
Contents
Stress Induced Neural Reorganization: A Conceptual Framework Linking Depression and Alzheimer’s Disease
要旨
慢性ストレスは、精神疾患や神経変性疾患だけでなく、心血管疾患、肥満、消化器疾患を含む多くの生理的疾患の危険因子である。慢性ストレスの過程で変化する分子や細胞のメカニズムは数多く存在し、うつ病などの精神疾患やアルツハイマー病などの神経変性疾患を発症する脆弱性を増加させる可能性がある。
これは、安静時のデフォルトモードネットワーク(DMN)を含む大規模な脳ネットワークへのストレスの影響、神経細胞の回路や構造へのストレスの影響、ストレスに対する細胞や分子の適応などから明らかであり、ストレスに関連した精神疾患を持つ人は、人生の後半に神経変性疾患を発症しやすくなる可能性がある。
これらの変化には、視床下部下垂体軸(HPA)軸の負帰還抑制の減少、前頭前野(PFC)と海馬における樹状突起のアーボレーションと棘密度の減少、神経発生を抑制し、神経細胞死を促進する可能性のある炎症性サイトカインの放出などが含まれる。これらの因子はいずれも、アルツハイマー病と同様にストレス関連精神疾患に関与していると考えられており、うつ病やアルツハイマー病患者の臨床研究や死後の研究でも観察されている。
本レビューの目的は、慢性ストレスが精神神経疾患と神経変性疾患との関連性を示唆する臨床的・前臨床的証拠をまとめることである。さらに、ストレスに関連した精神疾患の病歴を考慮に入れることの重要性について、層別化された患者集団におけるアルツハイマー病の病因に重要な役割を果たしている可能性があることから、臨床試験デザインの際に考慮に入れることの重要性についての根拠を示す。
キーワード:ストレス、アミロイド、ノルエピネフーリン、ドーパミン-β-ヒドロキシラーゼ、うつ病、アドレナリン受容体
I. 序論
1907年、アロイス・アルツハイマーはドイツのフランクフォード・アム・マインの精神病院に入院していた51歳の女性の観察結果を発表した。最初に報告された臨床症状は、見当識障害、妄想、幻覚、せん妄、記憶喪失を伴う気分の変化であった。彼女の意識状態は時間の経過とともに徐々に低下していったが、アルツハイマーはこれらの複雑な症状が強くなったり弱くなったりすることを指摘していた。死後の分析では、アルツハイマーは、巨視的な焦点変性はないが、神経線維とグリア細胞の異常な形態を持つ均等に萎縮した脳を指摘した。彼は、これは特徴的な病気であり、これまでに述べられてきた精神疾患の中で分類されるべきではないと結論づけ、個々の症例の組織学的分析は、特定の病気と一般的な疾患のカテゴリーとの臨床的な区別を容易にするかもしれないと結論づけた(Alzheimer 1907, Alzheimer, Stelzmann er al)。 1995)。
100年以上経った今でも、科学者たちはアルツハイマー病について謎に包まれている。多くの進歩がなされているが、500万人以上のアメリカ人に影響を与えているアルツハイマー病患者のための疾患修飾治療薬はまだ存在しない(Alzheimer’s 2015)。伝統的に、アルツハイマー病は、死後の脳組織の組織学的分析により、40(アミロイドβ40)または42(アミロイドβ42)アミノ酸の長さを有するアミロイドβ(アミロイドβ)ペプチドを含むシナプスタンパク質の凝集体を明らかにし、Tall(Tall、Rub et al 2002)およびBrak(Brak、Alafuzof et al 2006)によって概説された空間的に異なるパターンの異常リン酸化タウ(タウ-p)タンパク質を特徴としている。それらの病的形態では、アミロイドβ凝集体は老人斑(SP)として知られており、凝集したタウは神経原線維性タングル(NFT)として知られている。アミロイドβプラークは、びまん性のものから神経斑状のものまで、いくつかの形態をとることがあり、後者の形態では、ジストロフィー性の神経突起やタウ-p物質を含む(Thal, Rub er al)。 シナプスの喪失は疾患進行の主な相関関係であり(Selkoe 2002, Jack, Knopman er al 2010SPとNFTがシナプス機能を破壊する正確なメカニズムは不明であるが、シナプスの喪失を促進し、最終的には神経細胞の死を促進する相乗的な関係があるようである(Jack 2013)。バイオマーカーのステージングに関する現在の仮説では、正常な老化の産物であるタウ-pは、人生の早い段階で徐々に蓄積を開始するが、世界的なアミロイドβ負担がある閾値を超えると深刻に加速され、神経細胞の損傷と神経変性を開始することが示唆されている(Jack et al 2013, 2016)。一般的に、NFTの蓄積は経側頭葉皮質から始まり、側頭葉、海馬、側頭葉、頭頂葉、後頭葉へと進行すると考えられている(Braak 2006)。魅惑的なレトロスペクティブ研究は、典型的なアルツハイマー病と比較した場合のNFTの非定型分布に基づいて、神経病理学的に定義されたアルツハイマー病のサブタイプを同定した。889人の患者のうち、665例(75%)は典型的なアルツハイマー病に分類され、97例(11%)は海馬のNFTが3つの連合皮質に比べてまばらであるという観察に基づいて海馬スペアリングに分類され、127例(14%)は3つの連合皮質に比べて海馬のNFTカウントが高い大脳辺縁優位に分類された(Murray, Graff-Radford er al)。 海馬を温存する群のうち、男性(63%)の割合が高く、死亡時の年齢が若く、皮質領域のNFTカウントが高かった。大脳辺縁系優勢群は、主に女性(69%)で、死亡時年齢が高く、海馬を温存した場合と比較して、徐々に減少する傾向がみられた(Murray, Graff-Radford er al)。 発症年齢、教育レベル、および認知障害の評価に使用される質問票の結果、Mini Mental State Exam(MMSE)スコアは、これらの参加者の臨床病歴から記録されたが、うつ病の既往歴、または他のストレス関連精神疾患は考慮されなかった。では、層別化された患者集団におけるアルツハイマー病の病因に重要な役割を果たす可能性があるため、このような因子を考慮に入れることの重要性の根拠を提供する。
本レビューでは、臨床研究や前臨床モデルで観察されているアルツハイマー病と神経精神疾患の病態におけるいくつかの顕著な類似点について論じる。これらの異種疾患の間には重要な区別があるが、慢性的な曝露後に不適応となり、結果として大規模なネットワークの再編成や神経細胞の接続性の変化をもたらすストレスへの適応などの相同的な要素について論じる。さらに、うつ病やアルツハイマー病などの精神疾患で顕著な疾患の発現、発症、環境因子、ゲノム因子の不均一性は、患者の層別化、個別化治療の可能性が、効果的な疾患修飾治療法の出現に不可欠であるという考え方を支持している。神経精神疾患と神経変性疾患との関連を明らかにすることは、両疾患の知識を前進させる上で最も重要である。第一に、ストレスに関連した精神疾患に脆弱な個人の亜集団がアルツハイマー病の素因となる可能性があることを理解することは、アルツハイマー病の予防に有利なストレス関連の標的に向けた治療的介入の開発の機会を提供している。第二に、アルツハイマー病の進行を促進すると考えられているアミロイドβとタウ-pの異常を定義することは、うつ病を含むより広範な神経精神疾患状態の治療のための重要な研究領域であるかもしれない。
以下のセクションでは、慢性的なストレスが、うつ病患者に見られる根底にある神経精神病理に寄与している可能性があり、それが人生の後半で神経変性疾患に脆弱な人を作り出す可能性があるという仮説の枠組みを提案する(図1)。我々はまず、うつ病がアルツハイマー病の独立した危険因子であるという考えを支持する臨床研究をレビューすることから始め、慢性ストレスとうつ病とアルツハイマー病を結びつける追加の臨床的および前臨床的知見をレビューする。次に、うつ病とアルツハイマー病におけるアミネリック回路の重要性の基礎を提供し、ストレスによって誘発され、疾患の病態生理に寄与する可能性のある大規模なネットワーク適応、これらのネットワーク内のニューロンへの細胞および分子の変化について論じる。間違いなく、広範な研究は、気分に関連する精神疾患(Kendell、Krystal et al 2005,KugayaとSanacora 2005,McEwen 2005)とアルツハイマー病(PalopとMucke 2010,PalopとMucke 2010)における興奮性と抑制性の神経伝達のバランスの重要性、およびグルココルチコイド(グルココルチコイド)の影響(McEwen 2005,OakleyとCidlowski 2013)を実証している。これらのトピックに関する優れた以前に発表されたレビューがあるので、このレビューの焦点は、アルツハイマー病の文脈では過小評価されている回路である、青斑核(LC)-ノルエピネフリン(ノルエピネフリン)系に重点を置いて、ストレスに関連したアミネリック回路の機能不全になる(Ross, McGonigle er al)。
図1
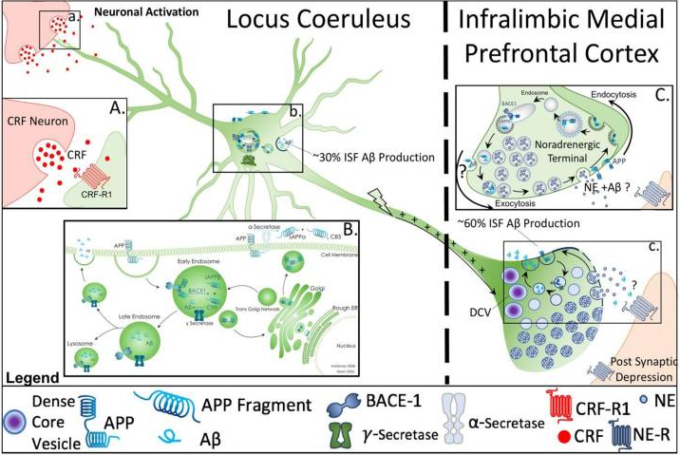
アミロイドβ(アミロイドβ)の産生と分泌の細胞機構を体液腺とシナプス部位から解明する。この模式図は、青斑核-ノルエピネフーリン(ノルエピネフリン)回路に関連して文献に記載されているアミロイドβ産生および分泌の仮説的および既知のメカニズムを反映している。図1a.は、コルチコトロピン放出因子(CRF)を放出するコルチコトロピン放出因子(CRF)末端のシナプス裂け目と青斑核樹状突起を指している。ボックス1Aは、ストレス条件下での青斑核ニューロンへのCRF放出の高倍率画像を示し、アミロイドβ産生および異常なリン酸化タウ(taup)に関与しているCRF受容体1に関与している。図1b.は、青斑核ニューロンのソーマ内でのアミロイド前駆体タンパク質(APP)の処理を示している。箱1B.では、細胞内でのAPPの処理のメカニズムを詳しく説明し、APPの細胞内局在が、α-セクレターゼを媒介する非アミロイド生成経路、またはβアミロイド切断酵素1(BACE-1)とγ-セクレターゼを媒介するアミロイド生成経路という2つの分岐した処理経路への参加を決定することを示している。一旦アミロイドβが形成されると、それは分泌経路に沿って継続し、その結果、細胞外空間への放出、またはリソソームでの分解をもたらす可能性がある(Haass, Kaether er al)。 マイクロダイアリシスを利用した研究では、内因性間質液(ISF)アミロイドβの約30%が分泌経路で産生され、体液腺突起から放出されると推定されている(Box 1B)が、約60%はシナプスで産生される(Cirrito、May et al 2003)(図1c)。ボックス1Cは、シナプスにおけるアミロイドβの産生および分泌が、エンドサイトーシスに依存した方法で、シナプス活性の変化に続いて起こる緊密に制御されたプロセスであることを示している(Cirrrito、Yamada et al 2005,Cirrrito、Kang et al 2008)。シナプス活性の増加は、小胞のリサイクルの増加と相関しており、APPのより大きな内包化とそれに続くアミロイド生成処理の増加につながるという仮説が立てられている(Cirrito, Kang er al)。 ストレスや外因性CRFの急性行動パラダイムを利用したマイクロダイアリシス研究では、アミロイドβのISFレベルの増加が実証されている(Kang、Cirrito et al 2007)が、青斑核ニューロンに関するこの相互作用の解剖学的基質はまだ調査されていない。
II. 臨床的証拠
a. 認知症の行動・心理症状
今日、アルツハイマー病の鋭い洞察の多くは、複雑な神経変性疾患の患者の臨床的・組織学的な違いに基づく層別化だけでなく、認知症の行動的・心理的症状(B脳卒中後認知症)に関する臨床研究の原動力となっている。McKeith and Cummingsは、伝統的に認知機能の低下が認知症の特徴とされてきたが、神経精神医学的評価は鑑別診断や症状の管理に不可欠であると指摘している(McKeith and Cummings 2005)。McKeithとCummingsは広範囲にアルツハイマー病の臨床集団におけるB脳卒中後認知症を研究し、優勢な3つの基本的な行動症候群を同定した。B脳卒中後認知症は、妄想、幻覚、激越、抑うつ、不安、多幸感、無気力、過敏、抑制、運動異常、睡眠障害、食欲の変化など、認知症の過程で起こりうる12の症状を検出する質問紙であるneuropsychiatric Inventory (NPI)を用いて評価されることがある。この評価を利用したレトロスペクティブ研究では、B脳卒中後認知症の発症は診断から45~50ヶ月の間に起こるとされている(Hart, Craig er al)。 同じ研究では、行動の変化が患者の88%までに見られ、うつ病がコホートの56%で最も一般的な心理的症状であったことが報告されている(Hart, Craig et al 2003)。
NPI不安サブスケールを利用したアルツハイマー病患者の不安の臨床研究では、不安は、年齢、発症年齢、教育レベル、MMSEスコアを調整した後、より重度の認知機能の低下と発症年齢が早い患者で最も一般的であることが明らかになった。介護者がアルツハイマー病患者の不安を報告したのは、調査した115人中30人(26.1%)であった。興味深いことに、不安は妄想、幻覚、多幸感、抑制とも相関しており、不安が報告された30人中19人(63.3%)の患者はうつ病も持っていた(Porter, Buxton er al)。 神経イメージング研究は、B脳卒中後認知症を持つ患者をB脳卒中後認知症を持たない患者と区別する神経生物学的変化があることを示唆し、さらに、アルツハイマー病におけるB脳卒中後認知症の存在は、セロトニン作動性、ノルアドレナリン作動性、およびドーパミン作動性関連のシナプス機械系、および炎症性カスケードのマーカーにおける遺伝的多型の存在によって推定される可能性があり、それはうつ病患者にも見られる(Klimek、Stockmeier et al 1997年、Ordway、Stockmeier et al 1997年、Ordway、Stockmeier et al 1997年、Ordway、Stockmeier et al 1997年、Ordway、Stockmeier et al 1997年、Ordway、Stockmeier et al 2003)。1997年、Ordway、Stockmeier et al 2003年、McCulley、Day et al 2004年、Szot、White et al 2006年、Borroni、Grassi et al 2009年、Grunblatt、Zehehetmayer et al 2009年、Combarros、Warden et al 2010)。)
b. アルツハイマー病の危険因子としてのうつ病
うつ病は、共存する神経変性疾患がなくても可逆的な認知障害を引き起こすことはよく知られており、いくつかの研究者は、大うつ病再発の臨床研究で海馬の萎縮を報告している(Sheline, Wang er al)。 一方、高齢者のうつ病症状は、横断的研究では臨床的なアルツハイマー病と関連し、縦断的研究では認知機能の低下や認知症やアルツハイマー病のリスクと関連している(Green, Cupples er al)。 これらの観察から、抑うつ症状がアルツハイマー病の危険因子であるのか、それともアルツハイマー病の病態の初期徴候であるのかを調べる臨床研究が行われるようになった。この目的のために、宗教勲章研究の一環として死後分析まで毎年身体的・精神的健康状態を評価された高齢のカトリック修道女、司祭、兄弟のコホートにおいて、抑うつ症状がアルツハイマー病発症のリスクと認知機能低下率の増加と関連していることが示されている。このコホートのさらなる研究では、抑うつ症状はプラークやタングルと有意に関連しておらず、SPやNFTと疾患の臨床症状との関連を有意に変化させなかったことが示されている。このことから、うつ病症状はSPやNFTとは独立したメカニズムを介して臨床的なアルツハイマー病のリスクと関連していると考えられる(Wilson, Schneider er al)。 これらの知見と一致して、抑うつ症状とアルツハイマー病の関係を検討した臨床研究の系統的なメタ分析とメタ回帰分析では、うつ病の既往歴はおそらく後にアルツハイマー病を発症するリスクの増加をもたらす可能性があり、したがって、疾患の前駆症ではなく、疾患の独立した危険因子を反映していると結論づけている。著者らは、しかしながら、彼らの知見は、うつ病がアルツハイマー病の遠隔的な危険因子とアルツハイマー病の近位の前駆的特徴の両方である可能性を排除するものではないことに注意を促している(Ownby, Crocco er al 2006)。これらの結果に沿って、4,046 人の被験者を含む Multi-institutional Research in Alzheimer’s Genetic Epidemiology (MIRAGE) 研究では、アルツハイマー病 の発症前のうつ病症状が アルツハイマー病 の発症と関連していることを発見した。驚くべきことに、記憶障害の25年以上前に発生したうつ病症状は、アルツハイマー病の発症と関連していた(Green, Cupples er al)。 これらのデータやその他のデータは、うつ病症状が後のアルツハイマー病発症の危険因子であることを示唆している。このように、臨床研究と疫学研究からの説得力のある証拠の集大成として、うつ病とアルツハイマー病の神経病理学において重要な役割を果たす可能性のある共通の基礎となるメカニズムを示唆するものが存在する。
i. うつ病とアルツハイマー病の関連性としての慢性ストレス
1. 慢性ストレス,うつ病,アルツハイマー病:臨床研究
うつ病患者、未治療の自殺者およびアルツハイマー病の臨床および死後の研究は、これらの神経学的障害の性質における顕著な類似性を明らかにした。その証拠は、神経精神医学と神経変性疾患が交差していることを示唆しており、それによって慢性的なストレスが特定の患者集団においてこれらの一見異なるクラスの障害を結びつけている可能性があることを示唆している。臨床研究では、正確なメカニズムは完全には解明されていないが、うつ病とアルツハイマー病の間には根本的な関係が存在することが明らかになっている。文献に蓄積された証拠に基づいて、慢性ストレスによって誘発される病理学的メカニズムは、しばしばうつ病の病因の重要な要因であり、慢性ストレスやうつ病などのストレス関連精神疾患を持つ個人を、アルツハイマー病のような長期的な神経変性疾患に素因づける重要な要因であるかもしれないという仮説が出されている(Pomara and Sidtis 2009, Aznar and Knudsen 2011)。
a. 多動性
慢性ストレスが神経精神医学と神経変性病理学の架け橋となる可能性があるという最初の証拠は、視床下部-下垂体-副腎(HPA)軸の過剰活性化と、うつ病患者や自殺願望のある患者、およびアルツハイマー病患者における負のフィードバック抑制の低下の指標を示す臨床研究に由来している。ストレスへの曝露は、うつ病およびいくつかの不安障害の発症および重症度と関連している(Kendler, Kessler er al)。 ストレスとの関連に加えて、不安障害とうつ病のメランコリックな特徴は、この病気の特徴であり、睡眠障害、集中力の欠如、落ち着きのなさ、および警戒心の増大によって特徴づけられる多動性の中核症状を共有している(Southwick, Paige et al 1999, Gold and Chrousos 2002)。動物モデルとヒトの両方において、コルチコトロピン放出因子(CRF)は、ストレスに対する末梢反応と中枢反応の両方を開始する神経ホルモンであり、うつ病患者では過分泌されることが示されており、うつ病患者の脳脊髄液(脳脊髄液)中のCRF免疫反応性のレベルが高いことからも明らかである(Nemeroff, Widerlov er al)。 CRF は、前頭皮質と海馬への ノルエピネフリン の唯一の供給者として認識されている第四脳室の基部に位置する ノルエピネフリン ニューロンのクラスターである 青斑核 を刺激することにより、ストレスに対する中枢反応に関与し、その広範な求心性神経はニューロン軸全体に ノルエピネフリン を供給する。青斑核-ノルエピネフリン系は、急性および慢性のストレス因子(認知的および身体的)の両方においてCRFに対して高い反応性を示し、メランコリックうつ病を経験している人はCRF/ノルエピネフリン緊張が高い状態を示している(Gold and Chrousos 2002)。また、慢性の治療抵抗性うつ病の入院患者では、1日中のコルチゾール出力が健康な対照群の2倍であることも臨床研究で明らかにされている(Juruena, Cleare er al)。 最後に、重度のメランコリック性うつ病患者のサブセットにおける症状の多くは、基底コルチゾール分泌の増加、リズムの鈍化、デキサメタゾン抑制に対する抵抗性など、ストレス軸の過剰活動に似ている(Gold 1988,Gold、Goodwin et al 1988,Gold and Chrousos 2002,Gold、Gabry et al 2002)。このように、ストレス系の亢進は、精神疾患の発症と重症度の両方を決定する重要な因子である。しかし、うつ病の固有の不均一性を認識することは必須であり、メランコリックうつ病と非定型うつ病では、それぞれCRF/ノルエピネフリンの高低に代表されるHPA軸活性の亢進や抑制など、相反する特徴を持つうつ病のサブタイプの特徴付けに反映されている。このような特徴は、治療上の意味合いが非常に大きく、患者集団を適切に層別化する努力の指針となりうるが、そうしないと、結果の解釈を混乱させる可能性があり、特に、うつ病および神経変性疾患におけるCRF/ノルエピネフリンストレス応答性の役割を明らかにしようとしている人には注意が必要である(Gold and Chrousos 2002, Gold, Gabry er al 2002)。
b. コルチゾール
アルツハイマー病患者の血漿コルチゾールとその代謝物に関する多くの臨床研究に基づいて、アルツハイマー病の発症と進行に寄与する因子としての高コルチゾール症の概念の支持もある(Weiner, Vobach er al)。1997, Csernansky, Dong er al)。2006慢性的なストレスがアルツハイマー病の危険因子であること(Wilson, Evans er al)。2003, Wilson, Schneider er al)。 高コルチゾール血症(Weiner, Vobach er al)。 1997)と負のフィードバック抑制の減少(O’Brien, Schweitzer er al)。 1994)は、腰椎穿刺を介して分離された脳脊髄液を利用したアルツハイマー病のいくつかの臨床研究で文書化されており、デキサメタゾン抑制試験の異常によって証明されている(Balldin, Gottfries er al)。 1983, Davis, Davis er al)。 1986)が、末梢および中枢のストレスシステムが亢進状態にあるという考えを支持している。高コルチゾール血症は、アルツハイマー病患者集団において広く記録されているが(Davis, Davis er al)。 1986, Masugi, Ogihara er al)。 1989, Oxenkrug, Gurevich er al)。 1989, Martignoni, Petraglia er al)。 1990, Franceschi, Airaghi er al)。 1991, Maeda, Tanimoto er al)。 1991, Weiner, Vobach er al)。 1991年、Weiner、Vobach et al 1997年、Lupien、de Leon et al 1998年、Carlson、Sherwin et al 1999年、梅垣、碇 et al 2000年、Peskind、Wilkinson et al 2001年、Rasmuson、Andrew et al 2001年、Csernansky、Dong et al 2006年、Huang、Luiら。2009,Arsenault-Lapierre、Chertkow er al 2010,Lara、Caramelli er al 2013,Zverova、Fisar er al 2013,Popp、Wolfsgruber er al 2015コルチゾールおよびHPA軸活性が加齢とともに増加する可能性があることを考慮することも重要である(Friedman、Green er al)。 1969年、Dodt、Dittmann et al 1991年、Raadsheer、Sluiter et al 1993年、Raadsheer、Oorschot et al 1994年いくつかの研究では加齢に伴うHPA活性の変化または低下がないことが報告されているため、これは依然として論争の的となっているが(Sharma、Palacios-Bois et al 1989)。おそらく、これらの違いのいくつかは、健康な高齢者を対象とした縦断的研究によって、あるサブグループではコルチゾールレベルが経年的に上昇し、別のサブグループでは低下し、第3のサブグループでは安定したままであることを特徴とする個人のサブグループの証拠を提供し、被験者の年齢はコルチゾールレベルまたはコルチゾール分泌の経年変化のパターンのいずれにも関係していないという証拠を提供することによって調整されるかもしれない(Lupien, Lecours er al)。 コルチゾールレベルとアルツハイマー病の進行との関係を評価するように設計された縦断的研究では、コルチゾールレベルは疾患の臨床的進行に有意に関連していたが、老化や生存には関連していなかったと決定された(Weiner, Vobach er al)。 1997)。しかし、別の研究では、横断的サンプリングで認知症の重症度と相関する高コルチゾール血症と関連するHPA軸機能障害は、アルツハイマー病患者の縦断的フォローアップサンプリングでは相関しないことがわかった(Swanwick, Kirby er al)。1998)。また、高コルチゾール血症だけでは、クッシング症候群患者(Starkman, Gebarski er al)。 1992, Forget, Lacroix er al 2000, Starkman, Giordani er al 2001)やストレスレベルのコルチゾール治療を受けた健康な成人(Newcomer, Selke er al)。 1999)で観察されたような、認知機能と海馬の体積に悪影響を及ぼすことにも留意すべきである。より最近の研究では、コルチゾールとアルツハイマー病の関係をさらに解明し、コルチゾールと記憶の関係は、アルツハイマー病患者がコルチゾールによって誘発された海馬の萎縮、またはMMSEスコア(Huang, Lui et al 2009)によって証明された認知機能によって不釣り合いに影響を受ける可能性があるように、個人の認知機能の低下の有無に依存する可能性があることを示している。2009)またはアルツハイマー病を診断するために使用されるスクリーニングツールであるBrief Cognitive Screening Batteryによって(Souza-Talarico, Chaves er al)。 Poppらが2015年に実施した研究では、アミロイドβ42と総タウの脳脊髄液測定値を含む交絡変数をコントロールした後、脳脊髄液コルチゾールの高レベルは、より速い臨床と認知機能の低下によって証明された疾患の進行の加速と関連していることが実証された(Popp, Wolfsgruber er al)。 さらに、ベースラインの認知状態によって評価された患者を層別化することで、本研究では、HPA軸の調節障害が軽度認知障害(MCI)の段階で、疾患の発症および発症の初期に起こることを立証することができた(Popp, Wolfsgruber er al)。 これをサポートするために、ストレスは、げっ歯類とヒト(パードン2011)でアルツハイマー病の病理学と非常に相関する危険因子と考えられているだけでなく、うつ病(ケンドラー、ケスラー et al 1995)を持つヒトである。興味深いことに、アポリポ蛋白質E(ApoE)対立遺伝子の状態上のストレスの影響を検討する研究は、ApoEのε4対立遺伝子の存在下で、高齢者、非認知症の個体の長期的なストレスが記憶力の低下につながることを実証している(ピービー、ランゲ et al 2007)。
c. 生体アミン回路の関与
アルツハイマー病患者の中で、大うつ病の存在は、中脳アミネリック核のニューロン喪失、およびノルエピネフリンおよびセロトニンの皮質レベルの低下と関連している(Zubenko and Moossy 1988, Zubenko, Moossy er al)。 1990, Forstl, Burns er al)。 1992)。アルツハイマー病の行動および心理的症状におけるノルエピネフリンの役割は、ノルアドレナリンニューロンの変性およびそれに続くノルアドレナリン伝達の変化が、うつ病、攻撃性、焦燥感および精神病に関連する行動の変化をもたらすという説得力のある証拠をまとめ、レビューされている(Herrmann、Lanctot et al 2004)。これらの知見に沿って、ノルエピネフリン 合成酵素の多型をリンクする遺伝学的研究がある(Combarros, Warden er al)。 さらに魅力的なのは、もともと1981年にBlessedらによって特定された、アルツハイマー病患者で示された青斑核神経細胞の損失は、個人の亜集団でより深刻になることができるという知見である(Cross, Crow er al)。 後に、1987年にBondareffらによって確認され、コリンアセチルトランスフェラーゼ(ChAT)活性の低下、ソマトスタチンとノルエピネフリンの濃度の低下、大脳皮質のより多くのプラークともつれに関連した青斑核のより大きな神経細胞の損失を持つアルツハイマー病患者のグループを同定した(Bondareff、Mountjoy et al 1987)。さらに、これらの患者では認知症の期間が長く、平均死亡年齢が高いことが指摘されたが、これらの差は統計的に有意ではなかった(Bondareff, Mountjoy et al 1987)。Bondareffらによる追加研究では、青斑核ニューロンの喪失は、Brodmann領域24(帯状突起)のノルエピネフリン濃度、ChAT活性、およびプラークおよびもつれの数、領域21(側頭)のChATおよびプラーク数、および領域10(前頭)のChAT活性と有意に相関していることが示された。さらに、青斑核のニューロン数、青斑核内のNFTの数は、認知症の重症度と推定期間と有意な相関があり、中枢ノルエピネフリン経路の変化がアルツハイマー病の病態生理に関連していることを示唆していた(Bondareff, Mountjoy er al)。 興味深いことに、うつ病とアルツハイマー病の遺伝的関連を調査したVienna-Transdanube Aging(VITA)研究では、うつ病とアルツハイマー病におけるChAT多型との有意な関連を発見した(Grunblatt, Zehetmayer er al)。 このように、青斑核神経細胞の喪失は、ノルエピネフリン伝達だけでなく、うつ病やアルツハイマー病の病因に重要な役割を果たしている前脳のアセチルコリン伝達にも影響を与える可能性がある。
神経精神疾患と神経変性疾患の両方におけるアミネール性脳機能障害の関与についての追加的な支持は、青斑核の神経細胞喪失を有する未治療の自殺者の研究から得られたものである。自殺した未治療者の青斑核におけるチロシンヒドロキシラーゼ(TH)レベルは、対照者のレベルよりも108〜171%高かった(Ordway, Smith er al)。 さらに、自殺した人の青斑核におけるニューロンの数が対照群と比較して23%少ないという知見から、重度のうつ病におけるノルエピネフリン伝達の変化は、より少ないノルエピネフリンニューロンの存在に起因するのではないかという仮説を立てた研究者もいる(Chan-Palay and Asan 1989)。さらに、自殺者の死後の脳組織ではα-2アドレナリン受容体(AR)の機能が変化していることが示されており、うつ病の病態におけるノルエピネフリンとARの関与をさらに支持する知見が得られている(Valdizan, Diez-Alarcia er al)。 死因としての自殺は、必ずしもうつ病が因果関係にあることを示すものではないが、米国自殺予防協会は、自殺する人の90%が死亡時に診断可能な精神疾患を有しており、そのうちの50%以上がうつ病と診断されていると報告している(Prevention 2015)。以前に議論されたように、青斑核ニューロンの顕著な喪失はアルツハイマー病のよく特徴づけられた特徴であり、アルツハイマー病患者の亜集団でより深刻に発生する(Bondareff, Mountjoy er al 2003年そして一般的な仮説では、青斑核のノルアドレナリン作動性ニューロンの部分的な喪失は、残存ニューロンにおけるTH mRNA発現の増加、青斑核周囲樹状帯への樹状突起の芽生え、および海馬への軸索突起の芽生えを介した生存ニューロンの代償的活性化を伴うとされている(Szot、White et al 2006年、Stefani、Olivola et al 2015)。これらの知見は、うつ病患者における脳脊髄液、血漿および尿中のノルアドレナリン活性の指標間の有意な正の相関を示す研究によってさらに支持される(Wyatt、Portnoy et al 1971,Roy、Pickar et al 1987,Roy、Pickar et al 1988)。しかし、他の研究では、うつ病患者が高レベルの不安も有していた場合を除いて、うつ病患者と対照の神経症患者との間で、脳脊髄液 ノルエピネフリンに有意な差を示さなかった(Post, Lake er al)。 現在のところ、うつ病患者における青斑核ニューロンの喪失を直接評価する臨床研究はないが、多様な薬理学的機序を持つ抗うつ薬の共通の治療効果が青斑核ニューロン発火の低下であり、それが神経保護につながる可能性があることを考えると、これは今後の研究の重要な領域であると考えられる(Szabo, de Montigny et al 2000, West, Ritchie et al 2009)。重要なことに、いくつかの研究では、アルツハイマー病を有する抑うつ状態の被験者において、より大きな青斑核ニューロン損失が認められている(Zubenko and Moossy 1988, Zweig, Ross er al)。 1988, Zubenko, Moossy er al)。 1990, Forstl, Burns er al)。 1992)。対照的に、ある研究では、アルツハイマー病患者における青斑核神経細胞の喪失は確認できたが、うつ病を有するアルツハイマー病患者、一過性のうつ病を有するアルツハイマー病患者、うつ病を有さないアルツハイマー病患者の間で青斑核神経細胞の喪失に有意な差は認められなかった(Hoogendijk, Sommer er al)。1999)。しかし、このような研究は、うつ病におけるメランコリックな特徴の有無が考慮されずに残されているため、うつ病の不均一性によって制限されていることに注意すべきである(多動性に関するセクションを参照)。このことは、結果の解釈に大きな影響を与える可能性があり、将来の研究デザインにも考慮すべきであろう。メランコリック性うつ病の患者や、より顕著なメランコリアの特徴を示す患者には、不釣り合いに影響を受ける青斑核ニューロンがあるかもしれないからである。したがって、うつ病の病態生理学的メカニズムが青斑核-ノルエピネフリン系の完全性に影響を与え、その結果、青斑核ニューロンの選択的な変性が起こり、選択された集団をアルツハイマー病の神経病理学的疾患に陥らせる可能性がある。これらの別々の研究はこの可能性を排除することはできないが、これまでの研究ではこの問題を直接研究しておらず、今後の研究の重要な領域である可能性がある。
II. ストレス関連精神疾患とアルツハイマー病に対する脆弱性の性差
ストレスは精神疾患の根底にある共通の特徴であり、特にうつ病のような女性に多く見られるものである。印象的なことに、アルツハイマー病患者の約3分の2は女性である(Alzheimer’s 2015)が、アルツハイマー病の発生率の性差に関する最新のデータは、男女でのアルツハイマー病の発生率が等値であることを示唆している。このような等値性にもかかわらず、危険因子や病理の分子ドライバーの性差は男女間で有意に異なる(Rasmuson, Andrew er al 2001, Beeri, Rapp er al 2009, Corbo, Gambina er al 2009)。ストレス応答を統合する脳領域、およびストレスに対する応答性を決定するシグナル伝達機構には、神経解剖学的および生理学的な性差がある。しかし、ストレス下では、雌ラットがCRFを放出して青斑核を活性化させるストレス因子に曝露された場合、雄ラットよりも雌ラットの成人ホルモンの状態に関係なく大きな応答を示した。さらに、青斑核のニューロンは、雌ラットの青斑核樹状突起は、雄ラットの青斑核樹状突起よりも長く複雑で、分岐点が多く、青斑核周辺部にまで伸びているなど、形態的な性差を示する(Bangasser, Curtis er al)。 最後に、そのトラフィッキングを変更する青斑核におけるCRF受容体1(CRFR1)のシグナリングに分子の違いがあり、雌はストレスへの応答を増強CRFR1を内部化しない一方で、雄はCRFR1の内部化によってストレスへの適応応答を示している()。これらの回路における協調的なCRF放出は、生命を脅かす可能性のある課題を持つダイナミックな環境での対処のためによく設計されているだろうが、これらの応答の開始は、ストレス因子が存在しない場合、またはストレス終了後に応答が持続した場合、特に女性では不適応であろう(Valentino, Van Bockstaele er al 2013, Bangasser, Wiersielis er al 2016)。さらに、コルチゾール産生および代謝は、アルツハイマー病初期の女性において変化しているようである(Rasmuson, Andrew er al)。
c. うつ病とアルツハイマー病の発達と進行における慢性ストレスの影響
i. アロスタシス、アロスタティック荷重、過負荷
ストレスの多い物理的または社会的刺激への適応には、コルチゾール、グルココルチコイド、CRFなどのホルモンおよび神経内分泌メディエーターを介した末梢神経系および中枢神経系の構成要素の調整が必要である。ストレス因子に適応する能動的なプロセス、HPA軸の関与、およびシステム生理学に組み込まれた負のフィードバック機構がストレス応答を終了させ、恒常性を維持することは、アロスタシスと呼ばれている(McEwen 2002,McEwen and Gianaros 2011)。これらの反応は必要なものではあるが、その正味の効果は結果が伴わないわけではない。この適応プロセスの累積的影響は、アロスタティック負荷と呼ばれる(McEwen 2002,McEwen and Gianaros 2011)。アロスタティック過負荷の状態は、ストレス応答を効率的にオフにすることができないことによって定義されたものであり、不適応応答と病態生理につながる慢性的なストレスへの曝露に起因する可能性がある(McEwen and Gianaros 2011,McEwen and Morrison 2013,McEwen、Bowles et al 2015)。(図2)のことである。
図2
ストレス関連精神疾患におけるアミロイドβ蓄積の仮説的帰結。我々は、慢性的なストレスがPFCにおけるアミロイドβの産生と蓄積につながり、シナプス後うつ病のメカニズムを引き起こすことを提案している。PFCニューロンの活動が低下すると、扁桃体などの他の大脳辺縁領域の活動が亢進し、樹状突起密度が増加する可能性がある。その結果、潜在的なフィードフォワードシステムが関与し、神経伝達物質や神経細胞の活動の不均衡が永続するという悪循環が生じ、不安や抑うつなどのストレス関連精神疾患として行動に現れることがある。LTD、長期うつ病。
アロスタティック過負荷の状態で観察される不適応反応は深遠であり、代謝および炎症性の変化を末梢および中枢の両方で引き起こし、神経細胞の遺伝子発現、神経細胞の形態、および大規模な神経細胞ネットワークのアーキテクチャの変化を促進する可能性がある。これらの変化は行動に重大な影響を及ぼすため、ストレス関連精神疾患の理解と治療には極めて重要である。最近の文献では、慢性的なストレスが人間の行動、大規模な脳ネットワーク、それらを構成するより小さなニューロンネットワーク、そしてそれらの活動を制御する個々のシナプスを大きく変化させるメカニズムが、精神神経疾患と神経変性疾患との関連性を提供している可能性があるという考えが支持されている。以下のセクションでは、アロスタティック過負荷、すなわち慢性的なストレスが、大脳辺縁回路の増強と皮質回路の減少をもたらすような神経回路の再編成をもたらし、その結果、認知症を含む精神症状によって証明される断絶症候群を引き起こすという仮説を支持する証拠をレビューする。
ii. ストレスと大規模な脳ネットワークの再編成。バランスをとるために
特定の作業中に信頼できる活動パターンを示す大規模な脳ネットワークの活性化や接続性は、脳の状態と呼ばれることがある。個人が外部環境に集中せず、むしろ内部の出来事や思考に集中しているときの脳状態は、デフォルトモードネットワーク(DMN)と呼ばれる解剖学的に定義された明確な脳システムに関与している(Buckner, Andrews-Hanna er al 2008, Buckner 2013, Raichle 2015)。DMNは、自伝的記憶の検索、将来の計画、他者の思考や知覚の構想など、内的な集中作業の際に活動する(Buckner, Andrews-Hanna er al 2008)。ヒト以外の霊長類とヒトを対象とした神経イメージング機能的磁気共鳴画像法(fMRI)の研究は、DMNに関連するコア領域を特定するために収束してきた。腹内側前頭前野皮質(vMPFC)(下肢下領域としても知られている後帯状皮質(PCC隣接前帯状皮質(pCu下頭頂葉(IPL側頭側皮質(LTC背内側前頭前野皮質(dMPFC海馬形成部(HF)である(Buckner, Andrews-Hanna er al 2008,Buckner 2013)。)
DMNの発見は、脳の状態の維持や、内心の状態の切り替えと外部環境との相互作用の神経的な相関関係についての仮説を立てる研究者たちを惹きつけている(Tang, Rothbart et al 2012)。DMNの抑制は、集中注意、ワーキングメモリ、その他の実行機能などの特定の認知プロセスの正常な動作に機能的に重要である(Anticevic, Cole er al 2012)。状態間のこの効果的な切り替えは、前島皮質領域と背側前帯状体領域からなるサリエンスネットワークの機能に少なくとも部分的に依存する(Sridharan, Levitin er al 2008, Bonnelle, Ham er al 2012)。
青斑核はこのメカニズムにおいて重要な役割を果たしていると考えられている(Corbetta, Patel er al 2008, Hermans, van Marle er al 2011) [広範なレビューは(Aston-Jones and Cohen 2005, Valentino and Van Bockstaele 2008)]を参照のこと。青斑核-ノルエピネフリン系は、注意、覚醒、認知の促進、ストレス刺激への応答に決定的に関与している。したがって、青斑核の活性化とノルエピネフリンの放出は、安静状態を終了し、皮質、皮質下、自律神経活動を含む調整を開始し、注意を増強し(Tang, Rothbart er al 2012, Buckner 2013タスク関連の刺激の行動出力を可能にすることが提案されている(Aston-Jones and Cohen 2005)(図2)。受動的なタスク状態では、青斑核-ノルエピネフリン系は高い強直性ベースラインを特徴とし、これはおそらく自発的な思考と関連している(Smallwood, Brown er al 2012)。タスクに集中した状態では、青斑核の強直活動が中程度のレベルまで低下することで、当面のタスクへの最適な関与が促進される可能性がある(Aston-Jones and Cohen 2005)。したがって、DMNと重なる特定の領域の不活性化は、少なくとも一部では、シータ振動によって反映される青斑核-ノルエピネフリン系に関連した高い強直活動によって引き起こされる可能性が示唆されている()。これを裏付けるように、モノアミン作動性伝達系に作用すると考えられている覚醒促進剤であるモダフィニルは、仕事で誘発されるDMNの不活性化を増強して、センサー運動の処理速度を促進することが示されているが、これはvMPFCの活動の変化に依存する効果であり、カテコラミン系の利得制御機能と一致する(Minzenberg, Yoon et al 2011)。
iii. 青斑核-ノルエピネフリンによるDMNの脱活性化の文脈でのアロスタシス
タスク指向行動のためのDMNの不活性化を開始する役割に加えて、青斑核-ノルエピネフリンシステムはストレス応答脳状態の重要な構成要素である。このことは、ストレスを受けた人は対照群に比べてDMNの活性化が大きく、安静時のネットワークの不活性化が損なわれていたという最近のヒトfMRI研究での証拠に照らして、特に重要である(Soares, Sampaio er al)。 具体的には、ストレスは内側前頭前野(mPFC内側眼窩前頭前野、PCC、pCuの機能的接続性を増加させた。Sousaらは、ストレスを受けた参加者で観察されたDMNの後領域、特にPCCと下側頭頂葉の増加は、感情的に重要な刺激のより長い処理とエピソード記憶の検索に関連している可能性が高いと結論づけている(Maddock, Garrett er al 2003, Wagner, Shannon er al 2005, Soares, Sampaio er al 2013)。さらに、この機能的接続性の増加は、DMNの接続性マップの収縮と関連しており、左PCCと左右頭頂下領域の特異的な減少を伴っていた。全脳解析では、ストレス群と対照群の相対的な頭蓋内容積に差はなかったが、ストレスを受けた患者ではDMNの総容積の有意な減少が観察され、特に左PCCと左右の頭頂下野に顕著な減少が認められた。Sousaはさらに、これらの減少は、ストレスによって誘発された皮質領域の萎縮効果を反映している可能性が高いと推測している(Liston、McEwen et al 2009,Soares、Sampaio et al 2012,Soares、Sampaio et al 2013)。本研究の参加者は、fMRIの他に知覚ストレス尺度(PSSハミルトン不安尺度、ハミルトンうつ病尺度についても調べた。その結果、ストレスを感じている人ほどハミルトン不安尺度のスコアが高いことが確認された。これは、不安を感じている患者ではpCuの不活性化が低いことを明らかにした過去のfMRI研究(Porter, Buxton er al 2003, Carey, Warwick er al 2004, Zhao, Wang er al 2007, Gentili, Ricciardi er al 2009, Soares, Sampaio er al 2013)やアルツハイマー病患者の不安を感じている患者(Zhao, Wang er al)。 このように、ストレスはDMN回路、特に前頭前野(PFC)領域における深遠な変化を反映した行動変化を促進するように作用する可能性があり、それは精神医学および神経変性疾患に重要な意味を持つ(後述)(図2)。
iv. DMNの不活性化の失敗:うつ病やアルツハイマー病への示唆
Bucknerらは、臨床疾患とDMNの破壊との間のおそらく最も説得力のあるリンクは、アルツハイマー病で起こることに注意してほしい(Buckner、Andrews-Hanna et al 2008)。代謝から、アミロイドβピッツバーグ化合物B(PIB)-陽電子放射断層撮影(PET構造と活性に基づくイメージング研究から、すべてのアプローチは、DMNが破壊されていることを示唆するために収束する(Buckner, Andrews-Hanna er al)。 アルツハイマー病患者と年齢を一致させた健康なコントロールにおける代謝の減少の神経解剖学的パターンは、DMNの解剖学的基質を反映しており、PCC、IPLとLTCが含まれている(ワグナー、シャノン et al 2005)。さらに、疾患の遺伝的リスクを持つ患者は、これらの領域で同様の代謝の違いを示している(Reiman, Caselli er al)。 アルツハイマー病患者の脳の局所的な萎縮を調査した追加の研究はまた、内側側頭葉の体積の顕著な減少を示している(Scahill, Schott er al 2002, Thompson, Patterson er al 2003, Buckner, Snyder er al 2005)、病気の前臨床段階でPCCと内側側頭葉の萎縮が加速されている(Buckner, Snyder er al 2005)。18F]フルオロデオキシグルコース(FDG)-PETを用いた研究では、グルコース代謝を介した神経細胞の活動を測定することで、DMNの正中線に沿った領域、特にPCCでは、他の脳領域と比較して約20%も不釣り合いに高い安静時グルコース代謝を示すことが示されている(Gusnard, Raichle er al 2001, Raichle, MacLeod er al 2001)。アルツハイマー病の病理学の説得力のある報告は、症状が現れる前であっても、アミロイドβが蓄積するネットワークとしてDMNを引用している(Sheline、Raichle et al 2010,Myers、Pasquini et al 2014,Koch、Myers et al 2015症状が現れる前(Klunk、Engler et al 2004疾患の経過の中でこのシステムの早期の調節障害を示唆している。これに関連して、認知的に正常な人を対象とした研究では、DMNと他のネットワークの内と内の間で、アミロイドβの蓄積が機能的接続性の変化を誘発し、おそらく局所的なネットワークの混乱と代償的な再編成を反映していることが示されている。さらに、これらの変化は、代謝低下のバイオマーカーよりも前に起こり、これらの深遠な変化が認知機能低下の発症前に起こっていたことを示している(Elman、Madison et al 2016)。記憶障害および睡眠障害を有する高齢者において、アミロイドβはまた、DMNにおける機能的連結性を乱した(Mander、Marks et al 2015)。これらの知見と一致して、アミロイドβ沈着のBraakステージングは脳の皮質領域で始まる(Braak and Braak 1991)。Bucknerらは、DMNの継続的な活動が、アルツハイマー病病理の形成を助長する活動依存性または代謝依存性のカスケードを増強することを提案しており(2005これは、アミロイド前駆体タンパク質(APP)処理およびシナプス活動依存性のアミロイドβ形成の既知のメカニズムと一致している(Cirrito、Yamada et al 2005)。
安静時状態-機能的結合性MRI(rs-fcMRI)を用いた研究が増えており、DMN活性の抑制の失敗が認知障害および神経精神疾患に関連する症状に関与していることが示唆されている(Benson, Kuhl et al 1983, Buckner, Andrews-Hanna et al 2008, Anticevic, Cole et al 2012)。うつ病ではDMN抑制の欠如が報告されている;Shelineらは、うつ病患者がネガティブなイメージを検討しているときにDMN活性を低下させることに失敗することを発見した(Sheline, Barch er al)。 うつ病では、DMN抑制の欠損は、mPFCの活動を反映していると考えられている効果である、うろ覚え(Hamilton, Furman er al)。2011)または否定的な内的思考と関連している(Hamilton, Furman er al)。2011, Anticevic, Cole er al)。 これを裏付けるように、大うつ病性障害患者の神経画像診断および死後分析では、dMPFC/外側PFC、前帯状体下、前帯状体前野、眼窩およびベントロラテラル、PCCにおけるグルコース代謝および脳血流の減少に対応する灰白質体積および細胞数の減少が明らかにされている(気分障害における構造的および機能的変化の優れたレビューについては(Drevets, Price er al)。 興味深いことに、うつ病患者では、dMPFC領域とDMNとの間のfMRI接続パターンが増加していることが報告されており(Kessler, Chiu er al)。 印象的なことに、Sousaらによる研究では、ストレスを受けた参加者においてmPFCとPCCの間の機能的接続性が増加していることが明らかになっており、ストレスによって前帯状皮質の安静時の活性化が増加したという知見は、うつ病患者で変化することが知られているネガティブ情報の情動的処理にも関連している(Surguladze, Brammer er al)。2005, Mogg, Bradbury er al)。2006)。
上記に示された累積的な証拠に基づいて、ストレスは、DMN活動を抑制する脳の能力を低下させる重要な病因因子である可能性がある。さらに、ストレス応答における青斑核-ノルエピネフリン系の役割は十分に確立されており、DMNをさらに調べることで、青斑核-ノルエピネフリン活性がDMN抑制にどのように関与しているかをよりよく理解できるようになるかもしれない。現在利用可能な文献によると、青斑核-ノルエピネフリン系はスイッチとして機能し、DMNの抑制を可能にすると同時に、タスクに集中した注意を可能にしていることが示唆されている。未知であり、今後の研究にとって重要なことは、慢性的なストレスと青斑核-ノルエピネフリン活動の下流の調節障害がDMN活動に及ぼす直接的な影響である。提示された証拠から推測されるのは、おそらく慢性的なストレスの結果として、青斑核-ノルエピネフリン系の調節障害が、少なくとも一部でDMNを抑制する能力の低下をもたらし、これがストレスに関連した疾患状態に重大な影響を及ぼすということである。さらに、DMNに関与する皮質領域の機能不全の結果として、神経内分泌、神経伝達物質、感情刺激やストレス因子に対する行動反応に関与する下流の回路の神経構造に影響を与え、悪循環を生み出す可能性がある(次項で詳しく説明する)。したがって、ノルエピネフリンがこれらの効果を発揮する可能性のある細胞および分子過程を理解することは、大規模ネットワークとそれに対応する脳の状態の維持と切り替えに関する理解を深めるだけでなく、ストレス関連精神疾患の神経基質とその行動的相関関係の理解を促進し、理解することができるかもしれない。以下のセクションでは、議論されている臨床所見のメカニズム論的裏付けを提供する可能性のある前臨床証拠をレビューする。
III. 前臨床のメカニズム
a. ストレス統合回路のアロスタティック再編成
ストレス反応の内分泌系、認知系、情動系の間の調整は、知覚されたストレス因子に対する適切な行動反応を促進するために必要とされる。この調整された反応において重要な役割を果たすのは、視床下部の傍室核(PVN)から放出されると、末梢および中枢のストレス反応系を並行して関与させる神経ホルモンCRFである。視床下部門脈系を介して前脳下垂体にCRFが作用すると、副腎皮質刺激ホルモン(ACTH)が放出され、コルチゾールとグルココルチコイドを産生するように副腎を刺激する。特に重要なのはグルココルチコイドであり、グルココルチコイドはPVN、海馬、PFCに豊富に存在する受容体に結合して活性化すると、ストレス応答を終了させる負のフィードバックループに参加するためである(McEwen 1988,Ahima and Harlan 1990,Ahima and Harlan 1991)。したがって、グルココルチコイドの循環血中コルチゾールレベルおよび間質液(ISF)レベルは、覚醒時のストレス応答状態の指標として使用される可能性がある。重要なことに、CRFは末梢ストレス応答に加えて、中脳核青斑核と扁桃体をそれぞれ刺激すると、ストレス応答の複雑に接続された認知的および感情的な四肢にも関与する(Valentino and Van Bockstaele 2008)。コルチコ肢神経回路の重要な構成要素であるmPFCは、扁桃体や脳幹など多様な領域への下流への広範な投影を有し(Sesack, Deutch er al)。 1989自律神経および神経内分泌バランスのトップダウン制御の実行メカニズムを提供し(Thayer and Brosschot 2005副交感神経(Thayer and Sternberg 2006)およびHPA活動(Diorio, Viau er al)。 特に重要なのは、mPFCからの興奮性突起が、基底側扁桃体(BLA)のガンマ-アミノ酪酸(GABA)エルゴ性介在ニューロンにシナプスし、扁桃体中心核(CeA)ニューロンの出力を抑制することである。mPFC からのトップダウン変調がない場合には、CeA の過剰活性化は、青斑核 ニューロンの活動と ノルエピネフリン の分泌の増加につながる可能性があるので、これは重要な意味を持っている、強化された感情と認知大脳辺縁系の興奮性とそれらの回路の減少皮質制御の悪循環を作成する(図 2)。
i. 慢性ストレスの前臨床モデルは、うつ病とアルツハイマー病を持つヒトの神経精神医学的欠陥を反映している
脳内のストレス反応領域は、機能的に統合されたネットワークとして構成されており、このネットワークが乱されると、認知機能や感情処理がグローバルなレベルで混乱する。慢性的で反復的なストレスの前臨床モデルは、気分障害の神経生物学的相関の調査を容易にするために開発されてきた(Drevets, Price er al 2008)。抑うつ状態にあるヒトで灰白質の減少が明らかな領域(mPFC、海馬)に相同性があると思われる領域では、反復ストレスの結果、樹状突起の萎縮とグリアの数と増殖の減少がげっ歯類で見られる(McEwen and Magarinos 2001,Radley, Sisti er al)。 ストレス条件下で樹状突起の萎縮を示すと報告されている領域の多く、特に、前臨床モデルでは右前辺縁下皮質および下辺縁下皮質、左前帯状皮質のII/III層錐体ニューロンが報告されている(Wellman 2001, Cook and Wellman 2004, Radley, Rocher er al 2006, Cerqueira, Taipa er al 2007, Shansky, Hamo er al 2009,Sousa and Almeida 2012)は、DMN構造における灰白質体積の減少の報告(Sheline, Raichle er al 2010,Soares, Sampaio er al 2013,Elman, Madison er al 2016およびヒトにおけるうつ病やアルツハイマー病に関与する他の領域(Chan-Palay and Asan 1989,Chan-Palay and Asan 1989,Soares, Sampaio er al 2013,McEwen, Nasca er al 2016)と一致している。図2)。これらの知見は、大うつ病性障害(MDD)におけるPFCと海馬構造における灰白質体積の減少の根底には相同的なプロセスがあるという仮説を立てる研究者もいる(McEwen and Magarinos 2001そして潜在的にうつ病や慢性的にストレスを受けた人のアルツハイマー病に対する脆弱性を高める可能性のある長期的な変化についての洞察を提供することができる(Pomara and Sidtis 2009)。
21日間の期間のための慢性的な拘束ストレスを利用したげっ歯類の研究は、地域的に特定の方法でPFC、扁桃体、および海馬の機能的および構造的な変化を示した(McEwenとGianaros 2011)。これらの変化は、mPFCの樹状突起が短くなるようなものであった(Wellman 2001, Cook and Wellman 2004, Radley, Sisti er al 2004, Cerqueira, Catania er al 2005, Radley, Rocher er al 2006, Cerqueira, Taipa er al 2007)一方、扁桃体では樹状突起の成長が明らかであった(Vyas, Mitra er al 2002, Vyas, Mitra er al 2002)。これらの結果と同様に、げっ歯類を用いた反復ストレスの他の研究でも、結果としてBLAニューロンの過剰興奮性が示されている(Shekhar, Truitt et al 2005, Vyas, Jadhav et al 2006)。下肢下層、前辺縁部、帯状皮質の第三層の錐体ニューロンを調査した他の研究では、雄性ラットにおいて慢性的なストレスにさらされると、樹状突起の先端部の遠位部分の長さが20%減少することが示されている(Hains, Vu er al)。 この樹状突起の収縮は、慢性ストレス後に、可塑性や認知パフォーマンスに関与することが知られている細い棘を中心とした30%以上の棘状突起の消失を伴った(Bloss, Janssen er al)。 これらの知見は、MDDの重症度が扁桃体の活動と正の相関があり、PFCおよび外側眼窩皮質領域の活動と負の相関があることを示すヒトの研究と一致している(Drevets, Price er al)。 このように、mPFCの活動が集団的に抑制またはダウンレギュレーションされ、同時に扁桃体の活動が増加することは、慢性的なストレスの条件下でストレス統合回路の変調と組織が変化するメカニズムとして機能する可能性がある。この推論に沿って、一部の研究者は、原発性およびいくつかの二次性気分障害におけるmPFCの神経病理学的変化が、感情表現に対するmPFCの調節的役割を損ない、ストレス因子や感情刺激に対する大脳辺縁反応を抑制または増強するのではないかと推測している(Drevets, Price er al 2008)。
これまでに述べてきた証拠は、PFCと扁桃体のストレス誘発性リモデリングが、樹状突起棘密度の低下によってPFCのトップダウンの実行制御が低下するようなモデルを支持するものである。さらに、PFCにおける樹状突起棘の後退は、樹状突起の成長と扁桃体ニューロンの過剰興奮性によって媒介される感情的な覚醒を高めることができる。このように、検討すべき次の重要な問題は以下の通りである。1. これらの変化は行動表現型にどのような影響を与えるのか?2. 2. 青斑核ニューロンへの同時作用はあるのか?これまでのところ、ストレス誘発性うつ病モデルにおける青斑核ニューロンを調べた研究はほとんどない。しかし、これまでの研究では、ストレス誘発性うつ病の条件下では青斑核軸索は退化し、抗うつ薬であるデシプラミンやイミプラミンの投与によって再生する可能性があることが示されている。このことから、青斑核ニューロンはうつ病では退化し、寛解では再生すると結論づけられている(北山、中村 et al 1994,北山、大谷 et al 2008)。より最近の研究では、神経毒6-ヒドロキシドパミン(6-OHDA)を青斑核に直接注入すると、強制水泳試験(FST)における不動時間の増加によって示される抑うつ表現型が得られ、これは行動評価の前にL-1-3-4-ジヒドロキシフェニルアラニン(DOPA)またはL-スレオ-3,4-ジヒドロキシフェニルセリン(DOPS)のいずれかを単回投与することで反転させることができるというように、この概念のさらなる支持を提供する(Szot, Franklin er al 2016)。研究者らはまた、6-OHDA後の生存青斑核ニューロンの数とFST不動との間に有意な正の相関があることを報告している。したがって、青斑核ニューロンのわずかな損失であっても、抑うつ症状を誘発するのに十分であり、MCI患者で観察されるような青斑核ニューロンの早期変性が、観察されたB脳卒中後認知症、特に抑うつ症状に寄与する可能性があると結論付けている(Szot, Franklin er al 2016)。このようなモデルにおけるタウ-pおよび内因性アミロイドβの異常の調査は、神経変性と神経精神疾患の交点の理解を前進させるために重要であろう。
II. 樹状突起棘密度のストレス誘発変化:グルココルチコイドと加齢の役割
McEwenとMorrisonは、生涯にわたる構造可塑性におけるストレスの役割に関する優れたレビューの中で、慢性ストレス下での樹状突起棘のターンオーバーにおけるグルココルチコイドの重要な役割について論じており、加齢がストレスに対するニューロンの回復力、および大脳皮質、海馬、扁桃体における可塑性のメカニズムに大きく影響することに言及している。グルココルチコイドは、急性・慢性を問わず、ストレス下での樹状突起棘密度の維持・調節に重要な役割を果たしている。さらに、前頭前野や海馬などの領域はグルココルチコイド受容体が豊富であり(Ahima and Harlan 1990, Ahima and Harlan 1991したがって、樹状突起の形態や萎縮におけるストレス関連の変化に対してより脆弱である可能性がある。
McEwenとMorrisonはさらに、ヒト、非ヒト霊長類、およびげっ歯類における年齢とともに低下するPFC依存タスクの脆弱性について論じている((Gallagher and Rapp 1997)でレビューPFCに依存した認知パフォーマンスの年齢による低下は、PFC錐体ニューロン上の軸索性シナプスのmPFCクラスの錐体層IIIの軸索性シナプスを構成する細くて高度に可塑性のある棘の損失に起因する可能性がある(Dumitriu, Hao er al)。 興味深いことに、若い動物では、慢性ストレス後に樹状突起の形態や認知能力が大きく変化したにもかかわらず、3週間の休息期間で樹状突起の完全な回復と部分的な脊椎密度の回復が見られ、完全な機能回復には十分である(Radley, Rocher er al 2008, Bloss, Janssen er al 2011)。対照的に、中年ラット(12ヶ月齢)や高齢ラットでは、慢性的なストレスを受けた後の安静時の神経細胞の回復は見られず、中年になると神経細胞の回復力が低下していることが明らかになった(Bloss, Janssen er al)。 さらに解析した結果、中高年ラットと高齢ラットでは、ストレスがない場合には30%の棘が失われており、この喪失は主に高齢ラットの細い棘の喪失に起因していると結論づけられた。これらの研究をまとめると、高齢ラットのmPFC錐体ニューロンは複数のレベルで可塑性を失っているという証拠が得られる。第一に、高齢動物のニューロンはPFC回路内で適切に機能するために重要な細い棘の集団を失っている。重要なことは、これら3つの加齢に伴う可塑性の変化はすべて中年と高齢動物の両方で観察されたことであり、このような可塑性欠損に対する予防策は、中年期に実施すると最適な効果が得られる可能性があることを示唆している。これらの観察は、慢性的なストレスにさらされた中年ヒトは、ストレスを受けていないコントロールよりもアルツハイマー病を発症しやすいという報告と一致している(Johansson, Guo er al 2010およびApoE ε4対立遺伝子の存在下でストレスに高齢で非認知症の個体を長期間曝露すると記憶力の低下につながるという報告と一致している(Peavy, Lange er al)。
iii. アロスタシスとシナプスのリモデリング
アルツハイマー病はシナプス障害の一つと考えられている。シナプスは、ニューロンとグリア細胞のためのコミュニケーションの主要なモードであり、局所的なニューロン回路と脳の異なる領域を接続するより広い回路の活動を指示する。シナプスは、様々な刺激に反応して頻繁にリモデリングやターンオーバーを行う動的な構造であり、経験によって形成される(Bloss, Janssen er al)。 このように、シナプスの完全性は脳機能に不可欠であるが、学習や記憶のプロセスや、常に変化する環境への適応のためにも可塑性が必要である。シナプスリモデリングには、棘の剪定による樹状突起の数の変化や、多様なタイプの樹状突起の除去や再生が含まれる(Sousa and Almeida 2012)。ストレスは同時にグルココルチコイド、CRF、ノルエピネフリンを含む神経調節物質の緊張に影響を与えるが、これらはすべて、地域的に特異的な方法で、受容体の結合に深遠な分子効果を持つ可能性がある。ストレスによって誘発される樹状突起の引っ込みと拡張、シナプスのターンオーバーのメカニズム、およびストレスによって誘発されるシナプス可塑性の行動的な影響についてレビューされている(McEwen and Morrison 2013)。シナプスのリモデリングはすべての脳領域でニューロン全体の健康に極めて重要であるが、複雑な認知プロセスが発生するPFCと海馬領域では特に重要である(Wellman 2001)。我々は、慢性的なストレス、うつ病、および アルツハイマー病 の過程で劇的に変化することが知られているシナプスの主要な構成要素を記述することによって、このセクションを開始する。このセクションで検討した細胞や分子の基質は、ストレスに関連した精神疾患におけるニューロンの構造、機能、および生存率に影響を与える基礎的なメカニズムの主要なメディエーターである可能性が高いので、慢性的なストレスとうつ病の既往歴がどのようにして個人をアルツハイマー病に対してより脆弱にするのかを推測している。
a. シナプスの細胞骨格構造:タウの役割
シナプスの構造的可塑性は、ニューロンやグリアのシナプスリモデリングの形で、機能的可塑性につながる(Sousa and Almeida 2012)。シナプスは、シナプス前ニューロン、定義されたシナプス裂け目、シナプス後密度(脳卒中後認知症)が顕著なシナプス後ニューロンで構成されている。タウは、微小管関連タンパク質(MAP)ファミリーのメンバーであり、主に軸索性微小管を安定化する可溶性のリン酸タンパク質であるが、細胞の形態を維持するために樹状突起性微小管も束ねて安定化する。軸索微小管のタウ結合と安定化は、細胞内小器官や小胞の輸送に重要である。タウ-微小管相互作用を制御する主要なイベントの一つはリン酸化であり、これはタウの結合と機能を変化させる。一方、シナプス後の樹状突起構造は、シナプス前末端構造との接続を行う樹状突起(樹状突起棘)を有し、膜成分と細胞質細胞骨格タンパク質との連結を担う多様な脳卒中後認知症タンパク質で構成されている(Kondo and Okabe 2011)。アクチンは、足場タンパク質の活性依存的な再分配を調節するシナプスの主要な細胞骨格成分の一つであり、シナプス活動の強さや脊椎構造の形態的拡大によって変化する可能性がある(近藤・岡部2011)。
b. シナプスにおける神経細胞の興奮性の調節:アミロイドβの役割
先に述べたように、アミロイドβペプチドはシナプスにおいて、シナプス活性とエンドサイトーシスに依存した方法で、約60%の時間で産生される(Cirrito, Yamada er al)。 アミロイドβの産生もまた、その前駆体であるAPPの細胞内局在によって大きく左右される(Haass, Kaether er al)。 典型的には、APPの内部化は、アミロイド原性処理およびアミロイドβの形成に有利であり、一方、表面上のAPPの存在は、非毒性の可溶性APPαおよびC83フラグメントを産生することが知られているα-セクレターゼに遭遇する可能性を高める(Hong, Huang er al)。 シナプスアミロイドβ産生がニューロンの活性化とエンドサイトーシスに依存しているという累積的な証拠に基づいて、ニューロンの活性化に続いて、シナプス小胞のリサイクルがAPPの内部化を増加させ、それによって細胞内コンパートメントに存在するβ-およびγ-セクレターゼの基質が増加することが提案されている。その結果、アミロイドβフラグメントは細胞外空間に放出される可能性がある(Cirrito, Kang et al 2008)。アミロイドβは、小胞放出の確率を高める結果として、シナプス前にカルシウム透過性を高める能力を有することが示されている(Abramov, Dolev er al 2009またはシナプス後に作用して、受容体の内部化を促進することによって神経細胞の活動を抑制する可能性がある(Snyder, Nong er al 2005, Wang, Yuen er al 2011, Ulrich 2015および長期うつ病(LTD)(Li, Hong er al 2009, Palop and Mucke 2010)。これらの知見に沿って、最適な濃度のアミロイドβは小胞放出と神経伝達を促進するが、低すぎる、あるいは高すぎる濃度のアミロイドβはシナプス活動を阻害することが実証されている(Abramov, Dolev er al 2009)。この証拠に基づいて、アミロイドβは中枢神経系において、神経細胞の興奮性を調節する負のフィードバックループに参加する、神経細胞の活動の調節因子としての生理的役割を持っていると推測されている(Palop and Mucke 2010, Palop and Mucke 2010)。神経細胞の活動が低いと、シナプスでのβセクレターゼの活性を促進することでAPPのアミロイドβへの処理が促進されるが、一旦アミロイドβが豊富に形成されると、シナプス後に作用して神経細胞の活動を抑制する。これは、先に議論したように、ニューロン間のシナプス接続が、通常の生理学的条件下で扁桃体や海馬などの領域をトップダウンで制御するPFCやそのグローバルネットワークのような下流の微小回路回路の青斑核のようなニューロンのグループの活動を決定することができるため、重要な意味を持っている。このように、アミロイドβのアンバランスは、神経細胞の機能、行動、気分にグローバルな影響を及ぼす可能性がある。
c. ストレスは、アルツハイマー病関連のシナプスタンパク質の異常の前駆因子である
ストレスは脳の複数の領域の活動を乱す可能性がある。シナプスでのアミロイドβ産生はシナプス活動によって決定され、慢性的な神経細胞の多動がアミロイドβ蓄積の原動力であることが実証されている(Yamamoto, Arima er al 2015)ので、ストレスがアミロイドβのシナプスレベルを上昇させ、おそらく機能障害のレベルまで上昇させる可能性があるという考え方は論理的である。アミロイドβペプチドの生化学的解析により、アミロイドβペプチドは高度に疎水性の約4kDaのタンパク質であり、安定な二量体、三量体、より高いオリゴマー、そして最終的には8nmのアミロイド線維へと自己集合する傾向が強いことが明らかになっている(Masters and Selkoe 2012)。このように、神経細胞の微小環境、および特定の条件によって示される活性のレベルは、神経細胞の微小環境に依存している。特に、慢性的な青斑核の過剰活性化を介して、ストレスは、その広範な突起を介して脳をレンダリングする可能性があり、アミロイドβペプチドの過剰な蓄積、およびその後の沈着、プラークを形成するために、より脆弱になる。重要なのはPFCにおけるアミロイドβの過剰蓄積であり、これは扁桃体を制御するGABA作動性介在ニューロンのシナプス後抑制を促進し、大脳辺縁部の興奮性を高めるための解剖学的基質を提供することになる。
正常な生理学的条件の下では、ニューロンのクラスターは同期して一緒に発火し、これは記憶と学習プロセスにとって重要なプロセスである(Jutras and Buffalo 2010)。しかし、何がアルツハイマー病を持つヒトで、そしてAPPを過剰発現し、その結果としてアミロイドβの産生が増加しているアルツハイマー病のマウスモデルで観察されている、回路やより広いネットワークにおけるニューロン活動の異常なパターンがあるということである(PalopとMucke 2010,PalopとMucke 2010)。これは、アミロイドβが通常参加している負のフィードバックループが、アルツハイマー病で変化することを示唆している。例えば、回路レベルでは、高レベルのアミロイドβは、ヒトAPPトランスジェニックマウスと非トランスジェニックコントロールの左右の頭頂皮質の脳波記録に示されているように、ネットワークの同期を増加させ、てんかん状の活動を誘発する(Palop、Chin et al 2007)。しかし、個々のニューロンを生体内試験でカルシウムイメージングによって調べると、非トランスジェニックコントロールから記録されたニューロンは中程度の活性を示すのに対し、プレセニリン1およびヒトAPP(PSAPP)遺伝子変異を発現するように遺伝子操作されたマウスのニューロンは、アミロイドβレベルが高く、低活性または高活性である(Busche, Eichhoff er al)。 最終的には、このことが、アルツハイマー病は断絶症候群、すなわちグローバルネットワークの非同期化であるという見方につながっている。統合失調症、自閉症、アルツハイマー病で観察される神経同期の障害は、基礎となる病態生理学的メカニズムとして切断症候群を強調する現在の理論と一致している(Palop, Chin er al 2007, Palop and Mucke 2010, Palop and Mucke 2010)。これらの理論によると、認知機能障害だけでなく、これらの障害の症状は、大脳皮質の機能的に専門化された領域間および領域内での分散した神経活動の協調性の機能不全から生じる(Elman, Madison er al)。 神経同期の低下は、断絶の結果であり得るが、神経応答の同期化は、疎結合ネットワークを横断するそれらの伝播に不可欠であるため、脳領域間の結合障害の原因でもあり得る(Abeles 1991)。
i. CRFおよびCRF受容体1
過去10年間で、多くの研究が急性および慢性ストレスの行動モデルやCRFの過剰発現(CRF OE)またはCRF受容体のアブレーションによってCRFR1シグナル伝達を調節する多くの遺伝モデルを利用することによって、アルツハイマー病におけるストレスの寄与を検討していた。アミロイドβおよび高リン酸化タウ蓄積に対する神経細胞活性化の影響を検討した先駆的な研究では、ストレス、またはCRF神経伝達の増加がアルツハイマー病神経病理を加速することが実証された(Kang, Cirrito er al 2007, Dong, Murphy er al)。 Rissmanグループの研究は、アミロイドβ蓄積に対するCRFR1アブレーションの効果を調べるために、CRFR1の遺伝子アブレーションを有するマウス系統を、アルツハイマー病のPSAPPマウスモデルと交配させて利用している。CRFR1をアブレーションしたマウスでは、PSAPPマウスと比較してアミロイドβ40, アミロイドβ42のレベルが有意に低下し、PSAPP-R1変異マウスではアミロイドβPPのC末端フラグメント(CTF)が同時に劇的に減少した。さらに、PSAPP-R1変異マウスでは、インスリン分解酵素(IDE)とアミロイドβPPのCTFが減少していることが示され、観察された変化の根底にあるメカニズムは、クリアランスではなくアミロイドβ産生が関与している可能性が示唆された。示唆されているメカニズムの一つは、CRFR1がβ-またはγ-セクレターゼのトラフィッキングのモジュレーターであることである。したがって、CRFR1はストレス応答性を司る主要な受容体であり、他の領域の中でも特に青斑核に豊富に発現しており、ADマウスモデルにおけるアミロイドβ産生の重要な寄与者であることが示唆される。
同様に説得力のある研究は、CRFR1の活性化またはアブレーションがタウのリン酸化に及ぼす影響を探る研究である。ある研究では、30 分間の急性拘束ストレスは、グルココルチコイド シグナルとは無関係に、海馬組織の可溶性画分の複数の アルツハイマー病 関連部位(S181,S199,S202/T205(AT8T212,T231,S396/404(PHF-1および S422)におけるタウ-p の急速かつ可逆的な増加をもたらしたことが実証された。さらに、CRFRはタウ-pを異なる形で制御しており、CRFR1がストレスによるタウ-pの増加に主に関与していることが明らかになった。これらの知見は、CRFR1の選択的アンタゴニストであるアンタラミンを用いた薬理学的研究によっても裏付けられ、CRFR1のアンタゴニストはストレスによるタウ-pの上昇を抑制するのに十分であることが示された。これらの結果は、ストレスにより AT8 と PHF-1 の tau-p が誘導されることに CRFR1 シグナルが特異的に関与していることを示唆している (Rissman, Lee er al 2007)。
慢性的な繰り返しストレスのタウ-pへの影響に焦点を当てたその後の研究では、CRFRがタウ-pだけでなく、その溶解性や凝集状態にも異なる影響を与えることが明らかになった。この効果が慢性/反復ストレスに一般化するかどうかを調べるために、CRFR1,CRFR2,または両方の受容体を欠失したマウスを用いて、拘束ストレスに1回暴露した後(急性ストレスまたは14回連続暴露した後(反復ストレス可溶性と不溶性の両方の海馬画分のAT8とPHF-1部位のレベルを調べた。急性ストレスに起因するtau-pは一過性であり、ストレスに関連した神経可塑性に関与している可能性があるのに対し、反復ストレスやCRFの過剰発現によって誘導されたtau-pはストレス曝露後も長時間存在し、明確な構造を持ち、洗浄性可溶性細胞画分に局在していた(Rissman, Staup er al 2012)。このように、CRFR1を介して慢性ストレスによって誘導されたタウ-pは、長期にわたってNFT形成をもたらす可能性のある前病理学的構造を形成している。
CRF OEマウスは生後3~6ヶ月で脳の萎縮を示し、生後9ヶ月では海馬依存性の学習障害を示す。さらに、CRF OEマウスを用いた研究では、野生型マウスと比較してタウ-pのレベルが増加していることが確認されている。CRF OEマウスは海馬のtau-pが増加しており、特にAT8,PHF-1およびS422は、CRF OEマウスではWTマウスと比較してそれぞれ350%、350%および170%増加していた。選択的CRFR1アンタゴニストであるR121919で30日間処理すると、CRF OEマウスのAT8およびPHF-1リン酸化部位におけるタウ-Pの上昇が完全に阻止された。対照的に、S262およびS422部位のレベルは、R121919の前処理によって変化しなかった。これらの結果は、CRFの過剰発現が海馬においてタウ-pを誘導しうることを示している。このプロセスは、特定の部位にはCRFR1を必要とするが、他の部位には必要としないことを示している。CRF OEコホートは、グリコーゲン合成酵素キナーゼ3β(GSK-3Bマイトジェン活性化プロテインキナーゼ(MAPK)p38,細胞外シグナル調節キナーゼ(ERK)1/2のリン酸化レベルが有意に増加していることを示した。サイクリン依存性キナーゼ5(cdk5)のレベルは、R12919で処理してもCRF OEマウスでは変化しなかった。活性化されたc-Jun N末端キナーゼ(JNK)のレベルのみがR12191919の前処理に敏感であったことから、JNKの活性化を介したタウ-pの誘導がCRF OEにおけるAT8およびPHF-1部位でのタウ-pの誘導の中心であることが示唆された。CRF OEはまた、球状のタウ凝集体の形成を増加させた。さらなる確認が必要であるが、所見は、生体内での病理学的前の海馬のタウ凝集体の証拠を表している可能性がある(Campbell、Zhang et al 2015)。重要なことに、これらのモデルでは青斑核は調査されておらず、CRFR1の豊富さとストレス誘発性うつ病モデルでの青斑核変性を支持する文献を考えると、タウ-pとアミロイドβ凝集体の研究が最も重要である。
Rissmanらは、ストレス誘発性のタウ-pはストレス適応に必要な必須のプロセスであり、加齢や慢性的な過剰刺激によって機能不全に陥ると仮説を立てている(Rissman 2009)。これを裏付けるように、CRF曝露がタウ-pに対する機能的意味合いを検討した研究では、ミトコンドリアの速度と移動距離が増加する一方で、CREB活性化とBDNF輸送が有意に減少することが明らかにされている(Le, Weissmiller er al 2016)。現象学的には異なるかもしれないが、アミロイドβに関連する毒性についても同様のケースが考えられる。アミロイドβは生涯を通じて脳内で産生されるが、おそらく年齢が進むにつれて、アミロイドβをクリアしたり処理したりする能力が変化したり失われたりして、蓄積を引き起こす可能性がある(Campbell, Zhang er al)。
II. ノルアドレナリンの影響
ストレス統合的な青斑核-ノルエピネフリンシステムの関与およびアルツハイマー病関連病理の増強におけるその仮定的役割は、最近レビューされている(Ross, McGonigle er al)。 ここでは、アミロイドβ蓄積におけるARの役割を簡単に再掲する。特に重要なのは、シナプス伝達とシナプスに存在するAPP処理機械に直接影響を与え、アミロイドβの生産と分泌に関与している青斑核の末端領域へのβ2とα2ARの局在である。電子顕微鏡による研究では、PFCの錐体ニューロンの樹枝状棘やGABA作動性介在ニューロンにβ2ARが局在していることが明らかになっている(Aoki, Venkatesan er al)。1998)。また、β2ARは、γ-セクレターゼ複合体のα1Aサブユニットと物理的に相互作用するβ-arrestin2との会合を介してアミロイドβ産生を調節することができ、その結果、複合体の触媒活性が増加することが推測されている(Thathiah, Horre er al)。 これらの知見を支持して、アルツハイマー病のトランスジェニック動物モデルにおける研究は、β2アンタゴニストがアミロイドβ40およびアミロイドβ42のレベルを減少させることを示しており(Scullion、Kendall et al 2011アルツハイマー病におけるβ-ARを標的とする治療の可能性が検討されている(Yu、Wang et al 2011)。α2ARはGi/cAMP系に結合しており、主にシナプス前に局在し、ノルエピネフリンの合成と放出を調節する自己受容体として作用する(Berridge and Waterhouse 2003)。加齢に伴う認知障害におけるカテコラミン神経伝達物質と受容体を研究した初期の研究では、α2-ARがPFCにおける認知機能低下の一因であることが確認されている(Arnsten and Goldman-Rakic 1985)。これに関連して、最近の研究では、α2aARアンタゴニストであるフルパロキサンを慢性的に投与することで、APP/PS1トランスジェニックマウスの空間作業記憶における加齢に伴う障害が、アミロイドプラーク負荷とは無関係に予防されたことが示された(Scullion, Kendall er al 2011)。α2aARはまた、APPの処理およびそれに続くアミロイドβの産生および分泌の調節に、内分泌系および分泌系を介して関与している(Chen, Peng er al)。 これは、自殺者の前頭前野におけるα2ARの機能の変化に関する上述の知見に照らしても興味深い(Valdizan, Diez-Alarcia er al)。 これらの証拠に基づき、β2およびα2aARがアミロイドβ産生および分泌の調節に役割を果たしていることは明らかである。さらに、ノルアドレナリン伝達後のβ2ARやα2aARの活性化とその結果としてのアミロイドβ産生は、ストレスによって誘発される青斑核の異常な活性化が、疾患の前段階や初期段階での大脳皮質でのアミロイドβ産生の増加に寄与している可能性を示唆している(図1,33)。
図3
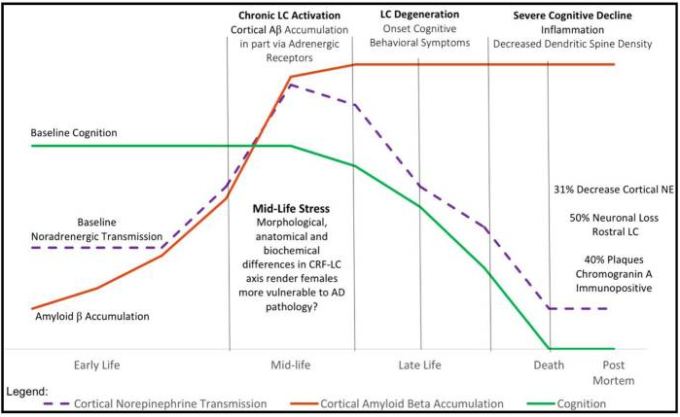
アルツハイマー病の進行に対する副腎皮質ネットワークの調節障害の影響を仮説化した。アルツハイマー病の認知症状が発症する15年から 20年前には、ストレスによって青斑核が慢性的に活性化され、それに伴ってノルアドレナリン伝達が増加することで、下肢内側大脳皮質などの皮質領域におけるアミロイドβの産生、分泌、蓄積が増加するという仮説を立てた。さらに、アミロイドβペプチドの異常な蓄積は、青斑核とその皮質の情報伝達突起との間のコミュニケーションの断絶、すなわち非同期化につながるという仮説を立てている。
本節で検討した所見は特に重要であり、アミロイドβペプチドは最初に大脳新皮質のII、III、IV、V層に蓄積し、海馬や扁桃体を含む大脳辺縁系の構造物へと進行し、最終的にBraak病期分類(1991)によれば中脳や脳幹構造物に蓄積すると考えられており、これらの領域の多く、特にアルツハイマー病の初期または前駆期に影響を受ける領域にはARが豊富に存在している。さらに、NFTの蓄積の古典的なパターンは、内葉と海馬形成で始まり、その後、連合体と一次皮質に進行することは、タウ-pの役割を果たすCRFR1の密度に関連している。より最近、Braakは、30歳未満で死亡した個人の死後脳組織の前絡み物質に反映されたタウ-pの異常を示す最初の領域が青斑核であることを提案した(Braak and Del Tredici 2011,Braak and Del Tredici 2011,Braak, Thal et al 2011,Braak and Del Tredici 2012,Braak, Thal et al 2013)。前述したように、青斑核はCRFR1が豊富に存在する領域でもあり、ここで述べたストレス条件下でのタウ-pのCRFR1制御のメカニズムは、これらの知見に関連している可能性がある(図1)。ここで検討された研究は、アミロイドβの蓄積とタウの過リン酸化におけるCRFとARの直接的な役割を示唆している。
V. 結論
ここで検討された研究は、ストレスが大規模なネットワーク活動や再編成から、アルツハイマー病神経病理の主要な構成要素であるアミロイドβやタウ-pを含むシナプス蛋白質の発現や機能の変化に至るまで、脳機能に広範な変化を誘発することを示唆している。あまり理解されていないのは、慢性的なストレスとうつ病との関係で、神経細胞の回復力の経時的、あるいは加齢との関係である。我々の知識の中で重要なギャップは、慢性的なストレスに対する神経細胞の回復力、またはアロスタティック過負荷が、うつ病患者では低下しているかどうか、もしそうならば、アミロイドβとタウ-pの蓄積に対する直接的な影響があるかどうかということである。神経細胞の活動を調節するアミロイドβの役割、ストレス反応の統合とDMNにおける安静時脳活動の両方に関与する重要なPFCネットワークの容積的減少を考えると、おそらくアミロイドβは見落とされていた神経ペプチドであり、うつ病などのより広範なストレス関連精神疾患との関連性があるのではないかと推測したくなるが、この点を説明するために、本研究では、アミロイドβがストレス関連の精神疾患の中で最も重要な神経ペプチドであることを明らかにする。この点を説明するために、我々は青斑核の慢性的な過活動がPFCなどの末端領域でアミロイドβの産生と蓄積をもたらし、シナプス後のうつ病のメカニズムを開始することを提案する。また、この時期には、慢性的な過活動化に伴い、異常なタウ-pが青斑核に蓄積し始め、可逆的な変性機構が活性化される可能性もある。PFCニューロンの活動が低下すると、扁桃体などの他の大脳辺縁領域が過活動化し、樹状突起の密度が増加する可能性がある。その結果、神経伝達物質や神経細胞の活動に様々なアンバランスが生じ、不安や抑うつなどのストレス関連精神疾患として行動に現れることがある(図2)。神経細胞の回復力のメカニズムは加齢とともに自然に低下していくことから、時間が経つにつれ、中年期になると、これらの変化の重症化が進むことが予想される。これは、後期のライフステージにおける不安や抑うつの発生率の増加、および特定のサブ集団におけるアルツハイマー病との併存率の増加のための仮説的な説明である可能性がある(図3)。現在の文献ではこの問題は取り上げられていないが、今後の研究の興味深い重要な領域である可能性がある。この仮説は、気分障害やうつ病などのストレス関連精神疾患の影響を受けることが知られている領域だけでなく、領域の体積変化を示すヒト臨床研究によって、限られた範囲で支持されるかもしれない。しかし、また、アルツハイマー病は、アミロイドβと異常なタウ-pが蓄積すると考えられている20年以上の前駆期から構成され、アミロイドβの最初の蓄積領域は大脳新皮質であるのに対し、異常なタウ-pは青斑核の最も早い段階に位置しているという事実によっても(Braak and Del Tredici 2011,Braak and Del Tredici 2011,Braak, Thal er al 2011, Braak and Del Tredici 2012, Braak, Thal er al)。) この仮説は、慢性ストレス下での皮質ネットワークの機能不全を永続させるだけでなく、DMNの活動を抑制することができず、ストレスに関連した精神疾患や神経変性疾患のより広い意味合いを持っている可能性がある(図2,33)。
ハイライト
- 慢性的なストレスが、うつ病患者に見られる根底にある精神神経病理に関与している可能性があり、それが人生の後半に神経変性疾患に罹患しやすい患者を生み出している可能性があるという仮説の枠組みについて議論する。
- ストレスは脳機能に広範な変化を誘発し、大規模なネットワーク活動や再編成から、アルツハイマー病神経病理の主要な構成要素であるアミロイドβやリン酸化タウを含むシナプス蛋白質の発現や機能の変化に至るまで、幅広い変化を引き起こす。
- アルツハイマー病神経病理の主要な構成要素であるアミロイドβやリン酸タウを含むシナプスタンパク質の発現や機能の変化に至るまで、大脳辺縁回路が強化され、皮質回路が減少し、認知症を含む精神症状によって証明される断絶症候群が生じるような、アロスタティック過負荷や慢性的なストレスがニューロン回路の再編成をもたらすという仮説を支持する証拠が提示されている。
- 提示された証拠から推測されるのは、慢性的なストレスが原因である可能性がある脊髄局(青斑核)-ノルエピネフーリン(ノルエピネフリン)系の調節障害が、少なくとも一部でデフォルトモードネットワーク(DMN)を抑制する能力の低下をもたらし、これがストレスに関連した疾患状態に重大な影響を及ぼすということである。
- さらに、DMNに関与する皮質領域の機能不全の結果として、神経内分泌、神経伝達物質、感情刺激やストレス因子に対する行動反応に関与する下流の回路の神経構造に影響を与え、悪循環を生み出している可能性がある。