
Contents
The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders
www.ncbi.nlm.nih.gov/pmc/articles/PMC7162839/
オンラインでは2020年3月13日公開
Sylwia Wójtowicz,corresponding author1 Anna K. Strosznajder,2 Mieszko Jeżyna,2 and Joanna B. Strosznajdercorresponding author1
概要
ペルオキシソーム増殖因子活性化受容体α(PPAR-α)は,リガンド制御核内受容体(PPAR)の一種である。これらの受容体は、レチノイドX受容体(RXR)とのヘテロ二量化後、標的遺伝子のプロモーターにあるPPAR応答要素(PPRE)に結合し、強力な転写因子として作用する。PPAR-αやPPAR-β/δ、PPAR-γなどのファミリーの他の受容体は、脳やその他の臓器に発現しており、酸化ストレス、エネルギー恒常性、ミトコンドリア脂肪酸代謝、炎症などに重要な役割を果たしている。PPAR-αは、脳内のグルタミン酸の恒常性維持やコリン・ドーパミン系のシグナル伝達に関わるタンパク質をコードする遺伝子の制御に関与している。また、PPAR-αは、アミロイド前駆体タンパク質(APP)の代謝に関与する酵素をコードする遺伝子の発現を制御する。PPAR-αは、APPの非アミロイド性分解経路を担うαセクレターゼをコードする遺伝子を活性化する。また、アルツハイマー病でアミロイドβ(Aβ)ペプチドの放出に関与する主要な酵素であるβセクレターゼ(BACE-1)をダウンレギュレートする。ADの脳では、PPAR-αおよびPPAR-γ coactivator-1α(PGC-1α)の遺伝子の発現が著しく低下している。PPARは、ADに限らず、他の神経変性・神経発達疾患や精神疾患でも変化していると言われている。PPAR-αの低下は、抗酸化・抗炎症プロセスを低下させ、アルツハイマー病患者の脳における脂肪酸輸送、脂質代謝、ミトコンドリア機能の変化の原因となる可能性がある。PPAR-αの特異的な活性化剤は、神経変性疾患や神経発達障害における脳細胞の代謝や認知機能の改善に重要であると考えられる。
キーワード PPAR-α、グルタミン酸シグナル、App/aβ代謝、ミトコンドリア機能、神経変性、神経保護
はじめに
ペルオキシソーム増殖因子活性化受容体(PPAR)は,PPAR-α,PPAR-β/δ,PPAR-γを含むリガンド制御型核内受容体である。これらの受容体は、それぞれ異なる遺伝子によってコードされている。PPAR-α(NR1C1)、PPAR-β/δ(NUC1またはNR1C2)、PPAR-γ(NR1C3)は、マウスでは第15,17,6番染色体に、ヒトでは第22,6,3番染色体に存在する。PPAR-γ遺伝子の代替プロモーターは、3つのアイソフォーム(γ1, γ2, γ3)を担っている。これらのPPARs遺伝子は、アミノ酸数468, 441, 475, 505,49-56kDaのタンパク質をコードしている[1, 2]。
PPARは、BergerとMollerのレビュー[3]やNierenbergらのレビュー[4]で述べられているように、複雑なメカニズムで転写を制御している。現在、PPAR-αとして知られている最初のPPARは1990年に発見された[5]。PPARは、RXRとヘテロ二量体化した後、PPAR応答要素(PPRE)と呼ばれる標的遺伝子の特定のプロモーター領域に結合し、転写因子として働く[4]。PPAR-γ coactivator-1 alpha (PGC1-α)のようなPPARs coactivatorは,PPARsだけでなく,エストロゲン受容体(ERs)や核内呼吸因子1および2(NRF1, NRF2)のような他の核内受容体との相互作用を通じて,遺伝子の転写に重要な役割を果たしている[6]。これらの受容体は,転写,エネルギー代謝,脂質代謝,さらには熱発生の調節に重要な役割を果たしている。PPAR-αは,脂肪酸β酸化経路を含むミトコンドリア代謝,エネルギープロセス,グルコース代謝,酸化還元状態,グルタミン酸系,コリン作動性/ドーパミン作動性神経伝達を制御している。また、PPAR-αは、脳内でアミロイドβ前駆体タンパク質(APP)の代謝に関与しており、Aβを介して直接的または間接的に、タウタンパク質のリン酸化にも影響を与えていると考えられている(図1)。(図1)PPAR-β/δは、中枢神経系(CNS)において、細胞の分化、脂質代謝、髄鞘形成プロセスを制御している[7]。PPAR-γとそのコアクチベーターであるPGC-1αは,神経変性や神経炎症における細胞の分化やミトコンドリアの生合成に重要な役割を果たしている[8-10]。
図1 脳におけるPPAR-αの役割
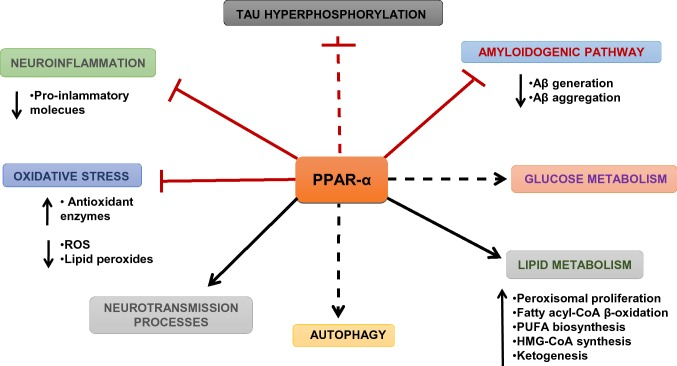
(D’Orioら[136]による、若干の修正を加えている。
この総説の目的は、神経変性疾患や精神疾患における脳内でのPPAR-αの役割を説明し、この受容体が新しい治療戦略の有望なターゲットとなりうることを示すことである。
PPAR-αと脳内の神経伝達におけるその役割
Wardenら[11]のデータは,脳内におけるPPARの分布を初めて明らかにしたもので,成体マウスやヒトの脳内の神経変性疾患に関与する特定の領域におけるPPARの細胞型プロファイルを定義した。二重免疫蛍光法を用いて、成体マウスと成体ヒトの脳において、PPAR-αだけがすべての細胞タイプと共局在することを示した。さらに、著者らは、強い神経免疫反応を引き起こすリポ多糖(LPS)注入後、すべてのPPARの顕著な神経細胞局在化が見られ、PPAR-γのミクログリアへの共局在化は非常に弱いことを指摘している。
さらに、Royら[12]は、海馬のさまざまな領域におけるPPAR-αの分布を調べ、PPAR-αタンパク質がマウスの脳のCA1,CA2,CA3および歯状回(DG)に局在することを確認した。その結果、PPAR-αはカルシウムの流入を制御し、シナプス可塑性の制御に関わる海馬のタンパク質をコードするいくつかの遺伝子の発現を制御することがわかった。PPAR-αは,N-methyl-d-aspartate(NMDA)受容体サブユニットNR2AおよびNR2B遺伝子[13],2-amino-3(3-hydroxy-5-methyl-isoxasol-4-yl)propanoic acid AMPA受容体関連サブユニットGluR1[14],さらにAMPA受容体関連活性関連細胞骨格タンパク質[15]の発現に関与している。これらの遺伝子は,いずれもシナプス可塑性に関連しており,サイクリックAMP応答要素結合タンパク質(CREB)を介してPPAR-αによって制御されている。Royら[12]のデータによると、PPARα欠損マウスは、CREBや記憶関連タンパク質が欠損しており、空間学習や記憶が低下している。
さらに、PPAR-αとそのリガンドが、脳内のグルタミン酸とコリン酸を介したドーパミン神経伝達の制御に関与していることが示されている[16-21]。PPAR-αシグナルは、グルタミン酸受容体の内因性アンタゴニストの代謝に関与する酵素の遺伝子転写を変化させ、Huangらが示したように、アストロサイトにおけるグルタミン酸トランスポーター-1 (GLT-1) のエンドサイトーシスを促進する可能性がある[16]。アストロサイトは,アスパラギン酸トランスポーター/興奮性アミノ酸トランスポーター1 (GLAST/EAAT1) およびグルタミン酸トランスポーター1/EAAT2 (GLT-1/EAAT2) を介したグルタミン酸の取り込みにより,中枢神経系におけるグルタミン酸のホメオスタシスを維持している。この2つは,アストロサイトにおける最も重要なグルタミン酸トランスポーターで,シナプスでグルタミン酸を除去する[22]。GLT-1は、成人中枢神経系におけるグルタミン酸の前脳への取り込みの90%を担い、その結果、脳内のグルタミン酸のホメオスタシスにも関与している[23]。アストロサイトのGLT-1の発現と機能が低下すると、グルタミン酸シナプスにおけるグルタミン酸のホメオスタシスのバランスが崩れてしまう可能性がある。Huangら[16]は、PPAR-α受容体のアゴニストがprotein kinase C (PKC)シグナル経路を介してアストロサイトにおけるGLT-1のエンドサイトーシスを増加させることを示唆している。ADの脳では、PPAR-αの発現と機能が低下しており、GLT-1の機能に影響を及ぼしている可能性がある(図2)。ADでは、他にもいくつかの要因がグルタミン酸のホメオスタシスを乱している。例えば、シナプス間隙からのグルタミン酸の除去に関与するいくつかのプロセスの変化は、シナプス後のグルタミン酸受容体を過剰に刺激する可能性がある。病的な状態では、グルタミン酸トランスポーターのスプライス変異体AEET2がグルタミン酸の取り込みを低下させる可能性がある[24]。さらに,Lauderbackら[25]は,GLT1が脂質過酸化産物である4-ヒドロキシ-2-ノネナールによって変化し,Aβ42によって増強されることを示した。Liら[26]は、数年前に、ADにおけるAPPの異常な発現がGLT1のダウンレギュレーションに関与している可能性を示唆していた。PPAR-αアゴニスト(GW7647とWY14, 643)および内因性アゴニストであるパルミチン酸(PA)の処理は、アストロサイトのGLT-1のレベルを有意に調節するが、その形態には変化がなかった[16]。
図2 PPAR-αとグルタミン神経伝達およびグルタミン酸ホメオスタシスへの関与
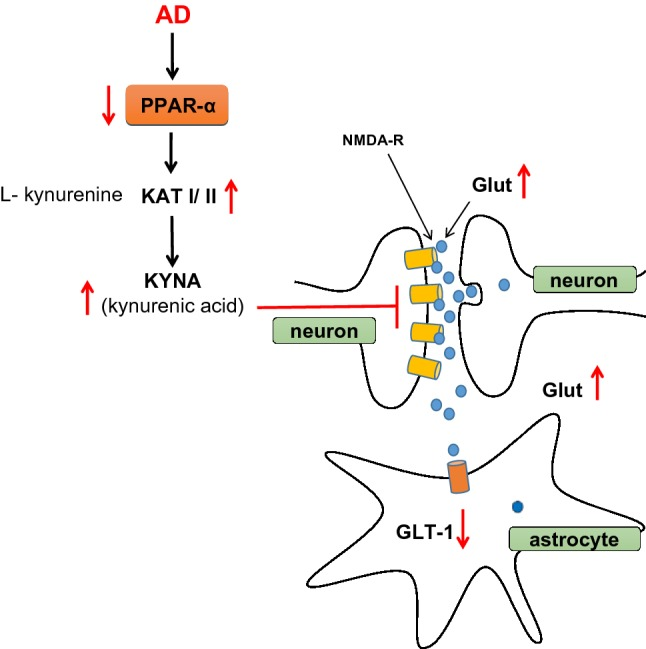
PPAR-αアゴニストであるフィブラートは、内因性のグルタミン酸受容体拮抗物質であるキヌレン酸(KYNA)のレベルを調節することにより、神経保護作用を発揮する。KYNAは、脳内でl-キヌレニン(l-トリプトファンの代謝物)から、主にKATIIアイソフォームを持つキヌレニンアミノトランスフェラーゼ(KAT)によって合成される。KYNAの脳内濃度が上昇すると、記憶障害や精神病症状の原因になると考えられている[27, 28]。KYNAとキノリン酸(QUIN)はトリプトファン代謝の主要産物であり、N-メチル-d-アスパラギン酸受容体(NMDA)とは異なる相互作用を示す。KYNAはNMDAアンタゴニストで神経保護作用があるが、QUINはアゴニストで神経毒性がある。どちらの化合物も学習と記憶に影響を与える[29]。
Zakrockaらのデータ[17, 18]によると、アンジオテンシンIIタイプ1受容体ブロッカーおよびPPAR-αのアゴニストであるgemfibrozilは、KATIIに対する阻害作用を介して大脳皮質におけるKYNA合成を減少させることが示されている。また、Zakrockaは、PPAR-αが記憶や精神疾患に有益な効果を発揮するのと全く同じ方法であることを示唆している。最近、AD治療のためのKYNAのいくつかのアナログが試験され、治療アプローチが示唆された[30]。これらのデータは、PPAR-αがグルタミン酸ホメオスタシスの制御に重要な役割を果たしていることを示唆している。グルタミン酸ホメオスタシスは、学習と記憶のメカニズム、神経可塑性における適応反応、およびいくつかの神経疾患の経過/結果において重要であることが知られている。Pierrotら[34]は,認知機能障害のあるTgマウスモデルにおいて,RXR刺激による海馬のシナプス可塑性の改善にPPAR-αが関与していることを報告している。特定の受容体アゴニストでPPAR-α受容体を活性化すると,α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)受容体のGluA1サブユニットの転写が促進され,これによりAMPA反応が起こり,RXRの活性化によりシナプス可塑性が改善されることがわかった。AD Tg認知機能障害マウスの海馬でPPAR-αをノックダウンすると、RXR活性化によるシナプス可塑性の改善効果がオスのみで消失した。GluA1サブユニットが長期増強(LTP)や認知に重要な役割を果たしていることは,Schmittらによって明らかにされている[35]。しかし、Pierrotら[34]は、ADのTgマウスモデルにおいて、RXR刺激によるLTPの改善は、GluA1の発現上昇と相関しており、PPAR-αがこの事象において、性別に関係なく重要な役割を果たしていることを示している。PPAR-αの発現が雌に比べて雄で2倍高いことが、ペマフィブラートによる雄の認知障害マウスのシナプス可塑性の改善に関与している可能性がある。PPARsおよびRXR受容体の活性化は、興奮性神経伝達に関与するタンパク質をコードするいくつかのシナプス関連遺伝子の発現を増加させる[12, 36]。おそらく、これらのメカニズムにより、PPAR-αのアゴニストは、加齢による認知障害のモデルマウスにおいて有望な効果を発揮すると考えられる[37, 38]。さらに、RXRの活性化は、神経細胞の樹状突起の複雑性や分岐を増加させ、分化を促進することが以前に報告されていたが[39, 40]、現在では、PPAR-αがこれらのプロセスにおいて重要な役割を果たしていると考えられている。
PPAR-αは、コリン作動性ニコチン受容体によるドーパミン作動性伝達の制御にも関与している可能性がある[19, 20]。PPAR-αは,ドーパミンニューロンのニコチン誘発反応を低下させることがわかっている。また,ニコチン性アセチルコリン受容体(AChR),特にβ2サブユニットを含む受容体の活性化は,ドーパミン神経細胞の活動を有意に制御する。Melisら[19]の研究では,電気生理学的,生化学的,行動学的手法を用いて,α7-nAchRの活性化が,動物の腹側被蓋野(VTA)におけるnAChRのβ2サブユニットと内因性のPPAR-αのアゴニストのレベルを高めることが示された。これらのデータは、ドーパミン神経細胞におけるnAChR/PPAR-αシグナルの重要な役割を示していると考えられる。要約すると、すべてのPPARの中で、PPAR-αだけが脳内の神経伝達プロセスや記憶機能の調節に関与していることを強調しておきたい。PPAR-αおよびPPAR-δの発現は、ADにおいて抑制されていることが観察されている[41]。PPAR-αの多型は、ADの危険因子と考えられている[42]。ADを含むいくつかの病態において、PPAR-αの転写は、PPAR-γとは異なる方法で制御されている[43-46]。最近では、PPAR-αが神経炎症におけるミクログリアの活性化に重要な役割を果たしていることが観察されており[47]、その活性化がスタチンの細胞保護効果を誘発することがわかっている[10]。
APP/Aβの代謝におけるPPAR-αの役割
PPAR-α受容体は、脳内でのAPPの代謝に重要な役割を果たしている。Corbettらの最新のデータ[48]によると、基本的な生理学的条件下では、PPAR-αは、APPαセクレターゼの活性化により、非アミロイド原性ペプチド(p3)や細胞保護効果が期待できる可溶性sAPPαの遊離をもたらすことで、APPの分解に関与していることが示されている。この非アミロイド性APP代謝経路の活性化は、BACE-1が重要な役割を果たすアミロイド生成経路によるAβの遊離から脳を保護する可能性がある(図3)。著者らは、PPAR-αアゴニスト(ゲムフィブロジル)が、海馬の培養でADAM10の発現を促進することを示した。この観察結果は、遺伝子組み換え動物(PPAR-αコーディング遺伝子をノックダウンしたもの)のデータでも確認された。別の研究では、PPAR-αのアゴニスト(WY14643)が海馬ニューロンにおけるADAM 10(αセクレターゼ)の発現を増加させるが、PPAR-αコーディング遺伝子を欠くニューロンではADAM 10が欠損することが明らかにされている[49]。Zhangら[50]の別のデータでは、PPAR-αアゴニスト(GW7647)がBACE-1の活性を阻害することでアミロイドβ(Aβ)の生成を制御することが示されている。さらに、PPAR-α受容体のアゴニスト(GW7647)は、ADの細胞モデルにおいてsAPPβの発現、BACE-1の活性およびAβ1-42のレベルを低下させたが、APPおよびプレセニリン-1(PS1)のレベルには影響を与えなかったことが確認された。Zhangら[51]の別の研究では、ホスファチジルイノシトール3キナーゼ(PI3-K)がPPARシグナルに重要な役割を果たし、Aβペプチドの産生を減少させることが示された。しかし、ADでは、PPAR-αシグナルが変化すると、アミロイド生成経路を介してAPPの代謝が活性化され、脳内でAβが遊離・蓄積される可能性がある。ADで過剰に放出されたAβペプチドがPPAR-αの変化に関与している可能性があり、病理学的事象の悪循環を締めくくっている(図4)。また、Kummerら[52]は、すべてのPPARs受容体を活性化する新規PPARアゴニスト(GFT1803)が、ADマウス(APP/PS1)のAβプラーク/Aβレベルおよびミクログリアの活性化を有意に減少させることを検出している。これらのデータから、PPARs受容体は、インスリン分解酵素(IDE)の発現を促進したり、その活性化によって、Aβの分解やクリアランスに関与していることが示唆された。さらに、pan-PPARアゴニスト(GFT1803)は、ADマウスの記憶機能に良い影響を与えた。しかし、アルツハイマー病患者を対象とした臨床試験では、ピオグリタゾンの効果は非常に限られたものであった。現在、PPAR-αはAD治療戦略の有望なターゲットであると考えられる。PPAR-αアゴニスト(フィブラート)の抗アミロイド作用は、臨床において長期的な患者の治療で観察された。これらの患者は、加齢に伴う非治療の健常者(対照群)と比較して、Aβ1-42濃度が低いことが特徴的であった[53]。Chandraら[54]およびChandraとPahan[55]によって発表された最後のデータでは、PPAR-αの活性化剤(桂皮酸とゲムフィブロジル)が、ADの動物モデル(5XFAD)の海馬と大脳皮質のアミロイドプラークを減少させることが実証された。さらに、神経炎症、ひいてはミクログリアやアストロサイトの活性化を減少させ、空間学習や認知機能を向上させることがわかった。活性化したミクログリアやアストログリアは、いくつかの炎症性サイトカインやケモカインを分泌するが、そのうちIL-1,TNF-α、IL-6,IL-8,TGF-βなどのいくつかは、アルツハイマー病患者で異常に発現していることが観察されている[56]。
図3 脳内のAPP分泌酵素の制御におけるPPAR-αの関与
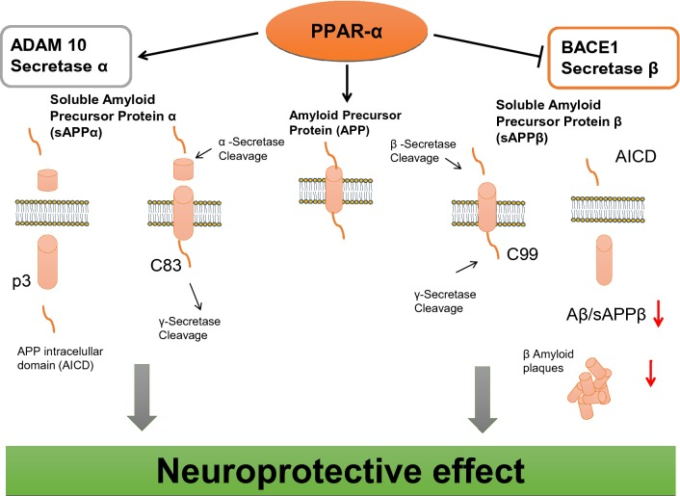
図4 AD脳のPPAR-αを阻害するとAPPの代謝が変化する
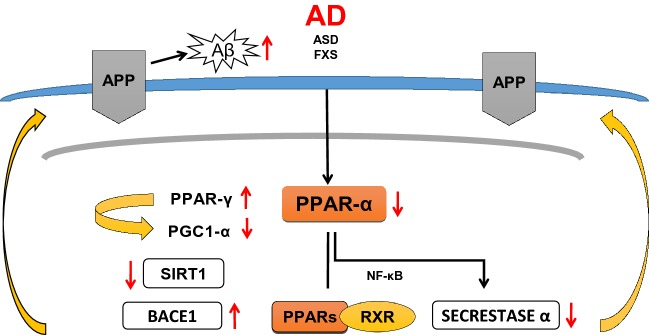
Aβのレベルは、PPAR-γや、BACE-1をコードする遺伝子の転写に関与する多くの転写因子によっても制御されていることが知られている。これらの転写因子の中には,NF-кB(活性化B細胞の核内因子),SP1(刺激タンパク質),YY1(陰陽1転写因子)のように,より重要な役割を果たすものがある[57]。さらに、この研究では、PGC-1αが、試験管内試験および生体内試験において、APP βセクレターゼやBACE-1の転写の制御にも関与していることが示された。PPAR-γ/PGC-1αとサーチュイン-1(SIRT-1)の相互作用は、BACE-1の制御に重要であり、空腹時を含むいくつかの環境条件によって調節される[57]。
これらの3つの因子は、BACE-1遺伝子のプロモーターにあるPPAR-RXR部位と相互作用する。同じグループは、PGC-1αの増強とSIRT1の発現上昇が、BACE-1遺伝子の発現を抑制することを示した(図4 BACE-1とγセクレターゼの複合体は、APPの逐次的なタンパク質分解、Aβペプチドの遊離、蓄積、アルツハイマー病患者の脳内における神経変性の病原タンパク質の拡散に関与している[58, 59]。さらに,Qiangら[60]は,SIRT 1が脱アセチル化によってPPAR-γを直接活性化することを示している。PPAR-γ/PGC-1αがアルツハイマー病のβセクレターゼの制御に重要な役割を果たしていることは,SweeneyとSong[61],Katsouriら[62]によって述べられている.
Qinら[63]のデータでは,アルツハイマー病の脳では,臨床的な認知症の進行によってPGC-1α遺伝子の発現が有意に低下することが示されている。さらに、PGC-1αタンパク質含量は、Aβ1-42アミロイド含量およびAD型Aβプラーク病理と負の関係にあることがわかった。また、Tg2576 ADマウスを用いた研究では、Aβ1-42レベルとPGC-1αの発現量に相関関係があることが示された。PGC-1αが欠損すると、不安、多動、後肢の痙攣などの行動上の変化が起こる。また,線条体やその他の脳の部分に海綿状の空胞化が見られた[64].また,PGC-1αが海馬ニューロンの樹状突起の維持に関与していることを示唆するデータもある.著者らは,PGC-1αを過剰発現させると樹状突起スパインが増加し,シナプスの分子分化が促進されるのに対し,PGC-1αをノックダウンするとシナプス形成が阻害されることを報告している[65]。図3は、生理学的条件下でのPPAR-αの、APP分解に関わる酵素に対する制御的役割を示している。PPAR-αが活性化されると,BACE-1が阻害され,Aβペプチドの遊離・蓄積が抑制されるとともに,αセクレターゼが刺激され,APPの非アミロイド性分解が行われる.
AD、パーキンソン病患者の脳、虚血脳、外傷を受けた脳で観察されるAβレベルの上昇は、PPAR-αシグナルの変化とミトコンドリア機能の役割に関連したグルタミン酸やコリン酸などの神経伝達プロセスの変化に起因すると推測される。
ミトコンドリア機能の制御におけるPPARの役割
ミトコンドリアの障害は、老化と神経変性疾患の両方において重要な役割を果たしている。Lamichaneら[66]は,PPAR-αが,カルニチンパルミロトランスフェラーゼ1(CPT1),中鎖アシル-CoAデヒドロゲナーゼ,アシル-CoAオキシダーゼ,脂肪アシル-CoAシンターゼなどの脂質代謝に関連するミトコンドリア酵素をコードする遺伝子の発現を増加させることを報告している。さらに、PPAR-αは、脂肪酸(FA)とその誘導体の輸送タンパク質をコードする遺伝子を活性化し、脂肪酸がβ酸化経路に入るようにする。また,PPAR-αは,遊離カルニチンと交換にアシルカルニチンエステルをミトコンドリア膜を介して輸送するタンパク質をコードする遺伝子の発現を誘導する[67]。さらに,PPAR-αは,RXRとともに,マロニルコアデヒドロゲナーゼ(MLYCD)をコードする遺伝子のプロモーターを活性化する[68, 69]。また,PPAR-αは,核内のピルビン酸脱水素酵素キナーゼ4(PDK4)をコードする遺伝子を活性化する。PDK4は,PPAR-β/δによっても活性化されるが,PPAR-γによっては阻害される[70]。PPAR-αに加えて,他のPPARs受容体も,アシル-CoAヒドロラーゼ酵素やω-水酸化酵素シトクロムp450 4Aサブファミリー(CYP4A)をコードする遺伝子の活性化に関与する可能性がある[38, 71]。PPAR-α,PPAR-γおよびそのコアクチベーターPGC-1αは,ミトコンドリア転写因子およびいくつかの核転写因子の活性化を通じて,ミトコンドリアの生合成に非常に強力な因子であることがわかっている。(TFAM-transcription factor A mitochondria, NRF1, NRF2, YY1, SP-1)を活性化する(図5)。ミトコンドリアの生合成は,ミトコンドリアの形成と組み立てを活性化するさまざまなシグナル伝達経路と転写複合体によって制御されている[4, 6, 8, 9, 72, 73]。
図5 ミトコンドリアの生合成と機能に対するPPAR-α、PPAR-γ、PGC1-αの役割
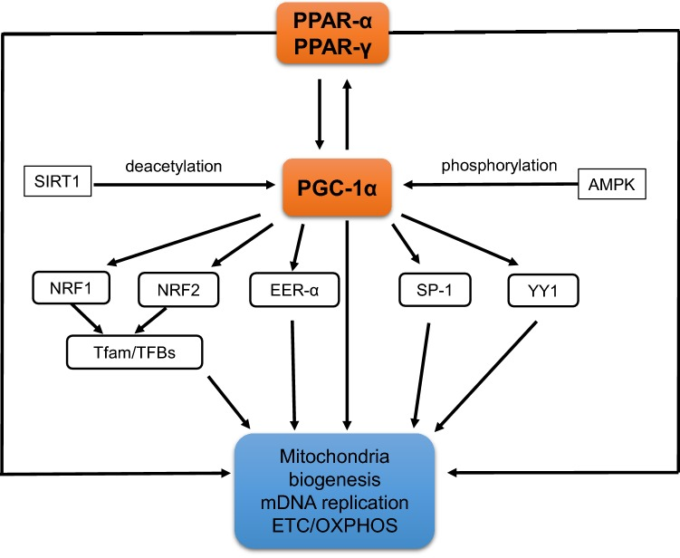
(Dominy and Puigserver [72]; Jornayvaz and Shulman [73]; Scrapullaら [6, 8]による。)
ミトコンドリアが核の直接的な支配下にあることは、よく知られている。ミトコンドリアのタンパク質の大部分は、核の転写因子やサーチュイン、ポリ(ADP-リボース)ポリメラーゼ(PARP)などの他のタンパク質や酵素と同様に、核DNA(nDNA)によってコードされている[72, 74-76]。さらに、PARP-1のように、これらのタンパク質の一部は、DNA修復や、核-ミトコンドリア間のクロストークに関与するシグナル分子であるポリ(ADP-リボース)(PAR)の合成にも関与している[77]。Huangら[16]は,PPAR-αがPARP-1の基質であることを明らかにした。PARP-1は、NAD依存性のDNA結合型酵素で、ストレス応答、細胞の生存と死の主要な媒介因子であり、いくつかの転写因子や他のタンパク質の分子制御因子でもある。Baiら[78]のデータによると、PARP-1の阻害は、Sirt1の活性化を介して著しい代謝効果を発揮することが示されている。Lapucciら[79]は,PARP-1がmtDNAの修復や転写に関わる核内遺伝子をエピジェネティックに制御することで,ミトコンドリアのホメオスタシスに重要な役割を果たしていることを示した。しかし,ミトコンドリアには独自のゲノムが存在し,PARP-1もミトコンドリアDNA(mDNA)の修復やミトコンドリアのエネルギープロセスの制御に関与している可能性がある[80, 81]。mDNAは、呼吸器系複合体I、III、IV、Vのサブユニットである13種類のポリペプチド、2種類のrRNA、22種類のtRNAをコードしていることがよく知られている[82]。その他、呼吸器系複合体IIのサブユニットや、mDNAの複製、転写、翻訳に必要なタンパク質など、これらのプロセスの制御に関与するすべての因子が核によってコード化されている[83]。比較的少数の核内因子が、すべての呼吸器系タンパク質の転写を調整している。転写因子の中には、上述の呼吸器系核内因子がある。NRF1,NRF2,SP1,YY1,エストロゲン関連受容体α(ERR-α)などである。さらに、ミトコンドリアには、PARP-1,SIRT-1,SIRT-3,SIRT-4,SIRT-5 などのサーチュインと呼ばれる NAD 依存性酵素が存在し、これらの酵素は、脱アセチル化、ポリおよびモノ ADP リボシル化、サモイル化などにより、多くの 転写因子 および他のタンパク質の翻訳後修飾に関与している可能性があることが示されている。[74, 75].
また,核内受容体スーパーファミリーのPPARや,DNAに結合したPARP,核内に存在するヒストン脱アセチル化酵素III型,サーチュイン(SIRT-1およびSIRT-6)など,制御ネットワークタンパク質の多くの構成要素が,ミトコンドリアの動態と機能に関与しており,神経変性疾患の発症と進行に重要な役割を果たしている可能性がある[75]。現在、ほとんどの神経疾患や代謝性疾患の発症・進行には、炎症が大きく関与していることが一般的に推測されている。PPAR-α,PGC-1α,およびその他の核内受容体ファミリーは,治療戦略における有望な標的となりうると考えられている[84]。
PPARの各核内受容体は、上述のように特定の機能を持っている。PPAR-αは、酸化ストレス、エネルギーホメオスタシス、ミトコンドリア脂肪酸代謝を制御しており、脳内の興奮性グルタミン酸神経伝達や、おそらくコリン作動性/ドパミン作動性シグナルにも影響を与えていると考えられている。PPAR-β/δとPPAR-γは主に脂質の代謝を制御しているが、脳内での特異的な役割はまだ完全には解明されていない[85, 86]。中枢神経系の髄鞘形成過程におけるPPAR-βの役割を示すデータもいくつかある[87]。
PGC-1αは、PPARを含む複数の核内受容体との相互作用により、神経炎症、神経細胞死、抗酸化防御に関連する遺伝子の転写制御に関与している可能性がある。さらに,PGC-1αは,ミトコンドリアの生合成,DNAレベル,ミトコンドリアの機能に関与するタンパク質をコードする遺伝子の発現調節にも重要な役割を果たしていると考えられている[73]。PGC-1αは,トリカルボン酸サイクル(TCA),酸化的リン酸化,脂肪酸のβ酸化に関与する遺伝子を活性化することで,ミトコンドリアの酸化的代謝を制御している可能性がある[88]。PPARはPGC-1αの発現の活性化に関与しており、PGC-1αはその後、これらの受容体のコアクチベーターとして、またPPARsの転写を調節する因子として重要な役割を果たしている。これらの関係は、ある種の分子的な悪循環をもたらす可能性がある。PGC-1αは,NAD制御酵素,SIRT-1を介した脱アセチル化,あるいは5′アデノシン一リン酸活性化プロテインキナーゼ(AMPK)や他のキナーゼ(p38)を介したリン酸化によって活性化される[64].PGC-1αの遺伝子発現およびタンパク質レベルは,中枢神経系の病態に応じて異なる影響を受けることが観察された。ADやハンチントン病(HD)の脳虚血や炎症などの他の病態では、PGC-1αの発現や活性が低下していることを報告するデータがいくつかある[52, 57]。
ハンチントン病患者ではPGC-1αのレベルが低下しており[89, 90]、PGC-1αが線条体細胞の機能に重要であることが示唆されている。パーキンソン病(PD)モデルマウスでは,PGC-1αの欠損が黒質のドーパミン神経細胞の変性に関与していることが示唆されている。PGC-1αの保護的役割は,Thauらによって筋萎縮性側索硬化症(ALS)マウスおよびALS患者においても明らかにされている[91]。
最近では,PGC-1αは,脳虚血性脳症,肝性脳症,1型糖尿病や2型糖尿病における脳の変化,外傷性脳症につながる可能性のある急性および慢性の脳損傷によって誘発される神経変性プロセスにおいても重要な役割を果たしていることが示唆されている[64, 92]。PGC-1αの合成は,環境条件の変化によって制御されており,この因子が環境からのシグナルを脳内の遺伝子発現に結びつけると考えられている。PGC-1αは,すべての脳組織とほとんどの脳領域で発現しており,酸化ストレス応答において重要な役割を果たしている[92, 93]。PGC-1αのシグナル伝達経路による神経保護のメカニズムは,Lvらによって最近明らかにされたように,非常に複雑である[92]。要約すると、PGC-1αは、PPARの機能に依存したメカニズムで、ミトコンドリア生合成、酸化代謝、脂肪酸酸化、およびグルコン生成を制御することにより、細胞の代謝および炎症反応に影響を与える。また、PGC-1αは、PPARsだけでなく、エストロゲン受容体などの他の核内受容体も制御することができる。神経保護におけるPGC-1αの役割に関する報告が増えてきているにもかかわらず、様々な病態における遺伝子発現やミトコンドリアの動的および機能の制御におけるPGC-1αのメカニズムは十分に解明されていない。
PPAR-αの天然および合成アゴニスト:実験から臨床試験まで
PPAR-αは、酸化ストレス、エネルギーホメオスタシス、ミトコンドリア脂肪酸代謝を制御しており、全てのPPARの中で唯一、脳内の興奮性グルタミン酸系神経伝達やコリン・ドーパミン系神経伝達に影響を与えている。PPAR-αや他のPARPsの発現は、脳の前頭前野、側坐核、扁桃体で顕著であり、神経細胞とグリア細胞の間で有意に高い値を示している[11]。
脳におけるPPAR-β/δおよびPPAR-γの具体的な役割は、まだ完全には解明されていない[85, 86]。中枢神経系における髄鞘形成過程にPPAR-βが関与していることを示すデータがいくつかある[87]。これらのデータに基づいて、PPAR-αの活性化が、いくつかの神経変性疾患や代謝性疾患の治療戦略において最も有望であることが期待されている。
Esmaeiliら[84]は、ALSモデルマウスにおいて、フェノフィブラートによるPPAR-αの優先的な活性化が、神経炎症を軽減し、神経変性を抑制することを示した。フェノフィブラートを投与した動物では、mRNAの解析により、抗炎症および抗酸化防御に関与するタンパク質をコードする遺伝子の転写に、フェノフィブラートが有意に作用することが示された。また、フェノフィブラートは、神経炎症に関与する遺伝子の発現を抑制した。PPAR-αのアゴニストを投与したPDモデルマウスでは、ミトコンドリアが維持され、運動能力が向上した。また、フェノフィブラートを投与することで、病状の進行を有意に遅らせることができた。
PD研究の一環として行われた他の実験動物モデルにおいても、Uppalapatiら[94]はPPAR-αアゴニストであるフェノフィブラート/フェノフィブリン酸の神経保護効果を観察した。この化合物は、認知障害に対して有意な改善効果を示した。パーキンソン病患者の約40%が認知機能障害や認知症を患っており、その頻度は健常者と比較して約6倍と言われている。過去のいくつかの研究では、PDにおけるPPARアゴニストの有望な効果が示されている[95-98]。Cartaら[99]は、進行性PDマウスモデルにおいてPPAR-γレベルを低下させ、TNF-α産生を抑制するロシグリタゾンの改善効果を報告している。また、ピオグリタゾンの無作為化試験では、PDに対する改善効果は確認されなかった[4]。PPARアゴニストの効果は、PDの前駆段階でもしばしば起こる抑うつ状態の治療においても認められ、PDの非常に重要な初期症状と考えられている。PDにおけるPPARの役割を評価するには、さらなる研究が必要である。
過去10年間の研究では、認知と感情におけるPPAR-αの重要な役割が指摘されている[100]。彼らのデータは、ストレス状態におけるPPAR-αの機能、その内因性アゴニスト、および神経ステロイドのレベルの関係を強調している。PPAR-αは、感情やストレス反応の調節に関与するこれらの脳領域で高くなっている。
PPAR-αの発現は、前頭前野、基底核、扁桃体、視床核で最も高く、海馬ではかなり低くなっている[11]。NisbettとPinna[100]のデータは、Hillard[101]によっても観察されたように、PPAR-αの活性化がストレス反応を媒介し、変調させることを示した。PPAR-αが情動やストレスに作用するメカニズムでは、アロプレグナノロン/プログナノロンなどの神経ステロイドが重要な役割を果たしているのではないかと提案されている[102]。PPAR-α刺激とアロプレグナノロン生合成の相関関係に基づいて、心的外傷後ストレス障害(PTSD)やその他の気分障害で観察される感情の変化に対する新しいバイオマーカー軸と治療戦略が提案されている[100]。PPAR-α受容体のアゴニストは、脳由来向神経性因子(BDNF)のレベルを高めることによっても神経保護効果を発揮する。Jiangら[103]は、海馬におけるPPAR-αを介したBDNFのシグナル伝達経路を介して、フェノフィブラートの抗うつ様効果をマウスで報告した。このマウスモデルにおけるフェノフィブラートの抗うつ様効果は、PPAR-α阻害剤と海馬のBDNFシグナル伝達経路の低下によってブロックされた。さらに、同じグループ[103]は、フェノフィブラートの抗うつ様作用は、カンナビノイド系を必要としないと推測している。うつ病の病態生理において、BDNFシグナルの変化が重要な役割を果たしていることを裏付ける研究がいくつかある[104, 105]。Kempら[106]やZeinoddiniら[107]が報告したように、ピオグリタゾンは双極性障害の抑うつ症状を有意に改善し、炎症性サイトカインのレベルを有意に低下させることが観察されている。また、Kempら[108, 109]やLinら[110]によるプラセボ対照二重盲検試験では、単極性うつ病においても、うつ病の重症度の改善が認められている。
これらのデータに基づき、PPAR-α、PPAR-γ受容体アゴニスト、さらにPGC-1αは、代謝性疾患、炎症性疾患、その他の神経疾患や精神疾患の治療における有望なターゲットとして提案されている。いくつかの神経変性疾患では、PGC-1αの発現が、その標的遺伝子であるNRF1やTFAMとともに低下している。このことは、PGC-1αとNRF1およびTFAMを介したシグナル伝達経路が神経変性や神経保護に重要であることを示唆している。PGC-1αとPPARは,生体エネルギー問題に対する神経系ミトコンドリアの適応反応に重要である[32]。さらに,PGC-1αがシナプス外グルタミン酸受容体(NMDAR)の活性や興奮毒性を負に制御していることを強調しておくことも重要だ[111]。ADに関するValleeとLecarpentierの研究[49]では、PPAR-γアゴニストがアルツハイマー病患者の学習と記憶の障害を軽減することが報告されている。
最近のいくつかの臨床試験では、PGC-1αがPPARと相互作用することで、気分の変化、特にうつ病の治療に有望な効果があることが示唆されている。Sepanjniaら[112]は、ピオグリタゾン治療がうつ病エピソードの気分を改善したと報告している。ピオグリタゾンの抗うつ作用のメカニズムは、よりよく解明されるべきである。脂肪酸やプロスタグランジンなどのPPAR-γの内因性リガンドのレベルが変化すると、シグナル伝達経路にさらなる効果を及ぼす可能性がある。
これらの内因性化合物は弱いアゴニストであり、生理学的および病理学的条件におけるPPARs受容体との相互作用の役割は完全には理解されていない。いくつかの化合物が合成されたが、中でもチアゾリジン系化合物(グリタゾンとも呼ばれる)は強いアゴニストである。これらの化合物は多くの疾患で研究され、肥満や他の代謝性疾患の臨床治療に使用された。現在までにロシグリタゾン(BRL-49653)とピオグリタゾンがII型糖尿病の治療に推奨されている。ロシグリタゾンを含むこのグループの化合物のほとんどは、心筋梗塞のリスクを高めるような危険な副作用のために、FDAによって米国市場から排除された[113]。
最近の研究では、PGC-1αとpan-PPARのアゴニストが、自閉症スペクトラム障害やその他の精神神経系の変化に重要な役割を果たしている可能性が指摘されている[114]。PPAR-αや全てのPPARのアゴニストが、双極性障害やうつ病に関与している可能性が指摘されている[4]。また、PPAR-αは、マウスの行動反復や認知的柔軟性にも影響を与える[115]。さらに、PPAR-αアゴニストの保護効果は、口腔内ジスキネジアのラットモデルや様々なタイプの脳症で示された[116, 117]。さらに、PPARsアゴニストは、アルコール消費行動にも影響を与える可能性がある[118]。レスベラトロールの脳卒中に対する作用は、フェノフィブラートの作用と同様に、PPAR-αの神経保護作用を介していることを示すデータがいくつかある[119]。PPAR-α受容体の刺激は、アストロサイトにおいてアロプレグナノロンの合成を誘導し、このホルモンが神経保護メカニズムに関与している可能性がある[120]。PPAR-αの神経細胞死や微小血管障害に対する治療効果については、MoranとMaが報告している[121]。PPAR-αとこのファミリーの他の受容体は、酸化ストレスと炎症反応の制御に重要な役割を果たしている。現在では、酸化ストレスが、神経変性疾患や神経発達・精神疾患の病因や病態の重要な要因であると考えられている。最近では、子供に多い自閉症スペクトラム障害(ASD)にミトコンドリアの酸化ストレスが関連していると考えられている。自閉症スペクトラムは、遺伝子や染色体の異常に加えて、出生前のストレスやホルモンレベルの変化、催奇形性化合物などの環境因子によって引き起こされる。この多因子性障害であるASDは、現在、80人に1人、あるいは41人に1人の割合で発症しており、適切な治療法はまだ見つかっていない。これまで主にADに関連して研究されてきたAPPを含むシナプス前のタンパク質の変化がASDでも起こることが観察され、示唆された。APPは、シナプス形成、神経突起の伸長、脳の発達、記憶形成、神経細胞の可塑性に非常に重要な役割を果たしていることが知られている。残念なことに、ASDの脳ではAPPの発現と分解が大きく影響されている。Amadeiら[122]は、ASD患者の脳にもβアミロイド斑が見られることを報告している。さらに、自閉症児の血液中には、可溶性sAPPαの上昇が早くから検出されていた。PPARのアゴニストであるピオグリタゾン、GFT1803(Pan-PPARアゴニスト)は、アミロイドβの沈着と認知障害から脳を保護することが観察された[52]。
2つの神経発達障害であるASDとフラジールX症候群(FXS)は、APP代謝の変化とAβプラーク形成に関連しており、学習と記憶機能に影響を与える可能性がある。sAPP、Aβ1-42,タウタンパク質、ApoE対立遺伝子は、ADだけでなくASDやその他の神経発達障害の有望なバイオマーカーとして考慮されるべきであることが示唆されている。PPARs/PGC-1αおよびSIRT-1が、Aβ生成の鍵となる酵素であるBACE-1の転写の制御に関与していることを示すデータがいくつかある。Wangら[57]のデータでは、BACE-1のプロモーターには複数のPPAR-RXRサイトが存在することが示されている。これまでの結果から、BACE-1の発現は、ADやASDではいくつかの転写因子によって大きく制御されており、炎症ではNF-кBやPPARによって制御されていることが示されている[123, 124]。
神経変性疾患の病態はまだ解明されておらず、治療も成功していない。PPARsアゴニストを含むいくつかの薬理学的化合物は、ASD、AD、PD、ASL患者の治療に適用されたが、PPARsアゴニストの多くの副作用が観察され、最近報告された[4]。
今日では、表11および表22に示すように、天然および合成化合物を含むPPAR-αアゴニストの可能性を探る新しい研究の方向性がある。さらに、いくつかの研究は、炎症や神経変性疾患の治療のための有望なデュアルおよびパンPARSアゴニストを見つけることに向けられている。パンPPARアゴニストであるベザフィブラートは、メタボリックシンドロームや糖尿病・高脂血症の患者の治療、抗コレステロール療法、心筋障害の予防に25年以上にわたって使用され、成功を収めている[125]。ベザフィブラートは、PDとHDのミトコンドリア機能と行動に良い影響を与えている[64, 126]。3種類の受容体を介して作用することで、PPAR-γが心血管系、代謝率、体重に及ぼす悪影響を排除できる可能性がある。残念ながら、神経変性疾患におけるPPARのアゴニストを用いた臨床試験の多くは、その効果がないことを示唆する研究があるため、議論の的となっている。最近のデータでは、PPARsアゴニストのポジティブな効果が期待できるものもある[127-135]。全体的には、遺伝子に関連した個別化された治療や、特定の薬理学的化合物の個別化された組み合わせが、より効率的であると思われる。PGC-1αとPPARsを標的とした薬理学的活性化合物のさらなる研究が必要である。
表1 PPAR-αのアゴニスト-天然および合成
(Adedapoら[127]、Singhら[128]、Rigano et al 2017[129]、およびContreasら[130]、Fournierら[2]によって発表されたデータによる。
| PPAR-αアゴニスト | |||
|---|---|---|---|
| Natural | 合成 | ||
| 内因性 | 外因性 | ||
| オレイルエタノールアミン(OEA) | モノテルペン | リナロール | 2-(4-クロロフェノキシ)-2-メチル-プロパン酸、クロフィブラートエチルエステル |
| パルミトイルエタノールアミド(PEA) |
セスキ テルペン |
トランスカリオフィレン | 2- [4- [2-[(4-クロロベンゾイル)アミノ]エチル]フェノキシ] -2-メチルプロパン酸ベザフィブラート |
| 8-ヒドロキシエイコサテトラエン酸(8(s)HETE) | ファルネソール | プロパン-2-イル2- [4-(4-クロロベンゾイル)フェノキシ] -2-メチルプロパノエートフェノフィブラート | |
| 8-ヒドロキシエイコサペンタエン酸(8(s)HEPE) | ジテルペン | フィトール | (5-(2,5-ジメチルフェノキシ)-2,2-ジメチルペンタン酸)ゲムフィブロジル |
|
アラキドン酸(ARA) (C20:4) |
トリテルペン | オレアノール酸 | |
| アントラコン | Norathyriol | (2-(4-(2-(1-シクロヘキサンブチル)-3-シクロヘキシルウレイド)エチル)フェニルチオ)-2-メチルプロピオン酸)GW 7647 | |
| ロイコトリエンB4 | プネニロプロパノイド | ロスマリン酸 | (2-[[4- [2-[[[(2,4-ジフルオロフェニル)アミノ]カルボニル]ヘプチルアミノ]エチル]フェニル]チオ] -2-メチル-プロパン酸)GW 9578 |
|
エイコサペンタエン酸(EPA) (C20:5) |
ベルバスコサイド | 2-メチル-2- [4- [3- [1-[(4-メチルフェニル)メチル] -5-オキソ-2H -1,2,4-トリアゾール-3-イル]プロピル]フェノキシ]プロパン酸LY518674 | |
|
リノール酸(LA) (C18:2) |
クマリン | クマリン | 2,2-ジクロロ-12-(4-クロロフェニル)ドデカン酸K 111 |
|
パルミチン酸(PA) (C16:0) |
リグナン | セサミン | (S)-3- [3-(1-カルボキシ-1-メチル-エトキシ)-フェニル]-ピペリジン-1-カルボン酸CP 900691 |
|
ステアリン酸(SA) (C18:0) |
ポリフェノール | プテロスチルベン | 4-クロロ-6-(2,3-キシリジノ)-2-ピリミジニルチオ酢酸、ピリニキシン酸WY 14643 |
| フラボノイド | ヒスピズリン | ||
| ワゴニン | 2-メチル-c-5- [4- [5-メチル-2-(4-メチルフェニル)-4-オキサゾリル]ブチル] -1,3-ジオキサン-r-2-カルボン酸(NS 220) | ||
| エピガロカテキン | |||
| イソフラボノイド | ゲニステイン | ||
| ダイゼイン | |||
| ビオカニンA | |||
| ホルモノネチン | |||
| テクトリジン | |||
| ビオフラボノイド | ビロベチン | ||
| アルカロイド | ピクラシジンC | ||
| ベルベリン | |||
| オキシマトリン | |||
| カプサイシン | |||
表2 神経変性・精神疾患の臨床試験および実験モデルにおける合成PPAR-αアゴニストの効果
| PPAR-αアゴニストの臨床試験 | ||||
|---|---|---|---|---|
| 薬名 | 疾患 | 主な効果 | 臨床試験 | 参考文献/臨床試験識別子 |
| ゲムフィブロジル | アルツハイマー病 | BACE1発現のダウンレギュレーション | IIフェーズ保留中 | NCT02045056 |
| フェノフィブラート | 薬剤耐性夜間前頭葉てんかん |
発作頻度の減少 運動行動発作への影響 |
IIフェーズ | [ 131 ] |
| Gemfibrozil | アルコール依存症 | 飲酒日あたりの飲み物と禁酒日数に影響します | IIフェーズが完了しました | NCT02158273 |
| フェノフィブラート | 週あたりの飲み物と飲み物への渇望を減らします | IIフェーズが終了しました | NCT03539432 | |
| ベザフィブラート | 双極性障害 | Montgomery-Åsbergうつ病評価尺度の前向きな変化 | フェーズ進行中 | NCT02481245 |
| PPAR-αアゴニストの実験モデルでの研究 | ||||
|---|---|---|---|---|
| 薬名 | 疾患 | 主な効果 | 参考文献 | |
| ゲムフィブロジル | アルツハイマー病 |
ADAM10を刺激します Aβ産生を減少させます |
[ 48 ] | |
|
Aβプラークの蓄積を減少させます 学習記憶を改善します |
[ 55 ] | |||
| Aβの蓄積を減らし、記憶障害と不安症状を逆転させます | [ 132 ] | |||
| WY-14643 |
タウタンパク質と炎症マーカーを減少させます モリス水テストの能力を向上させます |
[ 37 ] | ||
| GW7647 |
APPアミロイド形成プロセシングを調節します sAPPβの発現とBACE1の能力を低下させます Aβ放出とAβ産生を減少させます |
[ 50 ] | ||
| フェノフィブラート | 酸化ストレスの蓄積を調節します | [ 133 ] | ||
| フェノフィブラート | パーキンソン病 |
ラットモデルにおけるMPTPの損傷効果から保護します 炎症を軽減します |
[ 94、134 ] | |
| 脳損傷 |
アポリポプロテインE欠損動物の脳卒中の可能性を減らします 脳梗塞の量を減らす |
[ 135 ] | ||
総括
この総説では、神経変性疾患や精神疾患の際の脳におけるPPAR-αの役割と、PPAR-γ/PGC-1αとの相互作用に焦点を当てた。PPAR-αは、グルタミン酸のホメオスタシスの調節に関与しており、また、コリン作動性およびドーパミン作動性のシグナル伝達にも関与している。また、PPAR-α受容体は、脳内の神経伝達プロセスに関与するタンパク質をコードする遺伝子の転写に重要な役割を果たしている。しかし、PPAR-αがグルタミン酸系やその他のシグナル伝達経路においてどのような役割を果たしているのか、生理的な状態やADなどの神経変性・神経発達疾患においてどのような役割を果たしているのかを理解するには、さらなる研究が必要である。
最近の研究では、PPAR-αがアミロイド生成経路を介してAPPの代謝に関与していることが明らかになった。PPAR-αはαセクレターゼを活性化し、Aβペプチドを遊離させる主要な酵素であるBACE-1を阻害する。しかし、PPAR-αがAβペプチドの輸送に果たす役割や、ADにおけるAβペプチドの分解のメカニズムについては何も知られていない。
ADでは、PPAR-γとは逆のPPAR-αがPGC-1αと同様に制御されているが、ADにおけるPPAR-αシグナルの変化のメカニズムはまだ完全には解明されていない。
PPAR-αは、ミトコンドリアにおいて、脂肪酸やエネルギー代謝の調節に関与している。しかし、PPAR-αがミトコンドリアの生合成、電子輸送複合体の動態や機能を制御することの意義や、PPAR-αのアゴニストとしてII型糖尿病や肥満などの代謝性疾患の治療に長年用いられてきたフェノフィブラートの効果については、これまで十分に解析されなかった。現在、PPAR-αは、ADやその他の神経変性疾患、神経発達障害の新しい治療戦略のための有望なターゲットであると考えられている。しかし、PPAR-αが脳内でどのように作用するのか、そのメカニズムを詳細に解明する必要がある。