
Contents
まとめ
コロナウイルス病2019(COVID-19)の病態は、4つの悪質なフィードバックループが連鎖しているか、または同時に起こっているダイナミックな相互作用として想定されるかもしれない。これらは、ウイルスループ、高炎症性ループ、非冠状レニン-アンジオテンシン系(RAS)軸ループ、および高凝固ループである。重症急性呼吸器症候群(SARS)-コロナウイルス(CoV)-2は、肺胞上皮細胞(肺上皮細胞)に感染し、アンジオテンシン変換酵素2(ACE2)/アンジオテンシン(Ang-1-7)/Mas1R軸をダウンレギュレーションすることにより、心筋を照らす。ウイルスのフィードバックループは、宿主の自然反応の回避、制御されていないウイルスの複製、および過活動性の適応免疫応答のオンにすることを含む。炎症性ループは、実際のサイトカインストームで爆発するまで、高揚した炎症性反応がフィードバックすることで構成されている。ACE2/Ang-(1-7)/Mas1R軸のダウンレギュレーションは、重要な防御機構なしで肺を残し、RASの炎症性側にスケールを回する。凝固ループは、無限のフィードバックループの炎症と凝固の間の相互作用によって引き起こされる高凝固状態である。その結果、炎症性と凝固性が亢進した状態が急性免疫介在性肺障害を引き起こし、最終的には成人呼吸窮迫症候群を引き起こす。
1. 序論
2019年12月、中国湖北省武漢市の華南水産物卸売市場で新たなパンデミック性疾患が出現した。上気道感染症が急速に両側性肺炎に進展し、最終的には呼吸不全に至ることが特徴であった[1]。病原体はSARS-CoV-2と名付けられた新しいコロナウイルスであり、本疾患はCOVID-19と呼ばれていた[2]。この病気は湖北の元の核から急速に拡大し、2020年3月11日までにWHOはこの病気をパンデミックと宣言した。2020年6月23日現在、COVID-19は世界188カ国に感染し、全世界で9.131.445人の確定症例、472.856人の死者を出している[3]。
パンデミックの経過の初期に、臨床家や研究者は、本格的なCOVID-19が少なくとも3つの段階で進化していることに気づいた:咳、発熱、喘鳴、疲労、頭痛、下痢、呼吸困難を伴う第1の段階で、上気道呼吸器感染症を連想させる。第2期では両側性肺炎の急速な出現、程度の異なる低酸素血症を伴う浸潤、第3期では一部の患者が死亡に至る呼吸不全を発症したOmitがみられる[4]。約80%の人がSARS-COV-2感染症を無症状または軽症~中等症で発症し、そのほとんどが上気道と導気道に限定される。残りの20%は入院を必要とする症候性感染症を発症し、5%は集中治療室(ICU)での人工呼吸のサポートを必要とする [5]。感染の臨床段階は、ウイルスが肺に侵入することから始まる病原性イベントを反映している。あらゆる感染症、特にCOVID-19の臨床症状や病原性イベントは、いくつかの要因や力が宿主や病原体側にスケールを傾けるというダメージ反応の枠組みに照らして見なければならない[6]。したがって、時には病原体は実際の加害者というよりも、単なる開始者であることがあり、組織や臓器の損傷を引き起こすのは病原体の存在によって拘束されていない宿主の力である。
ここでは、COVID-19の病原体に関する現在の知見をレビューし、SARS-CoV-2感染と宿主の反応がCOVID-19の異なるシナリオをどのように描いているかを検討する。我々は、4つの悪循環ループ、すなわち、ウイルスループ、欠損した非カノニカルRASループ、炎症ループ、凝固ループが相互に作用していると考えている(図1)。
図1
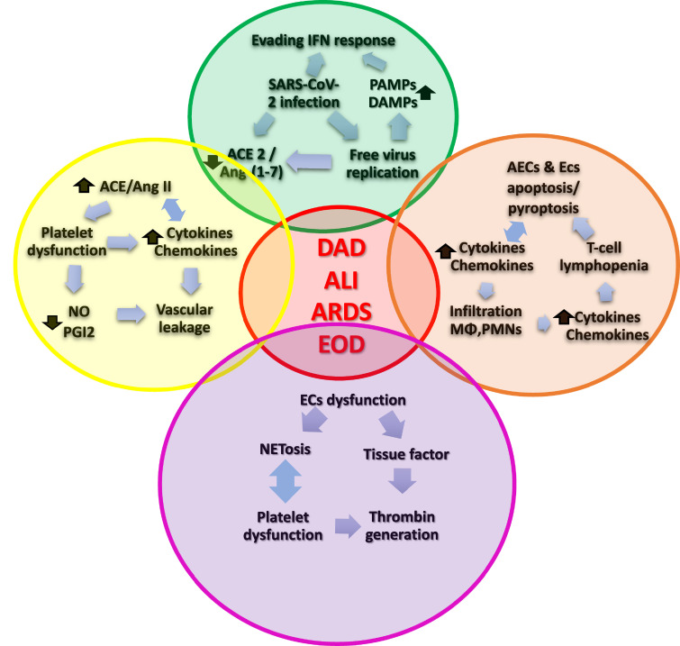
図1 COVID-19の病態生理における4つの有害なフィードバックループ。
炎症ループと凝固ループの間にある複雑な相互作用を示す。円間の交差はループ間の相互作用を表す。赤く着色された中央の円は生理学的カスケードの最終的なイベントを意味する。悪質なウイルスループは緑色で、高炎症性ループはオレンジ色で、ACE2/Ang-(1-7)ループは黄色で、高凝固ループは紫色で描かれている。
IFN=インターフェロン;PAMPs=病原体関連分子パターン;DAMPs=ダメージ関連分子パターン;SARS-CoV-2=重症急性呼吸器症候群コロナウイルス2;肺上皮細胞=肺胞上皮細胞;内皮細胞s=内皮細胞;ACE2=アンジオテンシン変換酵素2;Ang-(1-7)=アンジオテンシン1-7。ACE=アンジオテンシン変換酵素;Ang II=アンジオテンシンII;NO=一酸化窒素;PGI2=プロスタサイクリン;MΦ=マクロファージ;PMNs=多形核好中球;DAD=びまん性肺胞損傷;ALI=急性肺損傷;ARDS=成人呼吸窮迫症候群;EOD=内臓疾患。
β-コロナウイルスは、そのゲノムおよび構造の大部分を共有しているので、それらが病原性のメカニズムを共有することができ、これらの共有要素に対する宿主の応答がある程度同等であることは、非常に論理的であるように思われる。したがって、本レビューのいくつかの側面では、COVID-19で起こり得る病原性イベントを説明するために、他の人獣共通感染性コロナウイルスを参照する。
2. 最初の騎手:卑劣なウイルス
SARS-CoV-2は、これまで知られていなかったβ-コロナウイルスであり、2つのコウモリ由来のSARS様コロナウイルスの配列との同一性は88%、SARS-CoVとの同一性は79.5%、中東呼吸器症候群(MERS)-CoVとの同一性は約50%である[2]。SARS-CoV-2のゲノムは、29.9kbのサイズのポジティブセンス一本鎖RNAであり、少なくとも10個のオープンリーディングフレーム(ORF)を含む[7]。最近、非カノニカルなORFと、機能不明の少なくとも41個のRNA修飾が同定された[7]。最初のORFはウイルスRNAの3分の2を占める。これらは2つの大きなポリタンパク質に翻訳され、後に16個の非構造タンパク質(nsp1~nsp16)に加工され、ウイルス複合体の複製転写酵素を形成している[7]。これらのNSPは小胞体を二重膜小胞に再編成し、そこでウイルスの複製と転写が行われる [8,9]。ゲノムの残りの3分の1は、スパイク(S)、エンベロープ(E)、ヌクレオカプシド(N)、膜(M)の4つの主要な構造タンパク質と、現在のところ機能は不明だがウイルスの複製には関係のないいくつかの付属タンパク質をコードしている[10]。
SARS-CoV-2は、SARS-CoVと同様に、細胞内に入るためには受容体としてACE2を必要とする[11,12]。コロナウイルスSタンパク質は、エンベロープスパイク糖タンパク質を細胞内受容体ACE2に結合させることで、ウイルスの宿主細胞への侵入を決定する[13,14]。当初、SARS-CoVは膜融合によって侵入を達成したと考えられていたが、II型膜貫通型セリンプロテアーゼ(TMPRSS2)を媒介とするSARS-CoV Sタンパク質の重要なタンパク質分解的切断により、膜融合とウイルス感染がもたらされる [15]。ウイルスの侵入後、RNAゲノムは細胞質に放出され、2つのポリタンパク質と構造タンパク質に翻訳される[16]。
宿主細胞におけるSARS-CoVの生存は、免疫応答を回避する戦略によって緩和される。病原関連分子パターン(PAMP)と呼ばれる進化的に保存された微生物構造は、トール様受容体(TLR)、レチノイン酸誘導性遺伝子-I(RIG-I)様受容体、ヌクレオチド結合オリゴマー化ドメイン(NOD)様受容体、C型レクチン様受容体などのパターン認識受容体(PRR)によって認識される[17]。SARS-CoVは、PRRを欠く二重膜小胞の産生を誘導し、これらの小胞内で複製することができる[18]。さらに、SARS-CoVおよびMERS-CoVによってコードされるいくつかの構造およびnspsは、抗ウイルス自然免疫応答に拮抗する。インターフェロン(IFN)およびインターフェロン刺激遺伝子(ISGs)応答は、nsp1、nsp3マクロドメイン、nsps-deubiquitinase、およびORF3b、ORF6、およびORF9によって打ち消され、抗ウイルス応答を覆す[19]、[20]、[21]、[22]、[23]、[24]。nsp1は、宿主の翻訳機械の不活性化、宿主mRNAの分解、およびsignal transducer and activator of transcription 1 (STAT1)のリン酸化の阻害という3つのメカニズムによってIFN応答を阻害する[25,26]。nsp3の一部は、IFN調節因子3(IRF3)のリン酸化を阻害し、NF-ΚBシグナル伝達を阻害することにより、IFNおよびサイトカイン産生に拮抗するパパイン様プロテアーゼである[24]。Nsp7およびnsp15もまた、未知のメカニズムによるIFNアンタゴニストである[24]。ORF3bは転写因子IRF3およびNF-KBによるIFNβ誘導の阻害を介してIFNアンタゴニストを発揮するが、ORF6はJAK-STAT経路を介したシグナル伝達を阻害することによってIFNにアンタゴニストを発揮する[24]。MおよびNタンパク質は、TANK-結合キナーゼ1(TBK1)/IKBキナーゼε(IKKE)、およびTRAF3/6-TBK1-IRF3/NF-ΚB/AP1シグナルの負の調節を阻害することにより、IFNシグナルを平坦化する[25,26]。
IFN応答のアンタゴニズムは、さらにフリーウイルスの複製を促進し、その結果、さらにIFNシグナル伝達を減衰させ、異常な炎症性応答を誘導するためにPRRを刺激するウイルスPAMPおよびDAMPの増加をもたらす。SARS-CoV-2の複製能力は、感染したヒト肺組織において、I型、II型、およびIII型のIFNを有意に誘導することなく、SARS-CoVの複製能力の3.20倍以上であった[27]。自然免疫がSARS-CoV-2に対する最前線の防御であるため、遅くて協調性に乏しい応答は、より高いウイルス複製をもたらす可能性がある。この一連の事象、すなわち肺上皮細胞感染、IFNシグナル伝達阻害、および遊離ウイルス複製は、ウイルスの悪循環を描いている(図1)。
3. 第二の騎手:ギャザリングストーム
SARS-CoV-2 は、主に気道および肺胞 肺上皮細胞、特に肺胞界面活性剤を産生する細胞であり、I 型肺炎球の前身である II 型肺炎球に感染する。しかしながら、内皮細胞、周皮細胞、血管平滑筋細胞、マクロファージ、線維芽細胞、T細胞、心筋細胞、腸球、基底細胞表皮細胞、および上皮性尿細管遠位細胞など、受容体ACE2を発現するあらゆる細胞に感染する可能性がある [28]、[29]、[30]。SARS-CoV-2は、自然免疫が低下しているために複製が抑制されていない状況では、感染後早期に高濃度で複製することができる [28,31,32]。
肺上皮細胞における高濃度のウイルス複製は、多核化細胞(syncytia)、細胞質ウイルス包有物、およびアポトーシス(ウイルス複製を止めるための究極の細胞反応)の剖検所見で示されるように、細胞病理学的効果を誘導する[33,34]。これらの事象に続いて、肺上皮細胞による炎症性サイトカインおよびケモカインの増加レベルの産生が行われる [35,36]。さらに、SARS-CoVヌクレオカプシドは、核内因子κB(NF-KB)の細胞輸送を介して肺上皮細胞におけるインターロイキン-6(IL-6)の発現を活性化する[37]。肺への炎症性細胞の大量浸潤は、順番に、これらのサイトカインやケモカインによってマウントされている[32](図2)。肺の組織常駐型マクロファージは肺胞内の空隙に局在するが、この反応の優勢なサブセットではないようである[38]。SARS-CoV-2感染後の肺では、炎症性単球マクロファージと好中球の蓄積により、サイトカインとケモカインの追加放出が促進される[32](図2)。さらに、SARS-CoVのスパイクは、マクロファージにおけるIL-6および腫瘍壊死α(TNF-α)のアップレギュレーションを促進する[39]。
図2
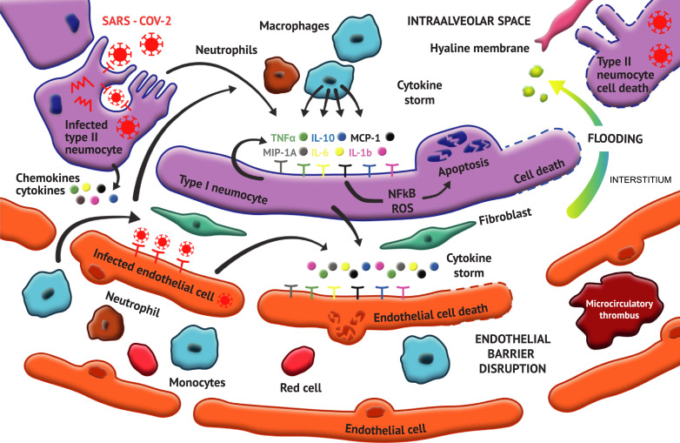
図2 SARS-CoV-2感染症(COVID-19)における急性肺損傷の生理学。
SARS-CoV-2は、ACE2受容体に結合して主にII型肺炎球に感染する。感染した肺炎球および周囲の肺炎球は、サイトカインおよびケモカインを分泌し、これにより単球-マクロファージおよび好中球が肺胞腔に引き寄せられ、追加のサイトカインおよびケモカインが分泌される。最終的に肺炎球はアポトーシス/ピロプトーシスを起こし、大量の炎症性因子を放出する。内皮細胞は感染し、接着分子を過剰発現し、ケモカインやサイトカインを放出する。内皮細胞はアポトーシスを受け、これは肺胞細胞のアポトーシスとともに、血管の漏出を増加させ、肺胞-毛細血管バリアを破壊する。高炎症性環境および内皮機能障害は、組織因子発現、血小板活性化、およびNETosisを介して凝固カスケードを活性化し、それらのすべてが微小循環血栓形成を促進する。内皮-肺胞関門の破断は、さらに間質性および肺胞空間の氾濫をもたらす血管漏出を促進する。ACE2/Ang-(1-7)/Mas1R軸のダウンレギュレーションは、血管収縮、炎症シグナル、内皮機能障害、血管漏出、プロトロンボティック状態の増加に寄与する。
SARS-CoV-2=重症急性呼吸器症候群コロナウイルス2;TNF-α=腫瘍壊死因子α;IL-10=インターロイキン10;MCP-1=マクロファージ化学吸引性タンパク質1;MIP-1A=マクロファージ阻害性タンパク質1A;IL-6=インターロイキン6;IL-1β=インターロイキン1β;NF-KB=核内因子κB。
サイトカインは局所炎症から全身循環にスピルオーバーする。COVID-19患者では、インターロイキン(IL)-2、IL-7、IL-10、顆粒球コロニー刺激因子(G-脳脊髄液)、インターフェロンガンマ誘導性蛋白(IP)-10、単球化学吸引性蛋白(MCP)-1、マクロファージ炎症性蛋白(MIP)-1A、およびTNF-αを含む炎症性サイトカインの血清レベルが高い。これらのサイトカイン/ケモカインレベルは、疾患の重症度と相関している[40,41]。COVID-19の重症患者は、軽症または中等症の患者よりもIL-6のレベルが高くなることが多い[42]。COVID-19では、ウイルス感染は目立った特徴ではなく、通常は短命であるが、SARS-CoV-2ウイルス感染の程度および期間は、疾患の重症度および血清中のIL-6レベルと関連している[43]。
内皮細胞(内皮細胞)は感染過程の非常に早い段階で感染する。ウイルスの迅速な複製と高揚性の炎症性サイトカイン/ケモカイン反応のために、アポトーシスを受けることがある[32,33]。このアポトーシス現象は、Fas/FasLやTRAIL-DR-5依存性のメカニズムを介して起こる[44]。また、炎症性単球-マクロファージはまた、肺内皮細胞と肺上皮細胞の両方のアポトーシスを促進するTNF-αを放出する[35]。内皮細胞sと肺上皮細胞のアポトーシスは、肺の微小血管床と肺胞細胞-毛細血管バリアの完全性を損なうため、血管漏れと肺胞浮腫を引き起こす[35](図2)。ペリサイトは毛細血管の内皮細胞機能を維持する上で重要な役割を果たしており、ACE2の発現量が最も高い細胞の一つである。SARS-CoV-2による感染は、内皮細胞の機能不全をさらに悪化させ、微小循環障害を引き起こす可能性がある[29]。
全身型COVID-19の顕著な特徴は、重度のリンパ球減少である。CoV特異的T細胞は、過反応性の自然免疫応答を抑制することができるため、ウイルスのクリアランスと宿主組織への追加的な損傷の制限に決定的な役割を果たしている[45,46]。しかし、SARS-CoV-2によって過剰な炎症反応が起こると、TNF-αを介した細胞のアポトーシスによってT細胞の反応が低下し、制御不能な炎症反応を引き起こす[35](図1)。さらに、正常なT細胞活性化はIL-6によって抑制され、リンパ球減少をさらに助長する[47]。リンパ球サブセットを検査した重症COVID-19患者では、激しいCD4、特にCD8リンパ球減少が認められ[42]、両者はTNF-αおよびIL-6血清レベルと負の相関を示している[41]。CD4細胞はT依存性B細胞を活性化することでウイルス特異的抗体の産生を促進するが、CD8細胞は細胞毒性があり、ウイルス感染細胞を死滅させることができる。CD8細胞は全炎症性肺間質浸潤の約80%を占めるため、細胞毒性の高いCD8リンパ球は免疫介在性の組織障害を仲裁することができる[34]。また、COVID-19患者では、これらの細胞は、プログラムされた細胞死タンパク質141などの機能的に疲弊したT細胞のマーカーを示す。
COVID-19患者のPBMCでは、アポトーシス、オートファジー、およびp53経路のアップレギュレーションが見られる[48]。MERS-CoVは、内因性および外因性アポトーシス経路の活性化を介してT細胞のアポトーシスを誘導することができ[49]、SARS-CoV Eタンパク質もまた、Cbl-XL結合を介してT細胞のアポトーシスを促進することができる[50]。SARS-CoV-2は非生産的にTリンパ球に感染することができるが、この感染がT細胞のアポトーシスを誘導するかどうかはまだ明らかにされていない[51]。あるいは、COVID-19患者の血清IL1-βレベルが上昇していることから、ピロプトーシスがリンパ球減少症の原因であることが示唆されている[52]。SARS-CoV感染では、ビロポリン3aは、NOD様受容体タンパク質3(NLRP3)の炎症ソームの活性化とマクロファージによるIL-1-βの分泌を誘発する[53]。膿栓症は大量の炎症性因子を放出することがある[54]。原因が何であれ、感染の後期には、T細胞の枯渇がウイルスの生存を促進し、結果的に感染を長期化させる可能性がある。
SARS-CoV-2感染の持続期をコントロールするために不可欠なのは、抗ウイルス中和抗体が重要な役割を果たす体液性免疫の出現である。しかし、動物モデルでは、抗Sタンパク質中和抗体(抗S-IgG)は、炎症反応を変化させて重篤な肺損傷を引き起こす可能性がある[55]。SARS-CoV感染では、急性呼吸器疾患の発症は患者の80%で抗ウイルスIgGの血清転換と一致しており[56]、死亡した患者はより早く抗S-中和抗体を発現していた[57]。抗S-IgGの存在は、抗炎症性単球-マクロファージ肺蓄積およびMCP-1およびIL-8の産生を促進する。このような炎症性サイトカイン放出は、ウイルス-抗S-IgG複合体の単球-マクロファージFcγRIIA受容体への結合を介して媒介されるであろうが、その遮断はIFN-γ、TNF-α、IL-1、およびIL-6の産生を減少させるので[55]。また、このような複合体が補体系の古典的な経路を活性化したり、抗体依存性の細胞毒性を誘導したりして、細胞障害を引き起こす可能性もあるだろう。したがって、考えられる根底にあるメカニズムは、ウイルス感染の抗体依存性亢進(ADE)であり、これは、ウイルスを完全に除去することができない初期の最適以下の抗体活性を持つ一部の患者で発生し、持続的なウイルス複製および炎症を引き起こす。
自然免疫反応が遅れているために、制御されていないウイルス複製は、過剰な免疫のキックバックと調節不全につながる細胞損傷を引き起こす。自然免疫応答と適応免疫応答に影響を与えるこの過剰反応は、組織や臓器の免疫介在性の損傷への道を開くことになる。この一連の事象は、炎症性の傷害フィードバックループに適合している(図1)。
4. 第三の騎手:無力な肺
RASは、血圧の恒常性と水電解質バランスを維持することにより、心血管系および腎機能の制御において重要な役割を果たしている[59]。当初、RASは、肝臓で合成されたアンジオテンシノーゲンがレニンの作用により活性ペプチドであるアンジオテンシンI(Ang I)に変換される線形ホルモン系として考えられていた[60]。その後、Ang IはACEを生成するAng IIによって切断される[61]。2つのGタンパク質共役型受容体(GPCR)が、アンジオテンシンII受容体1型(AT1R)および2型(AT2R)の作用を媒介している[62]。この正則的または古典的なRAS経路(ACE/Ang II/AT1R)の主要な役割は、交感神経系の緊張を高め、血管収縮を引き起こし、血圧を上昇させ、炎症、線維化および心筋肥大を促進することである[63]。
RASはまた、ACE2/Ang-(1-7)/Mas1Rからなる非カノニカルな対調節枝を持っている。システムの活性は、2つのブランチ間のバランスに依存する。ACE2は、Ang Iから1つの残基を除去してAng-(1-9)を生成し、Ang IIから1つの残基を切断してAng-(1-7)を生成することで、Ang-(1-7)の主な合成体となる[64,65]。Ang-(1-7)の機能的受容体はGPCR Mas1Rである[66]。陰性軸または対調節軸のコンフォメーションは、血管収縮/増殖ペプチドAng IIをダウングレードして血管拡張剤ヘプタペプチドAng-(1-7)を形成するだけでなく、Ang IをAng-(1-9)に分解し、それによってACEの基質の利用可能性を制限するためにも関連している。Ang-(1-7)はMas1Rに結合し、血管拡張、細胞増殖抑制、抗血栓作用、抗不整脈作用を誘導する[67]。ACE2活性は、ジスインテグリンおよびメタロプロテアーゼドメイン含有タンパク質17(ADAM-17、TNF-α変換酵素、TACEとも呼ばれる)によって制御される。ADAM-17はタンパク質分解的にACE2を切断し、ACE2の間質への脱落を引き起こし、組織内でのACE2活性の低下と循環ACE2活性の上昇をもたらする[68](図3)。ACE2レベルの血中および尿中測定が可能であることから、COVID-19の予後バイオマーカーとして使用できる可能性がある[69]。
図3
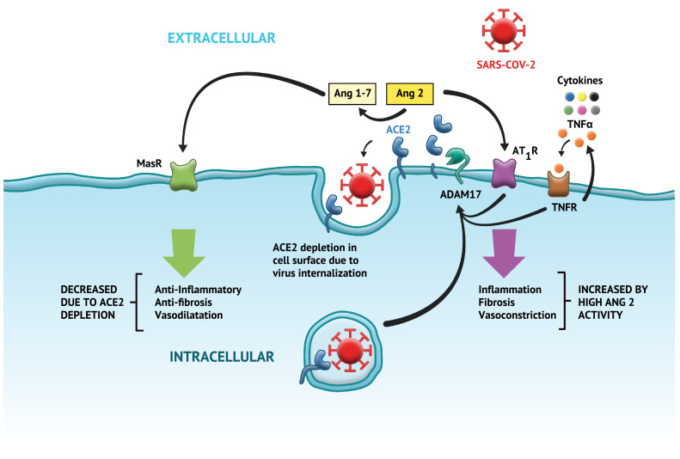
図3
コロナウイルス疾患2019の病態におけるアンジオテンシン変換酵素2(ACE2)/アンジオテンシン(Ang-1-7)/Mas1Rの含意。
肺などの臓器は、SARS-CoV-2誘導エンドサイトーシス後のACE2ダウンレギュレーションの結果として、非カノニカルRAS系の保護を失う。その結果、カノニカルACE/Ang II/AT1Rが優勢になり、Ang IIのレベルが上昇し、線維化、心筋肥大、活性酸素の増加、血管収縮、炎症、および内皮機能障害が促進される。AT1Rはこれらの作用のほとんどを媒介している。エンドサイト化されたSARS-CoV-2スパイクタンパク質は、ADAM 17を介してACE2のタンパク質分解的切断を媒介する。ADAM-17活性は、活性化されたAT1Rを介して、Ang IIのレベルの増加によって増強される。TNF-αはADAM17の主要な基質である。ADAM17はTNF-αを切断し、可溶性TNF-αを細胞外に放出する。TNF-αによるTNF-α受容体の活性化もまた、ADAM17の活性を高める。
ACE2=アンジオテンシン変換酵素2;SARS-CoV-2=重症急性呼吸器症候群コロナウイルス2;Ang II=アンジオテンシンII;ROS=活性酸素種;AT1R=アンジオテンシン1受容体;ADAM17=A disintegrin and metalloproteinase domain 17;TNF-α=腫瘍壊死因子α;TMPRSS2=膜貫通型プロテアーゼセリン2。
RASは、炎症性疾患の病態形成において重要な役割を果たしており、そのほとんどの炎症性作用はAng IIによって引き起こされる[67]。Ang IIは、組織傷害、炎症、および線維化に関連するいくつかの細胞機能および分子シグナル伝達経路を活性化する。それらには、カルシウムの動員、フリーラジカルの生成、プロテインキナーゼおよび核内転写因子の活性化、炎症性細胞のリクルート、単球および好中球の内皮細胞および中膜細胞への接着、接着分子のアップレギュレーション、サイトカインおよびケモカインの発現、合成および放出の刺激が関与している[68]。AT1Rはこれらの作用のほとんどを媒介する [70]。対照的なACE2/Ang-(1-7)/Mas1R軸は、白血球の遊走、サイトカインの発現と放出、および線維形成経路をネガティブに調節する。したがって、ACE2欠損は、血管接着分子、サイトカイン、ケモカイン、マトリックスメタロプロテアーゼの遺伝子発現を増加させることにより、血管炎症を増加させる[71]。ACE2の欠損は、炎症性因子のAng II誘発発現の増加、血管透過性の亢進、肺水腫の増加、好中球蓄積の増加をもたらす[72]。ACE2/Ang-(1-7)/Mas1R軸もまた、内皮細胞によるプロスタサイクリン(PGI2)産生と一酸化窒素(NO)放出を刺激し、付着性の少ない血小板活性を調節し、抗血栓活性を発揮するため、止血に不可欠な役割を果たしている[73](図1)。
RASはAng II合成の主要部位である肺組織で高い活性を示す。ACE2 は、亜鉛メタロペプチダーゼであり、N 末端を配向させた I 型のインテグラル膜糖タンパク質であり、触媒部位は細胞外空間に面している[74]。ACE2とSARSウイルススパイク蛋白質との結合は、酵素の内部化を誘発し、細胞表面からの活性を低下させる。SARS-CoVがその受容体に結合すると、細胞表面上の豊富さ、mRNAの発現、およびACE2の酵素活性が著しく低下する[75]。細胞外ドメインのタンパク質分解的脱落は、細胞表面での ACE2 の調節を低下させる第二のメカニズムである。SARSのSタンパク質は、一度ACE2に結合すると、細菌性エンドトキシンやリポ多糖類(LPS)と同様に、ADAM17(TACE)を活性化させることで脱落を誘導する[76](図3)。ACE2を細胞膜から放出することは、基質を触媒する上で重要なステップであり、ACE2活性の減衰が疾患発症に寄与する可能性があることを示唆している。最近報告されたヒト鼻上皮細胞におけるタイプIFNによるACE2発現の誘導は、このようにしてISGとして振る舞うことから、SARS-CoV-2によるACE2のダウンレギュレーションのさらなるメカニズムを強調している[30]。IFN誘導性ISGは宿主の抗ウイルス応答に重要であるため、IFN応答の阻害によるACE2誘導の欠如は、組織の保護の欠如をさらに引き起こすことになる。したがって、COVID-19では、ACE2は、肺上皮細胞への侵入の促進因子としての多面的な任務、および急性肺損傷(ALI)の病態形成における潜在的な役割のために、極めて重要な役割を果たしている[75]。SARSのマウスモデルでは、ACE2タンパク質発現のダウンレギュレーションは、肺炎の悪化、Ang IIレベルの上昇、血管透過性の上昇、肺水腫の増強、好中球浸潤、および肺機能のさらなる悪化をもたらした [72,77]。触媒的に活性化されたACE2タンパク質は症状を緩和し、活性化されたタンパク質は呼吸不全の転帰を改善した[72]。COVID-19患者では、Ang IIの血漿中濃度が健常者よりも有意に高く、Ang IIレベルはウイルス負荷および肺損傷と相関していた [78]。
ACE2が広く発現しているため、COVID-19は播種性感染症である。ACE2は腸内で高発現しており、SARS-CoV-2は腸球に生産的に感染することができる[28,79]。腸内でのACE2発現は肺よりも高いにもかかわらず、COVID-19患者の約10~12%しか消化器症状を経験しない[79]。粘膜バリアの障害および炎症性サイトカイン産生の増加を介したCOVID-19の病因への消化器系の寄与は、まだ決定されていない。MERS-CoVと同様に、近位尿細管のブラシ境界およびポッドサイトにおけるACE2の発現により、COVID-19の腎障害は、尿細管上皮細胞およびポッドサイトにおけるウイルス様粒子を伴うびまん性の近位尿細管損傷を特徴としており、これはSARS-CoV-2の直接感染を示すものである[80]。これらの所見は臨床的には急性腎障害および蛋白尿となり、COVID-19患者の0.9%から29%に影響を及ぼす [80]。
SARS-CoV-2感染による肺のACE2活性低下の結果は、Ang-(1-7)産生の減少である。Ang-(1-7)結合体である Mas1R は、肺におけるアポトーシス、血管新生、血管収縮、炎症などの Ang II 介在プロセスを打ち消すための一連の生物学的反応を促進する [81,82]。その結果、ACE2触媒機能の減衰は、次の猛烈なサイトカインストームに直面して無防備な肺を残して、強化された炎症と血管透過性をもたらし、肺のRASのバランスを乱す。また、II型肺炎球の感染は、その後、肺胞界面活性剤の生産を減少させ、肺の弾力性を減少させる。さらに、II型肺炎球の喪失はI型肺炎球の回復を減少させ、最終的にはガス交換と線維化に影響を与える[83]。上記のイベントシーケンスは、RASの悪循環のフィードバックループを描いている(図1)。
5. 第四の騎手:パンデミックの中の伝染病
COVID-19と凝固障害との関連は、中国の医師が主に低分子ヘパリンで治療された患者の28日死亡率が低下していることに気づいたパンデミックの初期に明らかになった[84]。この死亡率の改善は、敗血症スコアが4以上の患者、またはDダイマーが著しく上昇した患者であった[84]。COVID-19は、フィブリノゲンやD-ダイマーなどのプロコアグラント因子の増加を含む凝固障害と関連しており、どちらも予後不良と関連している[85,86]。ICUに入院した患者では、静脈血栓塞栓性イベントの発生率が25%から36%まで増加していた [87]、[88]、[89]。さらに、静脈血栓塞栓症に対する標準的な予防法は7.7%の患者で失敗した [90]。血栓性合併症の多くは静脈性であり、主に孤立性の肺塞栓症であったという報告もあり、これは塞栓性現象ではなく原発性肺血栓症である可能性を示唆している[89,91]。これに伴い、肺の病理学的研究では小循環血栓症が常に認められている[33,34](図2)。
内皮細胞の感染は、細胞浸潤とサイトカイン/ケモカインへの高暴露による異常と相まって、最終的に内皮細胞の機能不全とアポトーシスを引き起こす[33]。これらのすべてが微小血管プロトロンボティクス効果に寄与している[92]。止血と自然免疫の間には、血栓性炎症と呼ばれる激しい相互作用が存在する[93]。内在性と外在性の両方の凝固経路が炎症時に活性化することがある。内皮細胞およびマクロファージは、組織因子の発現を介して外因性経路を活性化する[94]。内在性経路は、NETosisと呼ばれるプロセスで多形核好中球(PMN)によって放出される好中球細胞外トラップ(NET)によって活性化される。NETsは、内皮細胞、血小板、および補体系を活性化し、内因性抗凝固剤を不活性化するプロテアーゼを放出する[95]。しかしながら、COVID-19におけるNETの役割はまだ議論の余地がある[96]。
血小板は二重の役割を果たす。第一に、IL-1βの不可欠な供給源であるPMNとマクロファージをリクルートするα顆粒を分泌することにより、炎症を促進する役割である[97]。その上、血小板はPMNを刺激して血小板を活性化し、フィードバックループを形成するNETosisを経る。血小板の第二の役割は、その露出した表面に酵素-因子-基質複合体を組み立てることによって凝固経路を活性化することである[98](図1)。
SARSのマウスモデルで見られた補体活性化は、免疫介在性病理に寄与する[99]。C3およびC5の活性化は、肥満細胞の脱顆粒およびPMNおよびマクロファージのリクルートを促進する[100]。活性化されたC3およびC5の血栓防止効果には、組織因子およびフォン・ウィルブランド因子発現の増加とともに、血小板および内皮細胞sの活性化が含まれる[95]。ループを閉じるために、トロンビン、および凝固カスケードの他の構成要素は、順番にC3およびC5を活性化することができる[95]。
トロンビンの主な機能は、血小板を活性化し、フィブリノーゲンをフィブリンに変換することによって血栓形成を促進することである[101]。しかしながら、トロンビンは多動性分子であり、プロテアーゼ活性化受容体(PAR)、主にPAR-1[101]を介して炎症を増加させることができる(図1)。トロンビンの生成は、負のフィードバックループおよびアンチトロンビンIII、組織因子経路阻害剤、およびプロテインC系などの生理的抗凝固剤によって制御される[101]。IL-1β、IL-6、およびTNF-αは、超大型フォンウィルブランドマルチマーの放出、および組織因子および第VII因子/活性化第VII因子の産生を促進し、内因性抗凝固剤のレベルを低下させながらトロンビン生成の増加を導く[101]。
ACE2/Ang-(1-7)/Mas1R軸は、血小板におけるMas1Rの活性化を介して抗血栓効果を発揮し、NOとPGI2を放出し、内皮機能障害から保護することによって[102,103]。COVID-19ではRASのこの分岐が正常に働いていないため、この保護機構が失われている(図1および33)。
重症COVID-19では、他の急性ウイルス感染症と同様に、抗リン脂質抗体の高い有病率が認められたが、SAR-CoV-2感染者の血栓症状態におけるこれらの抗体の役割については、まだ議論の余地がある[104]。
血栓性炎症の進行は広範囲の血栓症を引き起こし、低酸素血症、高体温血症、および低飲酒血症によってさらに増強される可能性がある[105]。低酸素血症は低酸素誘導性因子の発現増加を誘発し、これはさらなる炎症を促進し、血小板および凝固因子を活性化する可能性がある。それらは組織因子の発現を増加させ、プラスミノーゲン活性化インヒビター-1を増加させ、内因性抗凝固タンパク質Sを阻害する[106]。炎症性亢進状態および内皮損傷の設定では、対調節力ACE2/Ang-(1-7)/Mas1R軸が不活性であるのに対し、凝固の活性化が起こり、フィールドは高凝固性状態の完全な発現に委ねられる。この状態は、臨床的には、肺血栓症、静脈血栓塞栓症、または他の血栓性イベントに変換することができる。これらのイベントが微小血管肺床に影響を及ぼす場合、ALIをさらに促進し、ガス交換を損なう可能性がある。血栓性イベントの場所がどのようなものであれ、患者の予後を悪化させる。
高炎症状態と欠陥ACE2/Ang-(1-7)/Mas1Rの機能は、第四の有害なフィードバックループを活性化する。高炎症状態は高凝固を誘導し、その逆はACE2/Ang-(1-7)/Mas1R軸回避が両者を最大化する(図1)。
6. ダメージの広いスケール
CoVID-19の臨床スペクトルは広い。SARS-CoV-2に感染したすべての人が病気になるわけではなく,感染後に現れる状態は患者間で,あるいは同じ患者内でも時間の経過とともに変化する.したがって、ウイルス依存性、宿主依存性、環境依存性の要因がウイルス-宿主相互作用を変化させ、個人の感染しやすさだけでなく、臨床的な疾患で見られる広範囲の損傷を説明することが想定される。
気道内の初期のウイルス力価は、COVID-19の異なる進化のパターンを説明することができる。SARS-CoV-2は感染後非常に早い時期に大量に複製し、その結果、ウイルス複製の大きさが抗ウイルス反応の程度に影響を与える。ヒトでは、SARS-CoVとMERS-CoVの力価と疾患の重症度との間には強い相関関係がある[35]。
動物モデルでは、ウイルスが気道上皮細胞に感染するか、気道と肺上皮細胞(I型およびII型肺細胞)の両方に優勢に感染すると、疾患の挙動が異なる。感染を許容するマウスモデルでは、ウイルス抗原は気道上皮細胞に主に位置しているが、臨床疾患を発症しない。一方、高感受性マウスでは、気道と肺胞I型およびII型肺胞の両方に抗原が検出されている[35]。したがって、肺上皮細胞 の感染は、宿主の感受性と肺病理の発症の両方に重要であると考えられる。SARS-CoV-2の感染に影響を与える側面として、ヒト気道上皮の分化状態があり、これはこれらの細胞におけるACE2の発現レベルと正の相関がある[107,108]。小児ではACE2鼻遺伝子の発現が低いことは注目すべきことである[109]。この事実は、COVID-19の顕著な年齢分布と関連しており、小児はしばしば免れ、成人では年齢が上がるにつれて重症度および死亡率が高くなる。しかしながら、年齢が上がるにつれてますます予後が悪くなるのは、高血圧、心血管疾患、糖尿病などの一般的な併存疾患の存在に影響されており、これらの併存疾患はそれ自体が予後不良である [47]。さらに、これらの併存疾患は、高齢者患者におけるACE2の活性低下に関連しており、この欠損はSARS-CoV-2感染によってさらに悪化する [110]。ADEは、SARS-CoV-2に対する中和抗体の力価が最適でない場合に起こる、潜在的に有害な炎症促進メカニズムである。彼らは感染を制御することができないが、代わりにトロイの木馬のメカニズムによってマクロファージへのウイルスの侵入を促進する。ADEは、コロナウイルス感染の間の時間間隔が抗体低下のために十分に長い場合に起こる傾向があり、これは高齢者における重度のCOVID-19の可能性のあるメカニズムである可能性がある[58]。
COVID-19を有する女性は通常、男性よりも軽症である。女性は男性よりもPBMCからのIFNおよびIFN調節因子およびIL-10の産生が高く、TNF-αの産生が低く、PMNにおけるTLR4の発現が低く、NK細胞数が少なく、PMNの貪食活性が低いことを示す [111]。オエストロゲンはAng IIをダウンレギュレートし、Ang-(1-7)経路をアップレギュレートするため、RAS成分の発現、活性、組織応答性には明らかな性差がある[112]。また、Mas1Rの発現は雌ラットでは増加したが、雄では増加しなかった[113]。肥満の動物モデルでは、雌は循環中のAng-(1-7)レベルを維持し、アンジオテンシノーゲン、レニン、アンジオテンシンII、およびAT1R活性化によって誘導される高血圧および代謝性合併症から保護されているようである[114]。対照的なAT2Rの刺激は、雌のげっ歯類では代謝的に保護されるようであるが、雄では一貫性のない効果がある[115]。
英国では、ICU内のCOVID-19患者の72%が過体重または肥満であり[116]、北イタリア人の死亡者の99%が肥満、高血圧、糖尿病、心臓病、腎障害、または癌であった[117]。有害な転帰の要因としての肥満の頻繁な発生は、肥満の独立した役割の同定を急に作る肥満の人々における他の高い有病率の併存疾患によって頻繁に影を落とされている[118]。肥満とCOVID-19の関連は、慢性炎症状態、非効果的な免疫応答、またはRASシステムとの相互作用に起因している。
免疫経路はすべて、サイトカイン発現、抗原結合親和性、受容体ライゲーション強度、および下流シグナル伝達の変動などの機能的な結果を伴う遺伝的多型の影響を受けやすい[119,120]。高度な相互接続は免疫経路の顕著な特徴であり、そのため機能的な結果として生じる多型は、COVID-19に対する最適な免疫応答の成長を妨げる可能性がある。PAMPs認識および骨髄分化一次応答88(MDY88)のようなその下流分子によって誘発される応答は、TLR多型によって変化する可能性がある[121,122]。HLA遺伝子は極端な対立遺伝子多型を示す。これらの遺伝子は、適応免疫応答を誘発するために宿主HLA分子にウイルスペプチドを提示するため、それらの多型は抗原の結合および提示、そして結果として免疫応答に不均一性を引き起こす可能性がある。HLA-B*46:01は、SARS-CoVの発症および重症度の増加に関連しており[123]、SARS-CoV-2ペプチドの中で最も予測される結合親和性が少ない[124]。IL-6は、COVID-19患者における過活動性免疫応答において中心的な役割を果たしている。IL-6遺伝子には、そのタンパク質レベルの発現を修飾する機能的多型が存在するため、それらが疾患の重症度に影響を及ぼす可能性がある[125]。COVID-19の病態におけるRASの役割は必須である。ACE1遺伝子における多型A/DなどのACE遺伝子における一塩基多型およびハプロタイプは、ACEレベルの循環および組織濃度、ならびにACE2の発現低下と関連している[126,127]。興味深いことに、ヨーロッパにおけるCOVID-19の有病率はACE D対立遺伝子頻度と逆相関している [127]。IFN-induced transmembrane protein-3遺伝子の遺伝的変異はCOVID-19の重症度と関連している。IFN誘導性膜貫通タンパク質-3は、膜融合を制限するように作用する免疫エフェクタータンパク質である[128]。最近、呼吸不全を有するCOVID-19患者を対象としたゲノムワイド関連研究では、SLC6A20、LZTFL1、CCR9、FYCO1、CXCR6およびXCR1遺伝子を含む遺伝子座3p21.31に関連シグナルが同定されたが、HLA複合体には関連シグナルは認められなかった[129]。
最近、COVID-19患者の転帰におけるACE阻害薬(ACEi)とアンジオテンシン受容体拮抗薬(ARB)の有益な役割と有害な役割について議論されている。現在のところ、COVID-19患者におけるACEiまたはARBとの併用療法の有益または有害な効果を支持する証拠はない[130]。
7. 病理遺伝学に基づく治療的洞察
COVID-19は、ACE2を発現しているあらゆる組織や臓器に影響を与える可能性があるため、全身感染症である。しかし、最も恐ろしい、しばしば生命を脅かす病態はALIとARDSである。そのため、ICUへの入院や機械的な人工呼吸のサポートを防ぐために、これらの感染症の発症を回避することが主な課題となっている。我々はCOVID-19を、肺上皮細胞のウイルス感染とACE2のダウンレギュレーションが根源となっている樹木として想定することができる。木の幹は高炎症性および高凝固性の状態である。枝は、ALI、心筋炎、神経疾患、肝障害、消化管病変、皮膚病変などの末梢臓器疾患であろう。
SARS-CoV-2感染が引き金となった事象の連鎖は急速に進行するため、計画的な介入は可能な限り早期に行われなければならない。さらに、COVID-19の発症には非ウイルス性機序が関与しているため、計画された介入はすべて、これらの不均衡の修正または調節にも対処しなければならない。したがって、あらゆる治療的介入は早期に行われ、抗ウイルス療法およびアジュバント療法を組み合わせなければならない。しかし、診断の瞬間と最終的な入院は、介入のタイムフレームをマークする。
疾患の根源に取り組むために、COVID-19に対する潜在的な治療的介入は、まず肺上皮細胞へのウイルスの侵入に対処すべきである。SARS-CoV-2の肺上皮細胞への侵入は、スパイクが受容体ACE2に結合した後に起こる。具体的には、この結合は、Sタンパク質の受容体結合ドメインで行われる。したがって、このドメインに対する中和モノクローナル抗体を開発することは、ウイルスの結合およびその後の事象を防ぐための合理的な戦略である[131]。ACE2とSタンパク質との間の相互作用を標的とする別の可能性のある方法は、可溶性組換えACE2の使用であり、これは、表面結合した完全長ACE2へのウイルス粒子の結合を防止する可能性がある[132]。Vero-E6サル細胞株では、可溶性のACE2はSARS-CoVの複製を阻害し、SARS-CoV-2の回復を1000~5000分の1に減少させた[132,133]。また、SARS-CoV-2はACE2/Ang-(1-7)/Mas1R軸をダウンレギュレートするため、組換えヒトACE2(rhACE2)はCOVID-19におけるALIの発症を予防することができた。ARDS患者を対象とした第II相非盲検試験では、rhACE2の忍容性は良好であり、Ang IIレベルは低下し、Ang-(1-7)と界面活性剤プロテインDは増加した [135]。しかし、この試験では急性期の生理学的または臨床的転帰の変化を検出する力はなかった[135]。重度のCOVID-19患者におけるrhACE2を評価する無作為化比較試験がある(NCT 04287686)。ACE2とは別に、SARS-CoV-2の侵入にはTMPRSS2が関与しており、その阻害剤であるカモスタットメシル酸塩は、SARS-CoV-2の肺細胞株感染を有意に減少させた[136]。
エンドサイトーシスは、SARS-CoV-2感染において重要なステップである。AP-2関連プロテインキナーゼ(AAK1)はこのプロセスを制御している[137]。ヤヌスキナーゼ阻害薬であるバリシチニブは、AAK1を阻害することから、COVID-19の候補薬として主張されている[138]。アルビドールは、ウイルス膜と細胞膜の融合を阻害することで、ウイルスの侵入を阻害する[139]。しかし、小規模なレトロスペクティブ研究では、アルビドールは、アルビドールとロピナビル/リトナビル(LPV/r)の併用に対する非劣性を満たしていなかった[140]。クロロキンおよびそのより安全な誘導体であるヒドロキシクロロキンは、試験管内試験(in vitro)でSARS-CoV-2に対して有効である[141]。しかし、大規模なリカバリー試験からの最近のニュースでは、COVID-19で入院した患者にヒドロキシクロロキンの有益な効果がないことが示された;そのため、この試験は中止された[142]。Solidarityのような他の計画されている大規模試験では、ヒドロキシクロロキン群への患者の登録が中止され、国立衛生研究所がスポンサーとなっているORCHID試験も中止された[143,144]。
多数の抗ウイルス薬が臨床試験で試験されている。LPV/rはプラセボと比較しても十分な有効性を示すことができなかった [145]。IFN、LPV/r、およびリバビリンの組み合わせは、鼻咽頭スワブが陰性化するまでの時間が短く、症状の緩和においてLPV/rに比べて優れていることが示された[146]。2つの二重盲検プラセボ対照試験では、レムデシビルは統計学的に有意な臨床効果とは関連していなかったが、他の試験では入院した成人の回復までの時間を短縮した[147,148]。現在のところ、COVID-19患者の治療に有効性が証明された抗ウイルス薬はない。
別の戦略は、COVID-19における高揚した炎症反応を調節しようとするものである。コルチコステロイドの使用は議論の余地があり、SARSおよびMERSにおける以前の経験では支持されていない[149]。しかしながら、Recovery試験では、デキサメタゾンは、侵襲的機械換気を受けている患者では死亡数を3分の1、侵襲的機械換気を行わずに酸素を投与している患者では死亡数を5分の1に減少させた[150]。IL-6受容体拮抗薬であるトシリズマブは、IL-6が果たす病原性の役割のため、COVID-19の高炎症状態を治療するために推進されている。2件の観察研究では、高炎症性症候群を伴うCOVID-19肺炎におけるトシリズマブによる治療の臨床的有用性が示されている[151,152]。組換えIL-1受容体拮抗薬のアナキンラは、ARDSと高炎症を有するCOVID-19患者を対象とした小規模なレトロスペクティブ研究で有用性が証明されている[153]。トシリズマブ、アナキンラ、サリルズマブを用いた追加試験が進行中である。しかしながら、単一の分子または受容体を標的とすることでサイトカイン反応を修飾しようとする場合、サイトカインネットワークは高度な重複、冗長性、代替経路を持つ複雑な複合体であることを思い出すべきである。このことが、治療の逃避および最終的には反応の欠如を説明しているのかもしれない。
ALI、ARDSまたは血栓塞栓性イベントなどのCOVID-19に関連する高炎症性および高凝固性状態の結果に対する治療的介入は、本レビューの範囲を超えている。
8. 結論
病態生理学の知識は、疾患の管理に適切に対処するための第一歩である。ウイルスが宿主の免疫防御機構を回避するために使用するメカニズムや、害を与えるために使用するメカニズムを熟知していることは、ウイルスによって、あるいは感染の結果によって生じる機能不全や不均衡を中和するための適切な戦略を設計することを可能にする。集められた知識から、SARS-CoV-2が必要な開始因子であるにもかかわらず、重症COVID-19のほとんどの臓器損傷は免疫介在メカニズムによって行われるように思われる。疾患のスペクトルは包括的であり、すべての患者が同じ進行パターンを共有するわけではないので、生命を脅かす疾患に進行しやすい患者を迅速に特定するための予測因子の探索が最も重要である。重症化した場合には、疾患の急速な進展パターンは、少なくとも信頼できる予測因子が広く利用できるようになるまでは、早期の治療が必須となる。COVID-19の発症には、ウイルスおよび宿主依存性のメカニズムが関与していることが示唆されており、どのような治療戦略においても、抗ウイルス薬とアジュバント療法を組み合わせて宿主の反応を調節しなければならない。
これらの目標はすべて、基礎、トランスレーショナル、臨床の科学者と臨床医の幅広い努力によって達成されるものであり、患者とその家族、関連する専門家、そしてCOVID-19との闘いに携わるすべての人々の高度なコミットメントが必要とされる。その中でも、政治家や保健行政担当者はユニークな役割を果たすことになるであろう。
9.顕著な問題点
パンデミックに巻き込まれる中で、COVID-19の生理機能の解明には進展があった。しかし、宿主とウイルスのダイナミックな相互作用や、予後を改善するための改変の仕方など、ウイルス、環境、感染性の面でのギャップは少なくない。
β-コロナウイルスの中でも、構造や機能が似ているにもかかわらず、SARS-CoV-2が最も高い透過性に大きな差がある。無症候性のウイルス脱落が主な要因である。しかし、新たに記述されたORFやRNA修飾の役割や機能的な相関関係はまだ明らかになっていない。TMPRSS2はウイルスの宿主細胞への侵入に関与しているが、他の宿主タンパク質の関与についてはまだ議論中である。
気管支の木や肺胞腔に沿った異なる上皮細胞の役割を確認する必要がある。肺以外の他の臓器に侵入するウイルスのメカニズムは、すでにあまり知られていない。
臨床的な疾患の進行はやや予測不可能である。したがって、予後を予測する臨床的および生物学的マーカーを特定することは、患者のケアと資源消費を最適化することになり、パンデミック時にはこれが最も重要になるかもしれない。この努力には、併存疾患や性別がどのような役割を果たすかを含めなければならない。COVID-19に対する最適な治療アプローチの定義とタイミングは、無作為化臨床試験の実績によってのみ達成できる巨大な努力を意味する。これには、予防、診断、治療の進歩に貢献するための協調的な行動と多様な分野、資源、専門知識、技術の組み合わせが含まれるべきである。このセットは、欧州トランスレーショナルメディスン学会(EUSTM)が「ベンチサイド、ベッドサイド、コミュニティの3つの柱に支えられた生物医学分野の学際的な部門」と定義したトランスレーショナルメディスンのほぼ完璧な意味を構成している。
10. 検索戦略と選択基準
本レビューでは、1984 年から 2020 年までの欧米の情報源を中心とした雑誌や書籍の原本を検索した。これらの情報源から、特定された追加論文の参照リストを手探りで検索し、追加研究を検索した。最も関連性の高い研究を優先したが、トピックの幅広さを強調することにも熱心であったため、特定の関心分野を示す出版物をいくつか選択した。