化された。「内因性」成人海馬神経新生が有益であるにもかかわらず、なぜ外因性のNSCを海馬に注入したいのだろうか?もちろん、成人海馬神経新生は主に歯状回の顆粒細胞と一部の介在ニューロンの形成に限定されているため[205]、すでに重篤な影響を受けているAD後期患者において、生活習慣への介入によって生産性の高い成人海馬神経新生を活性化することは、疾患修飾の可能性が低く(脳の大部分が不可逆的に影響を受ける)、現実的に困難である(コンプライアンスが損なわれる)。これが、私が早期診断を主張する主な理由である。
一方、ADを予防し、あるいは治癒させるために、多面的介入を求める専門家も増えている。[636, 637]。彼らは、Aβの産生、クリアランス、凝集を阻害するだけの薬剤で、複雑なカスケードの初期の単一ステップを標的とする単剤療法では不十分であることを理解している。しかし、この総説で私が提案するようなUTADがなければ、彼らの主張は研究コミュニティにとって説得力を欠き、大多数はいまだに「魔法の弾丸」、つまりライフスタイルを変える必要のない治療法を見つけるという希望に駆られている。特に、介入に関する提案のほとんどが、ADにおいて多かれ少なかれ同時に進行する複雑なプロセスすべてに対する包括的な説明を欠いているため、どの分子標的および/または行動障害に対処する必要があるのか、どのような治療介入が必要なのかという疑問に対して、これまでのところ、むしろ恣意的に答えが出されている。しかし、これは有害なことである。というのも、どの研究者も通常、ADに関する自分自身の概念を支持するため、多くの矛盾した仮説が生まれるからである。そのため、一般の人々は、そのような部分的な説明の長所と短所についての安定した情報を受け取ることになるが、それは(LOMに沿って)ADを完全に予防したり治したりする根拠にはならないかもしれない。
ADと闘うことは、人間の本性と闘うことではない。ADは主に加齢それ自体が致命的な原因であると主張することは、一般大衆を不当に恐怖に陥れる。さらに、研究者がその影響を軽視すれば、誰が自分の生き方を変えようとするだろうか。したがって、私たちはむしろ健康的なライフスタイルへの転換を奨励し、ADの原因となる欠陥をできるだけ早期に修正するために、早期診断サービスを提供すべきである。
結論
提案されているアルツハイマー病の統一理論(UTAD)は、ADを行動欠乏症と定義している。ADは、長く続く人類の自然な生活史によって説明される自然要件と、人類のごく最近の文化的(技術的、社会的、経済的)歴史によって説明される現代のライフスタイルとの間の不一致から生じている。
これらの欠乏は、精神的健康を維持するために必要な神経細胞の若返りと海馬の成体神経新生を妨げるだけでなく、他の多くの生理学的プロセスやシステムも阻害し、精神的衰えを悪化させ、加速させる。UTADは、よく知られた複数の危険因子(環境的、遺伝的なものを含む)がどのように相互作用してADを引き起こすかについて包括的な説明を提供し、ADの病因と病態への影響を明らかにする。さらに、UTADは、1つまたは少数の危険因子の是正に基づく予防戦略がなぜ失敗するか、あるいは限られた成功しか示さないかを明らかにし、同様に、主要な病理学的原因とその是正を無視した治療戦略がなぜ効果がないままであり続けるかを明らかにしている。
逆に、UTADは、散発的なADを高い確率で予防するために、生涯の精神的健康に必要な本質的プロセスを支配する最小限の法則に基づく体系的な生物学的アプローチを主張している。さらに、UTADが提供する論理的枠組みは、ADの主要な病理学的プロセスを中断させることを目的とした、有害な欠損の個別化された矯正と多方面にわたるシステム生物学的アプローチに基づく治療レジメンの開発を可能にする。したがって、私の理論的発見に基づくこの総説は、現在の予防戦略を再考し、根本的に変える必要があると主張している。さらに、主要な原因や欠陥を無視した単剤的治療戦略は、認知機能低下を逆転させる闘いのチャンスを患者に与えるために、すべての疾患原因因子を排除することを目的とした、疾患経過のできるだけ早期に開始されるべきシステム生物学的アプローチに取って代わられるべきである。
資金提供
著者は独立した研究者であり、自己資金で行っている。本総説のデザイン、公開データの分析、データの解釈、原稿の執筆に影響を与えた資金提供団体はない。
著者情報
MNは独立した研究者であり、学術界と産業界の両方で科学的成功を収めた長いキャリアを持つ。
利害関係
著者は独立した研究者であり、金銭的または非金銭的な利害関係はないことを表明する。
略語
AA、アラキドン酸;アミロイドβ、アミロイドβ;AcAc、アセト酢酸;ACh、アセチルコリン;AChEI、アセチルコリンエステラーゼ阻害剤;ACTH、副腎皮質刺激ホルモン;アルツハイマー病、アルツハイマー病;AGEs、高度糖化最終生成物;成人海馬神経新生、成人海馬神経新生;α-リポ酸、α-リポ酸;α7-nAChRs、α7含有ニコチン受容体。ALS、筋萎縮性側索硬化症;BBB、血液脳関門;BDNF、脳由来神経栄養因子;D-β-ヒドロキシ酪酸、D-β-ヒドロキシ酪酸;CA、コルヌアンモニ;脳脊髄液、脳脊髄液;慢性的カロリー制限、慢性カロリー制限;CR、コルチコステロイド受容体;CRF、コルチコトロピン放出因子;歯状回、歯状回;DHA、ドコサヘキサエン酸。DHLA、ジヒドロリポ酸;DPA、ドコサペンタエン酸;EEE、濃縮環境;イチョウ葉エキス、イチョウ葉からの抽出物;EPA、エイコサペンタエン酸;EPO、エリスロポエチン;ERK、細胞外シグナル調節キナーゼ;F歯状回-PET/CT、2-デオキシ-2-[フッ素-18]フルオロ-D-グルコースとコンピュータ断層撮影との統合;FGF-2、線維芽細胞増殖因子2。GDNF、グリア細胞由来神経栄養因子;GH、成長ホルモン;おばあさん仮説、祖母仮説;GSK-3α、グリコーゲン合成酵素キナーゼ-3α;GSK-3β、グリコーゲン合成酵素キナーゼ-3β;HC、海馬複合体;HCAR2、ヒドロキシカルボン酸受容体2;HDAC、ヒストン脱アセチラーゼ;HMIT、海馬記憶指数理論。HPA軸、視床下部-下垂体-副腎軸;HPE、オトギリソウ抽出物;IGF-1、インスリン様成長因子1;IL-1、インターロイキン-1;IL-6、インターロイキン-6;間欠断食、断続的絶食;LDL、低密度リポ蛋白質;LEC、側頭葉皮質;LTP、長期増強;長期増強、最小値の法則。LRP1、低密度リポ蛋白質受容体関連蛋白質1、中鎖脂肪酸s、中鎖トリグリセリド、MEC、内側内耳皮質、地中海式ダイエット、地中海式ダイエット、MeHg、メチル水銀、MCI、軽度認知障害、MMSE、ミニ精神状態検査、MRI、磁気共鳴画像法、mTOR、哺乳類ラパマイシン標的、NFL、ナショナルフットボールリーグ。NGF、神経成長因子; NPD1、神経保護D1; NPY、神経ペプチドY; n-3 PUFA、オメガ3多価不飽和脂肪酸; n-6 PUFA、オメガ6多価不飽和脂肪酸; ニューロンの若返り、神経細胞の若返り; PGC-1α、ペルオキシソーム増殖因子活性化受容体γ共活性化因子1α; PRH、末梢皮質; PUFAs、多価不飽和脂肪酸。REM、急速眼球運動;ROS、活性酸素;SCD、主観的認知機能低下;SSRI、選択的セロトニン再取り込み阻害薬;SWS、徐波睡眠;TFAs、トランス脂肪酸;THH、テトラヒドロハイパーフォリン;TNF-α、腫瘍壊死因子α;UTAD、アルツハイマー病の統一理論;VEGF、血管内皮増殖因子;西洋型食生活、欧米式食事療法

略歴
ミヒャエル・ネールズはドイツの医師、作家、元競輪選手である。
1983年から1989年までフライブルク大学とハイデルベルク大学で医学を学ぶ。1997年、大学院で分子遺伝学の学位を取得した。50本以上の科学論文を執筆し、そのうちの2本はノーベル賞受賞者のポール・グリーンガードとマーティン・エヴァンスと共同で執筆した。2015年には、適応免疫系の発達に必要な重要な分子スイッチの発見で研究者をリードした功績が認められ、米国免疫学会から「免疫学研究の柱」として表彰された。
科学者、経営者としての長いキャリアの後、元自転車選手は20年間座りっぱなしの生活を送った後、スポーツに戻ることを決意した。2008年、彼はカリフォルニア州オーシャンサイドをスタートし、メリーランド州アナポリスをゴールとする3,000マイルのレース、レース・アクロス・アメリカを10日間、22時間56分で見事に完走した。参加者27人中7位でゴールした。このレースの経験を綴った本『Herausforderung Race Across America』を執筆し、DVD『You need no victory to be a winner』をリリースした。
2011年からは、進化史の観点から健康的な老化に必要な行動変容について数冊の本を出版している。最初は『メトセラ戦略』、次いで『アルツハイマーの嘘』、『アルツハイマーは治る』(2017年英語版出版)というアルツハイマー病に関する本で、この特殊な認知症の発症について、進化生命誌とシステム生物学の観点から持論を展開している。アルツハイマー病の発症、予防、治療に関する画期的な発見が評価され、2015年にロストック大学のハンス精神医学賞を受賞した。
(出典:https://en.wikipedia.org/wiki/Michael_Nehls) (設立者:icekafe)
www.cbdb.cz/autor-95145-michael-nehls