
The no-observed-adverse-eVect-level in drug safety
安全性評価における 使用、問題、および定義
Michael A. Dorato a,¤, JeVery A. Engelhardt b
2005年6月23日オンライン公開
概要
no-observed-adverse-eVect-level(NOAEL)は,非臨床リスク評価の重要な部分を占めている。NOAELは、試験デザイン、医薬品の適応症、予想される薬理作用、および標的効果のスペクトルに基づいた専門家の意見である。NOAELについては、一貫した標準的な定義はない。これは、何をもって有害事象とするかの定義が多様であることが一因である。毒性学者は、調査またはレビューのいずれにおいても、ある影響を有害または許容のいずれかとして定義することに一貫性がない。NOAELの一般的な定義は、「有害な影響を及ぼさない最高の実験点」であり、一般的な議論には適している。しかし,NOAELは毒性学的に関連した影響に基づくリスクの解釈や,期間や用量に応じた影響の進行を考慮していない.本論文では、毒性学的評価におけるNOAELの機能的な定義の問題点と応用について議論する。
キーワード
有害性;毒性的に重要;生物学的に重要;毒性的に関連性がある;安全マージン;ホルミシス;基準用量;無観察逆効果レベル
1. はじめに
ヒトでの有害事象を予測することができる動物での影響を特定することは、ヒトの治療目的で使用される医薬品の非臨床安全性試験の基礎である。動物における有害事象の同定は、一般的に発生率と重篤度に関連した用量反応パターンに従うことが予想され、実質的、重要または関連性のある事象が発生する用量レベルと、これらの事象が発生しない用量レベルを決定することができる。トキシコロジストの責任は、用量反応曲線上のどこに人間に有害となりうる影響が生じるかを、妥協することなく明らかにすることである。これは、複雑で反復的なプロセスのための簡単な説明である。有害・無害な影響がどこで発生するかについての最初の意見は、その分子に関連する追加情報が得られれば変わる可能性がある。長期的な研究から得られたデータは、短期的な研究では微々たるものと考えられていた観察結果が、有害な影響の初期指標であるかどうかについて、毒性学者に新たな、あるいは追加の視点を与えることがある。例えば、短期の非臨床試験で肝酵素の上昇が見られても、病理組織学的な相関関係がなければ、注意はされても有害とは考えられない。しかし、より長期の研究結果は、影響の進行や肝臓の組織学的変化を示し、反応の重要性や関連性、望ましくない反応の最初の兆候に関連する曝露期間について新たな解釈を提供する。
また、新薬候補の非臨床試験における有害事象は、種によって、あるいは同じ種でも試験によって用量反応が異なることが示されている。これは、通常の生物学的変動を表しているのかもしれないし、試験デザインの問題かもしれない。新規化学物質の安全性評価は、それが工業薬品であれ、新薬候補であれ、単純なものではない。客観性、細部への注意、実験モデルの知識、歴史的な視点、政府の規制要件に関する知識、データ解釈の経験などが必要となる。
新薬の安全性評価は、工業薬品の安全性評価とはアプローチが異なる。工業用化学物質の場合は、曝露期間や曝露量、意図などの定量的な詳細がよくわかっていないことがある。加えて、リスク/ベネフィットの安全性評価に役立つ治療上の適応症がない。新薬の可能性がある場合、最初の臨床プロトコルが承認された後、ヒトへの曝露がコントロールされ、慎重に測定される。非臨床試験および臨床試験では、望ましくない、あるいは有害な影響を検出することを目的とした一連の評価が行われる。新薬候補は、生体内で活性化するように設計されており、望ましい効果と望ましくない効果のスペクトルを持っている。したがって、リスク/ベネフィット分析では、その薬剤が対象とする疾患、治療を行わない場合の疾患の自然史、他の治療法の有用性などを考慮しなければならない。潜在的な新薬の場合、非臨床試験で見られたすべての効果が望ましくない、または有害であると考えることはできない。例えば、重度の肥満を治療するための薬剤は、非肥満の動物を用いた非臨床試験で有意な体重減少を示すことが期待される。
文献や規制ガイドラインから、多くの毒性学者が望ましくない反応の有無を示すために使用している用語、例えば、no-observed-eVect-level(NOEL)no-observed-adverse-eVect-level(NOAEL)adverseまたはnon-adverse、生物学的にsigniW-cant、毒性学的にsigniWcant/relevantなどの用語について、一貫した定義はないと結論付けられる。したがって、NOAELの性質や定義と同様に、医薬品の毒性学的プロールの性質も評価される。最後に、新薬候補の非臨床安全性評価に有用なNOAELの定義を示す。
2. 医薬品開発のための毒性学的プロール
毒性学は、リスクを特定し、評価し、管理するためのプロセスである。毒性学者は、対照群と治療群の間に実際の差があるかどうかを判断し、その差が実験的治療の影響であるかどうかを判断し、治療の影響が有害であるかどうかを判断しなければならない。これは、非常に複雑なプロセスのためのシンプルな表現である。以下は、毒性学のプロールに求められる基本的な質問
- どのような用量・曝露が動物に有害な影響を与えるのか、与えないのか?
- その動物モデルは、ヒトの毒性評価に適しているか?
- 毒性反応の兆候や変化はどのようなものであったか?
- 単回投与と複数回投与で効果は変わったか?
- 曝露期間に応じて症状は進行したか?
- 対象となった臓器・器官は何か?
- 毒性の影響は可逆的であったか?
- その物質は体内でどのように代謝・消去されたか?
- 毒性代謝物は生成されたか?
- 毒性作用への適応は見られたか?
- 毒性は実験モデル間で一貫していたか。
(Lewis et al 2002)は、治療関連および非治療関連の影響を判断するのに役立つ多くの要素を提示し、有害という用語の使用を明確にしている。
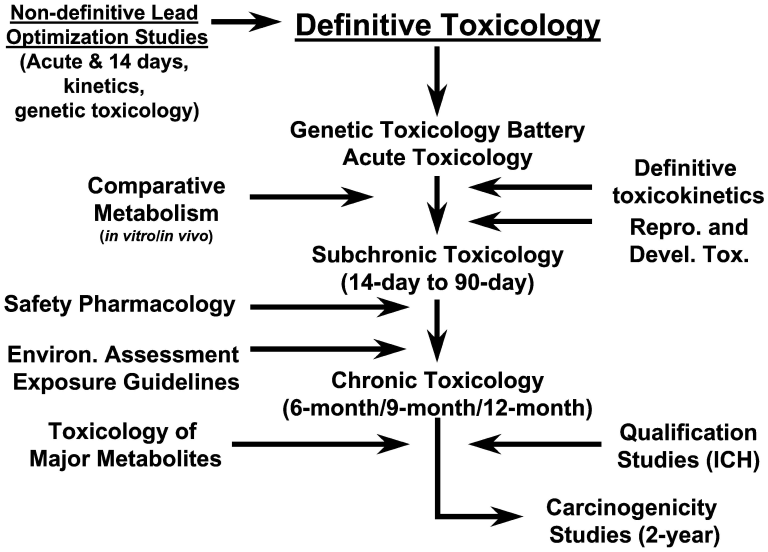
– 典型的な毒性学のプロール(図1)には、治験薬を安全に使用するために、毒性作用、用量依存性、曝露、標的臓器、バイオマーカーを特徴づける十分な期間と用量の研究を行うことが含まれる。標的臓器と関連するバイオマーカーを特定することは極めて重要である。「バイオマーカー」は現在、医薬品の安全性評価のための「バズワード」となっているが、毒性学研究では当初から、臨床的に影響を及ぼす可能性のあるバイオマーカーを常に取り上げ、特定していた。バイオマーカーの重要性は、パラケルススの「万物には毒があり、毒のないものはない:用量によって毒でなくなる」という言葉に象徴されている。善から悪を作ることができるなら、悪から善を作ることも可能である」(テムキン、1941)。他の科学分野と同様に、技術は進歩しており、非臨床試験で以前はモニターできなかった潜在的なバイオマーカーを決定する新しい方法が利用可能である。
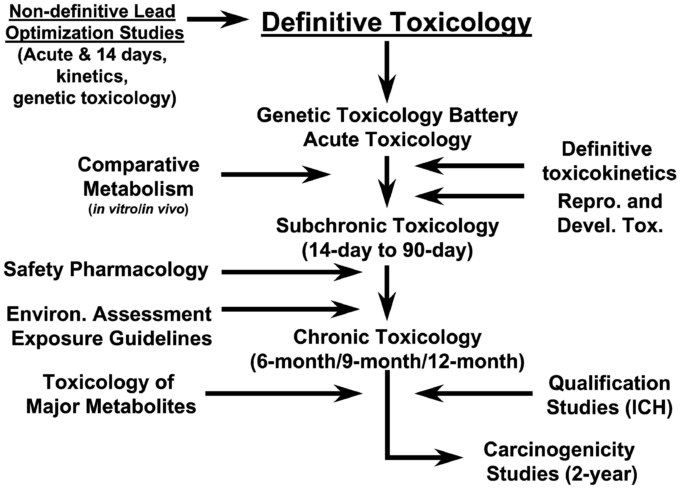
毒性学研究における様々な研究の段階的な進め方は様々である(図2)。毒性学試験と医薬品開発の各段階との関係は、ある程度、ケースバイケースである。各企業は、患者数、妊娠可能な女性の参加、規制当局とのやり取りなどを考慮して、異なる時期に試験を開始することができる。
図1. 典型的な毒物学のプロール
ICH(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceu-ticals for Human Use)では、欧州、日本、米国の新薬登録要件の調和を図っている。新薬候補化合物の非臨床安全性要件については、ICHのウェブサイトを参照されたい(www.ich.org)。特に、ICHトピックM3(M)(2000)及びICHトピックS6(1997)には、それぞれ低分子及びバイオ医薬品の試験デザイン及び試験期間に関するガイダンスが記載されている。一般的に、2週間の反復投与毒性試験は、世界のほとんどの国で初期臨床用量を裏付けるために必要とされる最小の試験期間である。米国食品医薬品局(FDA、1996)は、単回投与の臨床試験をサポートするための単回投与毒性学戦略を提案している。ヒト用医薬品委員会(CHMP 2004)も、低用量の単回投与をサポートするために必要な毒性試験の期間についてポジションペーパーを発表している。ICHガイドラインに対するこれらの地域的な差異の主な利点は、単回投与の臨床試験に先立って非臨床毒性試験を実施するために必要な薬剤の量が減ることである。
新薬の非臨床安全性試験に関する国際的な規制要件および既存の地域的差異については、Dorato and Buck-ley (1998)およびDorato and Vodicnik (2001)に記載されている。一般に、バイオテクノロジー製品の安全性試験に関する国際ガイドライン(ICH Topic S6, 1997)では、低分子化合物の開発を毒性学的にサポートするために必要とされるよりも、審査を行う規制当局とより多くの議論が行われている。
非臨床安全性評価の原則は、低分子でも大分子でも同じである。違いは、これらの原則をどのように実践するかにある。バイオテクノロジー製品では、適切な動物モデルを選択する際に、免疫反応や解放可能な生物学的受容体の存在と同様に、種の特定が大きな関心事となっている。新しいバイオテクノロジー製品は、2つの非臨床動物モデルではなく、1つの非臨床動物モデルで開発されることが多い。低分子化合物、バイオテクノロジー化合物のいずれを評価するにしても、毒性学的プロ グラムは継続的に行われ、通常、曝露期間が長くなるにつれて投与量は減少し、リスク/ベネフィットの 判断は反復的に行われる。低分子化合物と同様に、ICHトピックS6(1997)では、”毒性量と無毒性量(NOAEL)を含む用量反応関係に関する情報を提供するために用量レベルを選択すべきである “とされている。
図2.医薬品開発の各段階における毒物製剤の高度な関係
ヒトの毒性に対する動物モデルの予測可能性については、国際生命科学研究所(ILSI)が1999年に開催したワークショップで検討されている。Olson et al 2000)はそのワークショップの成果を発表した。このワークショップでは、非臨床の動物実験で見られた毒性が、実際のヒトの毒性をどの程度予測できるか、また、ヒトの臨床試験で潜在的な毒性を明らかにするために必要な動物での投与期間を検討した。2つの動物種で実施された毒性試験は、ヒトで見られる毒性を約71%予測した。げっ歯類以外の動物種で行われた毒性試験では、ヒトの毒性を63%予測し、げっ歯類で行われた毒性試験では43%予測した。また、期間61ヶ月の研究では、潜在的なヒトへの毒性の94%が予測された。今回の調査は、非臨床安全性データの使用とその適切性について貴重な見解を提供している。
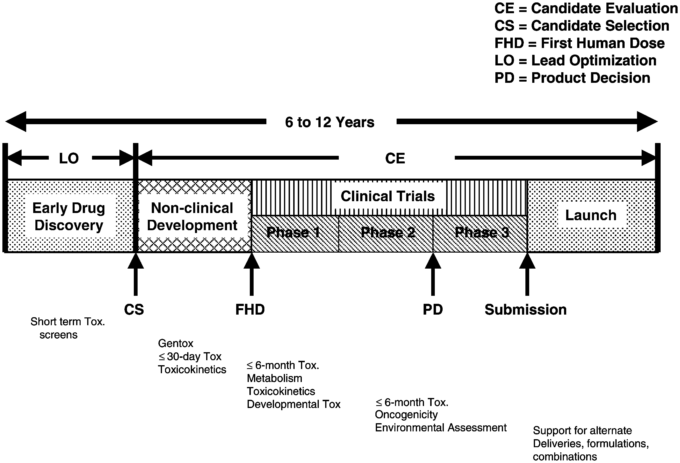
非臨床毒性試験の目的は、有効性と無効性のレベル、および最大許容量(MTD)の評価を行うことであるが、非臨床毒性試験の結果を最終的に応用するのは、安全性のマージン(MOS)の決定である。MOSの概念は、毒性学のプロールの重要な部分である。したがって、毒性試験で毒性を判定することは期待されることであり、望ましいことである。この情報を合理的に利用するには、絶対的な毒性の判定ではなく、臨床的な用量/暴露、臨床的な適応、利用可能な治療法、および潜在的な新薬の使用に関する総合的なリスク/ベネフィット分析との関連で行う。MOSと有効量およびNOAELの関係を図3に示する。NOAELは、MOSを設定するために、薬理学的に有効な用量と比較される毒性学的な用量反応曲線上の用量である。有効量は、多くの場合、臨床的に有効な用量であるが、創薬の初期段階では、有効量は、関連するeYcacyモデルで有効な用量として定義されることがある。
図.3. 安全性のマージンと毒性プロールの関係
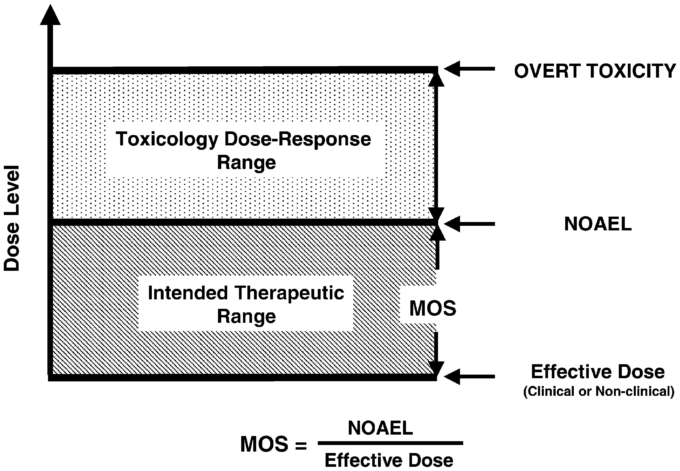
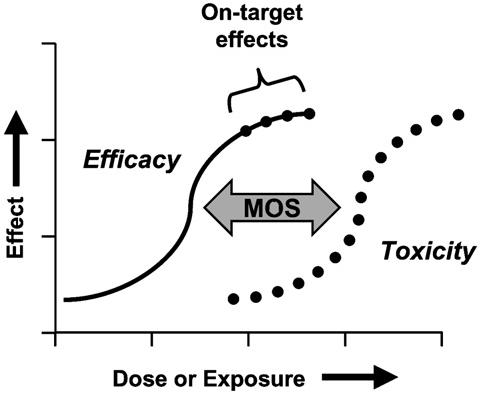
図4. MOSと、望ましくないと思われる高用量の無標的効果の発生との関係を示した、有効性と毒性の理想的な用量反応曲線。
様々な薬剤を用いた毒性試験では、動物モデルに薬理作用を及ぼさない用量を設定できないことがある。例えば、選択的エストロゲン受容体モジュレーター(SERM)と呼ばれる薬物群の化合物は、一般的に非常に低い暴露レベルで薬理作用を発現する。閉経後の女性を適応症として開発されたこのクラスの薬剤については、正常な成熟動物で見られるエストロゲン作用または抗エストロゲン作用が指摘されているが、閉経後の臨床集団にとっては必ずしも有害とは考えられていない。これは、潜在的な毒性を総合的に解釈する上で重要な考慮事項である。
第1回ICH会議(1991)の議事録では、毒性試験における観察結果の記述を助けるためにNOELとNOAELを使用することの意義について述べられている。”さらに、決定されるべき影響は毒性学的に相対的な影響、すなわち人の健康を危険にさらす可能性のある影響であることを念頭に置くべきである。したがって、NOELを特定の薬力学的「クラス」の影響の兆候と区別するために、「no-observed-adverse-eVect-level」という用語を使うことが好ましい」(Hess, 1991)。医薬品のリスク/ベネフィットの立場を構築する際には、観察された薬力学的な影響、すなわちインスリンおよびその類縁体によって生じる低血糖症を有害とみなすべきかどうか、またどのような患者集団に対して有害とみなすべきかを理解し、議論することが重要である。また、その影響をモニターし、回復させる能力を評価することも重要である。効能・効果と毒性の理想的な用量反応関係を評価することで、NOAELの性質をさらに理解することができる(図4)。1,50,99%など、任意のレベルでの曲線間の距離が、MOSのパラメータとして選択されている。パラケルススの言葉を借りれば(Temkin, 1941)薬理学と毒性学が本質的に「善」と「悪」であるかどうかという哲学的な問題は、ここでは議論しない。しかし、「高用量」と呼ばれる薬理学の発生は、非臨床毒性試験によるNOAELの評価を複雑にしている。実験化合物の低用量は通常望ましい効果をもたらし、高用量は通常望ましくない効果をもたらすことが予想される。このことは、図4に示すように、eYcacy曲線とtoxicity曲線に表されている。eYcacy曲線の上の部分は、望ましくないと考えられる過剰な薬力学的反応(on-target)を引き起こす可能性がある。これらの反応を先験的にNOAELの決定に用いるべきではない。むしろ、これらの影響が毒性学的に重要であるかどうかは、毒性学者、医師、規制当局の審査官の間で議論されるべきである。これらの議論は、リスクの認識、評価、および管理のための適切な視点を提供するものである。また、動物モデルと患者との関連性を理解することも、議論には重要である。例えば、非糖尿病の動物モデルにおける血糖降下剤の研究では、動物モデルが優血性であるために、有害な影響が生じる可能性がある。妊娠中の糖尿病女性に薬力学的に有効な量のインスリンを投与すると、先天性障害が減少することが示されているが、通常の健康な動物では逆の結果となる可能性がある(CPMP, 2002)。正常な健康動物にインスリンまたはインスリン類似物質を投与すると、ヒトのリスクには関係のない催奇形性が誘発される可能性がある。毒性評価における用量は慎重に選択し、被験物質の催奇形性の影響と、予告された低血糖症による影響とを区別する必要がある(CPMP, 2002)。毒性評価における疾患モデル動物の適切な使用については議論の余地があり、ここでの議論の範囲を超えている。非臨床毒性試験のデータは、対象となる人々の安全性への影響を考慮して解釈されるべきである。
病理組織学は、歴史的にNOEL/NOAELを設定するための最も一貫した基準である。影響のあるレベルと影響のないレベルを確実に決定するためには、病理学的評価の一貫性が重要である。そのため、すべての反復投与毒性試験において、病理学的ピアレビューを行うべきである。病理学的ピアレビューとは、独立した病理学者が一部の動物や組織について二次評価を行う品質管理プロセスである。このピアレビューでは、化合物に関連する全ての病変が試験で特定されていること、病変が正確に記録されていること、用語や重症度の等級が一次病理医によって一貫して使用されていることを確認する。さらに、ピアレビューの病理医は、一次病理医が書いた解釈の正しさを確認する。これらのプロセスを活用することで、有害事象の一貫した特定と医薬品のNOAELの決定が容易になる。
前述したように、非臨床毒性評価は繰り返し行われるプロセスである。このため、初期の短期試験における微妙な病理判定の解釈には多少の困難が伴う。間違いなく、非臨床毒性評価の目的は、臨床試験に移行する前に、妥協することなく分子の安全性を評価することである。その一方で、MOSの判定に必要な視点を持たない毒性データも同様に重要な問題である。毒性学者、臨床医、規制当局の間で行われるリスク/ベネフィットの議論の一環として、MOSの全体像を明らかにしながら、新薬の開発を段階的に承認することがある。すべてのWndingsを記録し、報告することは重要だが、潜在的に有用または重要な新薬の開発を中止する決定は、同等の注意と検討が必要である。
3. 観察不能な逆効果レベルの定義
リスク評価に用いられる一般的な用語の定義については合意が得られておらず、NOAELなどの用語の適用には一貫性がないことが一般的に認められている。表 1 は公表されている文献から抜粋した NOAEL の定義を示している。(Lewis et al 2002)および (Filipsson et al 2003)は、NOAEL は測定値/推定値であり、測定された NOAEL と測定された最低観察逆効果レベル(LOAEL)の間に位置する真の無害影響レベルとは異なる可能性があると指摘している。
Filipsson et al 2003)は、従来のNOAEL法に代わる方法を提示している。ベンチマーク用量法(BMD)は、対照と比較して有害反応の特定の変化に対応する用量に基づいている。この用量反応モデリング手法は、医薬品の評価には広く採用されていない。この方法は、従来のNOAEL評価の限界を解決するものであると主張しているが、従来のアプローチを使用すべき場合も認められている。Beck ら(1993)は、NOAEL と BMD の長所と短所の比較を行っている。
Calabrese ら(1999)および Calabrese and Baldwin(2003a)は、NOAEL の評価におけるホルミシスの議論を紹介している。ホルミシス反応では、毒性の閾値以下では生物学的作用が刺激され、閾値以上では抑制されることになる。低用量群は対照群よりも改善される可能性がある。これは、適応的または誇張された恒常性維持反応とその生物学的限界に関連していると考えられる。また、この反応は、ホルモンやホルモン様薬剤で見られるような、NOAEL以下の用量範囲で生じる、予想される薬力学的効果の範囲である可能性もある。NOAEL以下の用量で非有害事象が発生する可能性があることを認識することは、それらが被験物質に起因しないことを意味するのではなく(例えば、「正常な変動」)真に被験物質の用量反応の一部である。規制上の要求・期待により、新薬候補の毒性試験のほとんどは、ホルミシスが起こる可能性のある低用量よりもはるかに高い用量で実施されている。ホルミシスに関するデータの利用は,リスク評価におけるその位置づけがより明確になるまでは,NOAELを設定する従来の方法に取って代わることはないと思われる(Rodericks, 2003)。さらに、低用量の刺激領域では閾値とホルモン反応予測の間に重複が認められるため、ホルモンの用量反応曲線を予測する能力は、NOAELを設定するために使用した定義によって影響を受けることになる(Calabrese and Baldwin, 2003a)。実際、我々が定義で述べているように、統計的な有意差がないだけでは NOAEL とはならない。むしろ、NOAELは生物学的効果と統計学的効果の複合的な分析によって決定される。
一般的に、NOAELはMOSの重要な部分であり、毒性学的に関連した影響と予想されるpharma-cologyの解釈に関する専門家の意見であり、毒性学のプロールが発展するにつれ、繰り返し解釈される対象となる。ここでは安全係数や不確実性係数の適用については触れないが、NOAELはしばしば安全係数と組み合わされ、種を超えた外挿の変動性の問題に対処される(Bar-nes and Dourson,1988; Beck er al)。1993; Calabrese er al)。1992; Dourson and Stara,1983; Lehman and Fitzhugh,1954)。前述したように、毒性学プロールの開発初期には、病理組織学的な関連性がなく、安全性の問題とは解釈されない肝酵素などのパラメータの変化が見られることがある。後に、より長期間の研究において、これらの同じ影響が重度の肝障害の前兆として見られることがある。そこで、初期のバイオマーカー反応を有害と判断すべきかどうかが議論される。進行性の毒性を予測する反応としては、有害と考えるべきである。しかし、患者集団の適応、モニタリング可能性、可逆性、リスク/ベネフィット分析は、この観察結果が薬剤の継続的な開発にどのように影響するかに影響を与える。
NOAEL の性質は、おそらくその批判によって最もよく理解されるであろう(Calabrese et al 1999; Cala-brese and Baldwin, 1994, 2003a; Filipsson et al 2003; IPCS, 1999; Leisenring and Ryan, 1992; Woutersen et al 2001)。NOAEL を設定する従来の方法に関する懸念をまとめたリストを Table 2 に示す。NOAEL 推定値を改善する方法として、試験デザインの改善に関する提案が長年にわたってなされてきた。これらの提案には一般的に、より多くの用量群を追加することや、 微妙な変化の検出を向上させるために低用量群に多くの動物を追加することなどが含まれる。これらの提案は検討に値するものであるが、測定されたNOAELと真のNOAELの差異の可能性に関する懸念が、本当に臨床安全性に影響を与えるのかどうかを問わなければならない。動物モデルを用いた非臨床毒性試験から得られた情報に基づき、慎重に臨床用量を漸増させ、関連するバイオマーカーを評価することは、新薬候補を安全に試験するための有効なアプローチであることが長い間示されてきた。
表2 従来のNOAEL決定法の問題点
- 試験する実験用量の一つでなければならない
- 試験デザインに左右される
- 用量反応曲線の傾きを考慮していない観測値
- 推定値/派生値であり、推定値の精度は用量の広がりに依存する。
- 投与量の選択がNOAEL推定値の精度を左右する
- 値は実験系の固有の生物学的特性ではなく、実験デザインによって決まる。
- サンプルサイズに影響される
- NOAELは動物数/投与量が少ないほど高くなる傾向がある
- 実験ごとに異なる
- 有害事象の定義はレビュアーによって異なる
- リスクレベルはNOAELによる推定値よりも高い可能性がある
一貫したNOAELの定義がないという観察結果は、有害事象の定義が主観的な性質を持っていることで説明できる。非常に大まかに言えば、有害事象とは、非臨床毒性モデルの性能を損ない、一般的に成長、発育、寿命に有害な影響を及ぼす可能性のある変化(生化学的、機能的、構造的)と考えられる。より具体的には、非臨床毒性試験における有害事象は、ヒトの臨床試験で発生した場合に受け入れられないような事象でなければならない(FDA Guidance, 2002)。しかし、トキシコロジストは、非臨床毒性試験で観察された有害事象が有害か許容できるかを判断する際に、一貫した判断を下していない。実際には、非臨床毒性試験における観察結果の有害性に関する判断は、議論、異議申し立て、再解釈の対象となる。ある観察結果が有害であるという判断は、厳密な科学的解釈と同様に、公共政策によっても左右されることがある。Lewis et al 2002)は、非臨床毒性試験における反応が有害であるか非有害であるかを判断するための一連の手順を示している。このアプローチには、反応の適応性、一過性/持続性、進行性、他の影響との関連性、機能障害の発生、反応の一次性/二次性の評価が含まれる。このアプローチは「工業用」化学物質の扱いに重点が置かれているが、医薬品の安全性評価にも有効である。非臨床毒性試験で見られるあらゆる影響は、それが広く薬理学(オンターゲット)または毒性学(OVターゲット)として定義されているかどうかにかかわらず、望ましくない、つまり有害であると考えられる。新薬候補に関連するこれらのWndingsを解釈する際には、動物モデルの性質(正常と疾患)対象となる患者集団、および治療適応を慎重に評価する必要がある。
新薬候補の非臨床毒性試験の結果が有害であるか非有害であるかを判断するためのプロセスを簡単に図5に示した。最初の2つの質問は、非常に単純な形で示されているが、非常にわかりやすいものとなっている。大きな「グレーゾーン」は、統計的および生物学的な有意性の重要性に関するものである。毒性学者の専門的な意見が、観察された事象を有害か非有害かを決定する鍵となる。先に述べたように、適応症、臨床的見解、規制当局の見解、動物モデルの関連性などが考慮される。多くの場合、対照群と比較して統計学的な有意差を示す用量関連性のある反応は、有害事象として取り扱われる。しかし、新薬候補の評価では、治療目標も考慮しなければならない。活性医薬品の非臨床毒性試験では、治療上の適応に不可欠な薬理学的効果が期待される。これらは、臨床病理、行動、食物消費、体重増加などのモニタリングされたパラメータの、対照と比較した変化として示されることがある。期待される薬理作用に関連する薬力学的な影響は、有害なoV-targetの影響とは区別されなければならない(Williams, 1990)。Blackら(1999)は、非臨床試験のNOAELに対してMOSを行わないという効果的な規制決定戦略の例を示している。
異なる地域の毒性学者は、何が有害事象を構成するかについて異なる解釈を持っている。対照群からの非有害事象の統計的な差異の影響は、毒性の評価の差異につながり、医薬品の非臨床毒性試験においてNOELではなくNOAELを使用することを支持するものであり、第一次ICHで取り上げられた(Hayashi, 1991)。期待される薬理作用のうち、健常者では有害と考えられる非常に単純な例として、肥満治療薬の試験で非肥満動物に見られる重度の体重減少や、糖尿病治療薬の試験で非糖尿病動物モデルに見られる重度の低血糖などがある。これらはいずれもオンターゲットの薬理作用に関連するものである。これらは、臨床研究者にとっての問題点/警告として、使用上の見通しを立てるために留意すべきである。しかし、これらの問題は、潜在的な治療薬の臨床研究を妨げるものではなく、また、適切な対象者への使用を先入観から懸念するものでもない。統計的に有意であることは、それだけでは有害事象とはならない。この概念は、NOEL(有害事象だけでなく、あらゆる事象)とNOAEL(表1)の異なる定義に組み込まれている(FDAガイダンス 2002)。ある事象が統計的に有意でなくても、生物学的な有意性に基づいて有害とみなされる場合がある。例えば、我々の経験では、薬物による免疫介在性の細胞減少は、ほとんどの非げっ歯類の短期試験では必ずしも統計的に有意であるとは言えないが、確実に有害であると考えられている。同様に、統計学的に有意ではない臓器重量の増加も、生物学的には有意である可能性がある。用量反応性であっても、有害作用を判断するための単純な前提条件ではない。望ましくない受容体を介した影響は、Xatの用量反応を示すかもしれないが、有害と考えられるだろう。
さらに、NOAEL は個々の Wndings に対してではなく、各毒性試験全体に対して決定されるべきである。さらに、NOAELは試験の種類と期間ごとに設定され、特定の化合物に関する追加データの収集に伴って変化または成熟することが許容されるべきである。これにより、医薬品開発の過程で発見された化合物関連の影響の時間的・時間的関係を総合的にモニタリングすることができる。これにより、動物実験で得られた結果とヒトのリスク評価との関連性を確立することができる(Olson er al)。
4. 議論
潜在的な新薬の開発においては、重要な新薬の開発を進めようとする意欲と、患者の安全性に関連して保守的な判断を下す意欲との間でバランスを取る必要がある。そのためには、規制当局の毒性学者、スポンサーの毒性学者、臨床医が積極的かつオープンに議論することが重要である。この議論を進めるためには、非臨床毒性試験における有害事象の構成要素を明確に理解することが重要である。NOAELを特定することは、新薬の臨床試験を進める上での基礎となる。しかし、NOAELはリスクフリーではないことを明確に理解しなければならない。臨床的・非臨床的な反応のばらつきや感受性の高い集団の存在は、重篤な副作用の可能性を高める。我々は、潜在的な新薬の安全性評価に最も適したNOAELの機能的な定義を提案する。
慎重な生物学的・統計学的分析に基づき、曝露群と対照群の間で、副作用の頻度または重症度の、毒性学的に関連した増加を引き起こさない最高用量/曝露である。この用量では、微小な毒性の影響や薬力学的反応が観察されることがあるが、これらは人の健康を危険にさらすものではなく、また、継続的な曝露により重篤な事象の前兆となるものでもないと考えられる。
新薬の開発を継続するかどうかの判断には、前駆症状の判定と、より長期の試験による毒性プロールの追加的な洞察が非常に重要である。対照群との統計的差異は注目に値するが、それだけでは非げっ歯類およびげっ歯類の毒性試験による安全性評価には貢献しない。統計的なsig-niWcanceだけで、観察結果が有害であると判断してはならない。上記のNOAELの定義では、非臨床毒性試験の生物学的および統計学的結果を慎重に検討することが必要である。さらに、病理組織学的変化のスペクトル、標的臓器の特定、病変の可逆性の可能性は、NOAELの特定だけでなく、総合的なリスク評価にも役立つ。統計的に有意な結果のみではなく、入手可能なすべての毒性データを用いることにより、新薬候補の包括的な毒性プロールを合理的に解釈することが可能となる。この包括的なプロールは、新薬候補の潜在的な安全性に関する懸念を理解する上で、規制当局の評価者や臨床医にとって最大の助けとなるはずである。
我々は、毒性学試験における用量反応の評価において、NOAELの決定につながる伝統的なアプローチと呼ばれる方法をとってきた。別のアプローチについても検討した。ベンチマーク用量(Filipsson er al 2003)は、従来の NOAEL アプローチよりも強力な統計ツールであり、より正確なリスク評価を行うことができると主張している。Calabrese (2004, 2005) および Calabrese and Baldwin (1998, 2003b) は、数年前からホルミシスを毒性学的なリスク評価を改善するための手法として表現している。(Axelrod et al 2004)は、既存の毒性学的データでは、ホルミシスの概念を規制政策にまで一元的に拡張することはできないという議論を示している。毒性学者はホルミシスの概念をよく理解し、ホルミシスアプローチの有効性を判断する際には、専門的な判断と観察を行うべきである。過去40年間の医薬品安全性評価では、治療薬のリスク/ベネフィットを評価するための適切なツールとして、NOAELに対するより伝統的なアプローチが支持されている。しかし、毒性学者は、新しいアプローチや確立された慣行への挑戦を受け入れる必要がある。
病理学的なピアレビュープロセスの適用、有害の定義の一貫した使用、および受け入れられたNOAELの定義の実施により、製薬会社と医薬品規制当局は薬理学的なクラスの影響に関して共通の理解を得ることができる。さらに、一貫した定義が認識され、使用されるようになれば、世界の医薬品規制当局の間で、医薬品の非臨床部分に関する評価勧告の共有と受け入れが促進される可能性もある。しかし、そのためには、ここで提案されているような、ヒトの医薬品開発におけるNOAELの機能的な定義が受け入れられることが必要である。