
Contents
Tau in Alzheimer’s Disease: Pathological Alterations and an Attractive Therapeutic Target
pubmed.ncbi.nlm.nih.gov/33428128/
2021年1月11日
概要
アルツハイマー病は加齢に伴う神経変性疾患であり、主に以下の2つの特徴がある。
特徴:アミロイドβ(Aβ)からなる細胞外アミロイド斑と、異常にリン酸化亢進したタウからなる細胞内神経原線維のもつれ(NFT)。NFTsの数はアルツハイマー病患者の認知症の重症度と正の相関がある。しかし、アルツハイマー病の治療や予防に有効な治療法はまだない。
アルツハイマー病の病態をより深く理解することで、過去数十年の間に特定の治療法を生み出すための新たな戦略が明らかになってきた。いくつかの研究では、脳内でのタウ病理のプリオン様の播種と拡散が、アルツハイマー病の主要なドライバーである可能性が示唆されている。
タウタンパク質は、様々な神経変性疾患における病理学的な役割が大きいことから、治療的介入の開発のための有望なターゲット候補と考えられている。タウの高リン酸化異常は、神経変性疾患の原因となることが知られている。
本稿では,アルツハイマー病におけるタウ蛋白質の病理学的メカニズムに関する最近の研究成果を紹介するとともに,タウを用いた治療戦略について簡単に述べる。
キーワード
アルツハイマー病、タウタンパク質、高リン酸化、タウ病理の伝播
アルツハイマー病は、段階的な記憶喪失と認知機能障害によってマークされた年齢に関連付けられている認知症である。主に高齢者が罹患し、患者の脳組織が退化していく。現在、世界で約5,000万人が認知症を経験していると推定されており、2050年には1億5,200万人以上になる可能性がある。アルツハイマー病は米国では死因の第6位であり、中国では65歳以上の認知症有病率は5.14%となっている。
細胞外アミロイド斑と細胞内神経原線維絡み(NFTs)は、アルツハイマー病の2つの特徴的な病理学的特徴である。NFTsは、高リン酸化タウのペアらせん状フィラメント(PHF)を含む脳内線維性の集合体である[1]。NFTsの数はアルツハイマー病患者の認知症の重症度と正の相関がある[2, 3] 。タウはアルツハイマー病脳の多くの部位で異常に高リン酸化されている[4, 5]。
高リン酸化されたタウは正常な機能を失い、神経毒性を獲得し、NFTに凝集する[6, 7]。リン酸化に加えて、タウの切り捨てもその凝集を促進する[7]。アルツハイマー病脳では、タウの病理は、脊髄/脊髄下複合体と経耳介領域で始まり、大脳辺縁系に向かって徐々に進行し、時折、大脳辺縁領域と大脳皮質にまで進行する[8, 9]。タウ病理の地理的広がりは、アルツハイマー病の進行と相関しているようである[10, 11]。アルツハイマー病の脳からのタウ凝集体の適用は、注入部位にタウ病理を引き起こし、解剖学的に脳領域を結びつける可能性がある。組換えタウをヘパリンでインキュベートすると、アルツハイマー病脳内でのタウ病理伝播に類似したタウ病理が生じることが試験管内試験実験で示されている[12-14]。タウ病理のプリオン様伝播はアルツハイマー病の進化に大きく関与している[15]。
アルツハイマー病の病態生理学的基盤を調査するための共同研究にもかかわらず、その正確なメカニズムはまだ十分に理解されていない。さらに悪いことに、前臨床研究で有望と思われた治療法は、ヒト臨床試験では失敗している。ここでは、アルツハイマー病関連のタウの生物学と病態生理の進歩をレビューし、簡単にタウベースの薬物療法について説明する。
1 タウタンパク質とタウの病態生理
1.1 タウタンパク質
タウは、脳内で微小管内のチューブリンの集合をサポートし、微小管を安定化させる微小管に関連する神経細胞タンパク質である。ヒトのタウをコードする微小管関連タンパク質タウ(MAPT)遺伝子は、染色体17q21.31に局在し、それは16のエクソンで構成されている。2つの(2N)1つの(1N)またはゼロの(0N)N末端インサートと4つの(4R)または3つの(3R)C末端微小管結合リピート(MTBR)を含む352から441残基に変化する6つのタウのアイソフォームは、エクソン2,3,および10の代替スプライシングのために成人ヒトの脳で発現している[16,17](図1A)。4R-および3R-タウの約等モル量がヒト成人脳で発現している[16, 18]。げっ歯類のタウはヒトのタウと約90%の同一性を持ち、4R-タウは主に成体げっ歯類で発現し、3R-タウは胎児および新生児げっ歯類でのみ発現している[18, 19]。4R-tauは3R-tauよりも微小管との結合親和性が高く、以前に結合していた3R-tauを脱離させることができる[20]。タウは3つのドメインで形成されている:N末端部分のプロジェクションドメイン(微小管表面からはみ出している)中央部分のプロリンリッチドメイン(Srcホモロジー3(SH3)ドメインを含むタンパク質との相互作用を担う)C末端部分のアセンブリードメイン(MTBRとフランキング領域を構成し、微小管のアセンブリーとタウの凝集をサポートする)[21] (図1B)。タウは主に軸索に発現しているが、体幹コンパートメントでも発現している[22, 23]。
1.2 タウのリン酸化
タウは、そのリン酸化が微小管へのタウの結合を調節するリン酸化タンパク質である。アルツハイマー病患者からの可溶性高リン酸化タウはタウ1モルあたり5~9リン酸モルを保持しているが、健康なヒトの脳の正常なタウは平均1.9リン酸モルを含んでいる[4]。
タウは、複数のスレオニン(Thr, T)セリン(Ser, S)チロシン(Tyr, Y)残基でのリン酸化によって修飾される。ヒトの最長の脳タウアイソフォームであるタウ441(クローン番号、タウ40)は、それぞれ35 T、45 Sおよび5 Y残基からなる85の可能なリン酸化部位を保持している。アルツハイマー病脳のPHF-tauには複数のリン酸化部位が存在する[24-26](図2A)。S199, S202/T205, T231, S262, S396, S422 残基でのタウリン酸化は、アルツハイマー病 の進行を評価するために示唆されている[27]。タウリン酸化は微小管への結合親和性、細胞内分布、軸索輸送など多くのプロセスを制御している[28-30]。タウリン酸化異常は微小管の解体を誘導し、神経細胞の細胞骨格組織における微小管の機能を変化させる[28-30]。一方、リン酸化されたタウはニューロンの細胞質に蓄積され、タウのオリゴマーや凝集体(タウのもつれや線維状のフィラメントなど)が出現する[31]。
アルツハイマー病脳におけるタウの高リン酸化は、キナーゼによるリン酸化の増加および/またはホスファターゼによる脱リン酸化の減少に起因する可能性がある。タウは、プロリン指向キナーゼ(PDPKs)と非PDPKsによって異常に高リン酸化されている[32]。プロリン(Pro, P)残基が先行するタウのS/T残基は、サイクリン依存性キナーゼ5(cdk5)C-Junアミノ末端キナーゼ(JNK)を含むPDPKによってリン酸化される。グリコーゲン合成酵素キナーゼ-3β(GSK-3β)細胞外シグナル調節キナーゼ(ERK)二重特異性チロシンリン酸化調節キナーゼ1A(Dyrk1A)cdc2様キナーゼ1(CLK1)[33-36](図2B)。2B). プロリン非依存性タウS/T残基は、cAMP依存性プロテインキナーゼA(PKA)カゼインキナーゼ1δ/ε(CK1δ/ε)カルシウム/カルモジュリン活性化プロテインキナーゼ(CaMKⅡ)微小管親和性調節キナーゼ(MARKs)プロテインキナーゼR(PKR)およびアデノシン一リン酸活性化プロテインキナーゼ(AMPK)を含む非PDPKによってリン酸化される[36-40](図2B)。これらのキナーゼのそれぞれは、アルツハイマー病におけるタウの病態に関して異なる役割を持っている。アルツハイマー病脳では、cdk5はSer残基199,202,235,396,404,Thr残基181,205,212,217,231でタウ40をリン酸化する;GSK-3βはS235を除いてcdk5によってリン酸化された全ての部位をリン酸化する[41]。ERK-2は、Ser残基46,202,235,396,404,およびThr残基175,181,205,212,217,231でタウをリン酸化し[27]、Dyrk1Aは、Ser残基199,202,396,400,404,422,およびThr残基181,205,212,217,231で試験管内試験でタウをリン酸化する[42,43](図2A)。2A). これらのタンパク質キナーゼのいくつかによるタウのリン酸化は、他のキナーゼによる追加のリン酸化のためにタンパク質を準備する[42, 44, 45]。対照的に、プロテインホスファターゼ(PP)には、プロテインホスファターゼ1(PP1)2A(PP2A)2B(PP2B)および5(PP5)の他に、組織非特異的アルカリホスファターゼ(TNAP)カルサイクリン結合タンパク質およびSiah-1相互作用タンパク質(CacyBP/SIP)を含む異なるタイプのプロテインホスファターゼ(PP)が存在する[46-52](図2B)。PP2Aはヒトの脳内のタウの脱リン酸化活性の約70%を担っており[47]、高リン酸化されたタウに関するその役割はアルツハイマー病において偏見を持たれている[53, 54]。我々は最近、PP2AとGSK-3βの調節障害が、お互いの活動に影響を与えることで、間接的にも直接的にもタウのリン酸化に影響を与えることを報告した[55-57]。PP2Aは、cdk5,PKA、CaMKⅡなどの他のタウプロテインキナーゼの機能にも影響を与え、タウのリン酸化を制御することができる[17]。
1.3 タウの切断
タウは、いくつかのサイトで異常に切り捨てられている[58,59]、カルパイン、カスパーゼ、アスパラギニルエンドペプチダーゼ(AEPs)とカテプシン[60-62]を含む多くのプロテアーゼによって触媒される。アルツハイマー病脳のNFTsでは、少なくとも3つの特定のタウ切断部位(N368,E391とD421)が認識されており、Braakステージの進行に関連付けられている[63-65]。NFTsのタウはC末端が切り捨てられており、脳脊髄液(脳脊髄液)中のタウ優勢型は、MTBRを含むC末端部分の半分も持っていない。一方、SDSや還元剤に耐性のある高分子量(HMW-tau)のアルツハイマー病脳由来のタウ凝集体はN末端領域を持たない[66, 67]。切り詰められたタウは完全長タウよりも凝集しやすい[62, 68, 69]。我々は最近、いくつかのタウの切り捨てがリン酸化の特定の部位を調節し、タウの自己凝集を増加させ、アルツハイマー病脳(アルツハイマー病 O-tau)からのオリゴメリックタウへの結合とアルツハイマー病 O-tauによってシードされた凝集を促進することを報告した[70]。すべての切り捨て形態の中で、Tau151-391は最も重篤な病理学的障害を引き起こし、試験管内試験の培養細胞ではアルツハイマー病 O-tauによってテンプレート化された最も強力な凝集作用を引き起こす[70]。タウの切り捨てはまた、ミトコンドリア機能不全およびシナプス障害を促進する可能性がある[71, 72]。タウのトランケーションは、タウの病態形成に不可欠な機能を持っている。
1.4 その他の翻訳後修飾(PTM)
タウはまた、アセチル化、O-GlcNA シリル化、メチル化、S-グアニル化、ユビキチン化、SUMOy-lation、ニトロ化、カルバミル化および糖化を含む他の PTM によって多くの部位で修飾され得る。
1.4.1 タウのアセチル化
MTBRとタウのフランキング領域に位置する20個以上のリジン(Lys, K)残基は、CREB結合タンパク質(CBP)またはヒストンアセチルトランスフェラーゼp300によってアセチル化され、タウの脱アセチル化はヒストン脱アセチル化酵素(HDAC)6またはサーチュイン1(SIRT1)によって触媒される[73-75]。タウのアセチル化はタウの分解を阻害し、微小管へのタウの結合を阻害し、タウの凝集を亢進させる[76]。タウ症脳ではタウアセチル化レベルが上昇する。アセチルトランスフェラーゼp300はアルツハイマー病脳やADマウスモデルでアップレギュレーションされている[76]。本質的なタウの自己アセチル化は、タンパク質分解による切断とタウフラグメントの生成に関連している[74, 77]。アルツハイマー病脳ではSIRT1レベルが低下しており[78]、アミロイドβ治療は培養ニューロンでのSIRT1発現を低下させ[79]、アセチル化タウ蓄積を永続化させる。したがって、アセチル化の亢進または脱アセチル化の低下は、アルツハイマー病におけるアセチル化タウの病理学的な上昇に寄与している[76]。
1.4.2 タウのO-GlcNAシリル化
O-GlcNAシリル化は、単糖β-N-アセチルグルコサミン(GlcNAc)がS残基またはT残基のヒドロキシに結合していることを特徴とする核細胞質タンパク質の修飾であり、通常、タウはS残基またはT残基のヒドロキシに結合している。通常、タウはO-GlcNAcyaltionによって修飾される[80, 81]。O-GlcNAcylationはO-GlcNAcトランスフェラーゼ(OGT)によって行われ、O-GlcNAc修飾はO-GlcNAcase(OGA)によって剥離される[82-84]。OGTとOGAの相対的な活性と細胞内のUDP-GlcNAc量がO-GlcNA化レベルを制御する[85]。タウ部位特異的リン酸化とO-GlcNAシリル化は互いに競合する。O-GlcNAcylationレベルの低下は、異常なタウの過剰リン酸化とNFTs形成を促進する[86]。Ca2+過負荷に起因するカルパインIの過剰活性化は、ニューロン特異的グルコーストランスポーター3(GLUT3)をタンパク質分解し、結果としてアルツハイマー病脳におけるタウO-GlcNAシリル化の低下につながる[87]。したがって、脳のグルコース取り込みの改善は、アルツハイマー病の予防と治療のための魅力的なターゲットとなるかもしれない。
1.4.3 タウのユビキチン化
ユビキチン化は、ユビキチン-プロテアソームシステム(UPS)の一部としてのタンパク質分解やプロテアソームに依存しない機能など、タンパク質機能の多くの側面を制御する非常に汎用性の高いPTMである[88, 89]。タウタンパク質のいくつかのLys残基はユビキチンリガーゼ(E3リガーゼ)によってユビキチン修飾されており、その中にはヒートショックプロテイン70-インターアクティングプロテイン(CHIP)や腫瘍壊死因子受容体(TNFR)関連因子6(TRAF6)のC末端領域が含まれる[90, 91]。タウのユビキチン化は、アルツハイマー病脳の神経斑やNFT、パーキンソン病(PD)やピック病(PiD)に関連するレビー小体やフィラメントで見られた[92, 93]。
海馬はアルツハイマー病患者の初期段階でプロテアソーム活性の低下を示す[94]。プロテアソーム機能は凝集したタンパク質の存在下で低下し、そのレベルはタウとアミロイドβ凝集によって上昇する可能性がある[95]。アルツハイマー病脳由来の可溶性PHFは主にMTBRの残基K254/311/353でユビキチン化されている[96]。ポリユビキチン化ではなく、PHFにおけるタウのモノユビキチン化は、UPSによって制御されるタウ凝集体のタンパク質分解を促進するのに十分ではない[97]。したがって、タウのポリユビキチン化分解を増加させることは、アルツハイマー病治療のための有望な治療戦略となる可能性がある[98]。
1.5 タウ凝集
ネイティブにアンフォールドされた可溶性タウモノマーは、一般的に凝集する性質を持たずにランダムなコイル構造を呈している[99]。タウMTBR中の6つの残基(275VQIINK 280および306VQIVYK 311)によって形成される2つのモチーフは、タウフィラメントの凝集と形成のための基本的なものである。これらのモチーフは、タウモノマーの二量体への凝集、および不溶性PHFへの可溶性タウオリゴマーの病理学的凝集、最終的にはNFTsへの凝集に責任があるβシート構造にそのコンフォメーションを切り替える傾向を持っている[100]。アルツハイマー病のPHFは6つのタウアイソフォームのすべてを持っている[15]。
タウが媒介する神経変性には凝集が必要であるが、タウ凝集の基礎となる完全なメカニズムを明らかにする必要がある。タウの凝集は異常なPTMによって促進され、リン酸化がこの凝集の主な引き金になると考えられている。NTF の数は アルツハイマー病 患者の認知症の重症度と正の相関があり、タウ凝集はもともと NFTs の毒性を引き起こすと考えられていた[3]。興味深いことに、トランスジェニックマウスやアルツハイマー病患者を用いた研究では、ほとんどの神経細胞がNFTsの形成により死滅する可能性があることが示されている。NFTs形成のかなり前に機能障害、シナプス喪失、認知機能障害が発生することから、可溶性タウオリゴマーが神経変性の主な原因である可能性があるが、神経変性を引き起こす正確なオリゴマーの種は同定されていない[37]。タウアイソフォームの中で最大のものである4R2N(tau40)のリン酸化または擬似リン酸化は、タウの凝集を増加させ、4R0N(tau24)や3R2N(tau39)のリン酸化は減少させる[101, 102]。
切り捨てはタウの凝集に影響を与える。タウは様々なプロテアーゼによっていくつかの部位で切り捨てられており[58, 103],酸性末端はMTBRと相互作用してタウの凝集を防いでいる[104].我々は最近、報告されている切り捨て部位に従って、タウタンパク質のC末端とN末端の両方から 11の切り捨て部位を構築し、すべての切り捨て部位の病理学的活性を比較した[70]。我々は、タウタンパク質の最初の150または230残基の欠失は、その自己凝集、部位特異的リン酸化、およびアルツハイマー病 O-tauによってシードされた結合および凝集を増加させることを観察したが、最初の50残基の欠失はそれらの効果を有さなかった[70]。最後の 50 残基の欠失は、最後の 20 残基の欠失は有意な効果を示さなかったが、最後の 50 残基の欠失は、タウの部位特異的なリン酸化を調節し、アルツハイマー病 O-tau によって凝集に押収し、シードすることができるタウの自己凝集を促進した[70]。すべての切断形態の中で、Tau151-391が最も病理学的に活性であった。アルツハイマー病 O-tauは、培養細胞および試験管内試験でTau151-391の凝集を誘導した。最初の150残基と最後の50残基の欠失は、タウを病理学的特徴から保護し、タウの病理学的活性を促進した[70]。このように、タウの切り捨てを抑制することは、アルツハイマー病や関連するタウ症におけるタウ病理を抑制するための治療法として考えられる[70]。
他のPTMもまた、タウ凝集に影響を与えうる。リン酸化と同様に、K18/163/280/281/369のようないくつかのリジン残基でのアセチル化はタウの凝集を増加させる[74]が、他の多くのリジンでのアセチル化はタウの凝集を減少させる[105]。S400でのO-GlcNAシリル化[106]、Y18/394でのニトロ化[107]、複数の残基でのメチル化[108]、および特定のシステインでのS-グアニル化もまた、タウの凝集を減少させる[109]。K340でのSUMOylation[110]、K280および/またはK311でのカルバミル化[111]、および複数のリジン残基でのグリケーションは、タウの凝集を増加させる[101]。
PTMに加えて、突然変異もタウ凝集に影響を与える。G272V、P301L、V337M、R406Wを含むタウのミスセンス変異は凝集体を形成する傾向が強く、MTBR欠失変異ΔK280は微小管との結合親和性を低下させ、βシート構造を強化し、フィブリル化を促進する[112,113]。異常な線維化を防ぐためには、タウエクソン10の代替スプライシングと4R-tau/3R-tauの適切な比率が重要である。追加のMTBRを含む4R-tauは、3R-tauよりも結合しやすく、微小管の集合を促進する[114]。4R-tauは3R-tauと4R-tauの結合よりもヘパリンによって誘発される多量体化の引き金となる可能性が高いと考えられている[115]。また、デキストラン硫酸、ヘパリン、RNA、アラキドン酸などのいくつかの負に帯電した補因子もまた、試験管内試験でタウの凝集を促進する可能性がある[116]。14-3-3-3ζやFK506結合タンパク質4(FKBP4)を含むいくつかのタンパク質は、試験管内試験で凝集しやすい構造を安定化させることで、タウの凝集を誘導する可能性がある[117, 118]。
1.6 プリオン様増殖
タウ病変は、脊髄/脊髄下複合体と経胸膜領域で始まり、大脳辺縁系に向かって徐々に進行し、最終的には、大脳皮質に到達することができる[8, 9]。タウ病変の地域分布はアルツハイマー病の認知障害と正の関係がある。タウ病変は患者の予測可能なパターンに従って進行し、脳ネットワークに関与することが提案されている[9, 119, 120]。タウ病変の増殖には、非シナプス経路やシナプス経路によるテンプレート化された播種、細胞内への取り込み、分泌、細胞間移動などのいくつかの過程がプリオン様の方法で起こると予想されている[116, 121]。タウの蓄積が正確な経路を経て細胞に吸収されることは明らかではない。タウの蓄積はマクロピノサイトーシスを介して摂取され[122, 123]、ヘパラン硫酸(HS)プロテオグリカンが必要とされる[122]。細胞質と接触したエンドソーム内のタウの種は、その後、蓄積されていないタウの蓄積を促す。HSはアルツハイマー病脳におけるタウの過リン酸化に関与し、その後タウ蓄積に先行する[124]。
タウ病理学的プロパゲーションのプリオン様メカニズムは2009年に初めて提案された[125]。それ以来、いくつかの研究が、細胞外に投与された場合、タウの集合体が凝集体の生成を「種」とし、それが他の細胞に伝播することを示唆してきた。細胞内タウの伝播には、播種、凝集体の取り込みおよび放出が必要である。タウモノマーが細胞に吸収され、そこから放出される場合でも、凝集体をシードする能力はないと考えられる。発現された4R-tauは、275-280領域と306-311領域を含まない場合にはシードされない[123]。そこで、タウ凝集阻害剤(TAIs)は、タウ誘発の播種と拡散を減少させることができる。
正常なタンパク質が誤って折り畳まれたタウタンパク質の「種」と接触すると、それは病原性のある形に変換される可能性がある。この形質転換のメカニズムはまだ明らかになっていないが、テンプレートを介した構造変化を必要とし、その伝播はニューロンネットワークによって行われることが知られている[126, 127]。タウタンパク質は、それ自身が凝集するためのハブであり、その後、さらなるミスフォールディングのための「種」としての役割を果たす[40]。Dujardinらは、P301Sマウスの死後アルツハイマー病脳から得られたタウのいくつかの側面を調べ、シード活性は、タウの総量ではなく、オリゴメリック/過リン酸化タウのレベルと相関していることを発見した[128]。また、オリゴメリック/高リン酸化タウの豊富な形態は、疾患の進展にも関連しており、高リン酸化ホスホタウ(P-tau)レベルは播種活性と相関している。我々は最近、アルツハイマー病 O-tauが効果的に生体内試験および試験管内試験でプリオン様の方法でタウ凝集を誘導することを報告した[67, 70, 129, 130]が、アルツハイマー病脳全体のタウ病理の増幅と広がりの基礎となり得る。アルツハイマー病のO-tauは正常なタウを逮捕し、不飽和な方法で神経線維束を形成するためにテンプレート化することができる[31]ことから、タウ病理はタウの “種 “のプリオン様特性を介してヒトの脳内で伝播することができることが示されている[131]。
細胞間タウ病理転移の基本的なメカニズムの理解には、試験管内試験研究が重要な貢献をしている。例えば、培養細胞では、タウ “種 “はレシピエント細胞で同様のタイプのタウ凝集を促進し、タウ凝集の形態学的特徴を明確に再現することができる。病理的なタウは、細胞間転移の過程で様々な分子経路を関与している[132]。
2 タウを用いた治療戦略
タウの病理学的発達の各段階において、治療的介入のためのいくつかの機会がある。しかし、AD病理の理解に意味のある有意な進歩は、ヒトで有効であることが示されている治療法はまだ見出されていない。現在までのところ、アミロイドβに基づく治療法はアルツハイマー病症状の改善には効果がないように思われる。一方、タウに対する潜在的な治療戦略は、アルツハイマー病治療に非常に有望であると考えられている(図3)。タウを標的とした治療法は現在、開発の初期段階にあるが、進化の可能性は大きい。
2.1 タウの発現抑制
タウタンパク質は、アルツハイマー病病態の中心分子として、細胞に直接毒性があり、アミロイドβ毒性を媒介している[133]。したがって、タウの発現を阻害することは、アルツハイマー病治療のための有望なアプローチであると考えられる。内因性タウの発現量の低下は、アルツハイマー病 モデルマウスの行動異常や アミロイドβ誘発性認知障害に対する保護効果を示すことが示唆されている。タウのノックアウト(KO)はモデルマウスでは副作用が少ない[134]が、これはおそらく微小管に関連する他のタンパク質がタウの欠乏を十分に補うことができるからであろう。細胞内のタウモノマーレベルが低下すると、凝集体の形成を支配するバランスにより、タウの集合体が脱重合し、オリゴマー性のタウが減少し、PHFとして凝集することが決定される[135]。アンチセンスオリゴヌクレオチド(ASO)または小型干渉RNA(siRNA)は、タウの発現を減少させることができる。siRNAは、細胞および動物モデルの両方において、タウの病理および関連する機能障害を減少させる[71]。現在までのところ、siRNAは癌などの他の疾患には用いられているが、アルツハイマー病や他のタウ症に対する臨床試験では検討されていない[136, 137]。
図3 タウを標的とした治療アプローチ
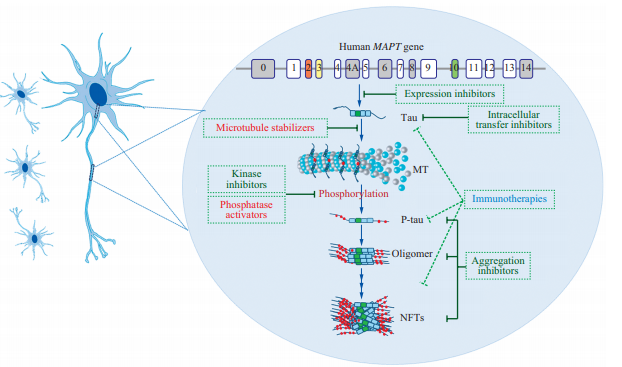
タウの発現とその病態と疾患修飾剤の可能性のメカニズム。MT、微小管、MAPT、微小管関連蛋白質タウ、NFTs、神経原線維タングル、リン酸化タウ、高リン酸化タウ
miR-106b、miR-125b、miR-132/122,miR-219を含むいくつかのマイクロRNAは、タウの発現とリン酸化を調節している[138-141]。ASOは約25年前から一般的な実験方法であったが、副作用のために好まれなくなっていた。最近では、ASOが脊髄筋萎縮の進行を緩和することが明らかになり、タウ症を含む様々な疾患に対するASO治療が復活する可能性がある[142, 143]。しかし、タウの発現抑制がアルツハイマー病治療に有効であるかどうかを評価するためには、さらなる研究が必要である。
2.2 タウリン酸化抑制
高リン酸化されたタウタンパク質が凝集体を形成し、タウ病理の伝播を決定する。タウの高リン酸化阻害は治療的アプローチであり、タウ蛋白質キナーゼと蛋白質ホスファターゼはアルツハイマー病の進化に重要な役割を果たしている。タウはcdk5,GSK-3β、ERK、Dyrk1Aなどのキナーゼによってリン酸化されている。GSK-3βに対する特異的な阻害剤である塩化リチウム(LiCl)とK252a(cdk5,ERK1,GSK-3βに対する非特異的な阻害剤)は、マウスモデルにおいて不溶性および高リン酸化タウのレベルを低下させる[40]。GSK-3βを標的とした低分子阻害剤としては、SRN-003-556,CHIR-98014,SB 216763などが現在前臨床段階にある。タウプロテインキナーゼの活性は、いくつかの方法で直接的または間接的にタウの病理に関連しており、それらを潜在的なターゲットとしている。
タウの主なホスファターゼはPP2Aであり、このPP2Aはタウに関わるホスファターゼ全体の70%以上に関与しているため、最も決定的なホスファターゼとみなすことができる[47]。PP2Aはタウの脱リン酸化を媒介しており、PP2Aとその活性化因子のレベルは、年齢をマッチさせた対照群と比較してアルツハイマー病脳ではダウンレギュレーションされていることがわかっている[53, 54]。我々は最近、PP2AとGSK-3のクロストークを報告した。PP2AはGSK-3βがS9残基で脱リン酸化されることでGSK-3β活性を増加させた。また、GSK-3βがアップレギュレーションされると、ロイシンカルボキシルメチル化酵素1(LCMT-1)やタンパク質ホスファターゼメチル化酵素1(PME-1)を介してPP2Aがメチル化され、PP2A活性が上昇することがわかった[55]。したがって、GSK-3βとPP2Aは相互に調節し、タウリン酸化に影響を与える。PP2A活性を標的としたいくつかの薬剤が現在開発されているか、または臨床試験で評価されている[24]。タウの高リン酸化はアルツハイマー病過程の予測イベントであり、タウタンパク質に対する戦略的な治療法の第一人者と考えられる。
2.3 タウ凝集抑制
いくつかのPTMは、タウの微小管結合能を低下させ、微小管からの剥離を促進する。この効果により、細胞内のタウ濃度が徐々に上昇し、タウタンパク質との相互作用の可能性が高まり、最終的にはタウの凝集を引き起こすことになる。タウオリゴマーは、アルツハイマー病における神経毒性と神経変性を引き起こす最も毒性の高い種である。タウの病理学的なプリオン様伝播の予防を目的としたタウ凝集阻害剤(TAIs)治療は、タウを標的とした重要な戦略である。
これまでのところ、ほとんどのTAIはメチレンブルーからの誘導体であり、その中には塩化メチルチオニウム(MTC)レンバーTM、LMTが含まれている。これらのTAIsは、タウトランスジェニックマウスのタングルおよびタウフィラメントを破壊し、タウトランスジェニックマウスの認知障害を妨げることが報告されている[144]。MTCはメトヘモグロビン血症の治療薬としてFDAから認可されており、アルツハイマー病や他の関連するタウ症の治療に再導入されている[40]。MTCや他のメチレン誘導体は、試験管内試験ではタウ-チューブリン相互作用を阻害することなく、タウ-タウ結合を阻害することでPHFのタンパク質分解安定性を逆転させることが広範な研究で実証されているが、その効果は試験管内試験/生体内試験や臨床試験では同じではない[145]。これまでのところ、これらの薬剤の効果は限定的であった[145]。例えば、LMTXは第二世代のTAIであり、第Ⅲ相臨床試験では軽度から中等度のアルツハイマー病患者の機能的および認知能力を改善することはできなかった[24]。一方、NPT088は、現在、第Ⅰ相臨床試験でTAIとして使用されている[146]。NPT088は、アミロイドβやタウを含む多くのミスフォールドされたタンパク質を認識し、再構築する融合タンパク質である。NPT088は、トランスジェニックマウスにおいて、アミロイドβプラークとリン酸化タウの病理を減少させ、認知能力を向上させることができる。タウの凝集を抑制する別の戦略は、2つのヘキサペプチドモチーフによって形成される立体ジッパーを阻害することである[147]。このアプローチは、タウに対する新薬の設計の基礎となる可能性がある。
2.4 タウの細胞間移動を阻害する。
タウ凝集体は、病理学的に誤ったタンパク質と同様に、細胞から細胞へと移動し、プリオンのような形で細胞外コンパートメントに放出され、タウ病理が脳の様々な領域に転移する可能性がある。これは、タウ症や他の神経変性疾患の進展を抑制するための治療の方向性である[148]。タウタンパク質の細胞間トランスフェクションを阻害することによりタウタンパク質を標的とすることで、アルツハイマー病患者のタウ病変を遅らせることができることが数多くの研究で示されている[148]。タウ病理は神経細胞に有害であるため、タウ凝集体の病理学的伝播を防止することで、疾患への影響を合理的に軽減することができる[40, 147]。タウの神経細胞間移動を阻害または減少させるためには、タウの放出を阻害する、タウの取り込みを阻害する、タウのオリゴマー化と細胞外タウ濃度を低下させる、という3つの異なる経路がある。タウ放出のブロックは、2つの異なる側面で標的とすることができる。タウ放出がブロックされた後、細胞外レベルとタウの利用可能性が低下し、それによって隣接する神経細胞によるタウの取り込みが阻害される[149]。
2.5 微小管の安定化
病的なタウタンパク質は微小管から剥離し、アルツハイマー病患者の微小管破裂につながる。したがって、微小管の安定化は、タウによる神経毒性を補うものと考えられている。微小管安定化を標的とすることは、タウを主眼としない治療戦略の可能性がある。この治療法の根拠は、微小管安定化剤がアルツハイマー病患者において有益な神経保護効果を有することである[150]。タクソール由来のエポチロンは、血液脳関門(BBB)を容易に通過できる低分子微小管安定化分子である。そのため、海馬ニューロンの欠損やタウの病理を軽減して空間記憶障害を回復させることができ、トランスジェニックマウスでは形態異常を伴う軸索数を減少させ、微小管数を増加させることが可能である[151-153]。アベオタキサンやダブネチドは最近発見された微小管安定化剤であり、微小管の安定化に向けた進歩をもたらしたが、動物モデルやヒト試験での有効性は不均衡である[135, 151]。また、TPI287やNAPのような他の微小管安定化剤は第Ⅰ/Ⅱ相臨床試験中である[135]。このように、この戦略的アプローチの開発には大きなギャップがある。このように、微小管安定化剤を標的としたこの治療戦略は、より効果的な臨床試験結果が必要であり、今後の評価が必要である。
2.6 タウ免疫療法
現在までに、最も有望なタウ標的化方法は、タウ免疫療法である可能性がある。P-タウペプチドまたはリン酸化タウに対する抗体を介した受動的または能動的な免疫化は、タウ病理および行動異常を減少させ、認知パフォーマンスを効果的に改善する[154]。最近、タウタンパク質のN末端領域に対する抗体(BIIB092)を用いた有望なタウ指向性モノクローナル抗体ベースのタウ免疫療法は、進行性核上麻痺(PSP)を対象とした第1b相臨床試験で優れた忍容性を示した[154]。しかし、第Ⅱ相臨床試験は無益であるとの分析から中断されており、比較的進行した病期であったことが失敗の理由と考えられている[https://www.alzforum.org/news/research-news/ abbvies-tau-antibody-flops-progressive-supranuclear-palsi]。血清から得られるものを含め、他の多くのタウ抗体は病理学的なタウに対して大きな選択性を示し、前臨床研究で有望視されている[135]。その中でも、最近のPSP試験で忍容性が高く、安全性が認められているUCB0107のようなタウ中心部を標的とした抗体が注目されている[135]。リン酸化タウに対する抗体(RO6926496,RO7105705)タウフラグメント(BMS-986168,C2N-8E12)タウコンフォメーション(抗タウオリゴマー特異的抗体)または全タウ(ABBV-8E12)は、現在臨床試験で評価されている[24, 25]。アルツハイマー病の神経変性を除去することは不可能であることは明らかであるが、脳の非障害領域へのタウの伝播を阻害することで、タウ病理の進行を遅らせたり止めたりすることは可能である。我々の最近の研究では、抗体43D(タウ6-18を標的とする)によるタウ受動免疫化が、アルツハイマー型高リン酸化タウ誘導病理の播種と伝播をブロックすることが示された。この結果、3xTg-ADマウスではアミロイドβ病理の減少と認知機能の向上が認められ、アルツハイマー病および関連するタウ症に対する有望な治療法であることが示された[155-157]。タウ免疫療法のメカニズムは未だ不明であり、この知見があれば、臨床試験の早期開発が促進され、アルツハイマー病患者に最も効果的な免疫療法を提供できる可能性が高まると考えられる。
3 結論と展望
タウはアルツハイマー病発症の中心的な分子である。タウの高リン酸化、トランケーション、凝集、およびプリオン様伝播は、アルツハイマー病病態の現在の理解に新しいパラダイムを提供している。最近の研究では、試験管内試験および生体内試験でのタウ介在性神経変性の前臨床モデルにおいて、タウオリゴマーはフィブリル状の集合体ではなく、シナプス機能を破壊することで細胞毒性を示し、神経細胞の死とタウ病理の広がりを引き起こすことが示されている[158-160]。しかし、アルツハイマー病を正しく理解し、新規の疾患修飾治療法や診断ツールを開発・改善するためには、基礎となる詳細なメカニズムと臨床アルツハイマー病症例との関連性についての知識が必要である。したがって、アルツハイマー病の初期段階でNTFやPHFが形成される前に、毒性のあるタウオリゴマーやシードコンピテントモノマーを検出して除去することは、タウ病理の予防に極めて重要である。タウ免疫療法を改善するためのエッセイは、同様の関連性を有する。現在臨床試験に提出されている抗タウワクチンやその他の受動的/能動的免疫療法は、将来の治療応用に大きな可能性を秘めている。