
Contents
Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease
www.ncbi.nlm.nih.gov/pmc/articles/PMC7246646/
オンラインで2020年4月30日に公開
Thuy Trang Nguyen,1,† Qui Thanh Hoai Ta,2,† Thi Kim Oanh Nguyen,3 Thi Thuy Dung Nguyen,4,* and Vo Van Giau5,6,*.
概要
アルツハイマー病(AD)と2型糖尿病の正確な関連性についてはまだ議論されている。しかし、血糖値のコントロールが不十分だと、アルツハイマー病の発症リスクが高まる可能性がある。この関係は非常に強く、アルツハイマー病を「脳の糖尿病」あるいは「3型糖尿病(T3D)」と呼ぶ人もいるほどである。最近の研究では、T3DとADを関連付ける証拠が次々と示されていることから、この総説では、アミロイドβ(Aβ)前駆体タンパク質の毒性処理とAβのクリアランスの両方がインスリンシグナルの障害に起因すること、インスリン抵抗性が生体エネルギーの調節障害を媒介してADに進行することを踏まえ、T3DとADの関係を示すことを目的としている。さらに、インスリン関連の治療戦略は、ADの進行を遅らせたり、将来の合併症を食い止めることで、ADの治療法の開発に成功することが示唆されている。
キーワード
アルツハイマー型認知症、代謝低下、2型糖尿病、3型糖尿病、インスリン抵抗性
1.はじめに
糖尿病は、世界中の個人、家族、社会の生活や幸福に大きな影響を与える重篤な長期疾患である。2019年の世界の糖尿病罹患率は9.3%(4億6,300万人)で、2030年には10.2%(5億7,800万人)2045年には10.9%(7億人)に上昇すると推定されている[1]。また、人口の高齢化は世界中で劇的に進んでおり、特に発展途上国では、医療システムだけでなく、社会保障サービスや政策にも圧力がかかっている。ベトナムでは、2030年までに糖尿病が死亡や障害の原因となる上位7疾患の一つになると予測されている[2,3]。糖尿病の増加に伴い、現在、ベトナムには約576万人の糖尿病患者がいる。ベトナムの人口における糖尿病の年齢調整済み比較有病率は 2017年には約6%であった[2]。現在では、多くの人が1型糖尿病や2型糖尿病に精通しているが、つい最近、3型糖尿病(T3DM)と呼ばれる別の形態の糖尿病が確認されている。あまり知られていないこのタイプは、脳内でインスリン抵抗性を示し、神経認知に影響を与える可能性が高く、アルツハイマー病(AD)の病因にもなると言われている。
アルツハイマー病は、米国における死亡原因の第6位、65歳以上の高齢者における死亡原因の第5位にランクされている[4]。現在のところ治療法はないが、症状に対する治療法はあり、研究も続けられている。神経伝達物質の欠損、変性した神経細胞、シナプス機能不全、細胞外に蓄積したβアミロイド(Aβ)と細胞内の神経原線維変化(NFT)が、ADに存在する主な粗大な醜形である[5]。アミロイド前駆体タンパク質(APP)は、γセクレターゼ複合体の活性酵素であるプレセニリン1(PSEN1)とPSEN2によって、Aβ38,Aβ40,Aβ42などの長さの異なるAβペプチドを生成し、膜内の複数の部位で切断される。
残念ながら、糖尿病は死亡原因の第7位としてADに続いており、2045年までに約5億人が罹患すると予測されている[1]。この2つの疾患は、環境と遺伝の両方が関与する多因子性の相互作用であると認識されている。しかし、インスリン抵抗性は、記憶障害、認知機能の低下、およびADに見られる特徴的な症状の多くと関連している。同時に、2型糖尿病は、AD発症の最も調整可能な危険因子の1つであり続けている。
糖尿病は、1型、2型、3型、4型の4つの臨床カテゴリーに分類される。1型糖尿病(1型糖尿病)は、主にβ細胞の破壊が原因で、ほとんどが絶対的なインスリン欠乏状態に陥る。2型糖尿病(2型糖尿病)は、インスリン抵抗性を伴ったインスリン分泌不全の進行によるものである。インスリン抵抗性は、肥満と密接に関連した一般的な現象であり、標的組織がインスリンに対して正常に反応できない状態と定義される。インスリン抵抗性は、通常、2型糖尿病の発症に数年先行している。2型糖尿病は、認知症や、認知症の中でも最も多いタイプであるADの危険因子とされている。1型糖尿病は主に小児や若年層に見られるのに対し、2型糖尿病は成人に多く見られ、世界の発症数の90%を占めている[6]。いくつかの疫学研究では、インスリン抵抗性が、糖尿病ではない集団においても、認知症やADのリスクを高めることが示唆されている。
最近発見された3型糖尿病(T3DM)は、科学者たちによって「3型糖尿病」と呼ばれるようになった。これらの科学者は、T3DMを、進行性の脳のインスリン抵抗性に関連した異常を引き起こし、その結果、中枢のインスリンシグナル伝達プロセスの障害、神経毒の蓄積、神経細胞のストレス、そして神経変性の経過につながるメタボリックシンドロームと定義しようとしている[7,8]。
In vitroおよび動物実験では、インスリン抵抗性が複数の異なる経路を介してADの病因に寄与することが示されている[7]。内分泌系の異常、特に糖尿病は、糖尿病の一種であるADによく見られる。糖尿病が記憶処理(認識・検索)脳の形態(脳萎縮)シナプス伝達に影響を及ぼすことは、ADの病態に影響を及ぼす危険な側面としてよく知られている[9]。
また、高インスリン血症によるインスリンシグナル伝達の障害とインスリン抵抗性は、遺伝子型にかかわらず、両病態の中心にインスリンを据える意味を持つ重要な因子である[10]。最近の多くの研究では、海馬のインスリンシグナルが低下すると、記憶やその他の実行機能が損なわれることが指摘されており、これは、インスリンシグナルの低下とインスリン抵抗性の同時進行が原因であると考えられている[11,12,13]。
この考察は、高インスリン血症とインスリン抵抗性、そしてその結果として生じるT3DやADなどの病態との間に強い関連性があることを提唱している[14]。末梢のインスリン抵抗性は、中枢神経系でのインスリンシグナルの減少につながり、続いて脳の代謝が変化する。Aβ毒性の増加、タウの過リン酸化、酸化ストレス、神経炎症は、中枢性インスリン抵抗性に起因し、神経変性を引き起こす。
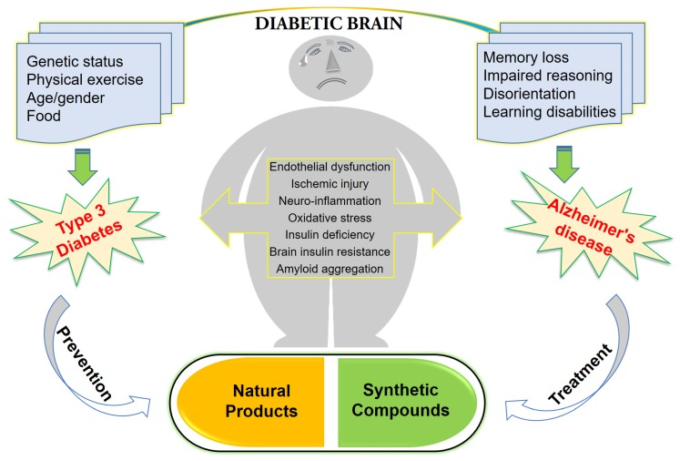
この研究では、アミロイドβ(Aβ)前駆体タンパク質の毒性の処理とAβのクリアランスの両方が、脳内のインスリンシグナルの障害に起因するという事実に基づいて、T3DMとADの関係を提示している。さらに、インスリン関連の治療戦略は、ADの進行を遅らせたり、将来の合併症を食い止めたりすることで、ADの治療法の開発に成功することが示唆されている。図1は、ADとその治療・予防に関するT3Dの概念を示したものである。
図1 アルツハイマー病に関する3型糖尿病とその治療・予防への取り組み
2. 中枢神経系(CNS)におけるインスリンとグルカゴンのシグナル伝達
インスリンは、血中のグルコース濃度を調節するホルモンで、膵臓のランゲルハンス島のβ細胞で産生され、ジスルフィド結合で結ばれた2本のポリペプチド鎖からなる。インスリンは,2つのαサブユニットと2つのβサブユニットで形成される膜貫通型糖タンパク質受容体に結合することで作用を開始する[14]。インスリンが受容体のαサブユニットに結合すると、インスリンが活性化され、βサブユニットの細胞質領域にあるいくつかのTyr残基が自己リン酸化されることが確認された[15,16]。自己リン酸化された残骸は、インスリン受容体基質(IRS)によって認識され、その中でもIRS-1とIRS-2は、インスリンシグナル伝達における2つの主要なプレーヤーであり、共通の中間体である。IRSは、細胞内のシグナル伝達経路を仲介する分子複合体を構成するのに適した理想的な存在である。インスリンとインスリン様成長因子(IGF-1)は、チロシンキナーゼ受容体であるインスリン受容体(IR)とIGF-1に接続する。インスリンの結合は、嗅球、大脳皮質、海馬で最も高い。さらに、インスリン受容体は、血液脳関門の内皮細胞にも発現しており、インスリンとIGF-1が血液脳関門(BBB)を通って中枢神経系に輸送される役割を担っている[17]。インスリンが脳に入る正確なメカニズムについてはまだ議論の余地があるが、血液中のインスリンは、受容体を介した能動輸送システムによってBBBを通過することができる[17]。この経路は、末梢からインスリンを注入すると、脳脊髄液(CSF)中のインスリン濃度が血中インスリンと比例して上昇するという研究結果と一致している[15,16,17]。しかし、脳内で産生されるインスリンの量や、このプールのインスリンが生理的に意味を持つかどうかについては、まだ解明されていない。脳内でのシグナル伝達には、中枢と末梢の両方のインスリンプールが重要である可能性がある。
インスリンとIGF-1には、神経細胞の生存と中枢神経系の健全性の維持に重要な機能が与えられている。インスリン受容体とインスリンのシグナル伝達は、カルシウムの流入、神経伝達物質の蓄積とシナプス結合、アポトーシス、神経新生など、受容体を介したいくつかのメカニズムに影響を与えることで、グルコースのホメオスタシス、神経細胞の統合性、認知に影響を与える [17]。また、インスリンは、長期増強(LTP)や長期抑圧(LTD)に強い影響を与えるGABA、NMDA、AMPAを介したメカニズムの発現やレベルを調節する。さらに、インスリンは、インスリンシグナルに不可欠なAKT-mTORおよびRas関連経路[19,20]の活性化を介して、興奮性シナプス[18]および樹状突起スパインの形成の拡大と保存に決定的に関与している[21]。また、インスリンは、アポトーシス経路やアポトーシスカスケードに関与する中間体を調節することで、細胞の生存にも影響を与えている[22,23]。
脳内にインスリンが存在することは、Havrankovaら[24]によって初めて報告された。彼らはラジオイムノアッセイを用いて、脳の抽出液中に高濃度のインスリンを検出した。また、その後、ヒトの脳だけでなく、いくつかの実験動物においても、高いインスリン濃度が報告されている[25]。最近では、中枢神経系におけるインスリンの産生についても広く研究されており、脳内でのインスリン生合成の可能性を示唆する様々な実験的証拠が得られている。ラットの視床下部の室周囲核にインスリンのmRNAが存在することが,in situ hybridizationによって明らかになった[26]。中枢神経系におけるインスリンの産生と分泌に関わる分子メカニズムは、特にATP感受性K+チャネルの脱分極に関連して、β細胞と神経細胞の間に類似性があることが明らかにされている[27]。この脱分極によるインスリンの放出は,シクロヘキシミドで抑制することができ,アストロサイトではなく,神経細胞に特異的であった[28]。興味深いことに、インスリン受容体(IR)を介したプロセスの機能障害は、IRの活性化の異常、インスリンの利用可能性の低下、IRをきっかけとした下流のメカニズムの低下などが原因となり、広範囲の脳疾患を引き起こす可能性がある[29]。また、IRは核内でRNAポリメラーゼIIと結合しており、ゲノム全体のプロモーターに顕著な濃縮を示すことが最近明らかになった[30]。これらの結果は、IRがプロモーターにおいて転写装置と相互作用することを明らかにし、インスリンの生理作用や関連する疾患に関連する遺伝子を制御する経路を特定している[30]。このように、インスリンは、これらの経路のいずれかに影響を及ぼすことで、神経細胞の性能や統合性を変化させ、その結果、学習や記憶などのADの特徴に障害をもたらす可能性がある。以前の研究では、アルツハイマー病患者と年齢をマッチさせた対照群では、脳内インスリンが同等に減少していることが示されており、脳内インスリンの減少は、ADではなく加齢の結果である可能性が高いことが示されている[31]。最終的には、健康な脳と病気の脳におけるインスリンの機能を完全に理解するためには、ADの重症度と年齢をマッチさせた対照群に関連した脳内インスリンについて、より深く理解する必要がある。このように、中枢神経系におけるインスリンレベルの低下は、抗アミロイド性タンパク質のレベルを低下させ、Aβの過剰産生とクリアランスの障害の両方を引き起こす可能性がある。
3. グルコースホメオスタシスにおける3型糖尿病の役割
糖尿病とこれらの他の領域との関係を理解する鍵は、糖尿病におけるエネルギーホメオスタシスの役割から始まる。エネルギー恒常性は、摂食行動とエネルギー消費の調整に依存する、よく調節されたプロセスである。近年、肥満や糖尿病などの発症による変化に伴い、ヒトのエネルギー恒常性の制御が注目されている。成熟した神経細胞には、神経細胞が死滅したり、神経変性や神経細胞消失などの病的状態に陥りやすい2つの特徴がある。1つ目の特徴は、完全に分化した(成体)神経細胞は永久的に分裂終了細胞であり、再生能力を持たないことである[32]。そのため、成体神経細胞がアデノシン三リン酸(ATP)部位の不足やエネルギー危機、酸化ストレスなどの細胞ストレスにさらされると、死滅するかアポトーシスを起こすか、退化するか神経細胞の変性・消失を起こし、神経変性疾患の素因となる[32]。第二の重要な特徴は、脳の神経細胞や組織は非常に要求の厳しい興奮性細胞であり、現在のATPの40%以上が神経細胞の生存または存続のために使用されているということである[33]。脳のグルコースの供給源は2つあり、基礎的なインスリンレベルによる皮質のグルコース代謝の刺激[34]と、グリアのβ-アドレナリン受容体の活性化によって刺激されるアストロサイトのグリコーゲンのグルコースへの変換がある。グルコース取り込みの増加は,インスリン感受性の高いグリアグルコーストランスポーター1型(GLUT1)によって細胞膜に輸送され,神経細胞で利用される。したがって,細胞内のバランスのとれたグルコース輸送は,脳に発現しているアストロサイトとグルコーストランスポーターに依存することになる[35]。
さらに,脳内のグルコース代謝障害の結果としてグルコースの取り込みが損なわれることにより,グルコースホメオスタシスの障害がT3DMの発症に重要であるかもしれない。グルコース輸送異常に関与するメカニズムとしては、脳のインスリン抵抗性と細胞内グルコース代謝障害が挙げられる。この2つの異常が、T3DMの脳内グルコース代謝低下、あるいは脳のインスリン抵抗性疾患状態の原因になっていると考えられる。神経変性疾患では、グルコーストランスポーターの減少がタウの異常な高リン化と相関していることが報告されている[36]。したがって、インスリンシグナルの障害は、全身の血糖値に影響を与えるだけでなく、さまざまな変性プロセスや神経細胞の死滅・消失を引き起こす[37]。また、2型糖尿病におけるインスリン抵抗性は、「インスリンの作用に対する体組織の感受性の低下」と定義されている[38]。同様に、脳のインスリン抵抗性は、脳細胞がインスリンとそれに対応するインスリン受容体に反応しないことと定義できる[39]。その結果、細胞膜に発現しているGLUTの数が減少するため、神経細胞内でのインスリン欠乏とグルコース輸送の障害が生じる。さらに、中枢神経系におけるインスリン抵抗性は、末梢におけるインスリン抵抗性と相関している。したがって、インスリンに対する反応性が失われると、神経細胞はインスリンの保護作用を受けられなくなるため、神経毒性の強い傷害を受けやすくなる可能性がある[40]。さらに、インスリン抵抗性の患者は、アポトーシス、神経変性、その結果としての認知機能の低下など、多くの病理学的特徴が増加している。
脳のインスリン抵抗性における神経細胞のインスリン受容体の脱感作は、2型糖尿病におけるプロセスと同様であり、T3DMとその将来の合併症を引き起こす上で重要な役割を果たしていると考えられる[41]。その上、2型糖尿病はインスリン抵抗性を特徴とするメタボリックシンドロームであり、これは神経変性症や神経内分泌疾患、あるいはT3DMの病理学的特徴でもある[34]。このように、グルコースのホメオスタシスは、T3DMの病因に一役買っている。T3DMでは、脳のグルコースの取り込みまたは代謝が損なわれる。したがって、2型糖尿病と脳の神経変性疾患の組み合わせは、このT3DMまたは神経内分泌障害と呼ばれる新しい糖尿病の分類として考えられるかもしれない。
4. 3型糖尿病とAβ蛋白質の病態
アミロイドーシスは、細胞外のアミロイド沈着を特徴とする線維性タンパク質の蓄積からなる病態であり、罹患する組織によって臨床的な差異がある。近年、ADの病態とインスリン抵抗性との関係について、新たな知見が得られている。2型糖尿病は、認知症患者の脳内にアミロイドβの沈着物が形成されるために不可欠な危険因子であると考えることが重要だ。インスリンへの継続的な曝露と神経細胞内でのAβ蓄積との間には、有害なサイクルが存在していた[42]。Farrisらによると,インスリン分解酵素(IDE)は,生体内のインスリン,Aβタンパク質,アミロイド前駆体タンパク質(APP)の細胞内ドメインのレベルを調節しているという[42].本研究では,変異型IDEの2型糖尿病モデルラットでは,2型糖尿病やT3DMの特徴である高インスリン血症や耐糖能異常,あるいは脳のインスリン抵抗性が認められた。このことは、IDEの機能低下がT3DMや2型糖尿病の原因となっている可能性を示唆しており、高インスリン血症、糖尿病、神経変性や神経細胞の喪失との間に最近認められた関連性のメカニズムを提供するものである[42]。したがって、健常者では、IDEはAβを減少させ、インスリンを調節し、さらにAPP細胞内ドメイン(AICD)を分解する。このように、インスリン、IDE、Aβの間には調節関係があった。脳のインスリン抵抗性の場合、インスリンがAβのクリアランスを促進できず、神経細胞内にAβが蓄積され、T3DMや脳のインスリン抵抗性の特徴である神経変性や神経細胞の損失を引き起こす可能性がある[42]。T3DMや脳のインスリン抵抗性については、Aβの発現やタンパク質処理の異常の結果なのか、原因なのかという議論がある[43]。T3DMが結果であるという概念からすると、Aβの毒性は脳のインスリン抵抗性を引き起こす可能性がある。Aβは、インスリンとその受容体で競合し[44]、IRの表面発現を低下させ、相対する受容体へのインスリンの親和性を低下させることで、インスリンシグナルを撹乱し、ホスファチジルイノシトール-4, 5-ビスリン酸3キナーゼ(PI3K)/Aktの活性化に直接干渉し、そのシグナル伝達の遮断を引き起こし、生存シグナルの障害、GSK-3β活性の活性化の増加、タウの過リン酸化の増加を引き起こす[45]。
一方、T3DMが原因という概念では、図2に示すように、酸化ストレスや神経炎症を伴う脳のインスリン抵抗性がAβ蓄積を引き起こす可能性がある。この概念を取り入れた研究では、インスリンの刺激がゴルジ網から細胞膜へのAβのトラフィッキングを増加または加速させる可能性があると主張している。したがって,インスリンはAβの細胞外への排泄を活性化すると同時に,インスリン分解酵素(IDE)による分解を活性化することで,Aβの細胞内への蓄積を抑制する可能性がある[46]。このように、インスリンのシグナル伝達が損なわれると、APPのプロセッシングとAβのクリアランスの両方が阻害されることになる[47]。その結果、神経細胞に対するAβの神経毒性が増大し、神経変性や神経細胞死を引き起こす可能性がある。2型糖尿病患者とアルツハイマー病患者は、膵臓と脳の両方で同様のアミロイドβ沈着を起こしている。何人かの研究者は、この新しい病理をT3Dとして扱うべきだと提案している[48,49,50,51]。インスリン様成長因子1(IGF-1)やペルオキシソーム増殖剤活性化受容体ガンマ(PPARG)など、2型糖尿病の標的受容体のいくつかは、タウタンパク質の発現やリン酸化の制御にも関与している[51]。
図2 脳のインスリン抵抗性とAβの凝集およびその毒性
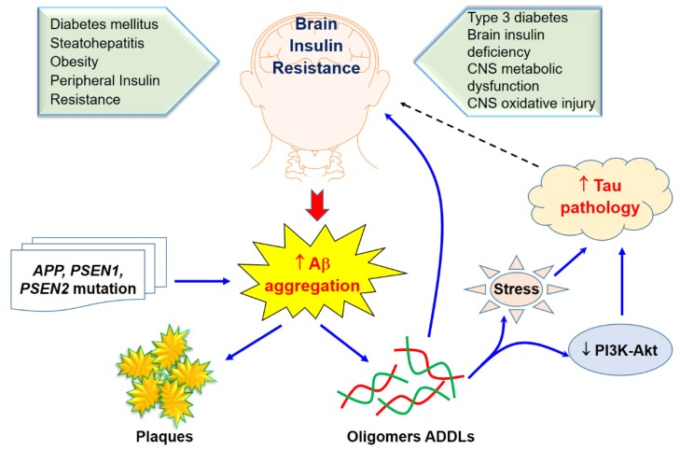
実線の矢印は、Aβの凝集が何らかの経路を介して脳のインスリン抵抗性に影響を与えることを示し、破線の矢印は、タウの病理が脳のインスリンに影響を与える可能性が高いことを示している。
5. 3型糖尿病とアルツハイマー病の関係
アルツハイマー病や糖尿病におけるインスリン抵抗性は、高インスリン血症を引き起こし、インスリンやAβの分解に必要なインスリン分解酵素(IDE)を飽和させる。近年、多くの研究が、2型糖尿病患者や肥満の人にADの発症率が高いことを示しており、これらの疾患を引き起こす共通のメカニズムを示唆している[10,52,53]。インスリン抵抗性は、糖尿病、肥満、ADに共通する主な特徴であると考えられる[54]。神経細胞のグルコース取り込みは、インスリンに完全に依存しているわけではないので、脳におけるインスリン抵抗性の概念は、インスリンのシグナル伝達経路の障害に関連していると考えられる。インスリンシグナル伝達経路の機能不全とその結果として観察される代謝低下の状態は、ADと2型糖尿病を結びつける生体エネルギーの変化の要因の一つと考えられている[55]。インスリン抵抗性の状態は、2型糖尿病の重要な予測因子であるインスリンの極端な上昇と末梢での相対的なインスリン活性の低下を伴って、神経細胞の機能と認知能力の低下をもたらす可能性がある[56,57]。その結果、神経斑の発生、海馬の萎縮、認知能力の低下、大脳皮質のグルコース代謝の低下につながり、記憶障害と密接な相関があると考えられる[50]。以前の研究では、p-Ser312IRS1の増加は、10年前にアルツハイマー病患者としてこれらの変化を維持していた前駆アルツハイマー病患者に現れたことが明らかにされた[58]。このことは、ADにおけるインスリン抵抗性は、臨床症状が現れる何年も前に発症することを示唆しており、神経由来のエクソソームは早期AD診断の可能性を秘めている。ADや2型糖尿病では、インスリン反応の欠如、インスリン受容体の機能低下、インスリン受容体の結合低下、インスリンシグナルカスケードの不適切な活性化などにより、脳内のインスリンシグナルが欠損している。このカスケードの変化による主な結果は、神経細胞のグルコース取り込みの低下であり、これは神経可塑性の低下、神経伝達物質の減少、生体エネルギー機構の崩壊、致命的な炎症カスケードの開始として現れている。全体として、インスリンシグナルの障害の結果は、脳の代謝障害に起因し、脳の機能不全を引き起こす可能性があり、表1に示すように、糖尿病、肥満、ADの関連性を説明する可能性がある[11]。
表1 T3DとADとの関連性を示す因果モデル
| 上流の危険因子 | 代謝前駆体 | 経路 | 無症候性病理学 | 病気の結果 | |
|---|---|---|---|---|---|
| 社会的要因:ストレス、低い社会経済的地位、特定の民族的および人種的グループ | 肥満 内臓脂肪症 |
血管プロセス 血圧と高血圧 高脂血症 アポリポタンパク質E |
脳血流 アテローム性動脈硬化症 |
アミロイド前駆体タンパク質 | アルツハイマー病 |
| 貧しい食生活:カロリー、脂肪、糖分が多く、5人が少ない | Inflammatory/Oxidative processes Inflammation Oxidative stress Endothelial function |
神経原線維 もつれ アミロイドB預金 |
|||
| 運動不足の 遺伝学と家族歴 |
高血糖高 インスリン血症 |
代謝過程 インスリン抵抗性 インスリン分解酵素 ペルオキシソーム増殖活性化 受容体 |
|||
| 子宮内および出生時体重における幼児期の曝露 | 脳と海馬の萎縮 白質の高信号 |
||||
インスリン抵抗性またはインスリンシグナルの機能不全は、グルコース代謝の変化による2型糖尿病の普遍的な特徴であり、その細胞死経路との相互依存関係は、図3に示すように、T3DとADを結びつける基礎となっている。T3Dは、脳の神経細胞が、記憶や学習などの基本的な作業に不可欠なインスリンに反応できなくなることで起こる。研究者の中には、インスリンの欠乏がADの認知機能低下の中心であると考えている人もいる。インスリン経路の機能不全やインスリン抵抗性は、受容体の機能不全、受容体の発現の変化、受容体結合の逸脱、リン酸化連鎖のイベントの誤作動やリン酸化に関わるキナーゼの活動の変化などの状態である。分子レベルでは、細胞はインスリン受容体を介してインスリンを感知し、そのシグナルはPI3K/Akt/mTORシグナル伝達経路と総称されるシグナルカスケードを介して伝播する。最近の研究では、ある種の細胞の生理的条件下では、この経路が双安定スイッチとして機能していることが示唆されており、インスリンの反応は閾値現象である可能性が高いと考えられている[13,59,60]。この経路のインスリンに対する感受性は、遊離脂肪酸などの多くの要因によって鈍化し、インスリン抵抗性を引き起こす可能性がある。また、インスリン抵抗性は、細胞をミトコンドリアアンカプラー(脱共益剤)、電子輸送鎖阻害剤、ミトコンドリアスーパーオキシドディスムターゼ模倣剤にさらすことで速やかに回復する可能性があるという知見にも基づいている[61,62]。
図3 インスリン抵抗性とアルツハイマー病をつなぐ分子経路を模式的に示したもの
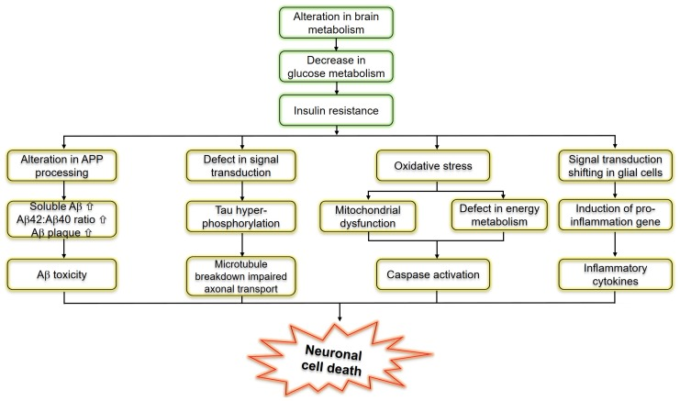
興味深いことに、インスリンシグナルの障害は、いくつかのトランスジェニックおよび非トランスジェニックのADモデルマウスで見られる。以前の臨床研究では、アルツハイマー病患者が耐糖能異常を有する可能性があることが報告されており、この2つの疾患の間には双方向の関係があることが示唆されている[63,64]。生後10カ月のマウスでは、海馬の抽出物の膜に結合しているIRS-1のレベルが低下しており[65]、海馬と大脳皮質におけるIRS-1とPI3Kの活性化が低下していることが観察された[66]。APP/PS1マウスの視床下部では、9ヵ月齢の海馬でセリン616にリン酸化されたIRS-1が対照群よりも高く[68]、13ヵ月齢の前頭皮質でセリン636および312にリン酸化されたIRS-1のレベルが上昇していることから、インスリン抵抗性のマーカーも報告されている[69]。また、末梢のインスリン抵抗性と併せて、5ヵ月齢のtg2576マウスの海馬では、IRS-1のセリン612でのリン酸化が抑制的に増加しているという報告もあった[66]。驚くべきことに、AβOの中枢注入は末梢のインスリン抵抗性をもたらし、これはAPP/PS1や3xTgADのADモデルマウスでもさらに観察された[70]。これらの概念を確認するためには、ADが糖尿病の表現型に影響を与えるメカニズムを調査するために、さらなる証拠が必要である。図2に示すように、天然の合成化合物を用いたADとその治療・予防のアプローチに関するT3D。
また、世界的な糖尿病の増加を受けて、この疾患に影響を与えるシグナル伝達経路を理解することが研究の大きな焦点となっている。インスリンのシグナル伝達は、主に肝臓、骨格筋、脂肪組織への作用を介して、グルコース、脂質、エネルギーのホメオスタシスを制御している。細胞シグナル伝達経路は、経路の構成要素間で起こる生体分子反応のリストで説明することができる。インスリンとインスリン様成長因子-1(IGF1)のシグナル伝達の障害(IIS)を伴う2型糖尿病は、認知機能障害とADを含む認知症の危険因子である[71]。重要なことは、全身のヘテロ接合でIGF1Rを不活化(IGF1R+/-)または神経細胞でIGF1Rを欠失(nIGF1R-/-)させると、ADのTg2576モデルマウスにおいて、行動障害やAβ蓄積を抑制しながら生存率を向上させることができたことである[72]。全身または脳内のIRS2シグナルの減少は寿命を延ばす[73]。IRS2の全身的な減少(IRS2-/-)は、認知機能を改善し、血糖値が正常なTg2576マウスのAβ沈着と早死にを減少させる可能性がある[72,74]。したがって、より最近の動物実験では、中枢神経系におけるIRカスケードではなく、IGF1R-IRS2シグナルを媒介とする細胞内シグナルの減少が、AD動物モデルにおいて神経保護効果を発揮することが明らかになっている[71]。
6. アルツハイマー病における3型糖尿病治療へのアプローチ
インスリン抵抗性は、T3Dの本質的な特徴としてよく知られており、したがって、T3Dの治療戦略、特にインスリン感受性の改善を目的とした治療戦略は、ADのリスクがある患者の初期段階にも役立つ可能性がある。糖尿病、インスリン抵抗性、および認知機能低下の間には、重複しながらも異なる病理学的特徴があるため、製薬業界の研究の観点から、栄養補助食品、抗酸化活性[76]、ポリフェノール、オメガ3脂肪酸、および脳と腸のつながり[77]を含む、ライフスタイル介入とともに、複数の標的を持つ薬物療法も検討されている[75]。
栄養補助食品の中でも、脳に浸透しやすい化合物であるクルクミンは、異常なタンパク質の凝集体を標的にすることができる[78]。クルクミンはまた、「初代海馬ニューロン培養におけるプロアポトーシス・シグナル伝達経路」を阻止する可能性がある。これまでの研究でも、マウスのメトホルミンをクルクミンやピペリンの補給と組み合わせることで、特にインスリン感受性やシグナル伝達の強化、全身の耐糖能の改善に関して、アルツハイマー病患者にとって有望な天然物質であることが示されている[78]。しかし、果物や野菜の抗炎症作用については、何十年も前から広く知られており、特に、炎症によるダメージを軽減する抗酸化作用については知られている[79]。齧歯類の研究では、カロテノイド、抗酸化ビタミン、ポリフェノール、フラボノイドなどの無数の生物活性成分により、様々な野菜や果物が「食事性酸化ストレスによる認知機能や脳の神経病理に対する」保護作用があるとされている[80]。様々な種類のフラボノイドが、生体内試験モデルを介して治療に貢献する可能性が示唆されている[81]。このことは、ADの予防や進行の抑制に向けた積極的なアプローチについての理解を深める上で、大きな可能性を秘めている。脳の発達と維持におけるオメガ3脂肪酸の重要な役割は、特にこの10年間でよく知られるようになったが、「脳の老化に対する影響が調査された」のはごく最近のことである[82]。オメガ3脂肪酸が豊富で、オメガ6脂肪酸が自然に少ない食生活は、アルツハイマー病患者の栄養療法の鍵を握っているかもしれない[83]。ケトジェニック食は、損傷したミトコンドリアを回復させ、普遍的な炎症を減少させながら、脳内のベータアミロイド斑を減少させ、クリアにする可能性さえある[84]。新しい研究では、糖化したAPOE4タンパク質とインスリン・シグナル伝達の不具合が、脳組織のエネルギー輸送障害だけでなく、主にコレステロールなどの脂質輸送障害にもつながることが明らかになった[84,85]。一般人口の約20%、アルツハイマー病患者の50%以上を占めるAPOE4は、脳がインスリンを処理する方法を阻害する原因となっている[86]。この遺伝子と、高脂肪食による末梢のインスリン抵抗性が相まって、脳のインスリン抵抗性を誘発した[87]。この遺伝子によって産生されるAPOE4タンパク質は、通常のAPOE3よりも神経細胞表面のインスリン受容体に積極的に結合することができる。APOE4はその後、脳細胞に永続的なダメージを与える。受容体をブロックした後、粘着性のあるAPOE4タンパク質は塊を作り始め、毒性を持つようになる[87]。さらに、タンパク質が神経細胞の内部に入ると、その塊は細胞の機械の中にロックダウンられ、受容体が神経細胞の表面に戻って仕事をするのを妨げてしまう。インスリンの信号処理はますます損なわれ、脳細胞を飢えさせてしまうのである。全身の血管系を改善する上で、運動以上に効果のある医薬的介入は、これまで存在しなかった [88]。このことは、生活の質の向上、脳内の神経化学物質の伝達、インスリン抵抗性の回復、特定の個人におけるAβプラークの除去能力など、アルツハイマー病患者や2型糖尿病患者にとっても大きな意味を持つ [88]。腸と脳の間の双方向のコミュニケーションである「腸脳軸」の概念は、多くの実験的および臨床的研究によって裏付けられているADの病因に大きく貢献している[77]。ADの進行に関するT3Dの治療または予防のためのいくつかの化合物および薬剤の代表的なものを表2に示した。
表2 抗糖尿病薬、インスリン増感薬のAD病態の様々な側面に対する有効性に関する前臨床および臨床研究の代表的なものをまとめたもの
| 化合物 | 潜在的な経路 | 研究デザイン | リファレンス |
|---|---|---|---|
| DA5-CH | タウのリン酸化を減らし、シータリズムを正常化します | ラットに注射された脳室内(ICV)、ストレプトゾトシン | [89] |
| DA-JC1 | Aβによって誘発される概日リズム障害を拮抗31-35 | ICV、アミロイド(31–35)ADモデル | [ 90 ] |
| DA5-CH | 海馬シナプス可塑性の改善とPI3K / AKTシグナル伝達経路の活性化 | ADのAPP / PS1マウスモデル | [ 91 ] |
| DA-CH3 | 小胞体ストレスとアポトーシスシグナル伝達の低下、脳内のアミロイドプラーク負荷の低下 | ADのAPP / PS1マウスモデル | [ 92 ] |
| インスリン | Aβオリゴマー誘発シナプス喪失およびインスリン受容体減少の予防、PKR媒介小胞体ストレスの改善 | ラット海馬ニューロン培養 | [ 93、94 ] |
| インスリン | ε4キャリアではないAD患者は、インスリンに対する感受性が低下し、認知能力に影響を及ぼします | ApoE-ε4対立遺伝子がホモ接合であるかどうかにかかわらずAD患者および静脈内注射された正常な被験者 | [ 95 ] |
| インスリン | 急性インスリン投与後のMCIADε4被験者の言語記憶の改善が、ε4キャリアでは改善されなかった | ApoE-ε4対立遺伝子がホモ接合であるかどうかにかかわらず、AD患者、MCI患者、および鼻腔内投与されたほとんどの被験者 | [ 96、97 ] |
| インスリン | 慢性的な鼻腔内インスリン投与量は、選択的注意、新しい情報の保持、およびMCIと初期のAD被験者の機能状態を強化しました | AD患者、MCI患者、および鼻腔内投与された正常な被験者 | [ 98 ] |
| インスリン | 治療後に改善された作業記憶を示したのは女性だけでした | 健康な男性と女性の鼻腔内投与 | [ 99 ] |
| リラグルチド | タウリン酸化の減少; c-AMP依存的な方法でのインスリン受容とシナプス喪失の保護 | Aβオリゴマーを含むカニクイザルICV | [ 100 ] |
| リラグルチド | 新規物体認識テストと恐怖条件付けにおける記憶障害の改善 | スイスのマウスにAβオリゴマーをICVに注射 | [ 100 ] |
| リラグルチド | 物体認識テストとモリス水迷路での記憶障害の回復。強化されたLTP; ミクログリアの活性化の低下; Aβプラーク負荷の減少 | APP / PSEN1マウス | [ 101、102 ] |
| エキセンディン-4 | 活性化Tyr465IRS1リン酸化を回復しながら、INKのSer312IRS1、Ser66IRS1の阻害的リン酸化を減少させる | ラット海馬神経培養 | [ 69 ] |
| エキセンディン-4 | モリス水迷路における空間記憶の改善; アミロイドプラークの減少負荷 | APP / PS1マウス | [ 69 ] |
| Exedin4-リラグルチド | eIF2α phosphorylation reduction | ラット海馬神経培養、APP / PS1マウス、カニクイザルにAβオリゴマーをICVに注射 | [ 94 ] |
| GLP-1エキセンディン-4 | 神経興奮毒性の減少 | ラット海馬神経培養、イボテン酸を基底核に注射したラット | [ 103 ] |
| ロシグリタゾン | 物体認識テストとモリス水迷路における記憶障害の逆転; Aβレベルの低下 | ADトランスジェニックマウスJ20系統 | [ 104 ] |
7.結論
T3DMとADの関係は、AβPPのプロセッシングとAβのクリアランスの両方が、脳内のインスリンシグナルの障害に起因しているという事実に基づいている。さらに、脳のインスリン抵抗性の分子メカニズムとして、IRS-1タンパク質のセリンリン酸化の増加(すなわちIRS-1の阻害)とIRSタンパク質の分解の上昇のいずれかが、毒性のあるAβプラークの凝集、タウの過リン酸化、オートファジーなどの共通の病理学的メカニズムとして関与している可能性に注目している。T3Dという言葉の知識や認知度を高めることは、病気の治療や予防への道を開く可能性があり、場合によっては治療法を提供することも可能である。現在のところ、認知機能の低下やADに対する有効性が確立された特定の治療法はない。そのため、ADがインスリンシグナルの不具合や不規則なエネルギー経路に根ざした病因を持つ疾患であると特定することは、疾患管理において重要な意味を持つ。ADとすべての糖尿病の具体的なメカニズムはまだ複雑ではっきりしていないため、社会経済的にも公衆衛生や医療システムに壊滅的な影響を与える可能性があるが、T3Dは現在の患者に多くの予防・治療戦略を提供する可能性がある。今のところ、認知障害に有効な効果を持つ、より多くの抗T3D薬の試験は、確実に有望な未来を持っていると思われる。