
Contents
The Role of Copper in Tau-Related Pathology in Alzheimer’s Disease
www.ncbi.nlm.nih.gov/pmc/articles/PMC7533614/
オンラインで公開2020年9月10日
Klara Zubčić,1 Patrick R. Hof,2,3,4 Goran Šimić,1,* and Maja Jazvinšćak Jembrek5,6,*.
要旨
アルツハイマー病を含むすべてのタウ症は、神経細胞やグリア細胞における異常な形態のタウタンパク質の細胞内蓄積によって特徴づけられ、微小管の安定性に悪影響を及ぼす。生理的条件下では、チューブリン結合単位(タウ)タンパク質は本質的に乱れており、二次構造をほとんど持たず、凝集することはない。アルツハイマー病では、それが集合し、さらに神経原線維のもつれ(NFTs)を構築するペアらせん状フィラメント(PHFs)を形成する。凝集体は、より凝集しやすい高リン酸化タウタンパク質で構成されている。アルツハイマー病の病理学はまた、酸化ストレス(酸化ストレス)を促進する銅のホメオスタシスの乱れにリンクされている。銅の不均衡は、アルツハイマー病患者で広く観察されている。銅イオンの調節障害は、タウの高リン酸化とβシートに富んだタウ線維の形成を開始し、悪化させ、最終的にはシナプス障害、神経細胞死、認知機能の低下に寄与する可能性がある。本レビューでは、タウ凝集のプロセスに影響を与える因子、凝集におけるシード部位として機能するために必要な微小管結合ドメイン内の小ペプチド配列の構造変化、酸化ストレス誘導、タウ高リン酸化およびタウ集合における銅の役割についてまとめている。酸化ストレス条件下でのタウ凝集に影響を与える様々な因子をよりよく理解することは、アルツハイマー病治療のための新たな標的や新たな薬理学的アプローチを明らかにする可能性がある。
キーワード:銅、タウ、凝集、酸化ストレス、アルツハイマー病
序論
Tubulin-associated unit (Tau)は、神経細胞、主に軸索に優勢に発現する微小管関連リンタンパク質である。タウは、微小管ネットワークの適切な組み立て、安定化、機能に不可欠である。タウはまた、軸索輸送を制御し、ニューライトの伸長を駆動し、ニューロンの形態を形成する(Choi et al 2009; Wang and Mandelkow, 2016)。
ヒトのタウは、1つの遺伝子に由来する6つのアイソフォームを持つ比較的大きなタンパク質である。これらのアイソフォームの長さは352〜441アミノ酸の範囲である。タウは、N末端部のプロジェクションドメインとC末端部の微小管結合ドメインという2つの機能ドメインに分かれている(レビューは、Šimić et al 2016; Barbier et al 2019を参照)。タウに関連する病理については、アイソフォーム間の大きな違いは、3つまたは4つの不完全な擬似リピートユニット(R1-R4)を含む微小管結合ドメインに位置している。各リピートは、18残基(オクタデカペプチド)の高度に保存されたフラグメントを含む31〜32アミノ酸配列からなる。したがって、これらのアイソフォームは、3R-tauまたは4R-tauアイソフォームと呼ばれている。これらのアイソフォームは、すべて第3の繰り返しR3(最長のヒトタウアイソフォームによると、残基306〜336)を含み、R2リピートの不在または存在によって互いに異なる。4R-tauアイソフォームは、より高い親和性で微小管に結合する。健康なヒトの脳では、3R-/4R-tauアイソフォームは同様の比率で発現しているが、この比率はアルツハイマー病(アルツハイマー病;レビューについては、Alavi NainiおよびSoussi-Yanic酸化ストレスtas 2015;Liu et al 2015;ChengおよびBai 2018を参照のこと)では乱れている。アイソフォーム間の他の違いは、プロジェクションドメイン(0N、1N、または2N)におけるN末端挿入部の数である。微小管結合ドメインと投影ドメインに隣接するプロリンに富んだ領域は、タウの微小管との相互作用を容易にする。タウのN末端部分は微小管間の間隔を決定し、微小管結合には関与しない(Schneider et al 1999; Kadavath et al 2015; Cheng and Bai 2018; Barbier et al 2019)。
タウは、進化的に保存された残基の小さなグループで微小管に強固に結合するが、介在する部分は柔軟なままである。したがって、タウは微小管に結合しているにもかかわらず、高度に動的なままである。これらの微小管結合モチーフの小群は、α-β-チューブリンヘテロダイマーの界面にある疎水性ポケットに結合することで、微小管のプロトフィラメント構造を安定化させている。このメカニズムは3R-tauや4R-tauのアイソフォームでも同様である。微小管への結合に関与する残基は、アルツハイマー病の特徴であるタウの凝集に決定的に関与している(Kadavath et al 2015)。
タウリン酸化とタウキナーゼのタウ機能への影響
タウは、その機能を修飾し、脳内に存在するタウアイソフォームの不均一性に寄与する多くの翻訳後修飾の影響を受けやすい。生物学的タウ機能の多様性は、主にアルツハイマー病病理学の伝播に特に関連する修飾として多くの注目を集めたリン酸化によって調節される(Jazvinšćak Jembrek et al 2013; Alavi NainiとSoussi-Yanic酸化ストレスtas 2015; Šimić et al 2016; Barbier et al 2019; Miao et al 2019)。アルツハイマー病では、タウは高リン酸化されており、微小管には付着していない。複数の残基でのリン酸化の全体的な増加として表される高リン酸化は、微小管に対するタウの結合親和性を低下させ、生理的なタウの機能の喪失の原因となる。高リン酸化は、タウと微小管との間の相互作用を乱し、その結果、微小管の不安定性、微小管束の減少、軸索輸送および神経細胞の構造の障害、そして最終的には細胞死をもたらす(Evans et al 2000;Alavi NainiおよびSoussi-Yanic酸化ストレスtas 2015;Barbier et al 2019)。
微小管との相互作用に加えて、そのN末端部分のタウは、非受容体チロシンキナーゼFynを含むSrcホモロジー3(SH3)ドメインを含むシグナル伝達タンパク質と結合する可能性がある。Fynは、特に高リン酸化されたタウを持つニューロンで、アルツハイマー病脳で上昇しており、タウの翻訳を促進することによってタウレベルの上昇に寄与している。Fyn自体もまた、Tyr18でタウをリン酸化する可能性があり、このリン酸化ステップは、アルツハイマー病の病理学にリンクされている(Lee et al 2004)。しかし、アルツハイマー病におけるFynの最も顕著な役割は、アミロイドβ毒性に対するタウの影響に関連している。軸索から樹状体コンパートメントへのリン酸化タウの誤局在化が、Fynの濃度を局所的に増加させることでアミロイドβの毒性効果を媒介していると考えられている(Li and Götz, 2017; Nygaard, 2018)。生理的条件では、タウは樹状突起に低量で存在するが、FynがNR2Bサブユニットを含むNMDA受容体とシナプス後密度(脳卒中後認知症)タンパク質脳卒中後認知症-95との間の相互作用を強化するシナプス後樹状突起膜へのFynの隔離に重要な役割を持っている。これは、可溶性アミロイドβオリゴマー(Alavi Naini and Soussi-Yanic酸化ストレスtas, 2015; Nygaard, 2018)によってトリガーされた興奮毒性シグナル伝達カスケードを駆動するためにアルツハイマー病において重要である。
最も長いタウアイソフォームは、リン酸化され得る85の残基を有する。それらのほぼ45は、健康な成人ヒトの脳から 10-18と比較して、アルツハイマー病脳でリン酸化されている。リン酸化残基の非常に少数のN末端領域と微小管結合ドメインで発生する。代わりに、リン酸化部位は主に微小管結合ドメインのフランキング領域に位置している(レビューについては、Alavi NainiおよびSoussi-Yanic酸化ストレスtas 2015;WangおよびMandelkow 2016を参照のこと)。タウのリン酸化は、少なくとも試験管内試験では、多くのキナーゼによって媒介される。それらの中で、グリコーゲン合成酵素キナーゼ-3β(GSK-3β)およびサイクリン依存性キナーゼ5(cdk5)は、特に重要であると考えられている。GSK-3βの活性の増加、cdk5とその活性化因子であるp35,p25のアップレギュレーションがアルツハイマー病脳サンプルの前頭皮質で認められた(Duka er al)。 これらのキナーゼは、アルツハイマー病に関与することが知られている複数のセリン残基およびスレオニン残基でタウをリン酸化し、それらの過剰発現または活性化は、タウ症に特徴的な表現型を誘導する(Terwel et al 2008; Kremer et al 2011; Jazvinšćak Jembrek et al 2013; Llorens-Martín et al 2014)。C末端で切り捨てられたGSK-3βは、アルツハイマー病脳で顕著に増加しており、切り捨ては、タウ高リン酸化とBraakステージと正の相関を示している。カルパインIによるGSK-3βのタンパク質分解的切断は、そのキナーゼ活性を増加させる(Feng et al 2013; Jin et al 2015)。両キナーゼは酸化ストレス(酸化ストレス)条件下では活性が亢進する(Shukla et al 2012; Alavi Naini and Soussi-Yanic酸化ストレスtas 2015)ことから、ROSの発生を減衰させることで活性が低下し、タウ過リン酸化のレベルが緩和される可能性があることが示唆されている。
アルツハイマー病および他のタウ症におけるタウ凝集
微小管を不安定化させるだけでなく、高リン酸化されたタウは微小管からの剥離時に脳内沈着物を形成する。これは、タウの溶解度が非常に高いため、やや予想外の結果である(Schneider er al)。 自己増殖性の凝集過程では、タウは最初に二量体を形成し、次にオリゴマーを形成し、それが蓄積してプロトンマーを形成する。ペアのプロトマーは、互いにねじれて、ペアらせん状フィラメント(PHF)を生成し、それが結びつき、神経原線維性タングル(NFT)の束に成長する;レビューについては、Mokhtar et al 2013;Liuu et al 2015を参照のこと)。胎児タウアイソフォーム(0N3R)は、高リン酸化状態でもPHFを形成しないため、その発現や翻訳後修飾を理解することは、アルツハイマー病治療や予防の開発に向けた今後の研究にとって重要であるかもしれない(Jovanov-Milošević et al 2012)。
高リン酸化に加えて、様々なプロテアーゼ(カスパーゼおよびカルパインなど)によるタウの開裂は、アルツハイマー病の病因形成に重要な別の修飾である(Kovacech and Novak, 2010; Zhou er al)。 タウの切断は、微小管結合ドメインの疑似リピートにまたがり、Glu391でC末端に切り捨てられるタウ断片を生成する。これらの切断された断片は、PHFの構造的なコアを形成する。コアからのタウフラグメントは、C字型のユニットからなり、3Rまたは4R-タウアイソフォームに由来するものとは独立して、長さが正確に3回繰り返される(Wishik et al 2014,2018)。このように、タンパク質分解抵抗性を有するPHFのフィブリルコアは、繰り返し配列であるタウのごく一部のみによって構築される。それは、C末端とN末端からの残基に由来する高度に動的なファジーコートに囲まれており、分子内の長距離静電相互作用のネットワークがコアをこのファジーコートとリンクしている(Bibow et al 2011)。アルツハイマー病の進行に関連して、切り捨てられたタウは、完全長タウの切り捨て形態への変換を誘発することによって、タウ凝集を播種する能力がなければならない(Wischik et al 2014,2018;Fitzpatrick et al 2017)。アスパラギンエンドペプチダーゼ、老化中およびアルツハイマー病において活性化されたシステインプロテイナーゼは、神経病理学的タウの変化の重要なメディエーターとして作用する。それは、タウを分解し、微小管アセンブリにおけるその機能を損なうとともに、凝集を促進する(Zhang et al 2014b)。
タウタンパク質全体は、試験管内試験で凝集する能力は低いが、偽リピート単位を含む断片は、分子間ジスルフィド橋を形成する能力に基づいて、はるかに容易に集合するが(Schweers et al 1995)タウタンパク質全体は、試験管内試験で凝集する能力を有している。核形成のための前提条件は、二量体の形成である(Friedhoff et al 1998)。したがって、実験条件下では、PHFの集合体は、しばしば、ジスルフィド結合形成を介して、ポリアニオン(例えば、ヘパリン)の存在下で二量体化を促進し、システイン残基を減少させたままにする還元剤の存在下では抑制され、二量体形成を妨げる(von Bergen et al 2000)。他のアミロイドについては、PHFにはα-ヘリックス構造とβ-シート構造の両方が存在するが、β-シート構造が凝集やフィブリル形成に重要であると考えられている(Sadqi et al 2002; Ma et al 2005)。しかしながら、アミロイド構造は、それらが完全に秩序化されていないため、構造-病理学的関係の正確な決定を複雑にするため、特性化には困難である(Fichou et al 2019)。それにもかかわらず、タウ凝集体は、アルツハイマー病におけるアミロイドβ(アミロイドβ)プラークの前に現れ、それらの蓄積は、アミロイドβ沈着物の形成よりもアルツハイマー病病理の重症度および認知機能の低下とより良い相関を示す(Bussière et al 2002;Wischik et al 2014;Šimić et al 2017)。アミロイドβカスケード仮説に沿った長年の研究努力がアルツハイマー病治療の改善をもたらさなかったため、近年、タウ病理を標的とした治療が注目されている(Šimić et al 2016; Cheng and Bai 2018; Jadhav et al 2019; Takeda 2019)。
他の翻訳後修飾がタウの機能および凝集に及ぼす影響は、はるかに特徴的ではない。グリカンを含まない正常なタウとは対照的に、アルツハイマー病脳からのPHFタングルは高度にグリコシル化されている。しかし、脱グリコシル化だけではタウが微小管重合能を回復するのに十分ではなく、微小管の集合を促進するためには脱グリコシル化と脱リン酸化の両方が必要である。脱グリコシル化後、PHFはまっすぐなフィラメントの束に変換され、PHF形成におけるグリコシル化の役割を示唆している(Wang et al 1996; Takahashi et al 1997)。タウの機能を制御するもう一つの重要な翻訳後修飾は、リジン残基のアセチル化である。リン酸化と同様に、アセチル化は、微小管との相互作用を損なうことにより、微小管集合体におけるタウの機能を阻害し、タウフィブリルの形成を促進する。微小管結合ドメインのLys280は、タウのアセチル化の主要な部位の一つと考えられている。Tgマウスモデルの脳組織において、およびヒトの様々な4Rまたは3R/4Rタウ症の皮質領域を形成する抽出PHFにおいて、特にLys274およびLys280におけるアセチル化の増加レベルが、病理学的に不溶性の高リン酸化タウ凝集体において観察された(Cohen et al 2011; Alavi NainiおよびSoussi-Yanic酸化ストレスtas 2015)。他の翻訳後のタウ修飾のタウ病理への寄与のより良い理解は、特に酸化ストレス中に、アルツハイマー病治療にいくつかの新しい機会をもたらすかもしれない。
ヒトの脳は、神経細胞のタウ蓄積のみに影響を受けやすいわけでも、アルツハイマー病に特異的なタウ凝集体であるわけでもない。その上、加齢自体が、主に内側側頭葉において、原発性加齢性タウ症として知られるタウのもつれの蓄積をもたらす(Crary et al 2014;Huang et al 2016;Demaegd et al 2018)。30以上の異なる形態のタウ症において、様々な形態学的外観を有する高リン酸化タウの介在物が、ニューロン、アストロサイト、およびオリゴデンドロサイトにおいて見出され得る(Alavi NainiおよびSoussi-Yanic酸化ストレスtas 2015;KahlsonおよびColodner 2016)。注目すべきは、神経細胞と比較してはるかに低いレベルではあるが、タウは通常、グリア細胞、特にオリゴデンドロサイトで発現していることである。タウ症のリストには、前頭側頭型認知症、進行性核上性麻痺、コルチコバサル変性症、アルギロフィリック・グレイン病、ゲルストマン・ストロースラー・シェンカー病、球状グリア内包物を伴う白色物質タウ症、および他のいくつかの形態の認知症が含まれるが、これらに限定されない。分子レベルでは、異なるタウ症の間の区別は、リン酸化に基づいており、凝集体に表示されるタウアイソフォームの数(レビューについては、Alavi NainiとSoussi-Yanic酸化ストレスtas 2015を参照してほしい)。例えば、アルツハイマー病では、6つのすべてのタウアイソフォームが凝集体で発見されている。
少なくとも理論的には、その標的化はアルツハイマー病のために何らかの治療的希望をもたらすかもしれないが、グリアのタウ病の病態への寄与はよく理解されていない。すなわち、グリア細胞におけるタウ病理は、神経細胞の機能を支えるグリアの正常な役割を破壊すると考えられている。これには、血液脳関門の維持や、特にグルタミン酸の分泌と取り込みを介したシナプス機能の調節におけるアストロサイトの役割や、オリゴデンドロサイトの骨髄化機能が含まれる(レビューは、Kahlson and Colodner, 2016; Leyns and Holtzman, 2017を参照のこと)。
タウ拡散
アルツハイマー病の進行性は、主にシナプス的に相互接続された神経経路を介して誤って折り畳まれたタウの広がりに基づいている。このように、細胞外のタウが細胞内に入り、細胞内のタウプールからタウフィブリルの形成を誘発する。タウの小粒で可溶性のオリゴマー型は、最も病原性が高く伝達性の高い部位と考えられている。したがって、低分子量のオリゴマー形成の阻害および低分子量のオリゴマー形態のクリアランスは、貴重な治療的アプローチを表し得る(Fichou et al 2019)。しかしながら、このための前提条件は、凝集プロセスを理解し、その動態に影響を及ぼす可能性のある様々なアルツハイマー病関連生物学的因子および細胞内環境の構成要素を同定することである。
小さなオリゴマーおよびプレフィブリラー種は、細胞間伝達様式(Lasagna-Reeves et al 2012;Alavi NainiおよびSoussi-Yanic酸化ストレスtas 2015;WangおよびMandelkow 2016)を介して、またはミクログリア細胞を介して、タウの病理学的伝播を可能にする(Španić et al 2019)。タウは、体細胞性コンパートメントと軸索性コンパートメントの両方で侵入し、前駆的および後駆的に進行し、したがって、トランスシナプス的および細胞間の両方で拡散する(Demaegd et al 2018)。伝播過程の2つの主要な構成要素は、テンプレート化されたミスフォールディングまたは「シーディング」(すなわち、自然に折り畳まれたタウタンパク質のコンフォメーション変化を誘導する異常なタウの能力)と細胞間の拡散である(Demaegd et al 2018)。タウは、トンネル状のナノチューブ(糸状チャネル)を介して細胞間で伝播するか、あるいは様々な分泌機構により、自由な形で、あるいは輸送小胞を介して細胞外に放出され、その後、近くの細胞に取り込まれると考えられる(Demaegd et al 2018)。しかし、より最近のパンデミック性拡散モデルでは、細胞外空間ではなく、神経細胞の突起を介してのみ細胞から細胞へのタウの拡散が予測されている(Vogel et al 2020)。タウはまた、特に内側側頭皮質において、正常な老化の間、コミュニケーション経路を介してユビキタスに拡散する。アルツハイマー病では、このプロセスは、アミロイドβ負荷を持つ領域で促進され、神経変性および認知機能障害のレベルと良好な相関関係で、内側側頭葉から近くの大脳新皮質へのタウの外部への拡散をもたらす。NFTは最初に脳幹と経胸膜皮質で形成され、そこから前海馬、側頭連合大脳皮質、最終的には二次的な播種イベントを経て一次感覚野に広がっていく(Šimić et al 1998, 2009; Vogel et al 2020)。
細胞間でのタウの拡散を支持する証拠があるが、これは死後のヒトの脳を調べても証明が難しい現象である。タウを過剰発現させた非神経細胞では、Asp421でのカスパーゼ-3介在性切断を伴う高リン酸化により、タウの細胞外排泄が促進された(Plouffe et al 2012)。病的タウの細胞型特異的な広がりは、病理学的タウを濃縮したアルツハイマー病脳抽出物をヒト変異P301Sタウトランスジェニックマウス(PS19マウス;Boluda et al 2015)の海馬と皮質に脳内注射した後に実証された。同様に、ヒトタウトランスジェニックマウスの海馬へのアルツハイマー病脳(アルツハイマー病 P-tau)由来のオリゴマーおよび高リン酸化タウの注入は、対側の海馬および同側の大脳皮質において、タウの高リン酸化と同様に、タウ凝集病理をもたらした(Miao et al 2019)。タウ凝集体を含む脳溶解液を正常な脳または若いトランスジェニックマウスの脳に注入すると、注入部位において、解剖学的に接続された領域において、重度のタウ病理が生じる可能性がある(LeynsおよびHoltzman 2017)ことから、誤って折り畳まれたタウタンパク質が、シナプス的に接続された回路に沿って、空間的および時間的に隣接する細胞に伝播することが示唆される(Lasagna-Reeves et al 2012;Liu et al 2012)。
自己増殖過程を定義することができる最小の配列を探索することにより、完全なR3リピートにまたがるペプチドがアミロイドフィブリルを形成し、自己増殖的に微小管結合ドメインのミスフォールディングを誘導し、疾患を拡散させる可能性があることが明らかになった(Stöhr et al 2017)。
タウの重合に必要な構造変化
タウの過リン酸化は、微小管からの剥離およびその重合および凝集を促進することが一般的に受け入れられている。PHFは、リン酸化部位の大部分を欠いたタウタンパク質の断片から形成され得る(Wille et al 1992)。また、プロテアーゼ消化を行わずに単離されたPHFは、タウのN末端部分のリン酸化エピトープに向けられた抗体で標識されるが、ファジーコートを除去すると免疫反応性が失われる。このことは、ファジーコートがリン酸化されたアミノ酸残基の大部分を含んでいることを示している(Šimić et al 2016; Wischik et al 2018)。さらに、特定のリン酸化部位でのタウのリン酸化は、PHF形成に拮抗する可能性がある。したがって、Ser214およびSer262でのリン酸化は、微小管へのタウの結合およびPHFの組み立てを防止し、リン酸化はタウの凝集を防止することによって保護的な役割を有することを示唆している(Schneider et al 1999)。別の研究では、NFTの形成は酸化的損傷の減少と関連しており、タウのリン酸化は抗酸化的機能を持ち、酸化的損傷からニューロンの構造と機能を最大限に保護することを意図した応答として機能しているという結論に至った(Smith et al 2002; Su et al 2010)。リピート領域に対応するコンストラクトの自己組織化は、架橋二量体の形成後に起こるので、神経細胞の酸化還元電位の上昇と過剰なタンパク質酸化が主にPHFs形成の原因であることが示唆されている(Wille et al 1992; Schweers et al 1995)。
したがって、タウのオリゴマー化の根底には、タウを凝集しやすくする構造変化がある。凝集は、ランダムコイル構造からβシート構造への局所的な転移を介して核生成部位で開始される。前述のように、ジスルフィド結合形成(架橋)を介した二量体化は核形成に不可欠であるが、二量体および初期のオリゴマー形態から成熟フィブリルへの移行には、多くのコンフォメーションおよび構造再配列が必要である(Friedhoff et al 1998; von Bergen et al 2005; Ganguly et al 2015)。α-ヘリックスおよびβ-シート構造の両方が、ex vivo PHFにおいて見られる(Ma et al 2005)。すでに述べたように、低リン酸化タウもまた重合することができるが、いくつかのコンフォメーション変化は、タウのリン酸化によって誘導され得る。したがって、酸化ストレスのような他のメカニズムがリン酸化と相乗的に作用し、PHFの組み立てに必要な構造変化を誘導するという仮説が立てられている(Pérez et al 2000)。
タウ凝集におけるヘキサペプチドモチーフの役割
PHFコアは主にリピートドメインから構築されており(Tomoo et al 2005; von Bergen et al 2005; Wischik et al 2018)、微小管結合ドメインはタウの凝集において重要な役割を果たしている。すべてのタウアイソフォームに存在するR3リピートユニットは、タウオリゴマー化の開始に重要な核形成コアとして機能し得る配列を含む(von Bergen et al 2000; Ganguly et al 2015; Stöhr et al 2017)。von Bergen et al 2000)は、タウの凝集を防止することを目的とした特異的に設計された阻害剤による治療的介入のためのコンフォメーションターゲットを表す可能性があるように、核生成を可能にする最小のタウ要素の同定が重要である。このフラグメントは胎児のタウアイソフォームに由来し、PHF43と命名された。PHF43はすぐに自己組織化してまっすぐな細いフィラメントになる。アルツハイマー病で見られるPHFに特徴的な周期的な超ねじれは、形成されたフィラメントには見られなかったが、これらの繊維に由来する種子は、より大きなタウフラグメントと完全長タウからのPHFの形成を核生成した。PHFsの集合は、β構造含有量の増加を伴っていた(von Bergen et al 2000)。PHF43内では、β構造形成の最も高い可能性に基づいて、ヘキサペプチド306VQIVYK311(PHF6)が、タウまたはタウ由来のフラグメント間の相互作用の根底にある最小の相互作用モチーフとして同定された。R2リピートは、同様の核形成モチーフ(PHF6*、275VQIINK280)を含み、それはまた、βコンフォメーションの傾向が高い残基を含むが、4つのリピートを有するアイソフォームにおいてのみ存在する(von Bergen et al 2001)。他の研究では、タウ分子間の分子間相互作用およびタウオリゴマーの形成を媒介するPHF6またはPHF6*モチーフの重要な関与が確認されており、2つの小さなヘキサペプチドモチーフが重合に必須であり、かつ十分であることが示唆されている(Goux et al 2004; Peterson et al 2008)。
PHFアセンブリ
PHFの組み立ては、これらのヘキサペプチド断片が、タンパク質全体のランダムコイル構造に埋め込まれたβ構造を局所的に形成することによって引き起こされると考えられる。システイン残基の二量化は、ヘキサペプチドモチーフを近接させ、βシート間の相互作用を促進する可能性がある(von Bergen et al 2000)。したがって、R3配列中のシステイン残基の変異は、アミロイドの形態およびそれらの物理的性質を変化させ、シーディング能力を低下させた(Stöhr et al 2017)。同様に、PHF6またはPHF6*からの残基のいずれかが、βシート形成を阻害する残基であるプロリンで置換されると、重合能力が阻害されることになる(von Bergen et al 2001)。 また、von Bergen et al 2000)は、βシート形成性が高いタンパク質全体のごく一部だけが、集合体のシードとして機能するのに対し、C末端およびN末端の尾は、ファジーコートを形成する秩序化されたコアから突出していることを報告している。しかしながら、さらなる研究により、PHF6およびPHF6*に加えて、微小管結合ドメインからの他のタウ由来ペプチドもまた、アミロイド形成性の高い素因を有し、一次核形成配列として作用し、フィラメント、チューブ、リボンまたはロール状シートのような様々なアミロイドの形成を促進する可能性があることが明らかにされた(Moure et al 2011)。
より大きなタウフラグメントにおいて、PHF6およびPHF6*は、互いにおよびそれ自身と相互作用し、分子間相互作用の結果としてオリゴマー種の不均一な混合物を生成し得る(PHF6-PHF6,PHF6-PHF6*およびPHF*-PHF*;Peterson et al 2008;Gangully et al 2015)。それにもかかわらず、PHF6を含む小ペプチドフラグメント(R3/wt)は、PHF6*(R2/wt)を含むフラグメントよりも凝集のためのより高い傾向を示し、R3/wtのアセンブリは、R2/wtの存在下で遅延した(Gangully et al 2015)。R3/wtペプチドは優先的にホモ二量体を形成したが、安定なR3/wt-R2/wtヘテロ二量体も存在し、R2/wtホモ二量体よりも安定であった。R3/wtホモダイマー内の個々の鎖は平行配向を好んだが、R2/wtペプチドからなるダイマーは反平行配向を採用しており、これは一般的に遅い凝集速度を示している。平行配向では、βシートはフィブリラリー凝集体の形成に重要な立体ジッパーを形成しやすくなる。また、R3/wtペプチド間では、形成された二量体の95%以上で複数の水素結合が形成されており、二量体の安定性に寄与していることがわかった。一方、R2/wtの二量体では、4個以上の水素結合が形成されていたのは10~20%に過ぎなかった(図1)。まとめると、タウ凝集を駆動する種としてのPHF6の役割は、PHF6*の役割よりも支配的であるように思われると結論づけられた(Ganguly et al 2015)。しかし、PHF6の構造に基づく阻害剤は、完全長タウによるシード化の防止には有効ではなく、最近の研究では、PHF6*がタウ凝集およびシード化のより強力なイニシエーターであり得ることが示唆された(Seidler et al 2018)。
図1
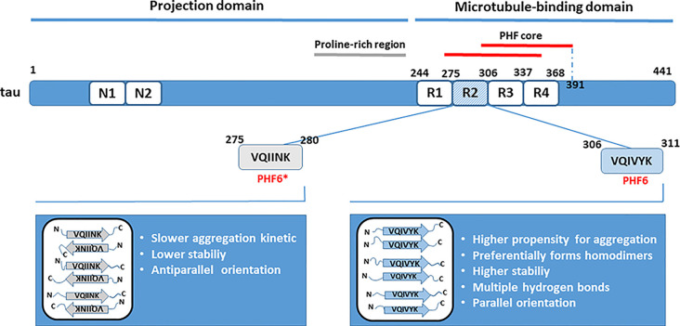
タウの二量体化におけるヘキサペプチドモチーフPHF6とPHF6*の役割 微小管結合ドメインに由来する残基は、タウの凝集に決定的に関与している。R2(PHF6*)およびR3(PHF6)リピートユニットの核形成部位がタウ凝集の種となることが、試験管内試験での研究で明らかになっている。PHF6モチーフは、その本質的に高い凝集性、水素結合によるより高い安定性、および二量体中のペプチド配列の平行配向性に基づいて、タウの二量体化の優勢な引き金となっている。ペアヘリカルフィラメント(PHF)コアを形成するアミノ酸残基の位置は、Wischik et al 2018)によれば示されている。
それにもかかわらず、さらなる研究は、ヘキサペプチド配列のみが自己増殖コアを表すかどうかを疑問視し、テンプレートのミスフォールディングおよび凝集を誘導することができる最小フラグメントの探索を続けてきた。PHF6が非常に迅速にアミロイドを形成することがわかったが、R3リピートの全長への5レシドステップでの緩やかな伸長はアミロイド形成を遅くし、繊維の形態を著しく変化させることがわかった(Stöhr et al 2017)。より重要なことに、内因性に発現したtau244-372フラグメントを疾患に関連した変異P301L/V337M[tau-RD(LM)と命名]を有する外部から誘導された凝集を研究することにより、これらの成長長ペプチドの凝集を誘導する能力に大きな違いが観察された。長さ31残基のR3配列から形成されたアミロイドは、フィブリル化したタウ-RDと同様にタウ-RD(LM)凝集を誘導するのに有効であった。一方、PHF6や20残基よりも短い成長ペプチドは、タウ-RD(LM)の凝集を誘導することができなかった。したがって、ヘキサペプチドから31残基を含むペプチドへの伸長において、結果として得られたペプチドは、よりゆっくりとアミロイドを形成したが、同時に、テンプレートのミスフォールディングを誘導するためにより強力であった(Stöhr et al 2017)。
試験管内試験でのタウ凝集のプロセスの解明に多大な努力が払われてきたにもかかわらず、多くの未解決の疑問が残っている。これらの実験は、主に翻訳後修飾がない状態で、任意のタウフラグメントを用いて行われたことを念頭に置かなければならない。したがって、組換えフィラメントの配列が、多くの因子がタウの組み立てに影響を及ぼす可能性がある、特に疾患を有する脳において、実際の生物学的システムに存在するものに似ているかどうかは、まだ明らかではない(Fichou et al 2019)。生体内での組み立てプロセスをよりよく理解するために、低温電子顕微鏡を用いて、アルツハイマー病脳からのPHFとストレートフィラメントのモニタリングを行った。分析の結果、それらは高リン酸化された完全長タウから構成され、培養細胞内でヒト完全長タウの凝集体を核形成しうることが明らかになった。試験管内試験での研究では、凝集のためのR2およびR3擬似リピートのより高い傾向が示されたにもかかわらず、アルツハイマー病脳から得られた両方のタイプのフィラメントの秩序化されたコアは、R3およびR4リピートユニット全体を包含する残基306から378までのC型サブユニットを含む同一のプロトフィラメントのペアで構成されていたが、N末端およびC末端は、すでに説明されているように無秩序なファジーコートを形成していた(Fitzpatrick et al 2017)。重要なことに、タウフィラメントの構造は、タウフォールディングの共通パターンを示すアルツハイマー病患者間で類似しているようである(Falcon et al 2018)。したがって、さらなる研究は、アルツハイマー病脳からのタウフィラメントを調べることにより、タウ症の分子メカニズムの理解を深めることに向けられなければならず、それはうまくいけば診断法を改善し、疾患を修正する治療法の開発を加速することになるだろう。
酸化ストレスとタウ凝集
酸化ストレスには、活性酸素および窒素種(ROSおよびRNS)のレベルの増加が、様々な酵素的および非酵素的抗酸化物質によって提供される抗酸化防御の内因性メカニズムを上回る状態が含まれる。ROSおよびRNSの増加した形成を介して、酸化ストレスは、神経細胞の構造および最終的な機能を脅かす本質的な高分子の酸化的損傷を誘導する(布村 et al 2001;Alavi NainiおよびSussi-Yanic酸化ストレスtas 2015;Jazvinshichak Jembrek et al 2015;Liu et al 2015,2017)。
他の細胞型と比較すると、ニューロンは、酸化ストレスおよびROSおよびRNS媒介傷害に対して特に敏感である(Cobley et al 2018;Wang et al 2020)。多くの証拠のラインは、酸化ストレスがアルツハイマー病神経変性の病因における重要な初期イベントであることを示している(布村 et al 2001;Mondragón-Rodríguez et al 2013;Alavi NainiおよびSoussi-Yanic酸化ストレスtas et al 2015;Liu et al 2015,2017;Wang et al 2020;Yeung et al 2020)。急性酸化ストレスは、タウのリン酸化に異なる影響を与え得るが、酸化ストレスへの慢性曝露は、タウの高リン酸化を促進し、タウをオリゴマー化およびNFTの形成をより容易にする(PatilおよびChan 2005;Su et al 2010;Mondragón-Rodríguez et al 2013;Liu et al 2015;Jazvinšćak Jembrek et al 2018)。したがって、亜致死的なグルタチオン合成の阻害によって誘導された酸化ストレスの延長は、分化したM17神経芽腫細胞におけるPHF-1エピトープでのリン酸化を増加させたが、これはおそらく、キナーゼJNKおよびp38の活性の増加およびプロテインホスファターゼ2A(PP2A)の活性の低下によるものであろう。特筆すべきは、JNKとp38の活性化とNFTへのコロケーションが生体内で観察されたことである(Su et al 2010)。PHF-1部位のリン酸化はタウフィラメントの形成に必要であることから、これらの結果は、高分子量バンドの存在によって確認されたタウの凝集を示唆している(Su et al 2010)。これらの結果から、酸化ストレスがタウの高リン酸化と凝集に先行していることが示唆された(図2)。HT22神経細胞では、酸化ストレス単独ではタウリン酸化を増加させることはできなかったが、タウリン酸化のパターンを変化させることができた。酸化ストレスとホスファターゼ活性の阻害を併用すると、タウは高度にリン酸化され、プロテアソーム分解の基質にもなりにくいことから、タウの酸化と高度リン酸化の両方がタウの凝集に重要であることが示唆された(Poppek er al)。
図2
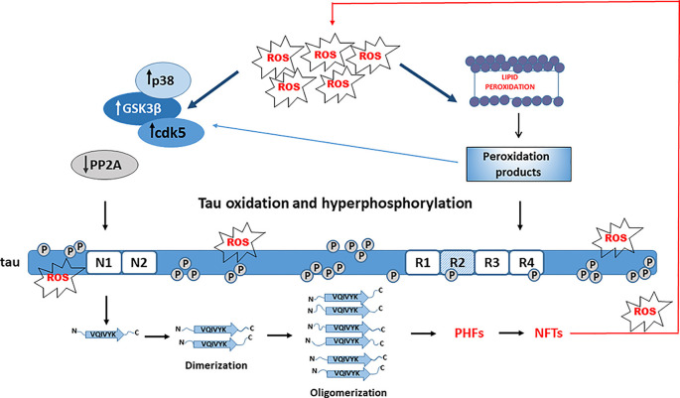
タウ凝集におけるタウの高リン酸化と酸化の役割 酸化ストレス(酸化ストレス)はアルツハイマー病関連キナーゼ(GSK-3β、cdk5,p38)を活性化し、PP2Aによるタウの脱リン酸化を阻害し、タウの高リン酸化を引き起こす。酸化ストレスが介在する脂質過酸化は、タウキナーゼをさらに活性化し、高リン酸化されたタウの構造変化を引き起こし、凝集を促進する毒性のあるアルデヒドをもたらす。
アクロレインは、アルツハイマー病脳で増加しているアラキドン酸からの過酸化産物であり、GSK-3βとp38を活性化することによってもタウの過リン酸化を促進し、タウのリン酸化における酸化ストレスと脂質過酸化の重要な役割を再び強調している(Gómez-Ram酸化ストレス et al 2003)。脂質過酸化の最終産物である4-ヒドロキシ-2-ノネナール(HNE)は、リン酸化された4R-タウの組み立て能力を増加させた(Pérez et al 2000)。リン酸化されたタウは、その重合とNFTsの形成をさらに加速するHNEとの反応における構造変化に対してより脆弱であり、NFTs形成におけるHNEの重要な役割を示唆していた(Liu et al 2005)。したがって、脂肪酸の酸化は、酸化ストレスとタウフィラメント形成の間のリンクとして、タウの高リン酸化を促進するための必須条件である可能性がある(Gamblin et al 2000)。興味深いことに、ステアリン酸およびパルミチン脂肪酸への神経細胞の曝露は、タウのリン酸化を増加させない。代わりに、飽和脂肪酸で処理されたアストロサイトからの条件付き培地は、アルツハイマー病特異的な部位でのタウリン酸化を増加させた。この効果は抗酸化剤で処理することで減少したため、アストログリアが介在する酸化ストレスはタウリン酸化に有意に寄与していると結論づけられた(Patil and Chan, 2005)。
酸化環境におけるタウ
タウは酸化ストレスに感受性があり、酸化ストレスはNFT形成に寄与する重要な因子と考えられている(レビューは、Liu et al 2015を参照のこと)。NFTは、アルツハイマー病患者の海馬組織における触媒的酸化還元反応の部位として同定されている(Sayre et al 2000)。高リン酸化タウ種の蓄積は、活性酸素の生産と酸化ストレス条件の誘導を刺激するが、酸化ストレスは順番に直接タウの高リン酸化を促進する。この悪循環の中で、活性酸素と異常タウの両方のレベルが徐々に上昇し、最終的には神経細胞死に至る(Alavi Naini and Soussi-Yanic酸化ストレスtas, 2015)。さらに、酸化ストレスが媒介する異常なタウのリン酸化は、興奮障害経路を介したFyn媒介およびアミロイドβ駆動のシナプト毒性におけるその役割に加えて、シナプス末端における生理学的なタウ機能の重要な調節緩和因子であり、シナプス障害の重要な基礎的なメカニズムである可能性が高い(Mondragón-Rodríguez et al 2013)。
酸化ストレスはタウの機能に影響を与えるだけでなく、タウのリン酸化に関与する酵素の発現と活性も調節する。H2O2に曝露された神経細胞分化したPC12細胞において、低濃度のGSK-3β阻害剤は酸化ストレスに対する保護を提供したが、高濃度では逆の効果を示した(Lee et al 2007)。有機溶媒1,2-ジエチルベンゼンの神経毒性代謝物である1,2-ジアセチルベンゼンもまた、活性酸素の産生を促進し、GSK-3βの活性を刺激し、雄性C57BL/6マウスの海馬においてタウのリン酸化を増加させた(Kang et al 2017)。アルツハイマー病ヒト脳からの海馬および皮質では、酸化ストレスによって誘導されることが知られているp38キナーゼの活性化および分布が、細胞内神経原線維病理と排他的にコロケーションしていた(Zhu et al 2000)。アミロイドβによって誘発される酸化ストレスの主な結果は、p38の活性化であり、それはさらにタウの高リン酸化につながることが、試験管内試験と生体内試験の両方で実証された。この知見は、アミロイドβとタウ病理を結びつけるものとしても重要である(Giraldo et al 2014)。さらに、初代皮質ニューロンにおいて、アミロイドβ誘発酸化ストレスは、カルシニューリンの調節因子(RCAN1;これは、Ser/Thrタウリン酸化酵素カルシニューリンの活性を阻害する)のレベルを上昇させたが、GSK-3βのレベルおよびタウリン酸化のレベルも上昇させた(Lorret et al 2011)。同様に、RCAN1-/-マウスからの神経細胞は、様々な酸化的課題に対してより耐性があった(Porta et al 2007)。
PP2Aは脳内の主要なタウホスファターゼであり、その活性はアルツハイマー病患者の脳では抑制されている(Taleski and Sontag, 2018)。PP2Aの不活性化は、GSK-3βの活性化と脂質過酸化の指標であるマロンジアルデヒドのレベルの上昇とともに、ラット海馬における低酸素誘導タウリン酸化において観察された(Zhang et al 2014a)。また、タウの脱リン酸化を刺激する神経細胞タンパク質ペプチジルプロリルイソメラーゼ(Pin1)は、アルツハイマー病海馬でダウンレギュレーションされ、酸化的に修飾され、活性が低下していることが確認された(Sultana et al 2006)。Pin1 は、ペプチド結合のシスからトランスへのコンフォメーションを変化させることで、大きな構造変化を引き起こし、タウの病理学的発達に影響を与える可能性がある(Sultana er al)。 興味深いことに、散発性アルツハイマー病に由来する変異型に類似したヒト切断タウタンパク質を発現するトランスジェニックラットモデルから得られた培養皮質神経細胞において、ヒト切断タウタンパク質の発現は、ミトコンドリアの数を減少させ、活性酸素のレベルを増加させ、神経細胞を様々な酸化ストレス誘導因子に対してより敏感にさせた。著者らは、タウの切り捨てはアルツハイマー病における酸化ストレスに先行し、酸化ストレスはアルツハイマー病におけるタウ病理の引き金ではなく結果である可能性を示唆した(Cente et al 2006)。
タウの異常な高リン酸化に寄与する酸化ストレス関連因子もまた、ミトコンドリアの分布を乱し、ミトコンドリア機能不全、ミトコンドリア酸化ストレス、ミトコンドリアスーパーオキシドジスムターゼなどの抗酸化酵素の欠乏を誘発する(Melov et al 2007; Cheng and Bai 2018)。ミトコンドリアの欠乏は、今度は酸化物質の生成を促進し、より厳しい酸化環境を作り出す(Prentice et al 2015)。誘導された酸化ストレスはまた、アミロイドβを含む様々なストレス因子に対するミトコンドリアの感受性を増加させ(Pérez et al 2018)アミロイドβは、活性酸素の産生を増強し、脂質過酸化を誘導し、レドックスバランスを乱し、ミトコンドリアの機能不全を導く(Diana et al 2008; Hu and Li 2016)。したがって、タウおよび神経細胞機能に対する酸化的損傷の誘発因子および結果、ならびにアルツハイマー病の進行における酸化的環境とタウの酸化および高リン酸化との相互作用をよりよく理解することは、抗酸化剤に基づく新規治療オプションの開発における将来の方向性にとって重要であるかもしれない。抗酸化療法の治療成績はまだ一貫性がないが、疾患進行の早期に酸化ストレスが出現することは、抗酸化剤が効率的な疾患修飾オプションである可能性を示唆している。
アルツハイマー病における酸化ストレス誘導因子としての銅
酸化ストレスの増加は、金属のホメオスタシスの調節の緩和によって生じる可能性があり(Sayre et al 2000)金属誘発酸化ストレスがアルツハイマー病におけるアミロイドβ沈着およびタウ病理を促進するという証拠がある(Greenough et al 2013;Birla et al 2020;Wang et al 2020)。少なくとも部分的には、アルツハイマー病における酸化ストレスは、銅のホメオスタシスの障害と関連している。銅は、脳機能の維持に不可欠な金属である。それは、多くの重要な生化学的・生理学的役割を持つ様々な酵素や構造タンパク質の不可欠な部分である(Stern er al)。 ミエリンの形成と保存、細胞呼吸におけるシトクロムc酸化酵素の活性、スーパーオキシド消去酵素である銅/亜鉛スーパーオキシドジスムターゼ(Cu,Zn-SOD)の適切な機能、カテコールアミン生合成に必要である(Gaetke et al 2014)。一方、銅は細胞内の電子移動反応に参加する反応性の高い遷移金属である。銅が結合していない場合、酸化状態(銅、Cu2+)と還元状態(銅、Cu+)の間の遷移により、酸化還元サイクルが始まり、危険な活性酸素が形成される可能性がある。生理的な状態では、銅の濃度は非常に厳密に調節されている(Gaetke et al 2014)。取り込み、細胞内輸送、タンパク質結合、および廃棄に関連する多くのメカニズムが、標的タンパク質への銅の十分な供給を確保し、銅イオンを結合した状態に保ち、酸化還元反応および活性酸素の形成を防止するために関与している。ヒト血漿中の銅の約95%はセルロプラスミンと結合し、残りはアルブミン、アミノ酸、および血液脳関門を通過する可能性のあるいくつかの複合体と結合している(Squitti et al 2008; Šimić et al 2019)。したがって、未結合銅の細胞質濃度は非常に低い(Rae et al 1999;KaplanおよびLutsenko 2009;Gaetke et al 2014)。しかしながら、多くの理由(環境汚染、過剰な食事摂取、銅代謝の先天的エラー、およびいくつかの特定の医学的状態など)により、多量の銅は、銅結合の内因性能力を圧倒し、酸化ストレス状態を促進する可能性がある(JomovaおよびValko 2011;EskiciおよびAxelsen 2012;Gaetke et al 2014;Hsu et al 2018)。
一般に、一価銅は、非常に低い濃度で発生する健康なニューロンにおいてより豊富であり、一方、Cu2+のレベルは、病理学的条件において上昇し、その毒性効果に大きく寄与する(Bagheri et al 2018; Kard酸化ストレス et al 2018; Ahmadi et al 2019)。内因性抗酸化物質(グルタチオンおよびアスコルビン酸など)の存在下では、Cu2+はCu+に還元される可能性がある。銅イオンは、過酸化水素の分解(フェントン反応)を介してヒドロキシルラジカルの形成をさらに開始する。このプロオキシダント反応で生成したヒドロキシルラジカルは、アミノ含有炭素との反応でタンパク質ラジカルを形成したり、不飽和脂肪酸を攻撃して脂質ラジカルを形成したりする。さらに、スーパーオキシドと一酸化窒素との反応で形成されたペルオキシナイトライトは、タンパク質複合体からの銅イオンの放出を促進し、より多くの活性酸素の発生を可能にする(Zielonka er al)。 したがって、活性酸素とRNSは最終的にタンパク質の酸化、神経細胞膜の過酸化的損傷、DNA損傷を誘発し、疾患の発症を促進する(Gaetke et al 2014)。また、Cu2+イオンとGSHとの相互作用により、ジスルフィド結合が形成され、Cu2+がCu+に還元される可能性がある。生成されたCu+はGSHと結合する可能性があるが、Cu(I)-[GSH]2複合体の形成は、分子状酸素を還元してスーパーオキサイドアニオンを生成する(Speisky et al 2009)。合わせて、銅の過剰なレベルは、神経毒性効果を発揮し、酸化ストレス状態を誘発することにより、神経細胞の機能および生存能力を危険にさらす可能性がある(Reddy et al 2008; Jazvinšćak Jembrek et al 2014; Sebio et al 2019)。
アルツハイマー病における銅の異常安定性
アルツハイマー病における銅のレベルは、血清、血漿、脳脊髄液、アルツハイマー病の脳領域からのサンプルを分析した際に、銅欠乏と銅過負荷の両方が観察されたことから、何らかの議論を呼んでいる(Ventriglia et al 2012;Pu et al 2017;Xu et al 2017;Bagheri et al 2018;Wang et al 2020)。例えば、Pu et al 2017)は、中等度および重度のアルツハイマー病患者において血清銅のレベルが上昇していることを発見している。逆に、誘導結合プラズマ質量分析法(ICP-MS)によって分析された銅の死後レベルは、内耳皮質および海馬を含む、検査されたすべてのアルツハイマー病脳領域において減少した(Xu et al 2017)。さらに、遊離銅、すなわち、セルロプラスミンに結合していない銅の濃度の増加が、セルロプラスミンの断片化とともに、アルツハイマー病患者において、病理学的変化および機能障害と良好な相関を示して観察された(Squitti et al 2008)。最近のメタ解析研究では、総銅と未結合銅はアルツハイマー病患者の血清/血漿中では上昇しているが、アルツハイマー病患者の脳では低下していることが明らかになった(Bagheri et al 2018)。まとめると、銅のホメオスタシスの異常がアルツハイマー病の病態に大きく寄与している可能性があると考えられる。大規模な集団研究はさらに、銅のレベルが認知機能の低下と負の相関があること、血中銅が低い被験者の方が短期記憶力と長期記憶力が優れていること、銅環境濃度が高い地理的な地域ではアルツハイマー病の発生率が高いことを明らかにした(Squitti et al 2016)。
銅イオンは一価(還元)状態でのみ輸送される(Macreadie, 2008)。しかし、銅のホメオスタシスのアンバランスは、銅の結合特性を変化させ、強固に結合した銅から細胞質内でほとんど遊離している銅へと変化させる。したがって、それはタンパク質に結合した銅のレベルの減少と緩く結合した銅または遊離銅の増加は、アルツハイマー病患者の血清や血漿中に観察される銅濃度の増加を説明することが示唆されている(Squitti et al 2008; Ventriglia et al 2012; Bagheri et al 2018)。銅代謝に関与する特定の遺伝子もまた、アルツハイマー病の発達と関連しているので、銅の代謝異常およびその結果としてのアルツハイマー病の影響を受けた脳領域におけるラビレ銅の増加は、アルツハイマー病の病理学的プロセスへの銅の寄与を考慮する場合、最も関連性の高い因子であり得る(James et al 2012;Squitti et al 2016;Bagheri et al 2018)。
銅の恒常性障害に加えて、アルミニウム、亜鉛、および鉄を含む他の金属イオンの代謝の摂動が、アルツハイマー病において報告されている(Hegde et al 2009; Kanti Das et al 2015; Rana and Sharma 2019; Wang et al 2020)。Al3+、Zn2+、Fe3+と比較すると、タウはCu2+に対して最も高い結合親和性を有する(Rane et al 2020)。これらの金属イオンはいずれも、活性酸素の発生をもたらす酸素移動が可能であるが、銅および鉄は活性酸素触媒として特に効率的である。しかし、鉄は大脳皮質に比較的微量に存在しており、アミロイドβとの相互作用は考えにくいとされている。一方、銅に注目する理由としては、銅が海馬に豊富に存在すること、アミロイドβ-銅複合体がアルツハイマー病におけるROSや酸化ストレスの重要な発生源であること、銅が脳機能に不可欠な多くの酵素の補酵素であることなどが挙げられる(Kanti Das et al 2015;Esmieu et al 2019;Wang et al 2020)。したがって、アルツハイマー病の進行における銅の役割を解読することは、銅が媒介する病理学的事象をよりよく理解することと同様に、アルツハイマー病のための新しい治療アプローチを開発するという文脈で有益なものとなるであろう。
タウと銅の相互作用-タウ凝集への影響
毒性の様々なモデルは、Cu2+結合に続くアミロイドβの毒性機能の獲得が、アルツハイマー病における銅毒性の最も重要なメカニズムの一つであることを示唆している。銅の除去は、試験管内試験でのアミロイドβの凝集を減衰させ、アミロイドβ沈着物の分解を促進し、アミロイドβ/Cu2+複合体の存在によって誘導されるROS産生を抑制する(Bagheri et al 2018)。タウの凝集およびタウ媒介毒性における銅の役割はよく理解されていない。前に説明したように、微小管結合ドメインからの疑似リピートは、タウの凝集に不可欠な役割を持っているが、銅の配位も可能である。したがって、銅がタウの凝集に関与している可能性が示唆された。銅がタウの凝集を促進するという証拠があり(von Bergen et al 2000;Soragni et al 2008;Kitazawa et al 2009;Kim et al 2018)銅の増加した量がNFTにおいて検出されている(Sayre et al 2000)。野生型ヒトタウを発現する二重トランスジェニックマウスモデルにおいて、多量の銅への曝露は、空間学習および記憶の障害とともに、アミロイドβの非存在下でのタウの高リン酸化および蓄積を増加させた(V酸化ストレスs et al 2014)。したがって、本研究は、タウリン酸化の増加がアミロイドβ病理の存在に依存しないことを実証し、銅レベルを標的とすることが、タウ病理の進行を修正する上で有益な戦略であり得ることを強調している。
タウは銅に対する1つ以上の結合部位を有しているが、結合親和性は、他の金属タンパク質に対して通常見られるほど強くはない。配位環境は、細胞内の酸化剤や還元剤との相互作用を可能にし、これらの相互作用は銅の濃度を局所的に増加させることがある(Sayre et al 2000)。銅配位錯体は、NFTの触媒的酸化還元活性の誘導剤として作用することが可能である。銅は、NFTにおいて還元性基質のH2O2依存性酸化を触媒しうることが示された(Sayre et al 2000)。さらに、R2ユニットからのオクタデカペプチドであるタウタンパク質のフラグメントは、銅を還元し、H2O2および非常に可能性の高いいくつかの他のROS部位を同様に生成する可能性がある。Cu2+還元の間、R2は酸化され、ジスルフィド結合によって連結された二量体を形成する(Su et al 2007)。上で説明したように、これらの二量体はPHFのビルディングブロックと考えられている。興味深いことに、別のレドックス活性金属イオンである鉄は、タウの酸化を触媒しない(Su et al 2007)。この発見は、銅と不適切に結合したタウが活性酸素の形成と酸化ストレス状態を引き起こす可能性があることを示している。さらに重要なことは、銅がタウの構造変化後のタウの酸化と二量体形成を触媒することで、銅はPHFの組み立てと疾患の進行を促進する可能性があるということである(Su et al 2007)。
銅は微小管結合ドメインからのR1-R4リピートユニットに結合する
銅と個々のR1-R4リピートユニットまたはその小さな断片との間の相互作用は、銅がタウ由来のペプチドに直接結合していることから、銅の結合が微小管結合ドメインの構造変化を誘導することを示唆している(Ma er al)。 二価の銅の存在下では、α-らせん構造含量とβ-シートの増加は、共通の空間的変化を表している。R2とR3の繰り返しでは、凝集のプロセスに先立って、さらに引き金となる他の構造変化が観察されている。4つの疑似リピートはすべて、Cu2+溶液で3日間インキュベートした後に凝集体を形成した。非晶質の凝集体は、R1およびR4リピート単位で観察されたが、Cu2+は、R2およびR3リピート単位でフィブリルの形成を促進した(図3A)。4日間のインキュベーション後、R2フィブリルおよびR3プロトフィブリルが形成された。さらに、R2リピートについては、活性酸素の産生が顕著であったのに対し、R3リピートについては、より凝集しやすかった。過酸化水素とのインキュベーション後にもオリゴマー化が観察されたが、凝集はCu2+の存在下でより顕著であり、内因性抗酸化物質である多量のアスコルビン酸によっても防止されなかった(Ahmadi et al 2019)。同じ研究において、神経細胞からの別の重要な抗酸化物質であるグルタチオンは、酸素への限られたアクセスの下で、微小管結合ドメインからの仮性リピートの凝集を防止しなかった。リピート単位を周囲酸素下でCu2+およびGSHとインキュベートした場合、R3についてより多くおよびより大きなプロトフィブリルが形成され、これはCu2+が存在する場合のR3凝集におけるGSHの重要な役割を示している(Ahmadi et al 2019)。まとめると、これらの知見は、Cu2+によって誘導された三次構造の構造変化と折り目が、その後の二量体化とフィブリル形成のための種とみなされる可能性があることを示している。
図3 ジスルフィド結合形成と二量体化におけるCu2+の触媒的役割
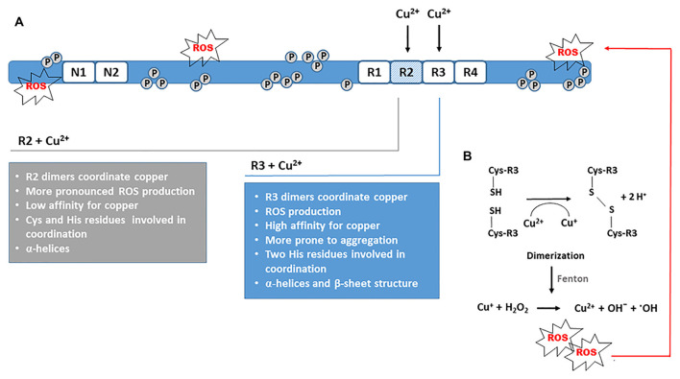
A)R2およびR3リピートユニットに由来するタウフラグメントの試験管内試験凝集は、PHF様フィブリルの形成を刺激する銅の能力を実証している。凝集は、酸化ストレス条件下で促進される。B)Cys残基間のジスルフィド結合形成を介した二量化は、タウのオリゴマー化とフィブリルの更なる形成を誘発する重要な開始ステップである。同時に、Cu2+はCu+に還元され、フェントン反応とヒドロキシルラジカルの生成を触媒する。発生した活性酸素は、タウタンパク質の重合を促進する酸化的損傷をさらに誘発する。
Shin and Saxena (2011)は、R1-R4擬似リピートのオクタデカペプチドのCu(II)配位を解析した。彼らは、3つの窒素ドナーと1つの酸素ドナーを持つ正方形平面のCu(II)配位ジオメトリを提案した。背骨アミドのカルボニル酸素とヒスチジンイミダゾール環の窒素原子は、おそらく直接、すべての4つのリピートユニットに由来するオクタデカペプチド断片のCu(II)配位に参加している(ShinとSaxena 2011)。他の研究はまた、オクタデカペプチドが銅を結合する可能性があることを示唆し、Cu(II)配位におけるヒスチジンイミダゾール基の重要な役割を強調した(Ma et al 2005; Zhou et al 2007; Ahmadi et al 2019)。注目すべきは、微小管とタウの相互作用に関与する残基は、銅の結合には関与していないようである。このことは、銅が微小管結合タウに結合し、多くのタウ分子の二次構造に影響を与えることを示唆している(Soragni et al 2008)。
いくつかの研究が、タウの凝集におけるR3リピートの重要な役割を示しているので、タウの凝集に対する銅の効果を考慮する場合には、R3ユニットに由来するより小さいペプチド配列が特に関心を持たれた(Ma et al 2005)。少なくとも、最長のタウアイソフォームのR3リピートに由来するオクタデカペプチド318VTSKCGSLGNIHHKPGGG3355については、銅の結合は非常に強く、高親和性である。Ma et al 2005)の研究では、このオクタデカペプチド由来の2つのヒスチジン残基のイミダゾール環がCu2+配位に決定的に関与しているが、生理的pHではバックボーンアミドや他のアミノ酸の側鎖が銅配位に関与している証拠は見出されなかった。彼らは、R3リピートからのオクタデカペプチドが、ヒスチジン残基の2つのイミダゾール環、C末端カルボキシル基、N末端アミノ基を介してCu2+の配位を四角い平面形状で行っていることを示唆した(Ma et al 2005)。R3リピートに由来するペプチドへの銅の結合特性には、様々な要因が影響していると考えられる。tau318-335ペプチドの場合、配位リガンドとコンフォメーションはpHに大きく依存する。酸性pHでは、水溶液中での主鎖の確認は、ランダム構造、αヘリックス、βターンの混合構造を採用していたが、生理的pHではαヘリックスの含有量が増加していた(Ma et al 2005)。0.25 mol eqのCu2+を添加した後、R3ペプチドは単量体のα-らせん構造を示し、1 mol eqのCu2+を添加した後にはβシートが存在した(Ma et al 2005)。βシート構造は、異常なタンパク質凝集に必須であることは一般的に受け入れられている。しかし、αヘリックスもまた、タンパク質の凝集体形成の引き金となる可能性があり、βシート構造に加えて、タウタンパク質にはヘリックスを破るアミノ酸が豊富に存在するにもかかわらず、αヘリックスはPHFにも存在している(Sadqi et al 2002)。これらの結果は、R3由来の18-merが低濃度のCu2+の存在下で自己会合を開始することを示唆している。αヘリックスとβシートターンがPHF形成に必要とされるように、銅は自己組織化の初期段階と後の段階でのPHF様構造の形成の両方に関与している可能性が高い(Ma et al 2005)。
R2リピートはフィラメント状ポリマーを形成することもできるので、R2由来のオクタデカペプチド287VQSKCGSKDNIKHVPGGG304の銅結合特性も調べた。R2リピート由来の18-merは、1:1のストイキオメトリと低い親和性で銅を結合した。生理的pHでは、システインとヒスチジンのイミダゾール基は、主鎖とは独立して銅の配位に重要である。Cu2+はR2フラグメントにおけるαヘリックスの形成を誘発し、銅濃度の増加に伴い、より多くのαヘリックスが形成された。R2ペプチドは、R3ペプチドと比較してα-ヘリックスを形成する能力が高かったが、より高い銅濃度で凝集した(Ma et al 2006)。同様に、R1リピートからのペプチド256VKSKIGSTENLKHQPGGG273は、1:1のストイキオメトリーで銅と結合する可能性がある。ヒスチジン残基は銅の主なアンカーであるが、二次構造のわずかな変化のみが銅結合時に誘導される(Zhou et al 2007)。興味深いことに、R1リピートからのオクタデカペプチドの凝集は、銅の存在下で阻害された。R3ペプチドの銅刺激による凝集は、配列中のヒスチジン残基の数に依存する可能性があると推測される。R3ペプチドが2つのヒスチジン残基を含むのに対し、R1では1つしか存在しない(Ma et al 2005; Zhou et al 2007)。Cu-R4複合体のCu2+誘発構造変化は、Cu-R1複合体と比較して小さかった(Ahmadi et al 2019)。Ahmadi et al 2019)は、R1およびR4リピートの両方が1つまたは2つの銅イオンと付加体を形成することを観察したが、R2およびR3モノマーとの銅錯体の証拠を見いだしていない。代わりに、彼らの結果は、R2およびR3二量体のみが銅と錯体を形成し、Cu2+錯体化における二量体化の重要な役割を示すことを示唆した。R1とR4の繰り返しでは、Cu2+がヒスチジンのイミダゾール基と相互作用していると考えられるが、これらの繰り返しは一次配列にシステインを含まないためか、二量化は観察されなかった。システインを含むR2とR3リピートでは、ヒスチジンが相互作用に関与していたが、Cu2+の存在下ではチオール基がジスルフィド結合を形成しやすいため、Cys残基の方が凝集に関与していたと考えられる。特に興味深いのは、R2およびR3リピートのCu2+との相互作用がROSの発生をもたらしたことである(Ahmadi et al 2019)。Cu2+の酸化還元活性により、Cys残基のチオール基の間にジスルフィド結合が形成される(図3B)。この酸化還元反応はまた、フェントン型反応を介して活性酸素産生をさらに触媒するCu+イオンを生成する(Ahmadi et al 2019)。別の研究では、Cu(II)-2His複合体がシステインに結合し、システイン酸化の間にCu(I)-2His複合体が形成されることが示された。同様に、この錯体はフェントン化学を促進し、活性酸素産生の増加をもたらした。悪循環の中で、Cu(II)-2His複合体は再生され、再びシステインを酸化することができるようになった(Zåbek-Addamska et al 2013)。R1およびR4擬似リピートについては、オリゴマー化もROS産生も気づかれなかった(Ahmadi et al 2019)。ジスルフィド結合形成も過酸化水素の存在下で開始される可能性がある。しかし、R2およびR3の二量体化は、Cu2+の存在下でより顕著であった(Ahmadi et al 2019)。すべての繰り返し単位(R1-R4)の存在下でCu2+を還元するアスコルビン酸の能力を調べたところ、R2およびR3はアスコルビン酸よりもCu2+に対して高い親和性を有しており、アスコルビン酸はR2およびR3の二量化を防ぐことができないことを示唆していることが判明した。以上のことから、R2とR3のリピートが銅の配位に関与している場合、ジスルフィド結合形成を介したオリゴマー化や活性酸素の発生にCu2+が重要な触媒的役割を果たしていることが示唆された。
より大きなタウフラグメントへの銅の結合
ヒトのタウの4つの擬似リピートモチーフと2つのフランキングドメインを含む198残基のK32フラグメントのような、より大きなタウフラグメントに関しては、酸化的条件下では、K32 C末端のペプチド287VQSKCGS293と310YKPVDLSKVTSKCGS324がCu(II)配位に関与していることが観察された。より小さい程度では、テトラペプチド306VQIV309,およびいくつかのヒスチジン残基(H299,H329,H330,H374,H388)もまた、相互作用に関与していた(Soragni et al 2008)。K32フラグメントについては、銅との結合によりβ構造への傾向が観察されたが、生理学的条件下での凝集速度が非常に遅いため、銅との結合後には少量の凝集体しか形成されなかった(Soragni et al 2008)。Soragni et al 2008)は、全長タウとK32フラグメントの両方について、モノマーごとにCu2+の結合部位が1つしかなく、重要な残基は主にR2とR3の繰り返しに位置していることを発見した。銅はR2とR3の繰り返しに同時に結合し、反応性基を空間的に近接させていると思われる。R2とR3の配列内では、2つのシステイン残基(C291とC322)が銅とタウの結合に必須であることが確認された。これらのシステインが銅によって酸化されることを考えると、チオール基間の分子内ジスルフィド橋は銅とタウの相互作用の結果として形成されていると考えられる(Soragni er al)。 したがって、本研究は、タウの凝集に対する銅の促進効果を示しており、オリゴマー化の過程において、R2とR3リピートのシステイン残基が重要な役割を果たしていることを確認した。
また、微小管結合ドメインの外側にあるタウのN末端部分に由来するペプチドを用いて銅の配位を調べた。残基1〜25または26〜44を含む2つのタウフラグメントは、2つの推定される結合部位、ヒスチジン残基のイミダゾール側鎖またはペプチド鎖のN末端アミノ基を介して銅を固定することができる。両方のペプチドの形態は、Cu(II)錯体の形成後に変化した。興味深いことに、両方のペプチドフラグメントは、Cu(II)媒介のアミロイドβの凝集を阻害し、その効果はtau26-44についてより顕著であった(Di Natale et al 2018)。タウ26-44ペプチドを含むN末端由来のタウタンパク質のフラグメントは、アルツハイマー病患者の脳脊髄液および脳組織中に検出される(Barthélemy et al 2016; Zhou et al 2018)。生理的条件下でのフラグメントtau26-44は、本質的に無秩序な構造のために凝集する傾向はないが、NMDA受容体媒介の神経細胞死を誘導することが可能な最小アミノ酸配列を表す(Amadoro et al 2006)。神経細胞培養物をタウ26-44フラグメントに慢性的に曝露すると、神経ジストロフィー、微小管の破壊、ミトコンドリアの喪失、酸化的リン酸化の障害など、アルツハイマー病病理の前症状段階に特徴的な多くの特徴が得られ、細胞外に分泌されたN末端切断されたタウフラグメントがアルツハイマー病病理の発症に大きく寄与している可能性があることが示された(Atlante et al 2008; Florenzano et al 2017)。
銅の効果は、タウ病理を模倣した動物モデルにおいても研究された(Sedjahtera et al 2018;石原 et al 2019)。例えば、3XTg-ADマウスにおいて、飲料水中の銅への慢性曝露は、タウの高リン酸化を悪化させ、cdk5アクチベーターp25をアップレギュレートするが、GSK-3βの活性は安定したままである。タウの高リン酸化はSer396/Ser404(PHF-1)エピトープで検出され、病理学的な後期タウの存在とタングル形成を示唆している(Kitazawa er al)。
タウのアセチル化が銅イオンと結合するタウの能力に及ぼす影響や、アセチル化されたタウの凝集に及ぼす銅イオンの影響については、ほとんど知られていない。最近、アセチル擬態変異K274Q(グルタミンに変異したリジン)がCu2+に対する結合親和性を増強し、Cu2+イオンが正常なタウと比較してK274Qの凝集をより強く促進することが示された(Rane et al 2020)。このことは、アセチル化が追加的にCu誘導毒性を増加させ、神経病理学的プロセスに寄与することを示唆している(Rane et al 2020)。タウのグリコシル化に対する銅の効果は、まだ決定する必要がある。いくつかのタンパク質では、その糖化形態が銅を介した酸化を受けやすいことが示されている(Kobayashi er al)。 これらの知見に基づいて、銅はタウのグリコシル化した形態の凝集を促進することが期待される。
最後に、グリアタウ症における銅の具体的な寄与については、まだ明らかにされていない。グリア細胞のタウ症では酸化ストレス関連マーカーが観察されており(Kahlson and Colodner, 2016)活性酸素の産生を増加させる毒性障害は、C6ラットアストロログリオーマ細胞株におけるタウの切断およびNFTの形成をもたらす(Means et al 2017)。銅の調節緩和がグリアタウ症に及ぼす影響を調べるためには、さらなる研究が必要である。グリア細胞の重要なニューロン支持的役割に基づいて、グリアのタウ凝集に関与する酸化ストレス主導のメカニズムをよりよく理解することは、アルツハイマー病および他のタウ症における効果的な治療のための新規ターゲットを同定するために重要である可能性がある。
アルツハイマー病における潜在的な治療アプローチとしての銅レベルの標的化
これらの知見は全体的に、天然または合成の銅キレート剤がアルツハイマー病の治療薬として考えられる可能性を示している。一般的に、金属を標的とした治療は、金属の除去または再分配を目的としている。キレート剤は、金属イオンおよび特定の分子との相互作用を破壊し、最終的にそれらの排泄を促進し、有害な結果を防ぐことを目的としている(Esmieu et al 2019)。しかしながら、キレーション療法は、議論の余地のある臨床結果を達成している。効果的で安全なキレーション療法の延長のためには、キレート剤の適用時期および投与量を慎重に最適化しなければならない(Hegde et al 2009)。また、一般的な金属枯渇や金属バランスを阻害するリスクがあるため、金属キレートの選択性も重要である。二価の陽イオンの増加はアルツハイマー病の初期に特徴的であるため、潜在的な銅キレート剤は疾患の初期段階の患者にのみ有益であると期待されている。以上のことから、長期的な使用を目的とした安全な銅キレート剤を開発することは非常に困難である。とはいえ、銅のレベルを標的とすることは、アルツハイマー病患者の血清中の特徴的な所見として、緩く結合した銅のレベルが上昇していることを考慮すると、アルツハイマー病において有望な選択肢であることに変わりはない(Buc酸化ストレスsi er al)。
アルツハイマー病の多因子性の病因および疾患の発症および進行に寄与する多数のメカニズムを考慮すると、効果的な治療アプローチは、マルチターゲット薬に依存する可能性がある(Sharma et al 2018)。様々な因子がタウ病理を支配しているため、病気の進行を緩和するための銅を標的としたアプローチは、タウ病理を標的とした他の介入、例えば、タウキナーゼの阻害、翻訳後のタウ修飾およびタウ凝集の阻害、能動的および受動的な免疫療法、およびタウ分解過程の調節などと組み合わせれば、おそらくより有用であろう。これに関連して、フラボノイドのような様々なポリフェノール化合物の神経保護の可能性が評価されてきた。これらの食事性抗酸化物質は、多機能効果を発揮する、すなわち、金属キレート剤、活性酸素スカベンジャー、およびレドックスシグナル伝達のモジュレーターとして作用する(Jazvinšćak Jembrek et al 2012;Ayaz et al 2019)。
ケルセチン
ケルセチンは、最も研究されている活性酸素の最も強力なスカベンジャーの一つである(Jazvinšćak Jembrek et al 2012,2014)。それは、組換えタウタンパク質(2N4Rアイソフォーム;Kumar et al 2019)の試験管内試験フィブリル化を防止する。R2ドメインおよび306VQIVYK311ヘキサペプチドモチーフを含むタウフラグメントの分子動力学シミュレーションにより、ケルセチンとの相互作用に伴う構造変化が明らかになった。ケルセチンは、最も凝集しやすい領域の近傍でタウタンパク質と相互作用し、特異的な水素結合および疎水性相互作用により、モノマーであるタウのネイティブランダムコイル状の状態を安定化する(Kumar et al 2019)。さらに、ケルセチンは、cdk5活性を阻害することにより、HT22マウス海馬細胞におけるオカダイン酸誘発タウ過リン酸化に対して有効であった(Shen et al 2018)。銅に対するケルセチンのキレート化は、ケルセチンが銅よりも過剰に存在する場合、ヒドロキシルラジカルの形成を抑制し、保護効果を示す可能性がある(Jomova et al 2017)。しかし、銅イオンの存在下では、ケルセチンは抗酸化活性およびプロオキシダント活性の両方を示す可能性があり、銅/ケルセチン相互作用の結果は一筋縄ではいかないことに留意すべきである(Filipe et al 2004; Zubčić et al 2020)。
EGCG
もう一つの検討されているフラボノイドは、緑茶由来のエピガロカテキン-3-ガレート(EGCG)である。スウェーデンAPP変異を持つマウスの飲料水にEGCGを経口投与すると、サルコシル可溶性リン酸化タウアイソフォームの形成が抑制され、認知能力が改善された(Rezai-Zadeh et al 2008)。ラット初代ニューロンにおいて、EGCGはリン酸化タウ種のクリアランスを増加させ(Chesser et al 2016年)サブソイ濃度でタウフラグメントHis-K18ΔK280の不溶性高分子量オリゴマーへの凝集を抑制した。このフラグメントは、最長のヒトタウアイソフォームの微小管結合ドメインにまたがっているが、Lys280を欠いており、毒性のあるオリゴマーのタウ凝集体の形成をもたらすβシート構造を容易に採用している。EGCGは、初期の凝集中間体と特異的に相互作用し、そのシーディング活性を阻害することにより、コンフォメーション変化を防止することにより、その効果を発揮すると考えられる(Wobst et al 2015)。EGCGは、活性酸素スカベンジャーとして作用し、酸化ストレス誘発DNA損傷およびアミロイドβ誘発脂質過酸化を防止し、Nrf2抗酸化経路を活性化することでGSHの産生を増加させる(Ayaz et al 2019)。さらに、EGCGは、パーキンソン病に特徴的な異常凝集であるα-シヌクレインのCu(II)誘発性フィブリル化・凝集を阻害し、Cu(II)誘発性活性酸素の産生を抑制する可能性がある(Teng et al 2019)。
クルクミン
植物Curcuma longaの根茎から抽出されたクルクミンは、健康を促進するポリフェノール系の栄養補助食品として高く評価されている。マルチターゲット化合物として、アルツハイマー病の予防と治療にも考慮されている。それは容易に血液脳関門を横断し、シナプス構造と機能を維持し、アルツハイマー病ラットの学習能力と記憶能力に積極的に影響を与える(Reddy et al 2018)。クルクミンは強力な抗酸化特性を持っている。それは、効果的な活性酸素スカベンジャーとして作用する酸化ストレスを減少させ、スーパーオキシドディスムターゼとカタラーゼの活性を刺激し、グルタチオンレベルを上昇させ、脂質過酸化を減少させる(Chen et al 2018)。天然のβ-ジケトンリガンドとして、クルクミンは銅を含む様々な金属を強くキレートする。したがって、クルクミンはアルツハイマー病における銅誘導性神経毒性に対して有効であると考えられる(Wanninger et al 2015)。N2a/APP695swe細胞およびAPP/PS1トランスジェニックマウスにおいて、クルクミンはタウのリン酸化とGSK-3βの活性を低下させた(Sun et al 2017)。同様に、アミロイドβを過剰発現しているTg2576マウスにおいて、ウコン抽出物を6ヶ月間投与すると、高リン酸化タウのレベルが〜80%減少した(Shytle et al 2012)。クルクミンは、4R0N成体タウに結合し、βシート構造の形成を阻害し、タウのオリゴマー化およびフィラメント化を阻害し、予め形成されたタウオリゴマーおよびフィブリルの解離を誘導することができることが、試験管内試験での研究で示されている(Rane et al 2017)。これら3つの例に基づいて、強力な金属キレート剤および抗酸化剤として作用する天然化合物は、Cu関連タウ病理の予防および治療における有望なアプローチであり得る可能性がある。したがって、動物モデルや臨床研究において、天然ポリフェノール化合物の銅誘発毒性に対する潜在的な有益な効果を探り、よりよく説明するためには、さらなる研究が必要である。
銅キレート剤として作用する小型の合成リガンドはすでに設計されており、主にアミロイドβ凝集体から銅を除去することを目的としている。前述したように、キレート分子はいくつかの特性を持っていなければならない。キレート分子は無毒で、血液脳関門を通過でき、銅と適度な親和性を持って結合するものでなければならない。また、キレート剤は、結合していない銅イオンをキレートし、銅イオンを沈降させることは重要であるが、様々な金属タンパク質から金属イオンを除去することは重要ではない(Sharma et al 2018)。おそらく、最も研究されている銅キレート剤は、アミロイドβから金属イオンを除去し、予め形成されたアミロイドβ凝集体を分解するのに効率的なクリオキノール誘導体である。それらはトランスジェニック動物において認知機能を改善することができるが、臨床研究におけるそれらの正の効果の証拠はまだ不足している(Budimir, 2011; Sharma et al 2018; Esmieu et al 2019)。銅錯体化とタウの病理学については、ほとんど研究が行われていない。過剰な銅と24時間インキュベートしたヒト神経芽腫細胞において、アルツハイマー病特異的部位におけるタウリン酸化の増加は、cdk5のp35/p25活性化因子の発現を減少させる銅錯形成剤によって減少した(V酸化ストレスs et al 2014)。同じ研究において、飲料水中の酢酸亜鉛の経口投与は、野生型ヒトタウ(hTau株)を発現する雄マウスの脳内環境銅レベルを枯渇させた。酢酸亜鉛は、タウタンパク質の全体的な発現を増加させ、Ser202/Thr205,Thr231,Ser396/Ser404(PHF-1エピトープ)でのタウリン酸化を減少させた。p35/p25アクチベーターおよびcdk5の発現は変化しなかったが、GSK-3βおよびSer9でリン酸化されたその阻害形態は枯渇しており、おそらく銅レベルが減少したときのタウリン酸化の減少に寄与していた(V酸化ストレスs et al 2014)。しかしながら、発現レベルおよびリン酸化パターンのこれらの変化は、18ヶ月齢のhTau動物の空間学習および記憶を改善しなかった(V酸化ストレスs et al 2014)。
別の研究では、ベンゾチアゾール含有化合物がCu2+と結合し、顕著なフリーラジカル消去活性を有する複合体を形成し得ることが示された。理論的には、Cu結合のためにタウと競合することにより、これらの化合物は、脳におけるCu誘発損傷を減少させる可能性がある(Geng et al 2012)。ビス(8-アミノキノリン)リガンドは、アミロイドからCu2+を除去し、活性酸素産生を減衰させることができる特異的かつ効率的なCu2+キレート剤の別の例である。還元環境下では、それらは脱金属化され、還元的条件下で銅の恒常性を取り戻すのに役立つかもしれない銅イオンを放出する(Nguyen et al 2014)。最後に、CuII(gtsm)と呼ばれるGSK-3β阻害剤を用いた興味深い研究が行われた。この研究は、より多くの銅を送達することが間接的にタウの病理学的に有益な効果を発揮することを示した。CuII(gtsm)は、銅を細胞内に送り込み、銅の細胞内バイオアベイラビリティを高める金属錯体である。還元環境にさらされると、CuIIはCuIに還元され、リガンドから解離し、細胞内の銅のレベルを増加させる(Crouch et al 2009)。SH-SY5Y細胞において、CuII(gtsm)はGSK-3βの活性を阻害し、Ser404およびSer396でのタウリン酸化を減少させた。CuII(gtsm)とネガティブコントロールである還元解離しにくい化合物CuII(atsm)の効果を比較することで、CuII(gtsm)が達成したバイオアベイラビリティの向上がGSK-3βの活性低下に必要であることが明らかになった(Crouch et al 2009)。
アルツハイマー病を修飾する治療法を探索することにより、金属の恒常性障害のより良い理解と、タウ関連の病理を遅らせることにおける抗酸化剤/キレート戦略の正確な可能性を、特に前臨床試験および臨床試験において、さらに探究しなければならない。いくつかの有望な知見があるにもかかわらず、アルツハイマー病の病態生理学的変化を改善したり、逆転させたりするために銅レベルを標的とすることの妥当性については、まだ議論の余地がある。アルツハイマー病 における異常な銅のホメオスタシスのために、脳内金属の再分配と金属のホメオスタシスの再確立は、金属の除去よりもむしろ、金属を標的とした戦略の第一の目標と考えられている (Hegde et al 2009)。今後の研究では、銅をベースとした戦略の保護能力を推し量り、タウ病変の進行に及ぼす影響をよりよく理解することが肝要である。これらの潜在的な銅修飾剤の薬力学的・薬物動態学的特性をよりよく理解するためには、より多くの薬理学的・毒性学的研究が必要である。その点、前述の天然化合物は、単独で、あるいは他の薬剤と組み合わせて使用することで、一般的な安全性といくつかの銅関連効果を減衰させる能力に基づいて、有望なアプローチとなる可能性がある。
おわりに
銅のホメオスタシスの調節障害は、アルツハイマー病 の初期イベントとして認識されている 酸化ストレス 状態を促進する。銅-タウ相互作用は、アルツハイマー病の病理学的変化に寄与する重要な分子因子である。最近の知見では、Cu2+が微小管結合ドメインに位置するタウペプチドの構造変化を誘発することで、凝集におけるCu2+の触媒的役割が明らかになっている。Cu2+は凝集を促進するだけでなく、活性酸素の生成を増加させ、酸化ストレス状態や神経細胞の損傷をさらに促進する。銅が介在する酸化的傷害とタウ凝集の伝播を減少させることができる金属イオンキレート剤や抗酸化剤は、アルツハイマー病の進行を緩和するための有望なアプローチとして提案されている。天然ポリフェノール化合物は、金属キレート作用と抗酸化作用を有しており、それらを有望かつ安全なマルチターゲット薬の候補としている。逆説的なことに、銅の供給量が少ない代わりに、より多くの銅を供給することが有益であるかもしれないことを示唆する報告がある。タウと銅の相互作用をさらに研究すれば、銅をキレートする新しいアプローチの開発に役立つかもしれない。アルツハイマー病の進行に寄与する多くの病理学的メカニズムに基づき、キレート剤は選択的ではなく、銅や他の金属のレベルを過度に低下させてはならないため、キレートアプローチには注意が必要であることを考慮すると、複数のプロセスを対象とした複合的な治療法がより良い効果をもたらすことが期待される。その点では、タウ凝集の阻害剤や解離剤(免疫療法など)は、キレート剤や抗酸化剤と併用することが考えられる。