
www.ncbi.nlm.nih.gov/pmc/articles/PMC5614326/
Aging Dis.2017 Oct;8(5):628-642.
2017年10月1日オンライン公開doi:10.14336/AD.2017.0103
pmcid: pmc5614326
PMID:28966806
概要
加齢は、時間依存的な生理的臓器機能の低下が避けられないものであり、がん発症の主要なリスクファクターである。医療、衛生管理、食料の入手の進歩により、平均寿命は延び、ほとんどの先進国の人口は、がん感受性年齢にある人々の割合が増加する方向に移行している。
老化のメカニズムは、最終的な結果を共有または乖離しているとはいえ、発がんにおいても生じることが分かっている。
現在では、加齢と発がんはいくつかの疾患メカニズムにおいて共通または分岐していることが明らかになっている。そのようなメカニズムには、ゲノムの不安定性、テロメアの消耗、エピジェネティックな変化、プロテオスタシスの喪失、栄養感覚の低下、代謝の変化、さらには細胞の老化や幹細胞の機能などが関与している。
また、がん細胞と老化細胞は根本的に正反対である。がん細胞は、有利な変異を持ち、細胞分裂が早く、エネルギー消費量が多い多動性細胞であり、老化細胞は不利な変異が蓄積し、細胞分裂ができず、エネルギー生産・消費能力が低下した低動性細胞と考えられるからだ。
しかし、加齢とがんは密接に関連しており、同じ戦略や薬剤が両者を標的として用いられることもあれば、拮抗的な多剤併用が有効で、一方を阻害すれば他方が活性化されることもある。
がんは老化病と考えることができるが、この2つのプロセスを支える共通のメカニズムは依然として不明である。老化と癌の共通する経路と分岐する経路について、より良い理解が必要である。
キーワード 老化、がん、ゲノム不安定性、エピジェネティック、テロメア、幹細胞、ゲノム不安定性、代謝
はじめに
加齢は時間依存的な生理的臓器機能の低下が避けられず、人間の罹患率と死亡率の最も大きな原因の一つである癌の主要な危険因子となる。米国国立がん研究所のサーベイランス疫学エンドリザルト(SEER)データベースによると、生涯で男性の43%、女性の38%が浸潤性がんを発症するとされている。このうち、男性の23%、女性の19%ががんで死亡するといわれている。がんの半数以上は70歳以上の高齢者に発生する[1]。
医療と栄養の改善により、過去1世紀で平均寿命は延んだ。世界保健機関(WHO)によると、現在、ほとんどの先進国で平均寿命は80歳を超えている。高齢化が進む中、がんは世界中でこれまで以上に重要な健康上の負担となってきている。
がんと老化の根底にあるメカニズムは、時間依存的な細胞傷害の蓄積である[2]。癌と老化は、癌細胞が「機能と体力の獲得」をするのに対して、老化細胞は「機能と体力の喪失」を特徴とする、正反対のプロセスのように見えるかもしれない[2]。しかし、この2つの特性には多くの共通点があり(表1)、それについて比較検討する。
表1 老化と癌に共通する、あるいは分岐するホールマーク
| 特徴 | エージング | 癌 |
|---|---|---|
| ゲノムの不安定性 | 増加 | 増加 |
| テロメア減少 | 短縮されたテロメア | テロメアは短くなるが、テロメラーゼは活性化する。 |
| エピジェネティックな変化 | ||
| DNAメチル化 | グローバルハイメチル化 | 腫瘍抑制因子の高発現と癌遺伝子の低発現 |
| ヒストン修飾非コード化DNA | 複合体 miR17-92のダウンレギュレーションなど、miRNAのデレギュレーション。 |
miR-17-92のアップレギュレーションなど、複合的なmiRNAのデレギュレーション。 |
| プロテオスタシス | ||
| 付き添い | 障害者 | オーグメンテッド |
| プロテアソーム活性 | 障害者 | オーグメンテッド |
| オートファジー-リゾソーム活性 | 障害者 | オーグメンテッド |
| 栄養センシングの乱れ | インスリンとmTORシグナルを阻害すると寿命が延びる | インスリンおよびmTORシグナルの阻害は抗腫瘍性である |
| 細胞老化 | 増加 | 前悪性腫瘍に好発するが、完全悪性腫瘍では回避される。 |
| 幹細胞 | 消耗品 | 腫瘍形成の起点となる可能性 |
1.ゲノムの不安定性
老化と癌の両方に共通する直接的な特徴の一つは、ゲノムの不安定性の発生である。ヒトのDNAは、外来放射線や内因性フリーラジカルなどの変異原に対して脆弱であり、私たちは一生の間に常にその危険にさらされている。実際、人体の細胞は何十億回となく細胞分裂を繰り返し、その間にDNAが複製されるが、そのたびに突然変異が起こったり、変異に苦しんだりする危険性がある。このような不可避の突然変異のほとんどは無害であり、大部分はDNA修復システムによって修正される。しかし、時間の経過とともに、ある程度のDNA損傷の蓄積が起こる[3]。ヒトゲノムにおける修復後の正常な変異率は、1回の細胞分裂で10億塩基に1個程度の変異であり、魔女は1,000 bpの遺伝子が細胞分裂ごとに100万分の1の確率で1個の変異を持つことを意味している[4]。重要なレギュレーター機能におけるゲノムの不安定性は、増殖シグナルの自己充足や転移能の増大など、がん細胞の駆動力となる多くの特性を獲得させる最も顕著な「実現可能な特徴」である[5]。ゲノムの不安定性は、ほとんどすべてのヒトのがんの特徴であり、腫瘍形成に必要なすべての変異を獲得するために、変異の割合は正常細胞よりも高くなる[6]。実際、多くのがんの全ゲノム配列解析から得られた知見や教訓から、変異率は全体的に高いものの、腫瘍の種類や部位によって異なることが知られており、一部の腫瘍はhypermutatedというラベルが貼られている[7]。
遺伝性症候群の教訓
DNA修復機構における様々な変異は、遺伝性非ポリポーシス大腸がん(HNPCC)[8]、BRCA1およびBRCA2の遺伝性変異を伴う乳がんおよび卵巣がん[9]、大腸がんの生涯リスクがほぼ100%となる家族性腺腫性ポリポーシス[10]といった遺伝性がん症候群の原因となっている。これらは、DNA修復機構の変異に起因し、変異率の上昇とゲノムの不安定性をもたらす、このような癌症候群の大きなグループのほんの一例に過ぎない。また、DNA修復機構の変異は、多くのプロジェロイド早老症候群を引き起こす可能性がある[11]。ウェルナー症候群やブルーム症候群は、二本鎖切断の修復やテロメアの維持に関わるRecQヘリカーゼの変異によって引き起こされるプロジェロイド症候群である[12,13]。これらの症候群は、非常に若い年齢での癌、早期老化の兆候(白髪、臓器機能および予備能の喪失)、および寿命の大幅な減少を示す[14]。コケイン症候群(CS)と色素性乾皮症(XP)はプロジェロイド症候群の他の2つの例で、どちらもヌクレオチド除去修復システム(NER)の変異の結果である。CSのNER変異は、活発に転写される遺伝子にのみ影響を与える(TCR-転写結合修復)のに対し、XPの損傷はゲノム全体に影響を与える(GGR-グローバルゲノム修復)[15]。両者とも早期老化の特徴を示すが、興味深いことにXPのみ皮膚癌への感受性が上昇する[15]。そのメカニズムはまだ不明であるが、CSでは変異細胞のアポトーシスが関与しているのではないかと推測されている[16]。
老化した細胞におけるゲノムの不安定性
ゲノムの安定性を維持することは、癌の発生や老化を防ぐための中核的な機能であるように思われる。ゲノムの不安定性と突然変異は、小さな点突然変異から大きな転座や欠失に至るまで、いくつかの方法で老化に寄与している可能性がある。体細胞においては、タンパク質コード配列の変化により表現型の崩壊をもたらすが、おそらくより一般的なのは、制御配列の変化であり、プロテオームとホメオスタシスの変化により、最終的には器官機能の衰退をもたらす[17,18]。実際、同じ組織の細胞間でも、加齢に伴って遺伝子発現に大きなばらつきがある。マウスの心筋細胞を用いた実験では、7つのハウスキーピング遺伝子、3つの心臓特異的遺伝子、2つのプロテアーゼ遺伝子を含む核遺伝子について調べたところ、すべての遺伝子で有意なばらつきが見られた[18]。加齢に伴う遺伝子の不均一性の増加は組織特異的で、例えば小腸細胞では主に点変異が蓄積し、心筋細胞では大きな再配列が見られる[19]。この遺伝子の不均一性は、隣接する細胞間の遺伝子発現を確率的に調節し老化に導くと提唱されている[18]。また、遺伝子の不安定性は、損傷が大きく、幹細胞のアポトーシスや老化を引き起こし、分裂能力のある細胞の枯渇につながる場合にも、老化を引き起こす可能性がある[17]。
ライバルのプロセス
がんと老化は、ある意味で相反するプロセスである[18]。一方、がんは、成長および転移に関して、腫瘍細胞に有利な変異の結果である(図1)。一方、老化は、細胞の生理機能に有害な変異や、老化、アポトーシス、そして最終的には幹細胞の枯渇につながるダメージの結果である[17,20,21]。p53のような細胞周期のゲートキーパータンパク質が高レベルになると、有害な突然変異や腫瘍形成から守るために老化とアポトーシスが起こり、逆に低レベルになると腫瘍形成性の突然変異を持つ細胞が増殖して癌を促進するという拮抗的多因子の考え方が登場する[17,21]。このようにゲノムの不安定性は、時間の経過に伴う細胞の消耗を反映しており、加齢に伴う細胞死の制御と組織の喪失につながる。しかし、それはまた、癌における制御されない細胞分裂と拡大を助長する、変化の蓄積による確率的リスクをも表しており、恒常性のために多くの細胞分裂を必要とする組織の生涯癌リスクが高いことからも明らかであり、組織の癌の変異の第一の原因が単なる「不運」であることを示している[22]。したがって、ゲノムの不安定性をより深く理解することは、プロセスとしての老化と発癌における役割の両方との関係を理解するのに役立ち、両分野における治療的介入の可能性をもたらすかもしれない。

図1 加齢とがんにおける幹細胞の生涯的な相互作用
幹細胞や前駆細胞における変化という観点から、老化や癌を見る単純化されたモデル。生物の寿命が延びると、長寿命の細胞(幹細胞など)は、通常の代謝から生じる細胞内酸化物質を含む多くのストレスによってDNA損傷を蓄積する。このような損傷を受けた幹細胞は、成長停止、アポトーシス、老化のいずれかを経るのが既定の経路である。より多くの幹細胞が増殖の場から退くにつれて、幹細胞や前駆細胞の全体的な数や機能が減少する。このような減少は、組織の恒常性や再生能力を損なう素因となり、老化や加齢に関連した病態の一因となる可能性がある。おそらく、一部の稀な細胞は、DNAが損傷しても増殖を続けることができるような変異を獲得することによって、この正常な既定経路から逃れることができるのであろう。このような、増殖はするが傷ついた細胞は、将来の悪性腫瘍の種となるかもしれない。このシナリオでは、癌も老化も、主として幹細胞や前駆細胞区画に蓄積した損傷に起因している。DNA損傷などの正常な成長停止シグナルがあっても幹細胞が増殖を続けるような突然変異(例えば、p16INK4aの欠損やテロメラーゼの再活性化)は、幹細胞プールを一時的に拡大し、その結果、加齢に関連する病態を遅らせることができる。長期的には、これらの変異は癌の可能性を高めるだろう。
正常な老化の過程で、幹細胞は損傷を蓄積し、その後ストレス依存的な変化、例えばCDKN2a(p16INK4a/ARF)遺伝子座の脱抑制やテロメアの短縮が起こる。その結果、分化した組織内に老化細胞(大きな六角形の細胞)が多くなる。幹細胞から直接、あるいはよりコミットした細胞から発生した前腫瘍性リージョンは、急速な増殖を行う(アスタリスクで示された小さな細胞)。これらの前がん細胞は、がん遺伝子の存在もあって急速に障害を蓄積し、老化細胞の割合が高くなる(白色で塗りつぶした六角形で示した細胞)。腫瘍細胞が老化プログラムを阻害する変異(例えば、Trp53やCDKN2aの変異)を獲得すると、完全な悪性腫瘍への進行が促進される。
イラストはFinkel T,Serrano M,Blasco MA.を基に修正したものである。がんと老化の共通の生物学。Nature.2007 Aug 16;448(7155):767-74.著作権©2007.
2.テロメア減耗
正常な人間の細胞の多くは、制御された細胞死(アポトーシス)を起こすまでに限られた時間しかない。細胞は、DNAのキャップエンドを使い切り、アポトーシスプログラムに入る前に、限られた数の細胞分裂しか行うことができない。アポトーシスは、細胞が永遠に生き続け、古い細胞が入れ替わることで突然変異が蓄積されるのを防ぐ。DNAのキャップエンドはテロメアと呼ばれ、各細胞の「命の時計」のような役割を果たし、限られた複製能力をカウントダウンしている[23]。テロメアの機能は、加齢と癌の両方において障害されている[24]。
テロメアとは、染色体の末端にある保護的な反復塩基配列のことである。DNAポリメラーゼはDNA鎖の末端を完全に複製することができないため、細胞分裂のたびに染色体末端は徐々に短縮されていく。真核生物のほとんどの体細胞は、テロメアを伸長させる酵素であるテロメラーゼを発現していないため、DNAのコード化が損なわれるまでの細胞分裂の回数に限りがある。幹細胞やがん細胞ではテロメラーゼの活性が高く、これらの細胞は不死である。体細胞のDNAが不安定にならないように、テロメアがある一定以上短縮して細胞分裂のヘイフリック限界に達すると、複製性老化に入る[23,25]。ヘイフリック限界(Leonard Hayflickにちなんで命名)は、細胞培養中のヒト胎児細胞が40-60回しか細胞分裂できず、それ以上の細胞分裂は停止するという観察に基づいている。この原因は、細胞分裂のたびにテロメアが徐々に短くなり、コーディングDNAを傷つけなければこれ以上細胞分裂ができない臨界点に達することにあることが、後に証明されている[25,26]。テロメアはまた、その制御に重要な保護タンパク質と関連している[27]。
テロメアの動態は、加齢、加齢性疾患、癌の重要な要因である。テロメアの維持は、遺伝的な要因と、ストレス、うつ病、喫煙、運動などの非遺伝的な要因の両方によって決定される[27]。
テロメア維持の構成要素に単一遺伝子の不活性化変異が起こる遺伝性テロメア症候群が知られている。これらの変異は通常、生体内で短いテロメアをもたらす。患者は通常、免疫機能の低下や特定の癌に対する感受性に加えて、糖尿病、心血管疾患、白髪、皮膚の色素沈着の変化など、老化を促進する表現型を示す[27,28]。
テロメアと老化
一般集団において、短いテロメアは、免疫機能の低下[29]、糖尿病[30]、心血管疾患[31,32]、変形性関節症[33]、心房細動[34]、アルツハイマー病[35]など多くの老化関連疾患と関連している。テロメア長が短いことは、60歳以上の加齢性疾患による早期死亡とも関連し[36]、テロメラーゼの過剰発現は老化を抑制するが、腫瘍形成の増加を犠牲にしている[37,38]。テロメアの長さは遺伝性が高く、ある研究では、テロメアの長さの50%以上を遺伝性が占め、どの年齢でも息子は娘よりテロメアが短いことが判明した。例外的に長寿でテロメアが長い家系の一親等は、二親等よりもテロメアが長く、また配偶者よりも長いことが示された[39]。
テロメアとがん
癌細胞におけるテロメラーゼのアップレギュレーションは、癌細胞が無限の複製能力を獲得する上で極めて重要なイベントであり、癌の特徴の一つである。ヒトの完全悪性腫瘍の80-90%では、正常組織と比較してテロメラーゼがアップレギュレートされている[27]。この酵素、ヒトテロメラーゼ逆転写酵素(hTERT)は、細胞分裂のたびに短くなるキャップエンドを元に戻す能力を持っている。hTERTによって短くなり続けるキャップエンドを回避することで、細胞はHayflick limitを回避し、アポトーシスに移行することはない。血液中の白血球のテロメアの長さと癌のリスクや死亡率との関連については、いくつかの研究やメタアナリシスで検討されている。いくつかの研究では、テロメア減少やテロメア短小ががん発症の危険因子であることが示されている[40-42]。膀胱がんの調整オッズ比(OR)は、テロメアが最も短い四分位の人は、テロメアが最も長い四分位の人と比べて1.88であった[40]。これは、肺がん(OR=2.39)、喫煙関連がん(OR=2.25)、消化器系のがん(OR=1.69)、泌尿生殖器系のがん(OR=1.73)にも当てはまる。10年間の人口追跡調査での発生率は、テロメア最長群で1,000人あたり5.1人、中間群で14.2人、テロメア短群で22.5人であった[43]。他の研究でも、がん死亡率との関連が示されている[44-46]。早期死亡の多変量調整ハザード比は、テロメアが最も短い4分位の人と最も長い10分位の人で1.31と1.43であり[44]、63,637人の7年間の大規模追跡調査では、癌死亡の多変量調整ハザード比は、最も短い人と最も長い10分位で1.35だった[46]。したがって、hTERTは、ある場合にはがん発症のリスク因子として作用し、別の場合にはがんの侵攻性のドライバーとして作用する可能性がある。この関連は、癌の種類によって異なり、ある癌は他の癌よりもテロメラーゼへの依存度が高いかもしれない。メタアナリシスでは、短いテロメアと膀胱癌、食道癌、胃癌、頭頸部癌、卵巣癌、腎臓癌との間に強い関連があることが示されている[47]。
老化と癌に共通する役割
最近の研究では、加齢に伴うテロメアの減少は、後に癌を発症する人ほど速いが、癌と診断される前に減少し、診断の3-4年前にテロメアが長くなることが示された[48]。これは、テロメア短縮が初期の発癌に関与し、その後癌に乗っ取られ、テロメラーゼ活性化などのテロメア伸長法が開始されることを表しているのかもしれない[48]。これは、がんの発生と進行の主要なプレーヤーである血中白血球にも影響を与えるかもしれない。このことが将来の研究で検証されれば、癌の早期発見のためのバイオマーカーとしての可能性を指し示すかもしれない[48]。また、hTERTを治療的に操作することで、癌治療としての可能性も期待できるかもしれない。さらに、これが老化プロセスにどのように影響するのか、治療的な観点からも追求されている。
3.エピジェネティックな変化
エピジェネティクスとは、DNAメチル化、クロマチンリモデリング、ノンコーディングDNAなど、DNA配列を変化させずに遺伝子発現を変化させること。
DNAメチル化
DNAメチル化は遺伝子制御の一般的なエピジェネティック機構であり、プロモーター領域のハイパーメチル化は遺伝子サイレンシングと関連し、ハイメチル化は遺伝子転写の増加と関連する。
メチル化は、多くのゲノムワイドなメチル化研究において、ヒトの年齢を予測する因子として示唆されている[49]が、その関係は複雑である。年齢が上がると、全体的にメチル化が進むが、多くの癌抑制遺伝子など、多くの遺伝子座がメチル化されている[2,50]。プロジェロイド症候群の患者やマウスの細胞も、正常な加齢に見られるようなメチル化パターンを示している[51]。13,000人以上の参加者を対象とした大規模なメタアナリシスでは、「エピジェネティック年齢」とも呼ばれるDNAメチル化ベースのバイオマーカーが、年代、人種、その他の危険因子とは無関係に全死因死亡を予測することが示された。これは、調査したすべての民族グループ(非ヒスパニック系白人、ヒスパニック系、アフリカ系アメリカ人)において一貫していた[52]。「エピジェネティック年齢」とは、Horvath[53]とHannum[54]によって報告された年代を強固に推定するもので、定義された数のCpGジヌクレオチドマーカー上のメチル化レベルに基づいている[52]。
がん細胞は、細胞周期制御因子p21、p16INK4a、Rb、BRCA1に代表されるDNA修復遺伝子やレチノイン酸受容体(RARB)などの腫瘍抑制遺伝子の高メチル化とサイレンシングによって広く特徴づけられている[55]。癌は癌原遺伝子の低メチル化と活性化にも関連しており、これは染色体不安定性を引き起こすことで発癌の原因となっているようである[56]。例えば、転移性大腸がんでは、特定のlong interspersed nuclear element-1(LINE-1)の低メチル化は、活性化した癌原遺伝子MET、RAB3IP、CHRM3と関連しており[57]、推定癌原遺伝子ELMO3の低メチル化および活性化は、肺癌発症と転移に関連している[58]。
異常なメチル化パターンは、ほとんどすべての新生物で観察され、がんの予防、予後、治療における分子マーカーとしての可能性を持っている[55]。レスベラトロール、緑茶、クルクミンなどのいくつかの天然化合物は、エピジェネティックな変化のメカニズムを通じて、がんの予防と治療における可能性を示している[59]。
ヒストン修飾
ヒストンは、共有結合かつ可逆的なアセチル化、メチル化、スモリエーション、ADPリボシル化、ユビキチン化、リン酸化によって変化させることができる[55]。これにより、ヒストン上のアミノ残基の電荷が変化し、負に帯電したDNA鎖と強くまたは緩く結合するようになり、活発に転写される緩いユークロマチンまたは強く沈黙するヘテロクロマチンになる。これらのエピジェネティックな変化は、転写制御機構のリクルートメントにも作用する[55]。エピジェネティックな変化は、ヒストン尾部の多くの異なる場所で起こり、メチルトランスフェラーゼ、デメチラーゼ、アセチルトランスフェラーゼ、デアセチラーゼなどの多くの酵素によって制御されている。この制御は非常に複雑で、多くの人が関わっているが、ヒストンの修飾が老化やがんに関係しているという研究は数多くあり、関与する酵素を阻害する薬剤の研究が進められている。これらの薬剤は、がん治療や健康的な老化を促進するために使用できる可能性がある。腫瘍のDNA配列を大量に解析した結果、ヒストン・メチルトランスフェラーゼはがんで頻繁に変異しており、乳癌の全ゲノム配列決定で同定されたドライバー遺伝子の5%を占めていることが明らかになった[60,61]。これはまた、薬物支配の重要なターゲットとなる可能性があり、多くの研究が進行中である[61]。
加齢はまた、ヒストンやクロマチン分布の特異的な複合的変化と関連している。ヒストン-3リジン-9トリメチル化(H3K9me3)は構成的ヘテロクロマチンの特徴であり、H3K9me2もまたヘテロクロマチンに関連している。加齢は、構成的ヘテロクロマチン遺伝子座に対する抑制の喪失と、他のゲノム領域における相補的ヘテロクロマチンの獲得に伴う、両者の減少または再分配に関連している[62]。H3Kme3は転写開始部位と関連しており、活発な転写と関連しており、レベルの上昇は高齢化と関連している[62]。これらは、加齢に関連したヒストン修飾の新規かつ刺激的な分野のほんの一例に過ぎない。この分野の理解が深まれば、将来的には、制御酵素に介入する薬剤が、健康な加齢のための薬剤の一部となるかもしれない。
非コード化DNA
マイクロRNAに代表される非コードDNAもまた、遺伝子制御に重要な役割を担っており、発現している全遺伝子の80%までを制御している可能性があることを示唆する証拠が登場している[63]。加齢と癌の両方において、多くの重要な経路の制御にマイクロRNAが関与していることを示唆する証拠がある。例えば、老化、IGF-1、mammalian target of rapamycin(mTOR)などである[49]。
多くの異なるmicroRNAの異常な発現は、様々な種類の癌の発生と予後の両方に寄与する可能性がある[55]。一例として、乳がんではlet-7ファミリーの多くのメンバーがダウンレギュレートされており、これはエストロゲン受容体(ER)のアップレギュレーションと関連している。ER陽性乳癌組織で実験的にlet-7を過剰発現させると、細胞増殖の抑制とアポトーシスの誘導が見られた[55,64]。また、ER陽性乳癌組織で実験的にlet-7を過剰発現させると、細胞増殖の抑制とアポトーシスの誘導が見られた[55,64]。
miR-17-92クラスターは、癌と老化の両方に関与する6つのmiRNAから構成されている。これらの欠失はマウスでは新生児致死であり、発生と老化の制御因子である。発現は加齢とともに低下し、過剰発現はマウスの寿命延長と関連している[65]。また、このクラスターは心臓病、神経変性疾患、骨粗鬆症など、加齢に伴う様々な病態に対して保護的である[65]。細胞の老化や加齢に関与していることから、多面的な役割が示唆されるが、発現量が増加すると癌化を促進し、血漿中濃度が幅広い腫瘍で上昇することから、潜在的なアラームマーカーとしての役割も示唆されている[65]。
アンチセンスRNAはmiRNAを妨害するために使用することができ、これらは肝細胞癌、膵臓癌、グリオブラストーマおよび乳癌の放射線化学療法との併用による癌治療において有望であることが示されている[66]。
CircularRNA(circRNA)は、約100塩基の円形のRNA配列で、古くから知られていたが、その機能についてはほとんど知られていなかった。近年、これらのRNAが、細胞の老化、アポトーシス、細胞周期の調節、老化など、さまざまなプロセスにおいて重要な機能を担っていることが明らかになってきている。また、細胞シグナル伝達にも関与しており、大腸がんにおいて保護的な役割を果たす可能性があり[67]、他の多くの悪性腫瘍においても発現が変化している[68]。
4.プロテオスタシスの喪失
プロテオスタシスとは、私たちのプロテオームの恒常性、すなわち健全なタンパク質の回転のことである。プロテオスタシスのネットワークは、シャペロンを介したフォールディング、プロテアソームによる分解、オートファジーから構成されている。プロテオスタシスが破綻すると、ミスフォールディングやタンパク質毒性を持つポリペプチドの凝集体が蓄積・沈着し、細胞の活力が損なわれてしまう。これは、多くの老化関連疾患、特にアルツハイマー病やパーキンソン病などの神経変性疾患や、主に高齢者が罹患する世界で最も一般的な失明原因である白内障の特徴である[69,70]。加齢に伴いプロテオスタシスの劣化が進行し、老化した生体のほぼすべての組織には、タンパク質の凝集体や封入体がある程度蓄積される[71]。プロテオスタシスは癌の発生にも関与しており、プロテオスタシスを標的とした薬剤の研究は、新しい抗腫瘍薬の可能性として検討されており、その多くが臨床に導入されている。
分子シャペロン(熱ショックタンパク質)
シャペロンとコ・シャペロンは、進化的に保存された小分子で、ポリペプチドのフォールディングと機能的なタンパク質へのリフォールディングを助け、そのようなフォールディングが達成できない場合は分解系に誘導する役割を担っている。進化の過程でシャペロンは、免疫系の調節、細胞分化、遺伝子発現、DNA複製、シグナル伝達、プログラム細胞死、細胞老化、発癌など、さらなるシャペロンの役割も獲得した[72]。これらのシャペロンの中で最も重要なものは、熱ショックタンパク質(HSP)であり、その転写は細胞ストレスの際にアップレギュレートされる。
加齢は、ストレスに対するシャペロンシステムの誘導の障害と関連しており、その結果、ミスフォールドしたタンパク質の蓄積が起こる[73]。シャペロンの過剰発現は実験動物の長寿と関連している[2]ので、HSPの誘導は損傷した蓄積タンパク質をリフォールディングすることで老化を止める可能性がある[74]。HSPのレベルは、神経細胞を含むほとんどの組織で加齢とともに減少し、熱ショックタンパク質の主要な転写因子であるHSF(heat shock factor)も同様である[74]。HSPは折りたたみの補助としてだけでなく、シャペロンを介したオートファジーと呼ばれるプロセスで、タンパク質をリソソームへ誘導することができる。このプロセスは、飢餓や酸化ストレスによって誘導される[74]。これは、飢餓がタンパク質のターンオーバーを増加させ、損傷したタンパク質を分解させることによって、寿命を延ばすメカニズムの一つであると考えられている[75]。また、加齢に伴うシャペロンシステムの調節障害は、両者が密接に関連していることから、免疫システムの調節障害を引き起こすことが提唱されている[76]。
シャペロン系は、癌の発生と進行にも関与しているが、老化のように活性が低下するのではなく、過剰な活性が常態化しているようである。大腸、子宮頸部、前立腺を含む試験されたほとんどのがんで、ほとんどの熱ショック蛋白質のレベルが著しく上昇している[72]。シャペロンは、細胞増殖、浸潤、新生血管形成の誘導、転移プロセス、免疫寛容の誘導など、様々な発癌プロセスに関与しており、様々な種類のヒト癌に関与していることが示唆されている[72]。これは、HER2、ALK、EGFR、BRAFなどの重要なシグナル伝達経路のメンバーとの関連などのメカニズムによって達成され、これらの調節障害はいくつかのがんと関連している[72,77]。
シャペロンはまた、腫瘍抑制因子(p53など)と結合して不活性化し、血管新生に関与するVEGFや一酸化窒素合成酵素を安定化し、シグナル伝達分子として作用して分泌されることができる[72]。腫瘍抑制性のアミロイド形成に対する保護もまた重要な特徴である[78]。いくつかのHSPの過剰発現は、多くの癌において予後不良とも関連している[72,78]。
HSPは、細胞の老化やアポトーシスの誘導を抑制し、事実上、化学療法から癌細胞を保護することができる[79]。その結果、癌の発生と進行におけるその重要性から、癌治療の標的としてのシャペロンに関する研究が盛んに行われるようになった。
現在、いくつかのHSP90の阻害剤が臨床試験中であり、臨床への道を歩んでいる。これらは単剤または化学療法との併用が可能であり、非小細胞肺癌のサブグループに対して顕著な可能性を示し、乳癌、消化管間質腫瘍(GIST)、メラノーマおよびいくつかの血液悪性腫瘍において有望な結果を示した[80](Non-Small Cell Lancer,Inc.前臨床試験において、HSP90阻害剤と放射線療法の併用は、通常放射線化学療法に抵抗性の肝細胞癌に大きな結果を示している[81]。HSP70阻害剤は前臨床試験で多発性骨髄腫に抗癌効果を示し[82]、HSP27阻害剤は去勢抵抗性前立腺癌に効果を示し、現在第2相臨床試験が進行中である[83]。シャペロン標的薬は、がん治療における新しいグループとして有望であり、現在、いくつかの研究路線が進行中である。
プロテアソーム
ユビキチン-プロテアソーム経路とオートファジー-リソソーム経路は主要なタンパク質分解システムであり、これらもまたプロテオスタシスに必須である。プロテアソームの活性化は、様々な実験生物において、生体内試験と試験管内試験で老化を遅らせる[84]。プロテアソーム活性は百寿者でも高く、彼らの健康的な老化のメカニズムの1つとして仮定されている[85]。食事性脂肪酸、花粉、藻類エキス、香辛料、いくつかの合成化合物など、プロテアソーム活性を高める化合物は、実験動物やヒト細胞株における健康寿命の延長につながった[85]。
プロテアソーム阻害剤は、主に多発性骨髄腫や異なるリンパ腫の治療薬として、すでに臨床に登場している。ボルテゾミブ(ベルケイド)は 2003年に承認された最初の阻害剤であるが、その後、いくつかの第2世代の阻害剤が登場した。ボルテゾミブは、疾患のあらゆる段階において多発性骨髄腫治療の定番となっており、一般に強力なグルココルチコイドや化学療法剤と併用されている[86](Partezomib is a standard in the multiple myeloma treatment)。ボルテゾミブは、現在、マントル細胞リンパ腫にも使用されており、他の多くのリンパ腫においても良好な効果を示している[86]。
オートファジー-リゾソーム
オートファジーは、リソソームを介したミスフォールドタンパク質、損傷したオルガネラ、細胞内病原体の分解方法である。オートファジーは、細胞質内の物質がリソソームに運ばれ、そこで分解される、細胞内の主要な分解システムである。細胞内物質の除去・分解は重要であるが、オートファジーの唯一の目的は、単純な物質の除去ではない。むしろ、オートファジーは、細胞の修復と恒常性維持のための新しいビルディングブロックとエネルギーを生み出す動的なリサイクルシステムとしても機能する[87]。オートファジーの後,結果として生じる分解産物は、新たなタンパク質合成,エネルギー生産,グルコネシン生成など、さまざまな目的に使用することができる。
オートファジーと老化-カロリー制限の役割
栄養失調を伴わないカロリー制限(通常、自由摂取量の20〜40%)は、長い間、最も確立された強力な延命方法の一つであり、この効果を媒介すると考えられるメカニズムの多くがオートファジーに収斂している。マウスを用いた研究により、55~65%のカロリー制限食を生涯にわたって与えたマウスは、非精製自由食を食べるマウスよりも平均寿命および最大寿命が35~65%大きいことが明らかにされている[88,89]。また、カロリー制限されたげっ歯類や非ヒト霊長類では、長期のカロリー制限は、糖尿病や癌などの加齢関連疾患の発症を遅らせることが判明している[88]。太り過ぎのヒトにおけるカロリー制限は、心臓の危険因子を減らし、インスリン感受性を改善し、ミトコンドリア機能を強化し、DNAの酸化的損傷を減らすことが示されている[88]。ヒトにおける延命効果について結論づけるのはまだ早いが、健康上の利点の証拠は現れている。カロリー制限にはいくつかのメカニズムがある。COX、NF-kB、MAPK経路の阻害による抗酸化作用と抗炎症作用がある[90]。カロリー制限はまた、グローバルなDNAメチル化を変化させ、ヒストン脱アセチル化酵素、中でもサーチュインをアップレギュレートし、クロマチン変化を引き起こす[90]。サーチュインはまた、まだ十分に解明されていない多くの転写因子との幅広い相互作用を通じて効果を発揮する[90]。カロリー制限もまた、上記と同じ経路を通じて、またmTORシグナルの減少、成長因子シグナルの減少、血管障害の減少を通じて、がんのイニシエーションと進行を抑制する強力な方法である[91]。カロリー制限は、mTORの抑制やサーチュインのアップレギュレーションなど、リソソーム-オートファジー活性の増加を媒介するいくつかのシグナル伝達経路をアップレギュレートする[92]。マウスを含むいくつかの実験動物において、オートファジーは寿命を延ばすのに十分であり、いくつかのオートファジー構成要素をノックダウンすると、この効果が消失する[93]。オートファジー遺伝子多型は、変形性関節症、老人性骨粗鬆症、神経変性疾患などの加齢関連疾患と関連している[49]。モデル生物において、オートファジーを増加させ、寿命を延ばし、加齢に伴う神経変性疾患の転帰を改善するいくつかの化合物が発見されている。その中には、mTOR阻害剤のラパマイシン、アセチルトランスフェラーゼ阻害剤のスペルミジン、赤ブドウの皮から単離され、フレンチパラドックスのワイン理論の中心的化合物として広まったレスベラトロールなどがある[49,94]。
オートファジーとがん-二重の役割
オートファジーはほとんどのがんでアップレギュレートされ、生存に重要である。特に治療後はほとんどの抗癌剤がアップレギュレーションを引き起こすため、オートファジーが化学療法抵抗性の方法として機能するという理論が生まれる[95]。オートファジーは、欠陥のあるミトコンドリアを分解し、また脂肪やタンパク質を分解して、高いエネルギーを必要とする腫瘍細胞にエネルギーとグルタミンを供給することによって機能する[96]。いくつかの証拠から、オートファジーは発癌において、一方では腫瘍の成長を促進するメカニズムを通して、他方では腫瘍抑制効果を誘導するという二重の役割を担っていることが示唆されている(図2)。NSCLCと膵臓癌の遺伝子操作マウスモデルでオートファジーをノックアウトすると、悪性腫瘍が良性の稀なオンコサイトーマ(欠陥ミトコンドリアが蓄積した腫瘍)に変化する[96]。
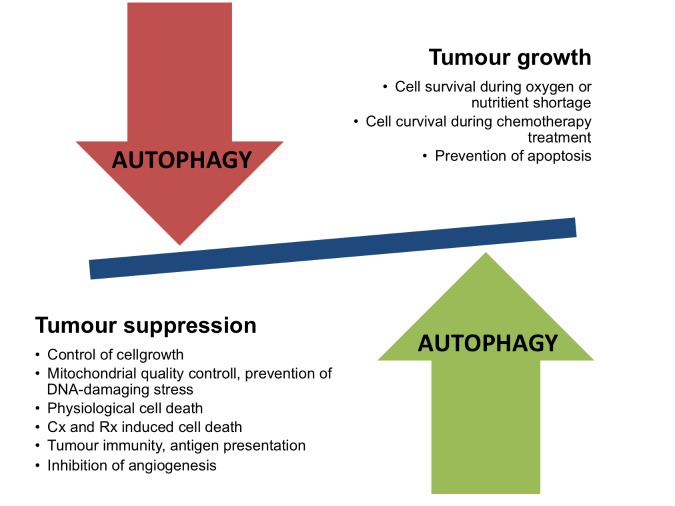
図2 がんにおけるオートファジーの二重の役割
オートファジーが関与する、腫瘍抑制または腫瘍増殖に関連するメカニズムの例。Cxは化学療法、Rxは放射線療法を表す。
抗がん剤による治療がオートファジーをアップレギュレートすることから、がん細胞株や実験動物において、抗がん剤とオートファジーを阻害する抗マラリア薬であるクロロキンやヒドロキシクロロキンを同時に投与した研究が数多く行われている。この組み合わせは、ほとんどの場合において、がん細胞に対する細胞毒性およびアポトーシスの増加を示している[95]。注目すべきは、これらの薬剤が、将来、抗悪性腫瘍剤に関する増大する兵器庫に組み込まれる可能性があることである。
5.栄養センシングの乱れとミトコンドリア機能障害
霊長類を含む様々な実験動物において、長寿を得るための最も早くかつ最も効果的な方法の1つがカロリー制限であり、栄養と代謝は長い間、がんとも関連してきた。人間の老化に共通する特徴として、内臓脂肪の増加と除脂肪体重の減少、インスリン抵抗性、筋肉のボロボロの赤線維の増加(筋肉細胞内の欠陥ミトコンドリアの蓄積)などがある[49]。これらの特徴は、がんを含む老齢期の疾患と関連している。
インスリン様成長因子
インスリン様成長因子(IGF-1)は、成長ホルモンに応答して発現が上昇し、インスリンと同じ経路でシグナル伝達を行う。このシグナル伝達経路を合わせてIIS(インスリンとIGF-1のシグナル伝達)経路と呼び、進化上最も保存された老化制御経路であり、AKT、mTOR、RAS、FOXOなど多くの標的を持っている[2,97]。このシグナル伝達経路のどのレベルでもシグナル伝達を減衰させる遺伝子多型や突然変異は、長寿の増加と関連しており、この経路はカロリー制限の有益な効果と関連している[98]。インスリンとIGFのシグナル伝達は、アポトーシスを抑制し、細胞増殖と分裂を促進することが示されており、遺伝的に変化した細胞の寿命を延ばす可能性があり、大腸がん、前立腺がん、乳がんに関連している[97,99]。IGF1およびIGFBP3の循環レベルは、前立腺がんの進行した男性における全死因死亡率および前立腺がんの発症リスクと関連している[97]。IGF1およびIGFBP3の循環レベルも、いくつかの報告では、全死因死亡率および乳癌の発症と関連しているが、研究は矛盾している[97,100]。循環血中IGF1の高値は、大腸癌の局所進行期および低分化症例とも関連している[97]。がんにおける全体的な役割は存在するようであるが、がんの種類やサンドステージに対する寄与の大きさや具体的な寄与はまだ不明である。
糖尿病
2型糖尿病は、膵臓がんの第3の危険因子であり、子宮内膜がんにも関連している[101,102]。大規模なメタ分析では、肥満手術の有無にかかわらず、意図的な体重減少もまた、がんリスクの有意な減少に関連していたが、IGFに関する結果が一貫していなかったため、これは主にエストロゲンおよび炎症性サイトカインの減少に起因していた[103]。現在進行中の研究では、がん治療におけるIIS阻害剤の可能性を探っているが、他の場所でレビューされているように、結果はまちまちである[104]。
mTOR経路とAMPキナーゼ(AMPK)経路
老化と癌の両方に関与する他の重要な栄養感知経路の参加者は、mTORとAMPキナーゼ(AMPK)である。mTORは、細胞の代謝、成長、増殖に重要な役割を担っている[105]。mTOR経路の遺伝的ダウンレギュレーションは、広範囲の実験動物において寿命を延長し、マウスにおけるラパマイシンによる治療は寿命を延長し、哺乳類における最も強固な化学的延命剤と考えられている[2]。いくつかのmTOR阻害剤は、主に免疫抑制剤および抗腫瘍剤として使用され、臨床に参入している。アップレギュレートされたmTORシグナルは、大腸がん、前立腺がん、乳がんを含む多くの異なるがんの発生、進行、転移に関与している[105]。mTOR阻害剤であるTemsirolimusは、現在、進行性腎細胞癌とマントル細胞リンパ腫の治療に使用されている。エブロリムスは、腎細胞がん、膵神経内分泌腫瘍、消化管や肺由来の非機能性NET(神経内分泌)腫瘍[106]、特定の星細胞腫、ホルモン受容体陽性進行乳がんに使用されている。新たな適応症を調査する研究が進行中であり、第二世代のmTOR阻害剤も進行中である。2型糖尿病の治療薬として一般的に使用されているメトホルミンは、AMPキナーゼを活性化し、他の効果とともにmTOR経路の阻害を引き起こす。メトホルミンは、試験管内試験で様々な癌細胞株の増殖を抑制し、様々な実験動物で寿命を著しく延長させる[2,107,108]。
6.細胞老化
細胞老化は、細胞が不可逆的に分裂を停止し、クロマチンの変化やセクレトームの変化を含む特定の表現型の変化を受けるプロセスである[49]。細胞老化は、テロメアの短縮、癌遺伝子の活性化、DNA損傷応答(DDR)経路、p16INK4aの活性化など、様々なストレス機構によって引き起こされる可能性がある。細胞老化の重要な実行者は、発癌に関与する腫瘍抑制タンパク質であるRetinoblastoma 1(Rb1)とp53のようである[109]。CDKN2Aの漸進的な抑制解除と細胞周期阻害剤p16INK4aおよびp19ARFの転写を通じて、細胞は分裂刺激と細胞分裂の履歴を記憶し、最終的にp53とRb1の増加によって細胞老化を引き起こすことがデータから示されている。この記憶には、ポリコーム複合体やヒストンメチル化を介したエピジェネティックな変化が関与しているようである[109]。
老化は、テロメア減少を起こした細胞や癌遺伝子を過剰発現している細胞の増殖を抑制することにより、抗癌機構として働く。この抗癌作用は、前癌病変に多くの老化細胞が見られることからも裏付けられている[110]。これは、突然変異を起こした潜在的に有害な細胞株の拡大を抑制する初期段階での保護効果かもしれないが、加齢に伴うこのような老化細胞の蓄積は有害となりうる[111]。老化細胞はまた、炎症性サイトカインや成長因子の熱心な分泌者であり、老化関連分泌表現型(SASP)として知られ、この分泌は癌の発生と老化の両方に関与しているため、老化は腫瘍促進効果を持つ可能性もある[112]。これらの中には、炎症性因子であるTNF-α、IL-6、マトリックスメタロプロテアーゼ、単球走化性タンパク質-1、およびFGF、EGF、VEGFなどの成長因子がある[111,113]。炎症は、肺の腫瘍形成を含む多くの悪性腫瘍のよく知られた危険因子であり、公害や煙によって開始され、おそらくmiRNAの調節が関与する[114,115]。老化細胞は通常、免疫監視と貪食によって除去されるが、加齢とともに徐々に蓄積されるようで、これは免疫老化による除去の低下と同様に産生の増加による可能性が高い[49]。免疫老化が老化に関与しているという証拠が増えつつあり、そのような老化はがん予防の代償であるかもしれない[112]。
p16INK4aとp53は共に老化の強力な誘導因子であり、癌に対する防御が老化を促進するという拮抗的な多面性を示す[112,116]。p16INK4aの発現はヒトおよび齧歯類において年齢と共に著しく増加し、老化の極めて優れたバイオマーカーとなる[109]。p16INK4aを欠損したマウスは、高い再生能力を示したが、その代償として自然発生癌や発癌物質による癌の発生率が増加した[112]。INK4a/ARF遺伝子座は、ゲノムワイド関連研究において加齢関連疾患に最も強く関連する遺伝子座であり[117]、マウスにおけるp16INK4aの条件付き発現は、加齢の生理学的特徴の多くをもたらす[118]。マウスでINK4aを発現する老化細胞を除去すると、老化と関連する疾患の発症を遅らせ、進行を停止させる[119]。
7.幹細胞
幹細胞は、私たちの生涯を通じて重要な役割を担っている(図1)。まず発生と成長期、そして成人期には維持と修復が行われる。幹細胞の枯渇や機能不全は、血液幹細胞の機能不全による貧血や免疫老化[120]、間葉系幹細胞の(喪失による)骨粗鬆症[121]、筋幹細胞の(喪失による)サルコペニア[122]など、老化に伴って見られる多くの病態生理過程を生じさせることがある。加齢に伴い、ホメオスタシスの維持や損傷後の組織修復の能力が低下することが研究により示されている。このような状態が続き、組織が恒常性の維持や適切な修復ができなくなると、生理的な衰えや老化が進行する[123]。
がんは老化に伴う疾患であり、がんの病因は複数の変異原性事象から構成されているが、そのようなリザーバーとして機能することができる唯一の細胞は、長寿命の幹細胞であると思われる。幹細胞は、前がん病変を蓄積するための理想的な細胞標的である[123]。
癌と老化は、十分な変異原性ヒットにさらされた幹細胞が迎える2つの結末と言えるかもしれない(図1)。生涯を通じて変異原性ヒットが起こり、それが遺伝的不安定性を引き起こすと、p16INK4aなどの腫瘍抑制因子が細胞周期を停止させ、アポトーシスや老化が引き起こされる。しかし、腫瘍抑制因子が破綻すると、癌が発生する可能性がある[109]。実際、老齢マウスの造血幹細胞は、若齢マウスの造血幹細胞よりも分裂回数が少なく、このことは、DNA損傷の蓄積とp16INK4aの発現に相関している[2,124]。
若いマウスの血液幹細胞は、年老いたマウスとは異なるT細胞やB細胞のサブセットを生み出すことができる。また、転写産物も老齢細胞と若年細胞では根本的に異なっている。最後に、若いマウスではリンパ系が優勢であるのに対し、加齢とともに骨髄系に偏っていく。このことは、老齢と若齢の血液学的悪性腫瘍でも反映されている[123]。
結論
老化は人生の避けられない道であり、がんは高齢になるほど多くなり、若年者では白血病を除いて比較的まれであることから、通常、加齢に伴う疾患と考えられている。したがって、両者に多くの共通点があることは驚くにはあたらない。しかし、がん細胞と老化細胞は基本的に正反対である。がん細胞は、有利な変異を持ち、細胞分裂が早く、エネルギー消費量が多い多動性細胞であり、老化細胞は不利な変異が蓄積し、細胞分裂できず、エネルギー生産・消費能力も低下した低動性細胞と考えられるからだ。このことは、両者の特徴にも表れている。
加齢とがんは、ゲノムの不安定性など多くの共通点がある。がん細胞は突然変異の恩恵を受けることが多いが、その他の細胞は有害な突然変異を蓄積し、生理機能の低下や老化を引き起こす。また、テロメアの減少も共通の特徴であるが、がん細胞はテロメラーゼを活性化することで細胞周期の停止を回避している。エピジェネティックな変化は両者に共通しており、その研究・知見は急速に蓄積されている。DNAやヒストンの共有結合修飾を担う酵素の阻害剤や、マイクロRNAを妨害するアンチセンス核酸には、大きな可能性がある。
プロテオスタシスは両者で変化し、タンパク質凝集体の蓄積とそれによる毒性は、多くの加齢性疾患の特徴である。がんはシャペロン、プロテアソーム、リソソーム活性をアップレギュレートすることでこれを回避している。すべての経路の阻害剤はすでに腫瘍学的治療で確立されており、新しい世代や新しい薬剤が継続的に試験・開発されている。栄養センシングの異常は両者に共通しており、IIS経路やmTOR経路、AMPキナーゼ経路の活性化を阻害することで寿命が延び、がんを食い止めることができるとされている。多くのmTOR経路阻害剤が臨床応用されており、AMPK活性化剤であるメトホルミンは、がん治療と健康な老化の両方に可能性を持っている。
細胞の老化は、がんと加齢が根本的に異なる特徴の一つである。通常、DNA損傷が蓄積すると、細胞周期阻害因子の発現が上昇し、老化やアポトーシスに至るが、悪性細胞は、p16INK4aやp53などの癌抑制因子の欠失などの変異を追加して、これを回避するからだ。
つまり、老化とがんは時間的にもメカニズム的にも相互に関連しており、同じ戦略や薬剤の多くが両者をターゲットにすることができる一方、拮抗的な多面性が作用し、一方の阻害が他方を活性化する場合もある。