
Contents
The Therapeutic Potential of Metformin in Neurodegenerative Diseases
www.ncbi.nlm.nih.gov/pmc/articles/PMC6060268/
要旨
神経変性疾患の治療法の研究は、高齢化が進み、個人、家族、社会への負担が増大している今日、大きな関心事となっている。この数十年の間に、これらの疾患の根本的な遺伝的・生物学的原因を理解するために大きな進歩があったにもかかわらず、一部の対症療法しか利用できなかった。
メトホルミンは長い間、2型糖尿病の治療に使用されていたが、他のいくつかの疾患にも有効であることが示されている。メトホルミンは、試験管内試験および生体内試験で十分にテストされており、ミトコンドリアのエネルギー産生およびインスリンシグナル伝達を含む多様な経路を標的とする承認された化合物である。
癌、心血管疾患、神経変性疾患などの加齢に伴う疾患に対抗するためのメトホルミンの有効性についてのエビデンスが増えてきている。我々は、ある種の神経変性疾患と糖尿病が明らかに関連していること、また、メトホルミンが他の糖尿病治療薬と併用することで、一部の患者の神経症状を軽減し、動物モデルや細胞モデルで疾患表現型を軽減できることを示すエビデンスについて議論する。
興味深いのは、メトホルミンが神経変性疾患と一般的に関連している経路を介して、細胞内の生存と死のシグナル伝達のバランスをとることができることである。健康な神経細胞では、これらの包括的なシグナルがエネルギー代謝、酸化ストレス、プロテオスタシスを抑制し、神経変性疾患を定義する機能不全や神経細胞死を回避している。これらの生物学的メカニズムと神経細胞の脆弱性との関連性、今後の臨床試験や治療法開発のための潜在的な困難性について議論する。
キーワード
メトホルミン、神経変性、糖尿病、パーキンソン病、アルツハイマー病、老化、ミトコンドリア
序論
ゲノミクスの進化は、神経変性疾患の遺伝的寄与についての理解を大きく前進させ、神経細胞の変性につながる生物学的カスケードを研究するための入り口を提供していた。また、バイオインフォマティクスやシステム医学の研究分野が拡大してきたことで、ターゲットを絞ったより良い治療や個別化された治療の機会も開かれていた。しかし、アルツハイマー病やパーキンソン病のように、これまで大きな進展を遂げてきた疾患でも、遺伝学、病態プロセス、臨床表現型の間に明確な関連性があることはほとんどない。神経変性疾患は、神経細胞の変性・死滅が進行することで起こる衰弱性の難病であり、現代社会においてもその罹患率は上昇している(1)。
そのため、アルツハイマー病やパーキンソン病(PD)の対症療法の開発が大きく進展しているにもかかわらず(図1)新規治療法や疾患修飾治療法の開発が求められている。
図1 前世紀のパーキンソン病およびアルツハイマー病の治療における主要な進歩の年表
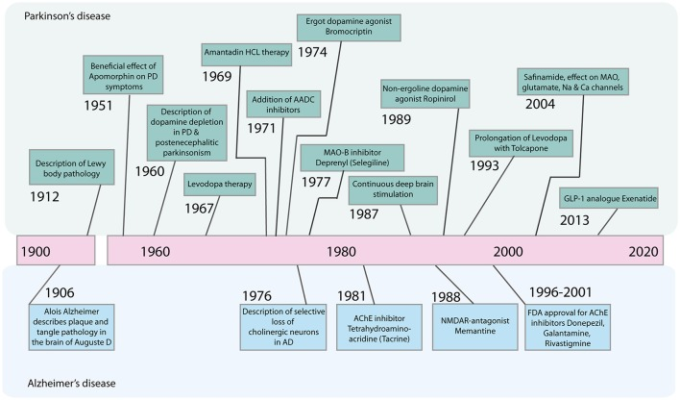
AADC、芳香族L-アミノ酸脱炭酸酵素、AChE、アセチルコリンエステラーゼ、GLP-1,グルカゴン様ペプチド1,HCL、塩酸塩、MAO-B、モノアミン酸化酵素B、NMDAR、N-メチル-D-アスパラギン酸受容体。
神経変性疾患の複雑で不均一な分子基盤を考えると、課題は圧倒的に見えることができ、過去数十年は、ほとんどが期待はずれの臨床試験の結果とその後の財政的投資の欠如を見ていた。
Gallega officinalis(フランスのライラック)は、グルコースを低下させるグアニジンを含み、何世紀にもわたって糖尿病の治療に使用されていた。誘導体のメトホルミンはビグアナイドで、1950年代にヨーロッパで、1990年代に米国で導入された(2, 3)。メトホルミンは最近、心血管リスクの低下、多嚢胞性卵巣症候群における卵巣機能の回復、肝性リポジェネシス、脂肪肝疾患の軽減、酸化ストレスの軽減などが報告されている(3,4)。メトホルミンが効果を発揮するメカニズムはまだ完全に定義されていないが、メトホルミンは肝臓でのグルコース産生を抑制し、末梢組織でのグルコース取り込みを増加させて血糖値を低下させることが知られている(3, 5, 6)。また、メトホルミンがミトコンドリアの呼吸鎖の複合体Iに直接作用してミトコンドリアの呼吸を遅らせることも認められている。
メトホルミンの治療の可能性:根拠
メトホルミンは、神経細胞の長寿メカニズムに干渉する可能性があり、すでにヒトでの使用が承認されているため、興味深い薬剤である。しかし、一般的にヒトの老化研究は、実験室で使用できる良好な老化モデルがないため、研究が遅れている。再プログラムされたニューロンの老化サインを保持することは、直接再プログラムプロトコルの使用によって可能になった(7, 8)が、これは研究グループによっては実行可能ではないかもしれないし、専門家ではない研究室で技術が確立されるまでに時間が必要とされている。ヒト細胞の老化の役割を調査するための新しい、簡単で手頃な方法はまだ非常に必要とされている。
それにもかかわらず、細胞の種類を問わず、ヒトおよび動物の研究から得られたデータは、インスリン機能の調節障害が老化および神経変性疾患の発症に寄与していることを示している(9)。特に認知症の分野では、インスリン抵抗性や糖尿病が疾患発症の一因であるとの認識が高まっている(10-12)。したがって、メトホルミンを使用する理由は、ミトコンドリアの代謝とインスリンシグナル伝達に作用して老化プロセスを遅らせる可能性があることにある。老化過程を遅らせることは、病気を遅らせることで老年期の生活の質が向上する可能性があるため、有益である。
糖尿病と神経変性疾患との関連性は、データが明確ではなく、正確なメカニズムは不明であるが、大部分は受け入れられている。神経変性疾患を有するヒトおよび動物におけるメトホルミンの使用に関する多くのデータが存在するが、メトホルミンの治療的使用は、結果がしばしば矛盾しているため、まだ受け入れられていない。これらの異なる結果は、疾患、モデル系、種、および関与する基礎となる生物学的経路に依存しており、ここでは簡単にレビューする。
認知症
認知症は、異質な起源を持つ一般的な神経疾患であり、最も重要な危険因子は加齢である。認知症は記憶やその他の認知機能に影響を与え、日常生活での活動に支障をきたする。国連の世界人口予測によると、世界の60歳以上の人口は今後30年間で約4倍に増加すると予測されている(13)。最も一般的な認知症はアルツハイマー病であるが、他にも血管性認知症、混合型認知症、前頭側頭型認知症、レビー小体型認知症、パーキンソン病型認知症などがある。
アルツハイマー型認知症
アルツハイマー病は最も一般的な神経変性疾患であり、世界で4500万人が罹患している(14)。アルツハイマー病の特徴は、進行性の記憶障害と認知機能の低下である。
神経原線維性タングル(NFT、異常なタウタンパク質からなる)やアミロイドプラーク[アミロイドβ(アミロイドβ)の細胞外凝集体からなる]は、アルツハイマー病脳の病理学的特徴である(15-17)。NFTタンパク質のタウは微小管と関連しており、その安定化に関与している(18)。タウの病理学的特徴とシナプス喪失は、アルツハイマー病患者の認知障害と相関している(19)。アミロイドプラーク成分アミロイドβは、β-セクレターゼBACE1(βサイトアミロイド前駆体タンパク質切断酵素1)とγ-セクレターゼ複合体による膜タンパク質APP(アミロイド前駆体タンパク質)の逐次切断に由来する(20)。これらのタンパク質構造の脳内での調節障害、異常修飾、および蓄積は、アルツハイマー病の基礎となる主要な病理学であると考えられている。
遺伝的観点から、アルツハイマー病のほとんどの形態は散発的で遅発性であるが、若年性アルツハイマー病の家族性形態が存在し、一般的にAPPまたはプレセニリンの変異によって引き起こされる(21-23)。散発性アルツハイマー病の基礎となる生物学的メカニズムはまだ定義されていない。炎症反応、ホルモン調節、ミトコンドリア機能不全、リソソーム機能不全などが関与している。ミクログリアの関与に関する遺伝的証拠も増えてきている(24-26)。それでも、アルツハイマー病を発症する主な危険因子は、加齢、APOE-ε4対立遺伝子キャリアであることを含む遺伝的危険因子、TREM2のバリアント、およびいくつかのGWAS遺伝子座、外傷性脳損傷、心血管危険因子、およびいくつかの環境的危険因子である(27-31)。
糖尿病と認知症:動物モデル
認知症におけるインスリンとグルコース代謝の役割を調査するために使用されるげっ歯類モデルのほとんどは、アルツハイマー病に焦点を当てている。高脂肪食を与えたAPP/PS1マウス(32,33)部分的にレプチン欠損(db/+)のAPP/PS1マウス(34)およびAPP23-(ob/ob)マウス(35)では、インスリンシグナルと耐糖能が変化している。したがって、APP負荷はエネルギー代謝の障害に対する感受性を高める可能性がある。
高脂肪食はインスリン抵抗性を誘導し、APPトランスジェニックマウスと同様に、アルツハイマー病のTg2576マウスモデルの両方でアミロイドーシスと記憶障害を促進する(36, 37)。高脂肪食や肥満は、野生型動物でも記憶障害に寄与する可能性がある。しかし、Agustiと同僚によってレビューされたいくつかの研究では、トピックはまだ矛盾するデータのために議論されたままにして、全く認知には効果がなかった(38)。糖尿病ラットでは、APP、アミロイドβ、リン酸化タウのレベルが上昇している(39)。これらのデータは、インスリンシグナルを介したエネルギー代謝の変化がアミロイドβの生成とタウのリン酸化の変化に関与している可能性を示唆しており、認知症に関連する2つのよく知られた生化学的イベントである。インスリンの調節は、有毒なアミロイドβオリゴマーからニューロンとシナプスを保護し、他のアルツハイマー病動物モデルにおいて認知を改善するための有効な戦略であることが証明されている(40, 41)。例えば、グルカゴン様ペプチド-1(GLP-1)インスリン様成長因子1(IGF-1)およびカロリー制限は、すべて神経保護効果を発揮することが示されている(42-44)。
メトホルミンと認知症:動物モデル
これまでメトホルミンの認知機能低下への効果を評価した動物研究はわずかであり、結果は一致していない(表(表1).1)。研究者が齧歯類のエネルギー代謝を変調させて認知障害を誘導しようとする方法は数多くあり、このことがこの文脈でのメトホルミンに関するデータの可変性を助長しているのかもしれない。いくつかの動物は高脂肪食を与えられているが、そのような(db/db)マウスのような他のものは、それらがインスリン抵抗性と肥満になる原因となる自発的な突然変異を持っている。そのような高脂肪食を与えた3つの研究では、メトホルミン治療により認知障害が軽減されたが(57,58,60)、1つの研究では改善が見られなかった(59)。db/db)マウスでは、1つの研究でメトホルミンが記憶力を改善することがわかった(53)が、別の研究では効果がないことがわかった(55)。野生型マウスの正常老化を調べたある研究では、メトホルミンは記憶障害に有害な影響を与えた(61)。この研究では、リン酸化によるAMPKの活性化は測定されていないため、これらの動物におけるメトホルミンの食事が最適であったかどうかは明らかではない。正常な老化におけるメトホルミンの効果を理解するためには、適切な対照を用いたより多くの研究が必要であることは明らかである。
表1 げっ歯類モデルにおけるメトホルミンの神経変性への影響を検討した研究
| 調査 | マウス系統 | 疾患モデル | G | 開始年齢* | 会った用量+アプリケーション | 研究/会った期間 | グループあたりn | 主な結果 |
|---|---|---|---|---|---|---|---|---|
| パーキンソン病 | ||||||||
| (45) | B6,Dat-Cre AMPKb1 / 2 KO | 20日目+ 22日目: MPTP:30 mg / kg |
M | 8〜10週間 | 100mg / kg /日 飲料水 |
27日 | n = 6〜10 | Metは、STではMPTPによるTH陽性ニューロンの喪失を減少させるが、SNは減少させず、アストログリオーシスを減少させる。TH 陽性ニューロンへの影響は、AMPK-KOとは無関係であるが、MetはWT動物でAMPKリン酸化を誘導する。 |
| (46) | C57BL | 1日目+ 2日目: MPTP:10 mg |
M | ? 20〜25 g |
150mg / kg /日 メタルプローブ |
7日 | n = 5 | Metは、SNにおけるTH陽性ニューロンのMPTP誘発性喪失には影響を与えないが、ミクログリアマーカーIba1のレベルを低下させる。 線条体におけるドーパミンの量を低下させる。 |
| (47) | C57BL / 6N | 7日目: MPTP:15 mg / kg、4x |
M | 8週間 | 200または400mg / kg /日、 飲料水 |
14日間 | n = 3/10 | MetはSNのTH陽性ニューロンのMPTP誘発性喪失を減少させる MetはSNとSTのpCrebとPGC1αを誘発する |
| (48) | C57BL / 6 | 1〜7日目: MPTP:30 mg / kg |
M | 10週間 20〜25 g |
200mg / kg /日 注射 |
14日 会った:8〜14日 |
n = 6 | MetはMPTP誘発性の運動障害を改善するMet はSNにおけるTH陽性ニューロンのMPTP誘発性喪失を減少させ、アストロ グリオーシスを減少させるMetはAMPKおよびAKTリン酸化を誘発し、リン酸化mTorのレベルを減少させ、SNにおいてBDNFを誘発する |
| (49) | C57BL / 6 | 5週間:3。5日ごと。MPTP:20 mg / kg + 250 mg / kgプロベネシド | M | 10週間 | 5 mg / ml 飲料水 |
5週間 会った:3〜35日目 |
n = 4–5 | Metは、MPTPによって誘発されるSNのTH陽性ニューロンの喪失を減少させ、炎症性サイトカインのレベルを低下させる |
| (50) | スイスアルビノマウス | 1〜5日目:MPTP:25 mg / kg + 250 mg / kgプロベネシド | M | ? 22〜25 g |
500mg / kg /日 強制経口投与 |
21日 | n = 12 | MetはMPTP誘発性運動障害の再生を改善するMet はSNにおけるTH陽性ニューロンのMPTP誘発性喪失を減少させ、BDNF発現を誘発する |
| (51) | C57BL / 6N | 無し | F | 10週間 | A:5 g / kgダイエット B:5 g / l飲料水 |
A:1ヶ月 B:6ヶ月 |
A:n = 20 B:n = 4 |
Metはマウスの脳内のリン酸化α-シヌクレインのタンパク質レベルを低下させる |
| (52) | C57BL / 6J | 1日目:MDMA 20mg / kg | M | 3ヶ月 | 200〜400 mg / kg / day i.p. 注入 |
3/8日 会った:400mg日1,200mg日2+ 3 |
n = 7〜12 | Metは、SNおよびCPuにおけるTH陽性ニューロンのMDMA誘発性喪失を減少させる |
| アルツハイマー病 | ||||||||
| (37) | P301SタウトランスジェニックC57BL / 6 | トランスジェニック、タウ突然変異 | m | 4週間 | 2 mg / ml 飲料水 |
4ヶ月 | n = 12–15 | MetはCXおよびHipでSer262-タウリン酸化を減少させるが、タウ封入体の数を増加させる MetはAMPKリン酸化およびPP2Aタンパク質レベルとCXおよびHIPを誘導する |
| (53) | db / dbマウス(BKS.Cg-m + / + Leprdb / J) | トランスジェニック、レプチン受容体変異 | M | 6週間 | 200mg / kg /日 強制経口投与 |
6週間 | n = 3〜10 | Metは減少125 I-AB 1-40のdb / dbマウスにおけるBBBに流入し、RAGE発現を MetはDB / DBマウスにおける記憶障害を改善する |
| (54) | 野生型 | 無し | ? | ? | 5 mg / ml 飲料水 |
16〜24日 | n = 6 | Metはマウス脳のSer202-およびSer262-タウリン酸化を減少させる |
| (55) | db / dbマウス | トランスジェニック、レプチン受容体変異 | m | 7週間 | 200mg / kg /日 i.p. 注入 |
18週間 | n = 6–11 | Metは空間学習と記憶に影響を与えないMetは 、ヒップのSer396リン酸化タウと同様に総タウタンパク質レベルを低下させ、JNKリン酸化を低下させる |
| (56) | C57BL / 6J | 無し | 5週間 | 2 mg / ml 飲料水 |
1週間 | n = 4 | MetはBACE-1およびAPPタンパク質レベルを増加させ、マウスの脳でAMPKリン酸化を誘導する | |
| 認知 | ||||||||
| (57) | C57BL / 6 | HFD(60%脂肪) | M | 12ヶ月 | 1%ダイエット | 6ヶ月 | n = 16 | Metは、HFDによって誘発される運動機能と記憶の障害を軽減する |
| (58) | NIHスイスマウス | HFD(脂肪45%) | M | 6〜8週間 | 300 mg / kgBW 飲料水 |
20日間 | n = 10 | MetはHFD誘発性認知障害を改善せず、アストログリオーシスに影響を与えない |
| (59) | ウィスターラット | HFD(脂肪45%) | M | ? 125〜150 g |
144mg / kg ダイエット |
10週間 | n = 16〜24 | MetはHFDに影響を与えない-位置テストへのマッチングで赤字を誘発する |
| (60) | ウィスターラット | HFD(59.28%脂肪) | M | 6週間HFD13 週間会った |
15 mg / kg 2x /日 強制経口投与 |
9週間HFD3 週間会った |
n = 8 | MetはHFDによって誘発された記憶障害を 減らするMetはHFDによって誘発されたミトコンドリア機能障害を減らする |
| (61) | C57BL / 6J | 無し | M | 4/11/22ヶ月 | 2 mg / ml 飲料水 |
3ヶ月 | n = 16〜18 | 会ったことは効果がないか、空間記憶を損なうことさえある |
| ハンチントン病と筋萎縮性側索硬化症 | ||||||||
| (62) | R6 / 2・B6CBAF1 / J | トランスジェニック、ハンチンチン変異(136-151 CAGリピート) | M / F | 5週間 | 2または5mg / ml 飲料水 |
約10週間(死ぬまで) | n = 5–9 | Met(2 mg)はオスのマウスの生存期間を延長するが、メスのマウスには影響しない |
| (63) | B6SJL-TgNSOD1 G93A | トランスジェニック、SOD1変異(G93A) | M / F | 5週間 | 0.5または2または5mg / ml 飲料水 |
約16週間(死ぬまで) | n = 6–15 | Metは、メスのマウスの神経症状の開始と病気の進行に悪影響を及ぼし、オスには影響を与えない。 |
BBB、血液脳関門、BW、体重、CPu、尾状突起、CX、皮質、高脂肪食、高脂肪食、股関節、海馬、KO、ノックアウト、MDMA、3,4-メチルエンドオキシ-N-メチルアンフェタミン、Met、メトホルミン、MPTP、1-メチル-4-フェニル-1,2,3,6-テトラヒドロピリジン、SN、黒質下層、ST、線条体、TH、チロシンヒドロキシラーゼ。
*肥満症の方には、体重が表示されていたが、元の記事に年齢が記載されていなかった場合は、体重が表示されていた。
また、メトホルミンは、同じ疾患モデル内の特定の生化学的イベントにマイナスとプラスの両方の影響を同時に与えることができるようである。例えば、P301Sタウ症マウスモデルでは、メトホルミン治療はタウリン酸化を減少させるが、タウ凝集を促進した(37)。著者らは、メトホルミンは脱リン酸化剤としては有益であるが、タンパク質の凝集を促進する可能性があることを示唆しており、後者は紛れもなくより広く受け入れられている神経変性疾患の病態である。同様に、短期間のメトホルミン治療は再びタウリン酸化を減少させたが、APPとBACE-1を活性化するため、負の効果があった(54, 56)。メトホルミンは再び総タウおよびセリン236でのタウリン酸化を減少させる正の効果があるようであるが、スルホニルウレア系糖尿病薬のグリベンクラミドは同様の試験ではるかに良好な成績を示した(53, 55)。
性別もメトホルミンの作用に影響を及ぼす可能性があり、これは動物データの解釈を複雑にする可能性がある。雄のげっ歯類が好まれることが多く、使用した動物の性別が見落とされたり、完全に省略されたりすることがある。すでに述べたメトホルミンの研究では、雄のマウスは認知機能の低下を示したが、雌のマウスは治療後に改善した(36)。
糖尿病と認知症:ヒトを対象とした研究
認知症の診断を受けていない2型糖尿病(2TDM)患者でも認知の変化が報告されており、メタアナリシスでは認知領域全体で中等度ではあるが有意な欠損が認められている(64-66)。また、2型糖尿病は軽度認知障害から認知症への転化や、無症候性軽度認知障害からアルツハイマー病への転化のリスクを高めるようである(67)。
2型糖尿病患者の脳画像研究では、非糖尿病患者と比較して、海馬体積を含む灰白質全体および局所灰白質体積の減少が示されている(68, 69)。これらの臨床データを総合すると、2型糖尿病患者は認知症を発症する可能性が高いことがほとんどである(10, 70-73)。糖尿病と認知症の関係は、逆にアルツハイマー病患者は2型糖尿病や耐糖能障害を発症するリスクが高いという報告によってさらに強化されている(74-76)。さらに、アルツハイマー病患者の死後脳病理では、インスリン受容体とIGFタンパク質レベルの低下が認められ、インスリンレベルとインスリンシグナル伝達のマーカーが脳内で変化している(77-80)。高血糖症や高インスリン血症もまた、アルツハイマー病の病態と正の相関を示している(75, 81, 82)。しかし、大多数の神経病理学的研究では、2型糖尿病または実際にグルコースレベルとアルツハイマー病病理の程度との間の関連を発見しておらず(83-87)、2つの研究では負の関連を示唆している(88, 89)ことを述べなければならない。
アルツハイマー病における臨床研究と神経病理学的研究の間の不一致の一つの説明は、血管病理の影響である。現在では、加齢に伴う脳の病理が例外というよりはむしろ規則であることが確立されている(90)。ほとんどの研究が2型糖尿病とアミロイドβ沈着との関連を示さないという事実は、したがって、アルツハイマー病の病理に2型糖尿病の大きな影響がないことを示唆しているように思われる。2型糖尿病とアルツハイマー病変を有する患者では、小血管疾患が認知機能に及ぼす影響は、このグループで認知症を発症する可能性が高いことを説明できるだろう。このことは、2型糖尿病がアルツハイマー病に大きな影響を与えないとしても、アルツハイマー病における糖尿病の適切な管理が重要であることを示唆している(67, 91)。2型糖尿病とアルツハイマー病の間の相互作用は、特に共有された病態生理学的特徴に関するエビデンスを考慮しても、血管病理学を超えて無視されるべきではない。
メトホルミンと認知症:ヒト研究
メトホルミン使用の認知機能低下とアルツハイマー病への影響を評価した臨床研究の結果は、ほとんどが正の影響を示している(表2).2)。メトホルミンの使用は、2型糖尿病における認知機能障害のリスクが有意に低いことと関連している(102, 107)。一般的な認知症の発生率は、経口抗血糖薬を投与されていない2型糖尿病患者と比較して、メトホルミン、スルホニルウレア、またはその両方を投与されている2型糖尿病患者の方が低い(96)。2つの研究では、メトホルミンを投与されている糖尿病患者では、スルホニルウレアまたはチアゾリジン系薬剤を投与されている患者よりもアルツハイマー病発症のリスクが低かった(97, 101)。しかし、1つの研究では、2型糖尿病に対するメトホルミンの長期使用(スルホニルウレアやチアゾリジン系薬剤は使用していない)は、アルツハイマー病発症リスクの増加と関連していた(103)。ある有益な研究では、潜在クラス分析を用いて、メトホルミンを投与されている2型糖尿病患者のうち、認知症を含む併存疾患のプロファイルが異なるグループを同定した。彼らは、メトホルミンの効果は、実際には薬剤を投与される患者のリスクプロファイルによって異なる可能性があると結論付けている(100)。
表2 神経変性疾患の発生および進行に対するメトホルミンの効果を評価した試験
| 調査 | 疾患 | 特徴 | 結果 |
|---|---|---|---|
| (92) | PD | 後ろ向きコホート研究、80万人のうち61,166人が糖尿病患者であり、後者の41,003人がOAA療法を受けた | コントロールと比較して、OAAなし(HR 2.18)およびOAAあり(HR 1.30)の2型糖尿病患者のPD発生率が高い。メトホルミン単独での治療のHRはスルホニル尿素単独(1.57)よりも低く(0.95)組み合わせは最低のHR(0.78)を示した。 |
| (93) | PD | メトホルミンを投与された93,349人の2型糖尿病患者(657,537患者年のFU)およびメトホルミンの有無にかかわらずグリタゾンを投与された8,346人の2型糖尿病患者(69,338患者年のFU)を対象とした人口ベースの後ろ向きコホート研究。 | メトホルミンを投与された患者と比較して、2型糖尿病のグリタゾンを投与された患者のPDの発生率は有意に低く(HR 0.72)まだグリタゾンを服用している長期のグリタゾン使用者ではPDの発生率はない |
| (94) | PD | メトホルミンのみを投与された41,362人の患者、シンバスタチンのみを投与された316,210人の患者、およびメトホルミンとシンバスタチンの両方を投与された52,311人の集団ベースの後ろ向きコホート研究 | メトホルミン単独と比較して、シンバスタチン単独(HR 0,64)またはメトホルミンとの併用(HR 0.74)を受けている患者のPDの発生率が低い |
| (95) | PD /認知症 | 後ろ向きコホート研究、メトホルミン治療を受けた2型糖尿病患者4,651人、メトホルミン治療を受けた2型糖尿病患者4,651人。> 21,000人年のFU | 非メトホルミン群と比較して、メトホルミン群のPD(HR 2.27)AD(2.13)およびVD(2.30)の発生密度が高い |
| (96) | 認知症 | 後ろ向きコホート研究、50歳以上の認知症のない127,209人の個人、うち25,939人が2型糖尿病、1,864人がメトホルミンのみ、9,257人がスルホニル尿素剤+メトホルミン | コントロールよりも2型糖尿病の認知症の発生率が高く、スルホニル尿素(HR 0.85)メトホルミン(HR 0.76)またはメトホルミンとスルホニル尿素の組み合わせ(HR 0.65)と比較して2型糖尿病 wo / OAAの発生率が高い |
| (97) | 認知症 | 65歳以上の67,731人の非認知症、非糖尿病患者が5年間観察され、2型糖尿病、抗糖尿病薬、認知症の発症が観察された。 | 非2型糖尿病と比較して新規発症2型糖尿病の認知症発症リスクの増加(HR 1.56)認知症を発症するリスクは、スルホニル尿素およびメトホルミンよりもチアゾリジンジオン使用者の方が高かった |
| (98) | 認知症 | メトホルミンを投与されている122,036人とメトホルミンを投与されていない67,822人の189,858人、1,000人年あたりの認知症発生率 | メトホルミンを服用している糖尿病患者は、メトホルミンを服用していない患者よりも認知症の発生率が有意に低かった(1,000人年あたり21.79対31.58,p <0.001) |
| (99) | 認知症 | 544,093人の参加者を含むメタアナリシス、インスリン感作物質を服用している2型糖尿病患者の認知症のリスク | 認知症の発生率は、インスリン感作物質を服用していないが有意ではないものと比較して、メトホルミン(RR 0.79)で減少した(p = 0.064) |
| (100) | 認知症 | 8,393人のメトホルミン使用者を伴う2型糖尿病の41,204人の男性における年齢関連の併存疾患のリスクに対するメトホルミンの異なる効果を持つサブグループを特定するための潜在クラス分析、 | 認知症を含むARCを発症するリスクに対するメトホルミンの異なる効果を示した患者の4つの潜在クラスを特定 |
| (101) | 認知症 | 後ろ向きコホート研究、17,200人の新規メトホルミンユーザーと11,440人以上の65歳以上の新規スルホニル尿素ユーザー、平均FU5歳 | メトホルミンを服用している75歳未満の人は、スルホニル尿素薬を服用している人よりも認知症を発症するリスクが低かった(HR 0.67,95%CI 0.61–0.73) |
| (102) | 認識機能障害 | 縦断的人口ベースの研究、2型糖尿病の55歳以上の365人のうち204人がメトホルミンを投与された | メトホルミンの使用は、認知障害(OR 0.49)と逆に関連し、より長い使用は、認知障害のリスクの低下と関連する |
| (103) | 広告 | 後ろ向き症例対照研究、7,086人のアルツハイマー病患者と対照をメトホルミン/他の抗糖尿病薬の以前の使用について比較した | メトホルミン(AOR 1.71)の長期使用者では、非使用者と比較して、スルホニル尿素(AOR 1.01)チアゾリジンジオン(AOR 0.87)またはインスリン(AOR 1.01)ではなくADを発症するリスクが高い |
| (104) | 広告 | 新たに糖尿病と診断された71,433人の患者と71,311人の非糖尿病対照、最長11年間のフォローアップ | 非糖尿病患者と比較して糖尿病患者のADの発生率が高く(0.48対0.38%)リスクに対する抗高血糖治療のプラスの効果はない |
| (105) | 広告 | ランダム化プラセボ対照クロスオーバー試験、MCIまたは軽度の認知症およびADの20人の非糖尿病患者がmgメトホルミンまたはプラセボを8週間投与され、その後8週間他の治療に切り替えられた | メトホルミンは脳脊髄液で測定可能であり、プールされた事後分析では、ASL-MRIでの8週間のメトホルミン曝露後の上眼窩前頭皮質と中眼窩前頭皮質の有意な増加、実行機能の尺度であるトレイルメイキングテストパートBの有意な改善 |
| (106) | HD | 観察研究; 4325人のHD患者、そのうち121人が2型糖尿病でメトホルミンを投与された | メトホルミンを服用しているHD患者は、言語機能と実行機能のテストではうまくいいたが、運動評価ではうまくいきないでした |
アルツハイマー病、アルツハイマー病、AOR、調整オッズ比、ARC、年齢関連併存疾患、ASL-MRI、Arterial Spin Label Magnetic Resonance Imaging、脳血流、Cerebral Blood Flow、FU、フォローアップ、HD、ハンチントン病、HR、ハザード比、MCI、軽度認知障害、OAA、経口抗高血糖薬、PD、パーキンソン病、2型糖尿病、2型糖尿病、VD、血管性痴呆。
Luchsinger氏らは介入試験で、無症候性軽度認知障害を有する過体重患者を対象に、プラセボと比較してメトホルミンを12カ月間毎日投与した場合の効果を検討した。メトホルミン投与群では選択的想起テストに改善がみられたが、他の認知機能またはバイオマーカーのアウトカムには改善がみられなかった(108)。結果はわずかに有意であり、複数の測定値に対する補正は行われていないことから、少なくとも観察された改善は独立した試験で確認されなければならないことが示唆されている。別の介入的短期メトホルミン試験では、アルツハイマー病による軽度認知障害または軽度認知症の非糖尿病患者にメトホルミンまたはプラセボを8週間投与した。メトホルミン投与群では、実行機能の指標では有意に改善したが、他の認知テストやバイオマーカーでは改善しなかった。ここでも、複数のテストは補正なしで行われた(105)。
2型糖尿病の有無にかかわらず、認知症におけるメトホルミンの使用に関するデータの大部分は概ね肯定的であるが、メトホルミンの効果は複雑な基礎となる病理学的プロセスに依存している可能性が高く、ある程度は神経変性プロセスではなく血管への効果に関連している可能性があることを考慮すべきである。場合によっては、メトホルミンは有害な影響を及ぼす可能性さえある。プロスペクティブな介入研究では、軽度の認知障害や軽度のアルツハイマー型認知症にメトホルミンの効果があるという説得力のある証拠を示すことはできなかったが、おそらく力不足か期間が短すぎたのであろう。大規模で明確な認知症コホートを対象とした、より長期的なメトホルミンの対照研究が必要である。
パーキンソン病
背景
パーキンソン病(PD)は一般的な神経変性疾患であり、60歳以上の人口の1%以上、85歳以上の人口の約4%が罹患している(109)。パーキンソン病は、徐脈運動と硬直、安静時振戦、姿勢不安定、および広範囲の非運動症状の組み合わせが特徴である(110)。他の神経変性疾患と同様に、パーキンソン病は臨床的にも病理学的にも異質であり、発症や進行に大きなばらつきがある。中脳に位置する黒質体前庭のドーパミン含有ニューロンの消失が進行すると、線条体のドーパミンが欠損する(111, 112)。主にα-シヌクレイン(αSyn)の凝集体からなるレビー小体と呼ばれる神経細胞内の不溶性タンパク質の介在は、パーキンソン病の主な神経病理学的特徴である(113)。レビー小体とタンパク質の凝集体は、複数の脳領域に認められ、疾患の進行とともに広がっていく(114, 115)。αSynの凝集と神経細胞の喪失につながる正確な生物学的メカニズムは不明のままであり、現在のところパーキンソン病の症状に対してはドーパミン補充療法や場合によっては深部脳刺激による治療が行われているにすぎない。
PD症例の約5~15%は疾患の原因となる遺伝的変異に起因しており、患者の約15%は一親等以内の親族にも発症している(116)。パーキンソン病の遺伝的構造はよく研究されてきたが、その構造は複雑である。これまでのところ、23の遺伝子座と19の遺伝子が家族性パーキンソン病と関連している(117)。ほとんどの神経変性疾患と同様に、大部分の症例は、リスクを修飾する遺伝的変異、環境因子、および偶然性の複雑な相互作用に起因していると考えられる。パーキンソン病に関与する遺伝子に関する知識は、基礎となる生物学的経路への洞察を可能にしてきた。複数の環境因子や疫学的データと合わせて、遺伝子データは、ミトコンドリア機能不全、リソソーム機能、炎症、凝集を起こしやすいタンパク質の蓄積、酸化ストレスなど、いくつかの細胞機能や経路を浮き彫りにしてきた(118, 119)。
パーキンソン病に対する神経保護化合物の研究に多額の投資が行われているにもかかわらず、今のところ臨床試験で説得力のある効果を示したものはない(120)。
糖尿病とPD:動物実験
げっ歯類の研究では、インスリン抵抗性とパーキンソン病の発症との間に関連性があることが示されている。高脂肪食は神経毒である1-メチル-4-フェニル-1,2,3,6-テトラヒドロピリジン(MPTP)と6-ヒドロキシドパミン(6-OHDA)に対する脆弱性を増強し、黒質神経変性と運動障害の増加によって測定された(121,122)。同様に、パーキンソン病のαSynマウスモデルでは、高脂肪食は運動器表現型の発現を加速させ、神経変性の早期発症をもたらした(123)。
インスリン抵抗性はドーパミンシグナル伝達を直接的に阻害する可能性がある。高脂肪食を与えられたラットでは、黒質突起のドーパミン機能が障害され(124)過体重マウスや糖尿病マウスではドーパミン神経細胞の変性が見られる(125)。
メトホルミンとPD:動物実験
これまでのところ、パーキンソン病における神経保護剤としてのメトホルミンの効果を評価した齧歯類を用いた研究はごくわずかである。これらの研究は主に急性MPTP誘発性パーキンソン病とメトホルミン治療の併用に焦点を当てている。これらの研究の実験デザインは非常に類似しているが、結果は可変的であり、メトホルミンの可変的な効果の主な原因はモデル化の違いであると主張している。しかしながら、MPTPとメトホルミン治療の用量と期間の違いが重要であるかもしれない(表(表11))。
げっ歯類を用いたほとんどの研究では、メトホルミンがドーパミン作動性ニューロンに対するMPTPの損傷作用を減少させることがわかっており、これは、黒質パーコンパクト(49,50)線条体(45)またはその両方(48)のチロシン水酸化酵素染色(ドーパミン作動性ニューロンのマーカー)によって示されている。2つの研究では、メトホルミンの保護効果は特異的ではないかもしれないことが示唆されている。しかし、Ismaielらの研究では、メトホルミンはSNにおいてMPTP誘発性のニューロン喪失に対する保護効果はなかったと報告しており(46)BaylissはSNにおいてドーパミン作動性ニューロンに対する保護効果はなく、線条体のみに効果があると報告している(45)。
神経毒であるMPTPによって誘発されるドーパミン作動性ニューロンの死から保護するメトホルミンの能力は、3つの研究でネズミの運動機能の改善と相関していると考えられている(48-50)。MPTPとメトホルミンはともに呼吸鎖の複合体1に作用することから、ミトコンドリアの生存に薬物が相互に影響を与えている可能性は排除できない。これらの研究では、メトホルミンは損傷を受けたニューロンを回復させるというよりも、主にMPTP自体の損傷作用を減少させた可能性がある。したがって、パーキンソン病の急性毒素モデルではなく、トランスジェニックマウスモデルでメトホルミンの作用を調べれば、その可能性についてのより良い洞察が得られるかもしれない。興味深い最初のヒントは、健康な非トランスジェニックマウスを用いた研究で、メトホルミンが脳内のαSynリン酸化を減少させることを示したことにある(51)。
糖尿病とPD:ヒトを対象とした研究
糖尿病患者におけるPD発症のリスクを評価した研究では、結果が非常に異なっている(126-132)。14件の症例対照研究からなる1件のメタアナリシスでは、2型糖尿病患者でPD発症リスクが低下していた(133)。逆に、Ceredaらは、4つの前向きコホート研究では糖尿病患者におけるPD発症リスクが増加したが、5つの症例対照試験ではパーキンソン病患者における糖尿病の有病率は増加しなかったと報告している(134)。最大の集団を有する症例対照試験では、対照試験と比較してパーキンソン病患者の糖尿病の有病率がさらに高いことが一貫して示されていることに注意しなければならない。さらに最近では、7つの集団ベースのコホート研究を含むメタアナリシスでも、糖尿病患者におけるPDリスクの増加が示されている(135)。これらのメタアナリシスを総合すると、2型糖尿病におけるパーキンソン病の発症率の増加を示唆しているように思われる。潜在的な落とし穴は、いくつかの研究に血管性パーキンソン病が含まれていることである。2型糖尿病は脳小血管疾患の一因となるため、血管病変を有する患者を除外しないと、パーキンソニズムの徴候を示す2型糖尿病患者が多くなるという結果に偏りが生じる可能性がある。この問題は、2型糖尿病におけるパーキンソン病の発生率の増加を示したいくつかの研究で取り上げられており、これらの相違を十分に説明することはできない。神経病理学的には、ある研究では血糖値の上昇と黒質質のレビー小体形成のリスクの増加との関連性が報告されており、さらにパーキンソン病の病態における2型糖尿病の役割を支持している(136)。
レビー小体を伴う認知症(DLB)とパーキンソン病性認知症(PDD)は、高齢者における認知症の一般的な原因である(137)。2型糖尿病を有するパーキンソン病患者では、グループは小さいが、認知機能の低下率が高く、灰白質・白質体積が低いことが報告されている(138)。PDD患者は、認知症のないパーキンソン病患者よりも経口グルコース耐性試験でインスリン抵抗性を示す可能性が高い(139)。スウェーデンのDementia Registryのデータを用いたある研究では、糖尿病患者ではDLBとPDDの頻度は低かったが(140)他の多くの研究では2型糖尿病とPDDの関連性は有意ではなかった(141-144)。
パーキンソン病におけるメトホルミン:ヒトを対象とした研究
臨床研究はメトホルミンのみを対象としたものではなく、メトホルミンを他の経口抗高血糖薬と比較したり、他の経口抗血糖薬と併用したりしたものである(表2参照)。すべての研究は異なる薬物を対象としており、比較することはほとんどできない。PDリスクに対するメトホルミンの正の効果を示唆する臨床データは不足している。Wahlqvist氏らは、2型糖尿病患者におけるパーキンソン病の発生率に対するスルホニルウレア、メトホルミン、または両方の薬剤の併用の効果を調べようとした。スルホニルウレアを投与された2型糖尿病患者は、経口抗血糖薬を投与されていない患者と比較してPDリスクが高かった。メトホルミン単独またはスルホニルウレアとの併用では影響はなく、メトホルミンがスルホニルウレアの有害な効果を救済する可能性が示唆された(92)。
Brakedal氏らは、Norwegian Prescription Database(NorPD)に登録されている2型糖尿病患者を対象に、グリタゾンをメトホルミン単独またはメトホルミン単独で服用している患者と服用していない患者でパーキンソン病の発生率を比較した。グリタゾンを服用している患者は、メトホルミン単独の患者と比較してパーキンソン病の発生率が有意に低かった。過去にグリタゾンを使用していた患者ではリスクの低下は認められず、リスクを低下させるためにはグリタゾンへの長期または恒久的な曝露が必要であることが示された(93)。スタチン、メトホルミン、またはその両方を投与されているNorパーキンソン病の患者を見ると、スタチンとメトホルミンを併用している患者では、メトホルミン単独と比較してパーキンソン病を発症するハザード比が低く、スタチンのみを投与している患者ではリスクが最も低かった(93)。スタチンの陽性効果は、抗炎症作用とミクログリア炎症反応の低下によってもたらされると考えられ、これは線条体ドーパミン活性に好影響を及ぼすことが示されている(145)。なぜメトホルミンはスルホニルウレアと併用するとプラスの効果があるように見えるが、スタチンと併用するとマイナスの効果があるのかという疑問に対処しなければならない。2型糖尿病と高コレステロール血症の併用は、高コレステロール血症単独よりもPD発症のリスクを高める可能性があり、このリスクはスタチンとメトホルミンの併用では十分に低下しない可能性がある。スルホニルウレアにメトホルミンを追加することで、スルホニルウレア単独での治療よりも2型糖尿病のコントロールが良好になり、それによってPDリスクを促進する作用が軽減される可能性がある。また、異なる薬剤間の複雑な相互作用も考慮しなければならない。
我々の知る限りでは、メトホルミンの使用と病勢進行に関するデータはない。また、インスリン抵抗性のない患者におけるメトホルミンの使用がPD発症に有益な効果をもたらすかどうかも不明である。
その他の神経変性疾患
筋萎縮性側索硬化症(ALS)ハンチントン病(HD)運動ニューロン病、非定型パーキンソン病など、他のまれな神経変性疾患を対象としたメトホルミン試験の報告は、私たちの知る限りではほとんどないか、あるいは全くない。ここでは、糖尿病との関連やエネルギー代謝を標的とした薬剤の使用に関する研究を簡単に紹介する。
筋萎縮性側索硬化症
ALSは進行性の神経変性疾患で、第一・第二運動ニューロンが変性し、痙縮や筋肉の萎縮を引き起こすのが特徴である。最終的には発話、嚥下、呼吸が困難になり、多くの場合、診断後数年以内に死に至る。神経化学的不均衡や遺伝子変異がALSの原因となることが知られているが、ほとんどの症例は散発的であり、高齢であることが重要な危険因子となっている。ALSに使用できるほとんどの薬物は症状の緩和のみであるが、リルゾールや最近ではエダラボンが病気の進行を遅らせることが示されている(146, 147)。
高齢の患者では糖尿病の保護効果があり、若年の糖尿病患者ではALS発症のリスクが高いことが報告されているが、これはALSと1型糖尿病Mおよび2型糖尿病との関連性の違いを反映していると考えられている(148, 149)。ほとんどの研究では、2型糖尿病患者ではALS発症のリスクが減少することが示されている(150, 151)。しかし、他の研究では、ALSのリスクや進行に有意な影響はなく、65歳以下の2型糖尿病患者ではALS発症のリスクが高いことさえ報告されている(152-154)。栄養状態はALSの重症度と負の関係があり(155)、高カロリー栄養はALSの潜在的な治療オプションとして示唆されている。PPAR-γアゴニストであるピオグリタゾン(インスリン抵抗性を低下させる)を用いた2つの試験(156,157)では、病気の進行に対する効果は示されていない(158)。
ハンチントン病
HDは、進行性の神経変性疾患であり、振り回される動作や精神症状、認知機能の低下などを引き起こす。最も一般的なのは早期発症で、通常は30~40歳頃に診断される。HDは、ハンチンチンというタンパク質をコードするHTTという遺伝子の欠損によって引き起こされ、その遺伝様式は常染色体優性である。HTT遺伝子のCAGリピートが拡大すると、ハンチンチンタンパク質の異常に長いバージョンが生成される。この結果、このタンパク質は細胞によって小さな有毒なフラグメントに分解され、これらのタンパク質フラグメントは凝集して疾患の原因となる神経細胞に蓄積される。
HD患者ではブドウ糖代謝の変化と2型糖尿病の割合の増加が報告されており(159, 160)、中国のHD患者の家族では2型糖尿病の有病率が高いことが報告されている(161)。しかし、他の研究では、HD患者と対照群との間で経口グルコース耐性試験や膵組織の違いを確認することはできなかった(162,163)。メトホルミンを投与されている2型糖尿病のHD患者は、メトホルミンを投与されていない糖尿病のないHD患者よりも認知検査の結果が良好であった。これは、メトホルミンを服用している2型糖尿病患者は、非糖尿病の対照群に比べて認知テストの結果が悪かった非HD対照群とは対照的であった(106)。
メトホルミン:神経変性疾患における作用機序
メトホルミンの効果に関して、これまでに実施された生体内試験試験では、相反する結果が得られている。研究デザインの大きな違いに加えて、これらの結果は、メトホルミンの影響を受ける多くの生物学的経路に起因していると考えられる。ここでは、神経変性疾患の治療薬としてのメトホルミンの可能性に最も関連する生物学的シグナル伝達経路および生物学的メカニズムのいくつかを議論する(図(図22))。
図2 神経保護剤としてのメトホルミンの可能性
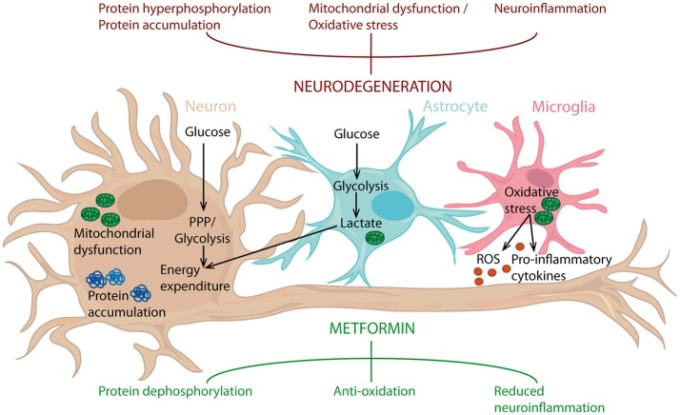
メトホルミンは、タンパク質の高リン酸化、酸化ストレス、神経炎症など、神経変性を引き起こすことが知られているプロセスを抑制することができる。メトホルミンはニューロンに作用するだけでなく、アストロサイトやミクログリアも標的とする。その結果、メトホルミンは、脳全体のグルコース代謝とともに炎症状態に影響を与え、それによって神経炎症を抑制し、抗酸化物質として作用し、タンパク質の脱リン酸化を導くことができる。PPP、ペントースリン酸経路。
中枢代謝とシグナル伝達
中枢代謝は、がんや神経変性を含むヒトの疾患に大きく関与している増殖、ストレス、生存に関わる包括的な細胞シグナル伝達経路と関連している。メトホルミンは、中枢代謝に作用し、エネルギーセンシング(グルコース代謝およびAMPKシグナル伝達)mTORシグナル伝達、炎症性シグナル伝達を含むいくつかの主要なシグナル伝達経路に作用する。ミトコンドリアのシグナル伝達については、別途検討する。
エネルギーセンシングと代謝
脳は全身のわずか2%に過ぎないが、総エネルギー消費量の約20%を占める人体の主要なエネルギー消費器官の一つである。脳細胞は、(i)ニューロン(脳のエネルギー消費量の70〜80%)と(ii)オリゴデンドロサイト、アストロサイト、ミクログリアからなるグリア細胞(残りの20〜30%を占める)から構成されている。ニューロンの高いエネルギー需要は、神経変性疾患における特定のニューロンサブタイプの選択的脆弱性を部分的に説明するいくつかの要因の一つである。エネルギー代謝が神経変性疾患の病因に関与していると考えられて久しいが、ここでは、神経変性疾患におけるメトホルミンの治療効果に関連するシグナル伝達経路および生物学的メカニズムのいくつかについて簡単に言及する。
AMPKシグナル伝達
AMPK は、細胞のエネルギー状態のセンサーとして進化的に保存されている。AMPKは、エネルギー不足の状態でAMPレベルを増加させることで活性化され、酵素は結果的にエネルギー消費を抑制し、異化経路を刺激する。AMPK の活性化は、mTor および PI3K-Akt シグナル(後述する 2 つの重要な経路)の阻害を含む幅広い効果を有する。
AMPKの調節障害は、インスリン抵抗性および2型糖尿病(164,165)および神経炎症(166-168)と関連している。AMPKはアミロイドβ生成とタウリン酸化の両方を調節することが示されているので、AMPKシグナル伝達はアルツハイマー病疾患の進行において主要な役割を果たしている。ニューロン培養におけるアミロイドβ生成およびタウリン酸化の阻害は、AMPK活性化に依存しており(169)AMPKの活性化は細胞外アミロイドβ蓄積を低下させる(170)。逆に、ニューロンでは、AMPKの活性化は、アミロイドβ毒性への応答としてタウのリン酸化に関連している(171,172)。
メトホルミンは、ミトコンドリアの呼吸に必要な電子輸送鎖の複合体Iを阻害し、それによってエネルギー不足を引き起こし、間接的にAMPK経路を活性化する(173-175)。このように、AMPKの刺激は、メトホルミン投与の重要な結果とみなすことができ、この薬剤の既知の効果の多くを説明することができる(図(図33))。
図3 メトホルミンの細胞標的
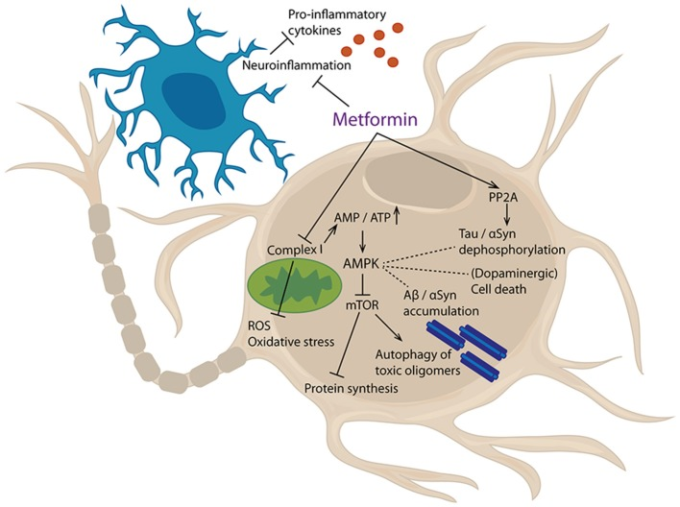
メトホルミンはミトコンドリア複合体Iを阻害し、それによってAMP/ATP比を増加させる。このエネルギー不足はAMPKの活性化につながり、特にmTorシグナル伝達を阻害する。さらに、メトホルミンは、PP2Aを活性化し、神経炎症性プロセスを阻害することができる。その結果、プロ炎症性サイトカインおよび活性酸素種(ROS)の産生が減少し、酸化ストレスが減少し、タンパク質合成が阻害され、有毒オリゴマーのオートファジーが増強される。さらに、タンパク質の脱リン酸化、タンパク質の凝集、および細胞死に影響を与える。
しかし、特に アルツハイマー病 の文脈では、AMPK シグナルの複雑な役割とメトホルミンの作用を理解するために、より多くの研究が必要とされている。ヒト神経幹細胞を用いて行われた研究では、メトホルミンを介したAMPKの活性化がアミロイドβに対する神経保護であることが提案され(176)、他の試験管内試験研究では、メトホルミンがmTOR/PP2A(Protein phosphates 2A)シグナル伝達を介してタウリン酸化を減少させることができることが示され(54)、アルツハイマー病に関連する分子病態を減少させることが可能であることが示された(177)。メトホルミンによるAMPKを介した更なるレベルの調節は、ニューロンにおけるBACE1タンパク質レベルを減少させるメトホルミンの能力に由来する可能性がある(178)。逆に、メトホルミンはまた、ニューロンのBACE1をアップレギュレートし、アミロイドβの生成を増加させることも報告されており(179)病気のニューロンでAMPKを活性化することによる有害な効果を示唆している。
PDマウスモデルでは、AMPKの関与は同様に多様である。神経毒MPTPの投与はAMPKシグナル伝達を活性化する(180)。興味深いことに、AMPKの過剰発現とAMPK阻害の両方が、神経毒で治療されたPDモデルにおいて生存を促進している(180)が、別の研究では、生体内のPDモデルにおいてAMPK活性化の保護機能を示す証拠が示された(181)。
細胞培養におけるαSynの過剰発現はAMPK活性を低下させ、AMPKの阻害はαSyn毒性に対する抵抗性を低下させた(182)。AMPKのサブユニットα1とα2は、低レベルではあるが継続的なAMPK活性により、ドーパミン作動性ニューロンの損失をほぼ完全に防止し、αSynの毒性に対する神経保護効果を有する(183)。したがって、げっ歯類のPDモデルでは、食事性メトホルミンはAMPKによって調節されたαSynのリン酸化状態を介してニューロン機能に影響を与えた(49, 51)。しかし、他のいくつかの研究では、異なる方向性を示している。過剰に活性化されたAMPKはαSynの蓄積を促進し(184)、AMPKの過剰活性化はαSynのGTPase PIKE-Lへの結合とドーパミン作動性細胞死を導く(47)。これらの研究は、AMPK活性の低下が、実際には少なくともパーキンソン病のαSynモデルにおいて有益である可能性があることを示している。
多くの神経変性疾患に見られるように、基礎となる遺伝的および生物学的原因は不均一であり、多くの場合、疾患スペクトルを越えて重なり合う複数の病態を引き起こしている。主にミトコンドリアを介したメトホルミンの作用は、各患者や疾患モデルにおけるミトコンドリアシグナル伝達の関与の程度や種類に応じて、AMPKに対して多くの反対の影響を及ぼす可能性がある。ここで考慮すべき重要な点の一つは、生物学的経路は、疾患の経過を通して必ずしも単一の状態で固定されているわけではないということである。特に神経細胞は、エネルギーの必要性に注意深く適応して生き延びるように進化してきた。ミトコンドリアの若返りと適応のために、洗練された代償メカニズムが開始される。このような複雑さが、ヒト神経変性疾患のモデル化を困難にしており、原因となる治療法や「すべてを治す」治療法が存在しない現状に貢献している。
グルコース代謝
グルコースは神経細胞の活動を維持するために必要な必須のエネルギー基質であり、脳内皮、アストロサイト、ニューロンに発現するグルコーストランスポーターを介して取り込まれる(185)。ニューロンはほとんどの場合、エネルギー源としてグルコースに依存しているが、空腹時にはケトン体を利用する。他の細胞型とは対照的に、ニューロンでは、速度制限解糖酵素ホスホフルクトキナーゼB3がプロテアソームによって高度にターンオーバーされ、解糖とは対照的にペントースリン酸経路(PPP)を介したグルコースの優先的な代謝をもたらす(119, 186)。
PPPの産物は電子供与体であるNADPHであり、同化反応に還元力を提供し、抗酸化力を維持するために重要である。PPPはニューロンが高いエネルギー需要を満たすのに役立つが、ニューロンは主に酸化性であるため、解糖とPPPの間の絶妙なバランスを維持することが、酸化的ダメージに対抗し、エネルギーを保存するために不可欠である。
一方、グリア細胞は主に解糖を介してグルコースを代謝して乳酸を生成し、ミトコンドリアの酸化率は非常に低い。グリアは軸索を代謝的にサポートしており、乳酸はグリアからニューロンへの勾配を横切ってシャトルされる(図(図2)2)(187,188)。興味深いことに、細胞培養では、ニューロンはグルコースよりも乳酸を好む(189)。ヒトの脳では、エネルギー需要は、酸化的損傷を相殺するために厳しく制御されなければならず、したがって、細胞培養および細胞培養培地の効果は、in situで実行されるメトホルミンに関する相反するデータを考慮する際に考慮されるべきである。
PPPおよびグルタチオン経路を阻害すると、神経変性の間に見られるのと同様の酸化ストレスおよび細胞死の増加レベルを引き起こす(119)。グルコースの低代謝はパーキンソン病脳で示されており(190)グルコース代謝の調節障害はパーキンソン病の病因の初期イベントとして提案されている(119)。Dunnらは、グルコース代謝の調節障害は、グルタチオンのリサイクル効率が低下するために酸化ストレスを引き起こすPPPの調節障害を介して起こり、このイベントこそがパーキンソン病で観察される酸化ストレスの増加レベルの根底にあると提案している(119)。
メトホルミンは、ミトコンドリアの複合体Iを阻害することで酸化性リン酸化を遅らせ、糖新生を阻害することで、これらの経路に作用し、NADHの利用を最小限に抑えることでニューロンの酸化的負担を軽減するのをさらに助ける効果がある。
インスリンシグナル伝達
インスリンは脳内で重要な役割を果たしている。体重、食物取り込み、代謝恒常性を制御するためのホルモンシグナルとして利用されている(191-193)。また、インスリンはドーパミン受容体の発現やドーパミン濃度に影響を与えることも示されている(194-196)。インスリンシグナル伝達の障害は、アルツハイマー病、PD、およびHDを含むいくつかの神経変性疾患に関与している(197-200)。インスリンは高血糖に反応して分泌され、脳を含む様々な器官で作用する。インスリン受容体の活性化とインスリン受容体基質を介したホスホイノシタイド-3-キナーゼ(PI3K)-Akt経路の活性化は、インスリンの代謝作用において中心的な役割を果たしている(201)。Aktの活性化は、mTOR、FOXO、Bアルツハイマー病などのタンパク質を調節する。全体として、Aktは100以上の既知の基質を有し、細胞増殖、細胞増殖、グルコース取り込み、タンパク質合成、グリコーゲン合成、アポトーシスに多様な影響を及ぼす(202)。AktはPP2A(203)、PHLPP1/2(204)、およびPTEN(205)によって間接的に阻害される。インスリン抵抗性は、Aktの上流および下流のシグナル伝達の障害と関連している(206-208)。
インスリンは、神経変性の症状を改善するために患者に投与され(209,210)、アミロイドβ誘導死から細胞を保護することが示されている(211-213)。インスリン分解酵素(IDE)は、もともとインスリンのターンオーバーにおいて重要な役割を果たすことが発見された(214)が、アミロイドβの分解に関与している。IDEは、ニューロンやミクログリアから分泌されたアミロイドβを分解し、そのクリアランスを媒介することができる(215)。さらに、IDEの機能低下は、生体内試験でのアミロイドβ蓄積に寄与する可能性がある(216)。ApoE4キャリアの海馬では、IDEの発現レベルが低下していることが測定されており(217)、IDEの発現と活性の遺伝的差異がアルツハイマー病発症に関与していることが示唆されている(218-221)。肝臓や脳におけるIDEの発現レベルの低下は加齢と相関しており(222)、IDEは酸化ストレスによるダメージを打ち消すことができ、神経保護的な役割を示唆している(223-226)。
メトホルミンは、AMPKを介して肝臓の糖新生を阻害することで血糖値を低下させる(227, 228)。AMPKは、インスリンおよびIGF1受容体の下流の重要な経路であるPI3K/Aktシグナル伝達を阻害する(229)。メトホルミンもまた、AMPKとは独立してインスリンシグナル伝達に作用することが示されている。メトホルミンは、インスリンおよびIGF-1受容体の発現をダウンレギュレートし(230,231)IRS-1を含むインスリン受容体(232)のリン酸化を減少させることが報告されている(230,233)。
メトホルミンの急性および慢性投与は、ヒトおよびマウスにおいて、インスリン分泌を誘導することが知られているインクレチンであるGLP-1のレベルを上昇させることがわかっている(234-236)。最近、パーキンソン病を対象とした無作為化二重盲検プラセボ対照試験では、GLP-1アゴニストがパーキンソン病の運動症状にプラスの効果を示すことが示された(237)ことから、神経変性におけるメトホルミンの作用機序に新たな可能性が生まれた。
mTORシグナル伝達
mTOR シグナルは、栄養や酸化還元状態などの上流のシグナルを統合し、細胞の成長、運動性、生存、死などの下流のプロセスを制御する、高度に保存された中心的なシグナル伝達経路(238)。mTOR経路は、多くの神経変性疾患で欠落しているミトコンドリアのバイオジェネシスとオートファジーの制御に重要な役割を果たしている。
mTORはセリン/スレオニンプロテインキナーゼであり、mTORC1とmTORC2のタンパク質複合体から構成されている。mTORシグナルは、主要なインスリンシグナル伝達経路であるPI3K/Akt経路によって標的とされている(239, 240)。PTEN(241, 242)とAMPK(243, 244)の両方がmTorシグナルを抑制し、ラパマイシンはmTORC1のよく研究された阻害剤である(245-247)。mTorシグナルは多くの下流イベントに影響を与えるが、作用の最も重要なメカニズムは、S6K1および4E-BP1のリン酸化および活性化、およびその後のRNA翻訳の制御を介してである(238)(図(図4)4.4)。興味深いことに、mTorシグナル伝達の欠乏は、インスリン抵抗性と糖尿病に関与している。S6K1の栄養依存性刺激は、インスリン抵抗性を誘導することができ(248,249)S6K1の欠乏は、高脂肪食誘発性インスリン抵抗性(250)から保護される。
図4 メトホルミンとラパマイシンの重複作用
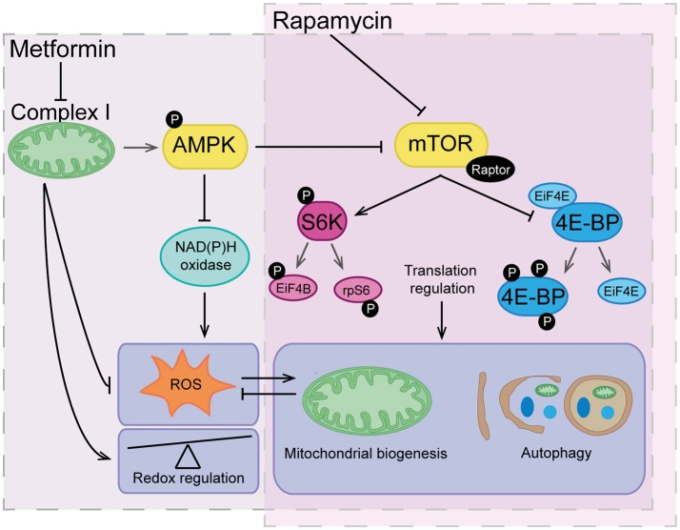
ラパマイシンは、mTOR、したがって翻訳調節を直接阻害することによって作用し、ミトコンドリアのバイオジェネシスやオートファジーなどの高度に調節されたプロセスに大きな影響を与える。メトホルミンは、複合体Iの阻害およびAMPKシグナル伝達の活性化を通じて、mTOR経路に間接的に作用する。また、メトホルミンは、複合体IおよびNAD(P)Hオキシダーゼの阻害作用を介して活性酸素種(ROS)を減少させ、レドックスレギュレーターとしての全体的な効果を発揮する。メトホルミンの作用の下流では、低レベルの活性酸素は、オートファジーを介してミトコンドリアの生合成や小器官やタンパク質のターンオーバーのためのシグナルを間接的に誘発することができる。逆に、オートファジーとミトコンドリアのバイオジェネシスを含む健全なミトコンドリアネットワークを維持することで、有害なレベルの活性酸素の蓄積をさらに減少させることができる。
mTOR阻害剤であるラパマイシンは、マウスの神経変性表現型を抑制し(251)、MPTPによって誘発されるドーパミン作動性ニューロンの喪失から保護する(252)。ラパマイシンはまた、L-DOPAの治療効果に影響を与えることなくジスキネジアの発症を抑制し、したがって、mTORC1シグナル伝達カスケードは、抗パーキンソン病治療のデザインのための有望なターゲットとなる(253)。
mTORシグナル伝達の亢進はアルツハイマー病患者で発見され、糖尿病や老化と関連している(254, 255)。ラパマイシンは、アルツハイマー病のマウスモデルで認知障害を廃止し、アミロイドβレベルを低下させる(256)。また、海馬の遺伝子発現シグネチャーを回復することにより、アルツハイマー病関連の表現型を改善する(257)。重要なことに、mTorはタウのリン酸化と分解を制御し(258)この経路はタウ症の治療のための興味深いターゲットにしている。
AMPKの活性化を介したmTORシグナル伝達のよく知られた阻害剤であるメトホルミン(259)と、この分野でより広く受け入れられているラパマイシンの治療効果を比較すると、明らかな違いは、メトホルミンのmTORに対する作用が比較的間接的であることである。ラパマイシンはFKBP12結合タンパク質と複合体を形成し、それがmTORC1を結合し、特異的に変化させる。メトホルミンは、複数の経路を介して間接的にmTOR経路に作用する。AMPKに依存しない経路には、転写因子の阻害(260)PI3K/AKT経路の阻害(261)およびREDDの誘導(262)が含まれる。これとは直接対照的に、ある研究では、メトホルミンはmTORC1を直接阻害することができ、AMPKではなくRag GTPasesに依存していることが示されている(263)。これらのデータは、メトホルミンが1つ以上の直接的な標的を有し、より多くの間接的な標的を有する可能性が高いという見解を支持しており、したがって、メトホルミンと研究ツールまたは治療法の使用がラパマイシンよりも受け入れられていない理由を説明している。
それにもかかわらず、mTOR経路は神経変性疾患の基礎となるいくつかの生物学的経路をリンクしており、したがって、このシグナル伝達カスケードを阻害するメトホルミンの能力は、メトホルミンを使用したより多くのメカニズム論的研究と臨床試験への組み込みが前向きに検討されるべきであるという議論を裏付けるものである。
炎症
神経炎症は神経変性疾患の進行における主要な推進力と考えられており、自然免疫機構の引き金となることが疾患発症の重要な要素として浮上してきている。ミクログリアや脳内の他の細胞型は、誤ったタンパク質や異常に局在した核酸に反応して活性化されることがある。これは、ミクログリアの生理的および有益な機能を転換させ、プロ炎症性メディエーターの持続的な放出につながる(264)。
非ステロイド性抗炎症薬(NSAIDs)の摂取は、人生の後半でアルツハイマー病の発生率を減少させることが報告されており(265,266)活性化されたミクログリアはアルツハイマー病患者の脳で発見されている(267,268)。
アルツハイマー病では、統合的なネットワークベースのアプローチにより、発症後期の自然免疫経路とミクログリア細胞に関連する遺伝子の摂動が同定された(269)。アルツハイマー病患者は、ニューロンとグリアにおいて誘導性一酸化窒素合成酵素(iNOS、神経炎症の産物)の発現増加を示し、一酸化窒素産生の増強につながる(270, 271)。活性化したミクログリアは、マウスの初代ニューロンにおいてタウのリン酸化をさらに誘導し、IL1β受容体とp38 MAPKストレスシグナル伝達を活性化させることができる(272)。
パーキンソン病では、患者は活性化したミクログリアとアストロサイトの数の増加を示し(273, 274)、ミクログリアの活性化は疾患の進行と関連している(273, 275, 276)。ミクログリアの活性化は、αSynの硝酸化を増加させ、神経細胞死をもたらす(278)。
免疫シグナルは転写イベントを誘発するだけでなく、ミトコンドリアを介した代謝フラックス、酸化還元バランス、代謝物バランスの変化も引き起こす(279)。ミトコンドリアの機能不全は神経炎症と関連しており(280)、中等度のミトコンドリアDNAストレスでさえも抗ウイルスシグナル伝達を引き起こす可能性がある(281)。
メトホルミンは、一般的な炎症パラメータを減少させ、異なる細胞タイプにおけるNF-κBシグナル伝達およびプロ炎症性サイトカインを阻害する(282-285)ことから、メトホルミンが神経炎症から保護する可能性があることが示唆される。興味深いことに、2つのMPTP誘発性PDマウスモデルでは、メトホルミンはミクログリアマーカーIba1のレベルを低下させ、黒質体前庭の炎症性サイトカインTNF-α、IL-1β、IL-6,およびiNOSのレベルを低下させた(46, 49)。ここでは、さらなる研究が必要であるが、メトホルミンは一般的な炎症に対して完全に肯定的な効果があるようである。神経炎症は神経変性疾患に関連する事象として認識されており、メトホルミンは有用なツールであり治療法であると考えられる。
ミトコンドリア
ミトコンドリアは、エネルギーを生産し、中枢代謝や細胞のシグナル伝達に必要な他の多くの機能を実行する重要な小器官である。ミトコンドリア機能不全は、すべての神経変性疾患を横断する現象であり、2型糖尿病におけるβ細胞機能不全の基礎を形成している(286)。神経疾患におけるミトコンドリア機能不全の重要な側面の一つは、ニューロンにおけるエネルギー代謝の厳密に制御された必要性が、ニューロンの終焉に関与する脆弱性の一部を部分的に説明できるということである。
パーキンソン病
パーキンソン病では、ミトコンドリアに決定的な影響を与える環境因子や疾患遺伝子の同定により、ミトコンドリア機能障害と疾患との関連性が証明されている。その結果、パーキンソン病におけるミトコンドリア機能障害の役割については多くの研究がなされてきたが、散発性パーキンソン病の基礎となる正確なメカニズムはあまり定義されていない。
PINK1またはパーキンの機能喪失変異はミトコンドリア機能障害の結果としてパーキンソン病を引き起こす(287-290)が、これは試験管内試験(291,292)および生体内試験(293-296)で解明されている。PINK1とパーキンは、ミトコンドリアの脱分極によって誘導されるマイトファジー(リソソームを介した損傷したミトコンドリアの除去)に重要な経路で作用する。ここでは、PINK1はパーキンの上流で機能している(295, 297)。ミトコンドリアが損傷すると、PINK1はミトコンドリア表面に蓄積し、パーキンをミトコンドリアに選択的にリクルートする(298, 299)。ミトコンドリア基質はユビキチン化され、損傷したミトコンドリアの除去につながる。PINK1は現在、ユビキチンキナーゼであることが知られているが(300)、まだ知られていない他の機能を持っている可能性がある。例えば、PINK1は、生体内での基底マイトファジーには必要とされず(301,302)複合体I(303)ミトコンドリアダイナミクス(304)ミトコンドリアプロテオスタシス(305)およびTRAP1を介したミトコンドリア代謝を調節することが提案されている(306,307)。
PINK1とパーキンは、肥満マウスと糖尿病マウスの血管壁の代謝ストレス下でアップレギュレーションされ、活性酸素種(ROS)産生とミトコンドリア機能不全を制限することで保護作用を持つ(308)。糖尿病マウスモデルでは、この場合、過酸化水素処理後に海馬におけるPINK1の発現が減少した(309)ことから、PINK1がストレスセンサーとしての役割を果たし、それに応じて多様な方法で機能することがさらに示唆されている。PINK1は、機能の損失がパーキンソン病を引き起こすので、一般に神経保護と関連しているが、PINK1は通常、ミトコンドリア外膜で高度にターンオーバーされており、したがって、過剰発現および/または発現の変化はまた、望ましくない下流のイベントを誘発する可能性があるためである。ある研究では、PINK1の過剰発現はMAPKおよびROSシグナル伝達を抑制し、細胞モデルにおいてインスリン抵抗性を緩和した(310)。逆に、PINK1の欠損は膵島およびβ細胞の機能を破壊し、グルコースの取り込み障害と血漿インスリンレベルの上昇を引き起こす(311)。PDタンパク質がエネルギー代謝に重要な役割を果たしていることを示す更なる証拠として、TP53INP1欠損細胞(TP53INP1は2型糖尿病の感受性遺伝子座である)がPINK1-パーキン経路を介してマイトファジーを損なう活性酸素の増加を引き起こすことを示す研究がある(312)。
αSynのパーキンソン病変異は、ミトコンドリアの完全性と機能の低下を含むいくつかの細胞障害と関連している。最近の研究では、ヒトおよび動物の脳においてミトコンドリアの損傷およびマイトファジーを誘導する非常に神経毒性の高いαSyn種が同定されている(313)。しかし、これらのミトコンドリアの変化が生体エネルギー機能に及ぼす影響はいまだに不明である。興味深いことに、αSynの毒性は、メトホルミンにリンクされているミトコンドリアATPアーゼであるTRAP1(314)によって緩和される。
この経路では、TRAP1およびミトコンドリアのセリンプロテアーゼHtrA2は、両方ともPDタンパク質PINK1の標的である(305,306)。HtrA2とTRAP1の遺伝的変異はパーキンソン病患者で発見されているが(307,315)、変異はまれであり、議論の的となっている(316-318)。疾患への遺伝的寄与にかかわらず、TRAP1は少なくともミトコンドリアにおいて、エネルギー代謝の微調整に関連する重要な制御的役割を果たしているようである。TRAP1の発現は腫瘍細胞において緊密に制御されているため(319)、TRAP1は、代謝再利用および炎症反応に重要なコハク酸脱水素酵素(321)を標的化し阻害することにより代謝スイッチ(320)として作用する(322)ことから、癌においてよく研究されている。
TRAP1の発現が変化した卵巣癌では、メトホルミンは化学療法に対して腫瘍を感受性にするのに有効であった(323)ことから、メトホルミンがTRAP1媒介のシグナル伝達に関連している可能性が示唆された。これに基づいて、メトホルミンはその後、パーキンソン病のTRAP1細胞モデルにおいてミトコンドリア機能障害の救済に成功した(307)。健康な人では、PINK1-HtrA2-TRAP1経路やその他の制御機構を介してミトコンドリアのエネルギー使用量を微調整することで、細胞がエネルギーを節約し、酸化的負担を軽減することができるかもしれない。メトホルミンが試験管内試験でこの微調整の役割を模倣する能力は、散発性パーキンソン病の1つのモデルにおいて有益であった(307)。しかし、メトホルミンが非疾患ニューロン、老化ニューロン、家族性および散発性パーキンソン病の他の形態において有益かどうかなど、まだ多くの疑問が残されている。一つの疑問は、メトホルミンが黒質体のドーパミン作動性ニューロンのエネルギー障害を特異的に標的にできるかどうかということである。選択的脆弱性はまだ完全には解明されていないため、この疑問にはまだ答えられていない。我々は、これらの細胞を特に脆弱にしているのは、経時的な酸化的負荷や代謝的負荷が原因ではないかと推測することができる。多くの酸化還元反応は、ミトコンドリアの活動の結果としてミトコンドリアで起こる。他の多くの細胞タイプと比較して神経細胞は高いエネルギー需要を持っており、黒質のドーパミン神経細胞では自律的なペースメーキングを行っているため(324)、これらの細胞はより高い酸化的負荷を持っていると考えられている。ドーパミン自体の代謝は高度に酸化的であり、いくつかの毒性種を形成する可能性がある。したがって、メトホルミンが、正常な酸化還元シグナル伝達を阻害することなく、ミトコンドリアでの酸化負荷を軽度に軽減し、時間の経過とともに効果が低下するオートファジーなどのプロセスを刺激することができれば、神経変性疾患の対策に非常に有用な薬剤と考えられる。神経細胞は、一度死んでしまうと、炎症が頻繁に起こり、めったに交換されないため、ストレスを補償し、あらゆるオッズに対して生き延びることを可能にする品質管理防御の洗練されたユニークなラインを持っている。それはちょうど必要な代償応答を妨害しないように適切なタイミングで介入するためにメトホルミン治療を使用することができるかどうかに依存し、むしろそれらを強化する。
アルツハイマー病
アルツハイマー病につながる正確なミトコンドリアイベントはパーキンソン病に比べて定義されていないが、加齢は依然として最大の既知の危険因子である。エネルギー代謝とミトコンドリア機能不全は、シナプス変性、アミロイドβ沈着、神経原線維のもつれの形成など、アルツハイマー病の基礎となるメカニズムの主要なイベントとして提案されている(325)。ミトコンドリア機能不全は、アルツハイマー病の初期の細胞イベントの後に発生し、さらなる変性の進展に寄与しうるという膨大な証拠があるが、ミトコンドリア機能不全が本当に単なる二次的イベントであるのか、あるいは一次的な病態に関与している可能性があるのかは、しばしば不明である。例えば、タウの場合、異常なタウは酸化ストレスを引き起こし、ミトコンドリアの脱分極、ミトコンドリア複合体活動の障害、エネルギー出力の低下などのミトコンドリア障害を引き起こす(326, 327)。タウはまた、アダプタータンパク質の助けを借りてミトコンドリアが移動するトラック、微小管に局在し、欠陥ミトコンドリアの動きは、アルツハイマー病のいくつかのモデルで示されている(328,329)。
ミトコンドリアの代謝がアルツハイマー病脳で変更されているという証拠もある((330でレビュー)。トリカルボン酸(TCA)サイクル酵素であるピルビン酸デヒドロゲナーゼ、イソクエン酸デヒドロゲナーゼ、およびα-ケトグルタル酸デヒドロゲナーゼは、アルツハイマー病脳組織および患者由来の線維芽細胞において影響を受けている(331)。これらのチェックポイントTCAサイクル酵素の変化は、ストレスおよび酸化還元の変化に応答して、しばしば代謝再配線に関連している。マトリックス酵素に加えて、酸化的リン酸化(OXPHOS)の欠乏が報告されている[(332)でレビューされている]。
アルツハイマー病研究では、ミトコンドリア機能不全のメカニズムモデルはほとんどなく、主にアルツハイマー病の原因遺伝子がミトコンドリアに存在しないことが原因である。認知症とアルツハイマー病におけるメトホルミンの作用機序は、メトホルミンがミトコンドリアの生合成とエネルギー保存を介してミトコンドリアの質の制御に作用するという点で、パーキンソン病においても同様である可能性が高い。
複合体Iのパラドックス
メトホルミンの作用の多くは、複合体Iの阻害の間接的な結果であると考えられている。メトホルミンの複合体Iに対する正確な阻害機構は完全には解明されていない。MPTPやロテノンのような他の複合体I阻害剤の阻害機構は、特に疾患における結合部位および毒性のメカニズムの点でよりよく知られている。
コンプレックスIの欠乏は、長い間、ミトコンドリア機能不全およびパーキンソン病リスクと関連してきた[レビューは(333)を参照のこと]。コンプレックスIの欠乏は、アルツハイマー病、HD、ALSでも報告されている(332)。神経毒であるMPTPとロテノンは複合体Iを阻害し、毒性レベルの活性酸素を発生させ、神経細胞死を引き起こす。活性酸素を発生させない(または発生量が少ない)亜致死濃度のミトコンドリア阻害剤が有益である可能性があるが、ほとんど知られていない。
メトホルミンは危険なレベルの活性酸素を発生させないことが一般的に認められている。あまりにも多くの活性酸素を発生させることなく、薬理学的に酸化的リン酸化を減少させ、その結果、酸化的負荷を(適切なタイミングで)減少させることは、確かに課題である。我々は、特定のミトコンドリア複合体V阻害剤であるオリゴマイシンの亜致死濃度が、TRAP1欠損PDモデルにおいて、メトホルミンと同様の範囲でミトコンドリア機能障害を救済できることを発見した(307)が、メトホルミンはヒトでの消費が承認されている化合物であるため、我々はメトホルミンの保護効果のみを追跡調査した。異なる部位に作用するいくつかの呼吸鎖阻害薬の滴定を用いて、神経保護作用と毒性の可能性を評価することは興味深いことかもしれない。例えば、ミトコンドリア複合体IIIの阻害剤であるアンチマイシンAは多量の活性酸素を発生させることが知られているが(334)、オリゴマイシンや他の呼吸鎖阻害剤はほとんど、あるいは全く活性酸素を発生させないことが示されている(335)。
老化
Lopez-Otin氏は、老化の主な特徴として、ゲノムの不安定性、テロメアの萎縮、エピジェネティックな変化、プロテオスタシスの喪失、栄養感知の低下、ミトコンドリアの機能不全、細胞の老化、幹細胞の枯渇、細胞間コミュニケーションの変化を挙げている(336)。これらの特徴はすべて、何らかの形で神経変性疾患の発症に関連している。ここでは、これらの特徴に関連して、ミトコンドリアにおけるメトホルミンの作用機序に最も関連していると考えられるいくつかの特定の側面に注目する。
ミト核タンパク質の不均衡
ヒトのミトコンドリアDNA(mtDNA)は、ヌクレオイドの束の中に結合しており、高いコピー数を持ち、母性遺伝し、高い突然変異率を持つ(337)。ミトコンドリアの損傷および/または枯渇は、ストレスシグナリングおよび適応的代謝応答を誘導する。MtDNA の不安定性は、多くのヒトの疾患や老化で観察される生理学的に関連するストレスである(281)。ミト核タンパク質の不均衡は、核内タンパク質とミトコンドリア内にコードされたタンパク質の間の化学量論的な不均衡であり、多くの種で長寿の鍵となる応答として活性化されている(338)。mtDNAの変化は散発性パーキンソン病患者の呼吸鎖機能障害に直結しており、最初に複合体Iが影響を受け、次に複合体IVが影響を受けることが示されている(339)。ミトコンドリアで翻訳されたタンパク質と核内でコードされたタンパク質の間の化学量論の不均衡は、細胞シグナルであると同時に、ミトコンドリアの適応のマーカーでもあると考えられている。mTOR阻害剤であるラパマイシンは、ミトコンドリア核タンパク質の不均衡を引き起こすツールとして使用されており(338)、メトホルミンはヒト細胞におけるミトコンドリア核タンパク質の不均衡を調節することが可能である(307)。
酸化ストレスと老化
活性種の産生は、通常、細胞の抗酸化防御によってバランスが保たれている。抗酸化防御に対する活性種の量のバランスが崩れると、酸化ストレスが生じ、タンパク質、脂質、ヌクレオチドに損傷を与えることになる。
ミトコンドリアは、電子輸送鎖を介してエネルギー生産に酸素を使用するため、活性酸素の主要な供給源となる。電子は、電子輸送鎖の複合体に沿って移動している間に漏れる。漏れた電子は、分子状酸素と反応してスーパーオキシドラジカルを形成することができる。スーパーオキサイドは、Mn-SODと反応して過酸化水素、活性酸素、シグナル分子を形成することができる。過酸化水素は分解されて水を形成するか、金属と反応して反応性の高いヒドロキシルラジカルを形成する。ミトコンドリアでは、主な漏出部位は、複合体 IV の酸素への 4 電子の移動にあるが、複合体 I、複合体 III、およびミトコンドリアマトリックス内の TCA サイクルの特定のデヒドロゲナーゼを介しても漏出する。酸化ストレスの結果は、細胞の種類と重症度に応じて、増殖、適応、損傷、老化、または死を含む[レビューは(340, 341)を参照のこと]。神経細胞は、高エネルギーバーストとカテコラミン神経伝達物質代謝のために、大量の活性酸素に対抗する必要がある。
酸化的損傷は神経変性疾患の主な原因である[レビューは(342)を参照]。酸化的ストレスと酸化的損傷の両方がストレス適応につながる可能性がある。ミトコンドリアにおけるそのような適応メカニズムの一つは、呼吸器複合体の微調整された阻害、またはミトコンドリアのアンカップリング蛋白質を介したアンカップリングであるかもしれない。ミトコンドリアのアンカップリング蛋白質が神経保護作用を持つことを示す証拠は数多く存在する[レビューは(343)を参照]。細胞の老化は、適応応答が、細胞がもはや分裂できなくなるほどの損傷から鍵となる分子を適切に保護できない場合に起こりうる。
PDタンパク質DJ-1は、神経変性とエネルギー代謝との間のリンクを提供する。DJ-1は、ミトコンドリアを安定化させ、酸化ストレスから細胞を保護するためのシャペロンおよびプロテアーゼとして作用する(344)。他の細胞機能としては、転写共活性化因子としての Ras の結合(345)PTEN の阻害によるホスホイノシタイド-3-キナーゼ(PI3K)/AKT シグナルカスケードの負の制御(309, 346, 347)シャペロン機能(348, 349)RNA 結合(350)などがある。議論の余地はあるが、DJ-1はまた、グリオキサラーゼ(351)およびデグリーケース(352)酵素活性を有すると主張されている(353)。DJ-1はまた、膵臓のβ細胞の生存率と同様にインスリン分泌にも影響を与え、DJ-1ノックアウトマウスは膵島細胞の活性酸素レベルの増加、耐糖能の障害、インスリン分泌の減少を示している(354)。
研究のギャップ
メトホルミンを用いたヒトや動物の神経変性疾患の治療や予防のための試験では、ほとんどの場合、相反する結果が得られている。データによると、細胞培養、動物、ヒトの神経変性過程に対するメトホルミンの効果は、肯定的なもの、全くないもの、あるいは有害なものであることが示されている。
その結果は、種、細胞タイプ、あるいは基礎となる代謝状態に依存している可能性がある。しかし、2つの有望な研究分野は、神経炎症と老化であるが、さらなる研究が必要とされている。神経炎症におけるメトホルミンの役割を直接調べた研究はほとんどないが、この分野では研究の焦点が高まっているため、より多くのメトホルミン研究が行われる可能性がある。老化におけるメトホルミンの正確な役割は、神経変性疾患に対するメトホルミンの可能性をさらに理解する前に、少なくとも部分的に理解する必要がある問題である。そのための大きな障害となっているのが、主に試験管内試験だけでなく生体内試験でのヒト老化モデルが不足していることである。
もう一つの知見のギャップは、非糖尿病患者におけるメトホルミンの使用による潜在的な副作用があるかどうかである。例えば、メトホルミンの長期使用がビタミンB12欠乏症につながることはよく知られている(355)。ビタミンB12と葉酸は、神経伝達物質の生合成に重要なアミノ酸からのトランスメチル化反応と水酸化反応に必要である。神経伝達物質の代謝が障害されている患者および/または他の薬剤を投与されている患者において、これがどの程度の影響を及ぼす可能性があるのであろうか。
メトホルミンの治療可能性:実現可能性
神経変性の治療にメトホルミンを使用することで、臨床医や科学者が治療や研究ツールとしての可能性に疑問を抱く理由はいくつかある。
第一に、メトホルミンは多くの生物学的経路に作用しているようであり、そのメカニズムを特定することは非常に困難であるという点である。
第二のポイントは、一般的に「抗加齢」薬について議論の的になっていることである。老化の生物学的基盤についてはほとんど知られておらず、実験室で効率的に老化をモデル化する方法についてもほとんど知られていないため、「抗老化」薬の宣伝は答えよりも多くの疑問を投げかけてしまうことが多い。
研究者の間では、メトホルミンがミトコンドリアの呼吸を阻害することで作用するという事実があり、これはパーキンソン病の分野で長年の研究により、実際に病気の発症に寄与することが示されている。
これとは正反対に、メトホルミンが実現可能で有用な薬剤であることにはいくつかの議論がある。第一に、グルコース代謝は、ニューロンの酸化還元状態、したがってニューロンの長期的な生存にとって中心的な重要性を持っている。第二に、人口として我々はますますインスリン抵抗性であるため、メトホルミンは特に適している。メトホルミンは安価で安全な薬剤であり、副作用も少ないため、試験管内試験、生体内試験、試験での研究の進展は歓迎されるだろう。
アルバート・アインシュタイン医科大学の加齢医学研究所の所長であるNir Barzilai氏は、メトホルミンやその他の関連薬は、健康で無病息災な生活を何十年も延ばすことができると示唆している(356)。他の科学者はメトホルミンについては特に言及していないが、ニック・レーンは2005年のミトコンドリアに関する著書の中で、もし私たちが老年期の病気から身を守るために長生きするのであれば、より多くのミトコンドリアと、より洗練されたフリーラジカル検出システムが必要であることを示唆している(357)。メトホルミンが有害な影響を与えることなく、適切な生理学的瞬間に検出システムを変更できるかどうかは、少なくともエキサイティングな可能性を秘めている。
この分野の今後の展開
メトホルミンは、老化に対抗するという課題において有益である可能性があり、臨床研究では、メトホルミンが2型糖尿病患者の認知機能にプラスの効果をもたらす可能性が示唆されている。メトホルミンがどのように作用するかをよりよく理解することは、神経変性分野の研究者が将来の研究や試験を成功させるために役立つだろう。TAME(358)のような今後の研究はこの点で役立つだろう。
メトホルミンのアンチエイジング効果は、有害な量の活性酸素を発生させることなく、エネルギー生産の多段階プロセスを妨害する能力によって要約することができる。この作用だけでも神経保護作用があると考えられ、メトホルミンは他の生物学的経路を活性化することでさらに保護することができる。例えば、ミトコンドリアのエネルギー産生を遅らせることは、肝臓でのシグナル伝達イベントのカスケードを誘発し、結果としてグルコースとインスリンの減少をもたらす。成長と増殖と生命維持のバランスをとる栄養感知におけるインスリンの重要な役割は、メトホルミンを興味深い薬剤にしている。老化研究の分野は発展しており、生体内試験や試験管内試験の老化モデルが進歩している。
おそらく、メトホルミンの作用が複雑であるために、さまざまな神経変性疾患の形態における加齢の役割を理解するためには、より多くの研究が必要であるため、この薬剤は現段階では神経変性疾患の潜在的な治療薬としての役割を果たすことはできないだろう。今日のメトホルミンの最大の価値は、神経変性疾患の根底にあるメカニズムの解明に役立つ可能性を秘めているかもしれない。